

ABSTRACT
Normal development requires tight regulation of cell proliferation and cell death. Here, we have investigated these control mechanisms in the hyaloid vessels, a temporary vascular network in the mammalian eye that requires a Wnt/β-catenin response for scheduled regression. We investigated whether the hyaloid Wnt response was linked to the oncogene Myc, and the cyclin-dependent kinase inhibitor CDKN1A (P21), both established regulators of cell cycle progression and cell death. Our analysis showed that the Wnt pathway co-receptors LRP5 and LRP6 have overlapping activities that mediate the Wnt/β-catenin signaling in hyaloid vascular endothelial cells (VECs). We also showed that both Myc and Cdkn1a are downstream of the Wnt response and are required for hyaloid regression but for different reasons. Conditional deletion of Myc in VECs suppressed both proliferation and cell death. By contrast, conditional deletion of Cdkn1a resulted in VEC overproliferation that countered the effects of cell death on regression. When combined with analysis of MYC and CDKN1A protein levels, this analysis suggests that a Wnt/β-catenin and MYC-CDKN1A pathway regulates scheduled hyaloid vessel regression.
KEY WORDS: Hyaloid, Vascular regression, Wnt signaling, MYC, CDKN1A, P21, Endothelial cell
Summary: A model for developmentally scheduled regression of vascular endothelial cells reveals a link between the Wnt/β-catenin pathway and the cell-cycle regulators MYC and CDKN1A.
INTRODUCTION
Much progress has been made in our understanding of the mechanisms that regulate development of the vascular system. Angiogenesis – the de novo growth of vessels – has received much attention, and we know, for example, that integration of the Notch, VEGF and Wnt signaling pathways is crucial (Benedito et al., 2012; Chappell et al., 2013; Cheng et al., 2008; Corada et al., 2013; Masckauchán et al., 2005; Phng et al., 2009; Udan et al., 2013; Yang et al., 2008). Vascular remodeling, which involves vascular pruning (defined here as the segment-by-segment re-shaping of a vascular bed) and vascular regression (defined here as the complete involution of a vascular bed), has also been investigated, and we know that the two processes involve different types of Wnt pathway response. In vascular pruning, autocrine non-canonical Wnt responses play an important role (Franco et al., 2016; Korn et al., 2014), whereas in vascular regression the Wnt/β-catenin pathway is required (Lobov et al., 2005; Rao et al., 2007).
One model for the study of vascular regression is the hyaloid vessel system of the eye: a transient vascular network that serves a nutritive function for the developing retina in mammals (Ito and Yoshioka, 1999) and teleosts (Alvarez et al., 2007). In mammals, the hyaloid vessels and pupillary membrane undergo a scheduled regression immediately after birth in a process that is driven by endothelial cell apoptosis (Ito and Yoshioka, 1999; Lang et al., 1994; Poché et al., 2015). Apoptosis of hyaloid vascular endothelial cells (VECs) is known to be regulated by the Wnt (Glass et al., 2005; Gong et al., 2001; Lobov et al., 2005; Poulter et al., 2010; Toomes et al., 2004, 2005; Xu et al., 2004), angiopoietin (Gale et al., 2002; Rao et al., 2007) and VEGFA (Stalmans et al., 2002) signaling pathways. A previously proposed model aims to explain how the Wnt and angiopoietin pathways are integrated to control hyaloid regression (Rao et al., 2007) (Fig. 1). This model incorporates analysis showing that macrophages are required for hyaloid regression (Lang and Bishop, 1993) because they produce Wnt7b and elicit a Wnt/β-catenin response in VECs (Lobov et al., 2005). The model also suggests that the Wnt/β-catenin response is required for hyaloid regression because it stimulates cell cycle entry (Rao et al., 2007) and, when combined with pericyte-derived angiopoietin 2 (Ang2)-dependent suppression of Akt (Datta et al., 1999; Peters et al., 2004; Rao et al., 2007), can result in VEC apoptosis. The inputs of the Wnt and angiopoietin-Akt pathways to β-catenin stabilization are finely balanced: Lrp5 co-receptor heterozygosity can switch the influence of Ang2 on β-catenin target genes from positive to negative compared with wild type (Rao et al., 2007) (Fig. 1). More recent work has suggested that a melanopsin-dependent light response pathway normally suppresses levels of VEGFA and that this diminished level of VEGFA is permissive for hyaloid regression (Rao et al., 2013).
Fig. 1.

Hyaloid regression model and vessel expression of the Wnt/β-catenin pathway and target genes. (A,B) Schematic describing the previous hypothesis for integration of Wnt and angiopoietin signaling during hyaloid regression. The model suggests that Ang2 produced by pericytes has the dual functions of suppressing Akt to permit cell death and of upregulating Wnt7b expression in macrophages. In turn, activation of the Wnt/β-catenin (CTNNB1) pathway by macrophage Wnt7b promotes cell cycle entry, which is a prerequisite for cell death. This model explains how the hyaloid vessels are maintained (A) and how they regress (B), and why the macrophage is an essential mediator of regression. DVL, disheveled; Fzd, Frizzled. (C,D) Charts showing hyaloid vessel expression levels in FPKM for all Gene Ontogeny-classified Wnt/β-catenin pathway genes (C) and for Wnt/β-catenin pathway target genes (D). In C, the FPKM scale is contracted between 20 and 25. Arrowheads indicate the genes assessed functionally in this study. In D, the green vertical line indicates the point above which the linear FPKM scale is presented in different increments. In D, feedback target genes are highlighted in red. All transcripts with an FPKM of at least 1.0 are shown.
The Myc gene is a target of the Wnt/β-catenin pathway (He et al., 1998). It is also widely accepted to influence the regulation of cell cycle progression and cell death (Askew et al., 1991; Evan et al., 1992). One mechanism central to the regulation of cell proliferation by MYC is its suppression of the Cdkn1a gene, which encodes the cyclin-dependent kinase inhibitor CDKN1A. MYC-dependent suppression of Cdkn1a can occur through SP1 and SP3 (Gartel and Shchors, 2003; Gartel et al., 2001), and through MIZ1, although the latter mechanism appears to apply only when there are aberrantly high oncogenic levels of MYC (Walz et al., 2014; Wolf et al., 2013). The regulation of apoptosis by MYC is believed to be a mechanism of intrinsic tumor suppression (Lowe et al., 2004). Evaluation of the biology of MYC has understandably focused on the tumor systems in which Myc mutations play a very prominent role and so there are very few studies assessing the role of MYC in developmental responses. One such study was conducted by Potente and colleagues, in which MYC was identified as an essential regulator of endothelial metabolism and proliferation (Wilhelm et al., 2016).
In the present analysis, we have addressed the role of MYC and CDKN1A in the Wnt/β-catenin response that is required for developmentally scheduled hyaloid vessel regression. This has shown that the Wnt/β-catenin pathway co-receptors LRP5 and LRP6 both have activity in VECs that promotes hyaloid regression. We also show that in Myc and Cdkn1a VEC conditional mutants, the hyaloid vessels persist, but for different reasons. In the absence of CDKN1A, levels of apoptosis are modestly reduced, but dramatic VEC over-proliferation is enough to produce hyaloid persistence. In the absence of MYC in VECs, both cell death and proliferation are reduced to 10% of that observed in control samples. Finally, we show that the absence of MYC results in elevated expression of CDKN1A. This suggests that an established MYC-Cdkn1a regulation mechanism (Gartel and Radhakrishnan, 2005) is relevant to a developmental setting where decisions for proliferation and cell death are coupled. These findings are also consistent with the previous model for hyaloid regression (Rao et al., 2007) (Fig. 1) in which Wnt/β-catenin-driven cell cycle entry is a prerequisite for programmed cell death.
RESULTS
The hyaloid vessel complex expresses a broad selection of Wnt/β-catenin pathway genes
Lrp5 encodes one of two Wnt/β-catenin pathway co-receptors (Joiner et al., 2013). Lrp5 mutation either in mouse (Kato et al., 2002; Lobov et al., 2005) or human (Gong et al., 2001) results in hyaloid vessel persistence. The phenotype is robust and, unlike some examples of hyaloid persistence (Rao et al., 2013), lasts into adulthood. Lrp5 is known to be expressed in hyaloid VECs and hyaloid-associated macrophages (Kato et al., 2002; Lobov et al., 2005). To obtain gene expression data for other components of the Wnt/β-catenin response pathway, we performed an RNAseq analysis of hyaloid vessel preparations from postnatal day (P) 5 wild-type mice. From the RNAseq data, we displayed the FPKM (fragments per kilobase of transcript per million mapped reads) for all Wnt/β-catenin pathway-associated genes (Fig. 1C) according to Gene Ontology criteria (Ashburner et al., 2000). This analysis showed that with an FPKM of 7.0, Lrp6 was expressed at a level higher than Lrp5 (FPKM of 3.1). As the majority of cells within the hyaloid tissue preparation are VECs, this suggested that LRP6 might function alongside LRP5 to mediate Wnt/β-catenin signaling. Furthermore, display of Wnt/β-catenin pathway target genes (Fig. 1D), some of which are feedback targets (Fig. 1D, red), revealed robust expression levels of many. This is consistent with expression of the Wnt/β-catenin reporter TOPGAL (Lobov et al., 2005) and TCF/Lef:H2B-GFP (Fig. 2C) in hyaloid VECs.
Fig. 2.
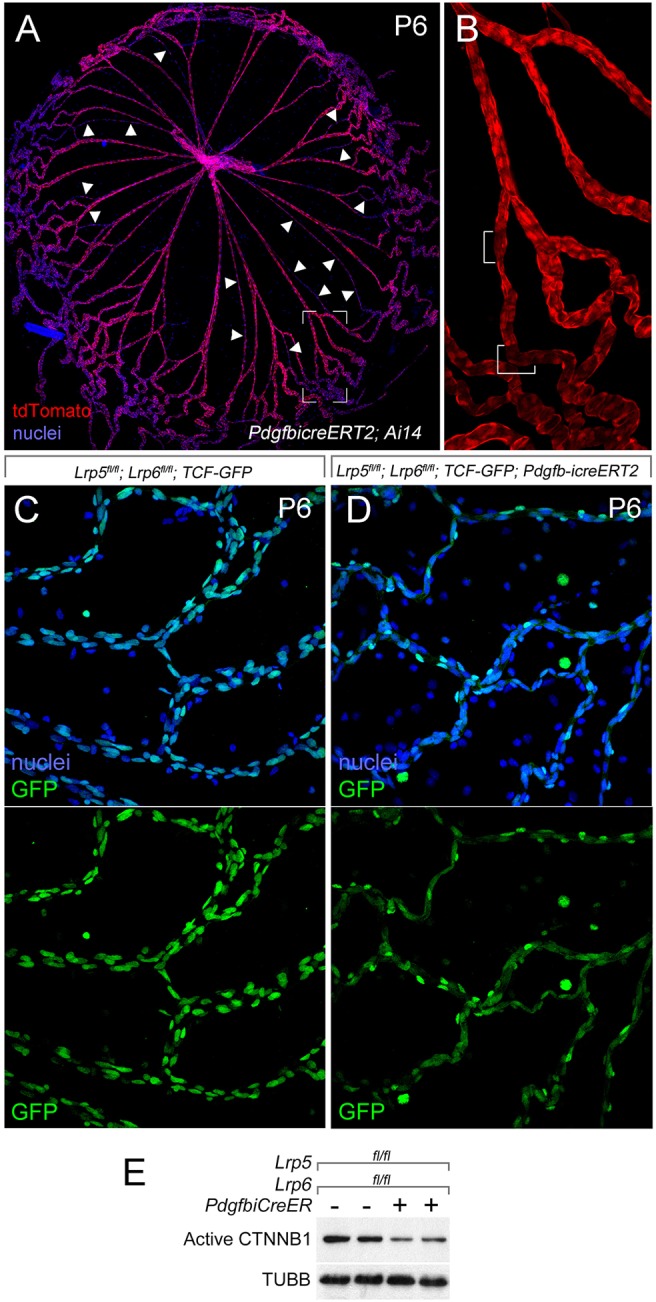
The Wnt/β-catenin pathway in hyaloid VECs is LRP5 and LRP6 dependent. (A,B) P6 hyaloid vessels from a Pdgfb-icreERT2; Ai14 mouse after tamoxifen injection at P2. A is labeled with Hoechst 33258 for nuclei (blue) and for tdTomato of Ai14 (red). Arrowheads indicate regressing capillary segments. B shows the red channel (tdTomato) of a magnified region of A (outlined). In B, the white brackets indicate a few cells that do not appear to express tdTomato. (C,D) P6 hyaloid preparations from mice of the indicated genotypes visualized for nuclei (with Hoechst 33258, blue) and for GFP from the Wnt/β-catenin reporter TCF-GFP (upper panels), or for GFP alone (lower panels). The cytoplasmic GFP signal from Pdgfb-icreERT2 is evident in D (lower panel). Consistent with compromise of the Wnt/β-catenin response, the number of TCF-GFP-expressing cells is dramatically lower with Lrp5 and Lrp6 conditional deletion (compare C with D, green channel). (E) BMVECs isolated from Lrp5, Lrp6 VEC conditional deletion mice also show diminished levels of active β-catenin (CTNNB1) compared with the loading control, β-tubulin (TUBB).
Lrp5 and Lrp6 combine to mediate the VEC Wnt/β-catenin response in hyaloid regression
To address the issue of whether the LRP5 and LRP6 co-receptors had overlapping activities in hyaloid VECs, we combined the Lrp5fl and Lrp6fl alleles (Riddle et al., 2013) with the VEC-specific Pdgfb-icreERT2 (Claxton et al., 2008) [Lrp5/6 conditional knockout (CKO)]. Pdgfb-icreERT2 expresses a cre activity that is induced by tamoxifen (Claxton et al., 2008). To assess the efficiency of Pdgfb-icreERT2-mediated gene deletion in the hyaloid vessels, we used Ai14, a tdTomato-expressing ROSA26-based allele as a cre reporter. After tamoxifen injection of Pdgfb-icreERT2; Ai14 mouse pups at P2, we found that most hyaloid VECs showed tdTomato fluorescence at P6 (Fig. 2A,B). Macrophages were negative. The fluorescence intensity of tdTomato varied somewhat across the hyaloid preparations, and was reduced in regressing vessels (Fig. 2A, arrowheads) but absent from some cells (Fig. 2B, brackets).
As additional validation, we determined whether Lrp5/6 CKO diminished Wnt/β-catenin pathway activity. First we took advantage of TCF/Lef:H2B-GFP, a reporter for this pathway (Ferrer-Vaquer et al., 2010). Lrp5fl/fl; Lrp6fl/fl; TCF/Lef:H2B-GFP pups served as controls and Lrp5fl/fl; Lrp6fl/fl; Pdgfb-icreERT2; TCF/Lef:H2B-GFP as the experimental genotype. After tamoxifen injection at P2, hyaloid vessels were harvested at P6. Visualization of nuclear GFP signal in the control and experimental hyaloids (Fig. 2C,D) showed that there were fewer positive nuclei when Lrp5 and Lrp6 were deleted (Pdgfb-icreERT2 incorporates a cytoplasmic GFP and this is evident in the images from cre-positive hyaloids). However, the number of positive nuclei was still substantial and probably reflected cell types not targeted by Pdgfb-icreERT2 (pericytes) or incomplete deletion of the four Lrp5 and Lrp6 alleles in VECs. As a second validation, we harvested brain microvascular endothelial cells (BMVECs) from control and Lrp5/6 CKO mice after cre recombinase was activated in vivo. BMVECs and hyaloid VECs can be considered similar as they both belong to vascular systems that supply the central nervous system (CNS) and are also similar in terms of pericyte investment and presence of macrophages. We then assessed, in cultured BMVECs by immunoblot, the level of active β-catenin. The diminished level of active β-catenin in Lrp5/6 CKO cells (Fig. 2E) indicated the expected change to Wnt/β-catenin pathway activity.
In control, Lrp5fl/fl; Lrp6fl/fl mice, the P8 hyaloid has an average of 23 major vessels (Fig. 3A,F). Lrp5 homozygote conditional deletion hyaloids showed a vessel number significantly elevated compared with the control (Fig. 3B,F). This indicated, as might be expected (Lobov et al., 2005), that Lrp5 activity within hyaloid VECs is required for hyaloid regression. The homozygous conditional mutant for Lrp6 also showed hyaloid persistence (Fig. 3C,F). Quantitatively, the contribution of Lrp6 to the regression pathway is indistinguishable from that of Lrp5 (Fig. 3F, compare the control gray bar with the third and fifth bars). However, when combined in a compound homozygote, the phenotypic effect is greater than the addition of each individual co-receptor (Fig. 3E,F, dark-blue bar). In homozygote-heterozygote combinations, Lrp6 heterozygosity does not enhance the Lrp5 homozygote phenotype (Fig. 3F, bar 4). By contrast, Lrp5 heterozygosity on the Lrp6 homozygous background significantly exacerbates the phenotype (Fig. 3D,F). The degree of hyaloid persistence in the Lrp5; Lrp6 conditional homozygotes was significantly lower than that observed in the Lrp5 germline mutant (Lobov et al., 2005) (Fig. 3F, blue line). This may be because of incomplete deletion of the four co-receptor alleles in VECs but equally leaves open the possibility that macrophage LRP5 is an important contributor to the phenotype (Kato et al., 2002; Lobov et al., 2005).
Fig. 3.
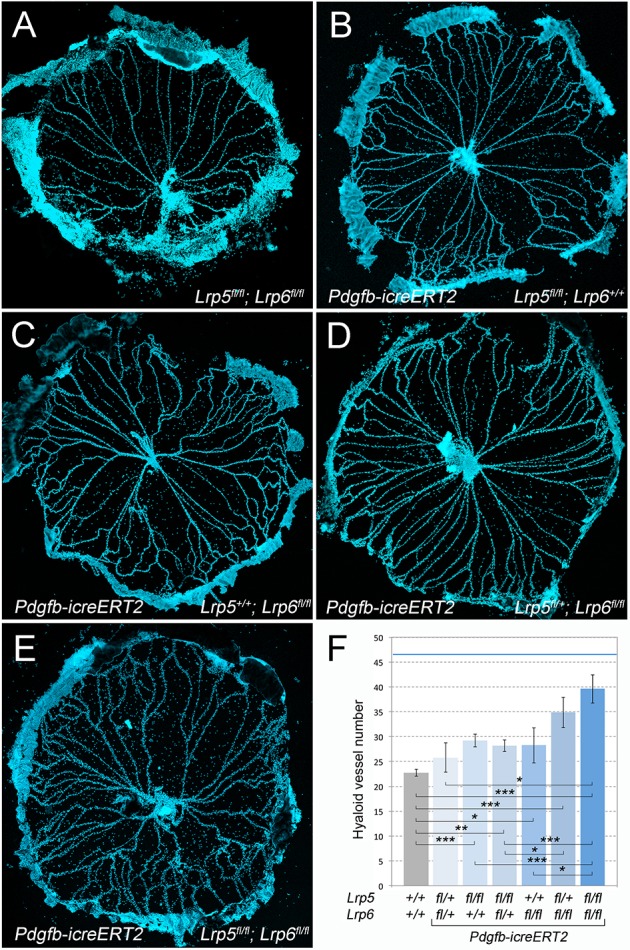
Lrp5 and Lrp6 are both required in VECS for normal hyaloid regression. (A-E) Hoechst 33258-labeled hyaloid preparations from P8 mice of the labeled genotypes. (F) Quantification of hyaloid vessel number in an allelic series of Lrp5fl, Lrp6fl deleted with Pdgfb-icreERT2 as labeled (n=67, 4, 19, 17, 8, 8 and 10 from left to right). The blue horizontal line is the number of hyaloid vessels observed in an Lrp5 germline mutant mouse (n≥4). Data are presented as mean±s.e.m. *P<0.05>0.01, **P<0.01>0.001, ***P<0.001 (one-way ANOVA, Tukey post-hoc test).
Lrp5 and Lrp6 deletion in VECs suppresses proliferation and apoptosis
Based on previous analysis (Lobov et al., 2005; Rao et al., 2007), we might anticipate that Lrp5/6 CKO would result in reduced cell proliferation and reduced apoptosis. We assessed hyaloid preparations for EdU incorporation (Salic and Mitchison, 2008) and for activated caspase 3 (Namura et al., 1998) at P5. Lrp5fl/fl; Lrp6fl/fl pups served as controls and Lrp5/6 CKO as the experimental genotype. Visualization of EdU incorporation in hyaloids showed that labeling in control animals was largely restricted to vascular cells (Fig. 4A,B), although a few EdU-labeled extravascular cells were detected (Fig. 4B, green circles). By contrast, in Lrp5/6 CKO animals, the distribution of EdU labeling was distinct (Fig. 4C), and was found in both vascular (Fig. 4C,D) and many extravascular cells (Fig. 4D, green circles). Labeling with the myeloid marker Iba1 confirmed that, for both genotypes, the vast majority of extravascular cells were myeloid (Fig. 4E,F). When we quantified EdU incorporation in vascular cells and normalized for vessel number (Fig. 4G), we found that the EdU labeling index was lower when Lrp5 and Lrp6 were deleted from VECs. Labeling of control and experimental hyaloid vessels for activated caspase 3 and the quantification of apoptotic vessel segments (Fig. 4H-J) revealed that Lrp5 and Lrp6 conditional deletion resulted in lower levels of VEC programmed cell death. When combined, the data presented in Figs 2-4 indicate that Wnt/β-catenin signaling mediated by the combined activities of LRP5 and LRP6 is required both for cell cycle entry and for cell death in VECs of the hyaloid vessels.
Fig. 4.

Loss of Lrp5 and Lrp6 from hyaloid VECs suppresses proliferation and apoptosis. Flat-mounted hyaloid vessels from Lrp5flox/flox; Lrp6flox/+; Pdgfb-icreERT2 or cre-negative littermate controls labeled for EdU and Hoechst (A-D), isolectin and Iba1 (E,F) or activated caspase 3 and Hoechst (H,I) at P5. (B,D) Enlarged images of A,C showing EdU-positive macrophages (circled in green) that were excluded from quantification. (E,F) Isolectin- (IB4, red) and Iba1- (green) labeled hyaloid preparations of the indicated genotypes. In the right half of each panel, the red channel is removed to show only the Iba1 labeling. In H,I the boxed region is enlarged on the right to show an example of an apoptotic segment. Quantification of EdU-positive nuclei (G) or caspase 3-labeled apoptotic segments (J) (n≥4). Data are mean±s.e.m. P values were calculated using Student's t-test.
VEC deletion of Myc results in hyaloid vessel persistence
We have previously proposed a model for the regulation of hyaloid regression in which the Wnt/β-catenin and angiopoietin pathways are integrated (Rao et al., 2007). One central component of this model (Fig. 1A,B) is that cell cycle entry is a prerequisite for the cell death program that is the driving force behind vascular regression (Rao et al., 2007). In turn, this implied that MYC, a key regulator of cell cycle entry and a Wnt/β-catenin pathway target gene (He et al., 1998), might also be required for hyaloid regression.
To test this possibility, we performed a conditional deletion of a Mycfl allele (Wilson et al., 2004) using Pdgfb-icreERT2 (Claxton et al., 2008) and tamoxifen injections at P1, P2 and P3, and then performed hyaloid vessel preparations at P8. Control mice with the genotype Myc+/+; Pdgfb-icreERT2 (Fig. 5A,D) showed a normal average number of hyaloid vessels. By contrast, both heterozygote and homozygote conditional deletion mice showed an elevated number of hyaloid vessels at P8 (Fig. 5B-D). These data indicate that Myc expression in VECs is required for hyaloid regression. The degree of hyaloid persistence was dependent on Myc gene dose (Fig. 5D). Gene dose-dependent hyaloid persistence is also a characteristic of mice in which the signaling ligand required for this response, WNT7b, is compromised (Wnt7bd1 mutant mice) (Lobov et al., 2005; Rao et al., 2007).
Fig. 5.
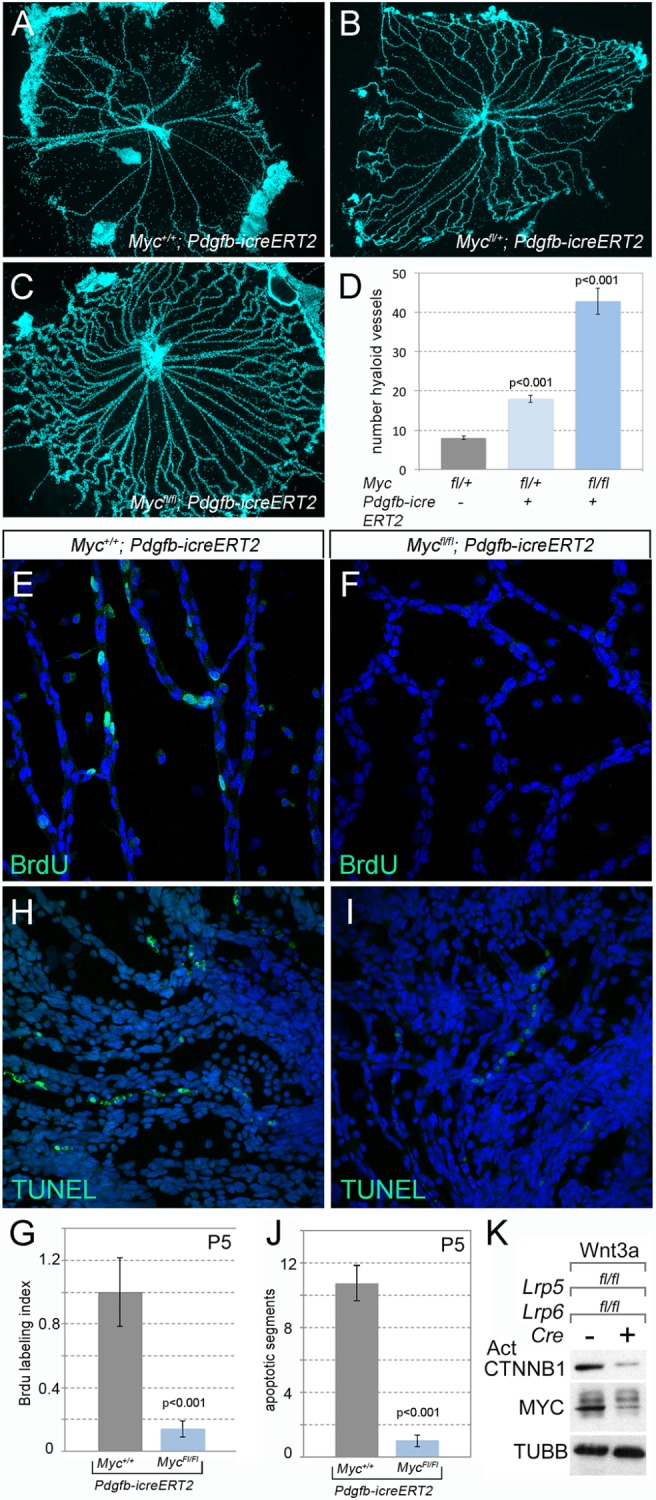
Myc is required for cell proliferation and apoptosis in hyaloid VECs. Flat-mounted P8 hyaloid vessels from VEC conditional Myc heterozygote and homozygote mice as labeled. There is dramatic hyaloid persistence in homozygous Mycflox/flox; Pdgfb-icreERT2 mice (C) and mild persistence in heterozygous Myc (Mycflox/+; Pdgfb-icreERT2) mice (B) compared with wild-type littermate controls (Myc+/+; Pdgfb-icreERT2) (A). (D) Quantification of hyaloid vessel numbers in mice of the labeled genotypes. (E,F) Flat-mounted hyaloid vessels from P5 animals were labeled for BrdU (n≥6) (E,F) or TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) (H,I) along with Hoechst. Quantification of BrdU-positive nuclei (G) and TUNEL-positive apoptotic vascular segments (J). Data are mean±s.e.m. Statistical significance was tested using Student's t-test, n≥4. (K) Immunoblot showing active β-catenin (CTNNB1), MYC and β-tubulin (TUBB) in Lrp5/6 control and CKO BMVECs stimulated with Wnt3a.
To establish the cellular cause of hyaloid persistence in Mycfl/fl; Pdgfb-icreERT2 mutant mice, we performed BrdU and TUNEL labeling to detect S-phase cells and programmed cell death, respectively. This showed that MYC-deficient hyaloid VECs proliferated at very low rates compared with controls (Fig. 5E-G). Similarly, the number of apoptotic vessel segments was very low (Fig. 5H-J). These data show that MYC is required both for cell cycle entry and for cell death in hyaloid VECs.
To determine whether, as would be expected (He et al., 1998), Myc was regulated in a Wnt-dependent manner in microvascular endothelial cells (MVECs), we performed immunoblotting on BMVECs from control and Lrp5/6 CKO mice after cre recombinase was activated in vivo. This showed that Lrp5/6 CKO resulted in low levels of the active form of β-catenin and a lower than normal level of MYC (Fig. 5K). This shows that expression of MYC in MVECs is dependent on LRP5/6 activity, and is consistent with data showing that Myc is a Wnt/β-catenin pathway target gene (He et al., 1998). As MYC promotes cell cycle entry, these data are consistent with a model in which programmed cell death in hyaloid VECs is dependent on cell cycle entry (Fig. 1) (Rao et al., 2007).
VEC-specific Cdkn1a deletion results in hyaloid persistence
A model for programmed hyaloid regression in which cell cycle progression is required for cell death would suggest that a modulation of cell cycle progression might also result in a hyaloid vessel phenotype. The cyclin-dependent kinase inhibitor CDKN1A is expressed in VECs and is known to suppress cell cycle entry (Brühl et al., 2004; Rössig et al., 2002). To investigate the function of CDKN1A in hyaloid regression, we used an available Cdkn1a germline-null allele (Deng et al., 1995) and generated a new conditional allele (Fig. 6A). The conditional allele was designed to simultaneously delete exon 2 of Cdkn1a and place in the transcript an artificial exon that expressed the fluorescent protein reporter dsRed. Although we could demonstrate the expected pattern of recombination, we could not detect dsRed expression either directly or by antibody labeling. As the unrecombined Cdkn1afl allele displayed no phenotype when homozygous, the absence of dsRed expression might reflect an unanticipated absence of transcription after recombination. Despite this, the allele has proven valuable for assessing the function of Cdkn1a in hyaloid regression.
Fig. 6.
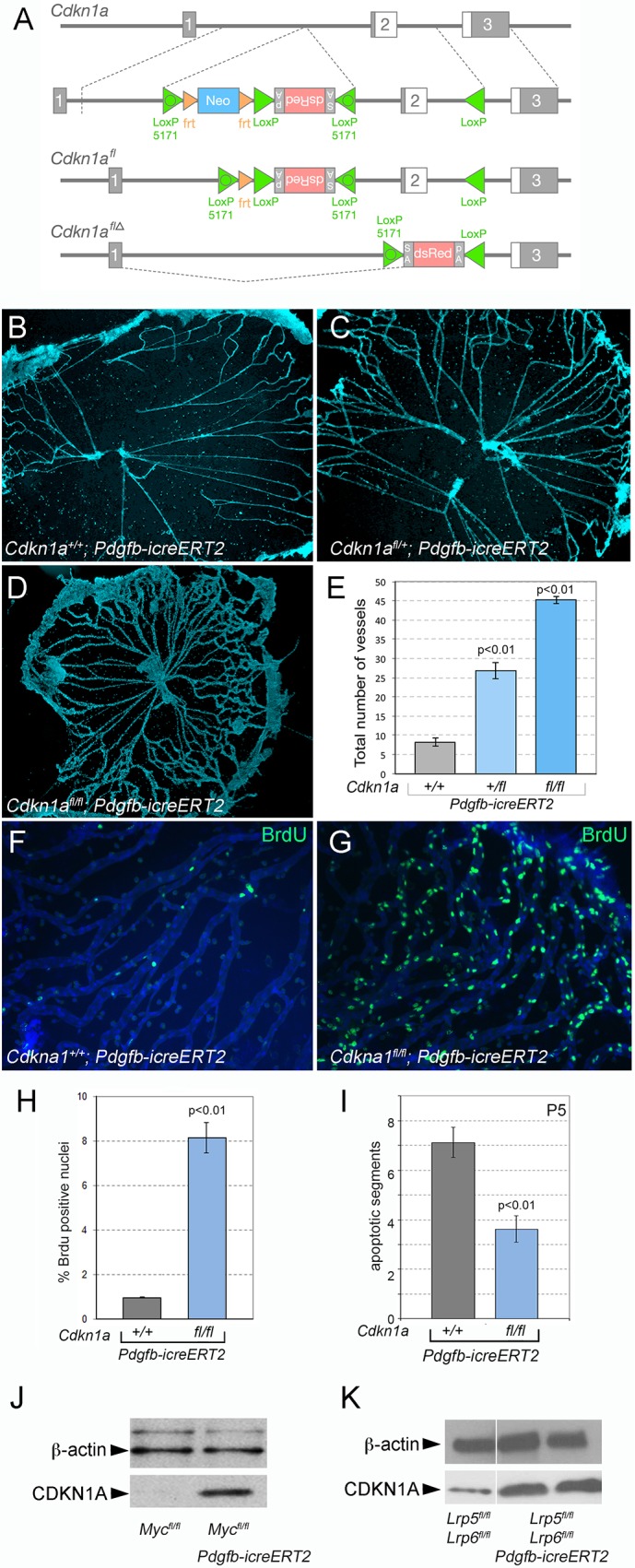
Loss of Cdkn1a induces proliferation that results in hyaloid persistence. (A) Schematic describing the generation of the Cdkn1a flox mouse line (see Materials and Methods for details). An FRT-flanked neo gene followed by an inverted DsRed cassette was inserted in the intron upstream of exon 2. The DsRed gene is preceded by an internal ribosome entry site (IRES) and flanked with a splice acceptor (SA) and a poly-A signal (pA), all of which are in the inverted orientation. In addition, wild-type and mutant LoxP (LoxP 5171) sites were inserted in the locations shown. Upon expression of flip recombinase, neo was excised from the gene to create Cdkn1afl. Upon expression of cre recombinase, head-to-head LoxP sites mediate inversion of the intervening DNA using either the wild-type (left) or mutant (right) LoxP sites, thus converting the DsRed cassette to the sense orientation and deleting exon 2 to create Cdkn1aflΔ. (B-D) Flat-mounted P8 hyaloid vessels labeled with Hoechst show dramatic hyaloid persistence in Cdkn1a conditional null VECs (Cdkn1afl/fl; Pdgfb-icreERT2) (D) and milder persistence in Cdkn1a heterozygous (Cdkn1afl/+; Pdgfb-icreERT2) (C). (E) Quantification of hyaloid vessel number in mice of the labeled genotypes (n=5). (F,G) Flat-mounted hyaloid vessels from mice of the indicated genotypes labeled for BrdU at P5. (H,I) Quantification of BrdU-positive nuclei (H) and TUNEL-positive apoptotic vessel segments (n=8) (I). Data are mean±s.e.m. Statistical analysis was carried out using Student's t-test. (J,K) Immunoblots of hyaloid tissue lysate prepared from animals with the indicated genotypes that were labeled with CDKN1A antibody. β-Actin served as a loading control.
In germline Cdkn1a mutant mice, we could show loss of CDKN1A immunoreactivity in hyaloid vessels (Fig. S1A,B) and hyaloid vessel persistence (Fig. S1C-F). With an average of 49 capillaries remaining, the phenotype is equal to that of the most severely affected Lrp5 germline null (Lobov et al., 2005). As it was possible that this reflected Cdkn1a function in many cell types, we also performed deletion of the Cdkn1afl allele using Pdgfb-icreERT2 (Claxton et al., 2008) and determined whether deletion of Cdkn1a in VECs had any consequence for hyaloid regression. As with the experiments for Myc deletions, we injected mice with tamoxifen on P1, P2 and P3, and then analyzed hyaloid vessel preparations at P8. This showed that, compared with Cdkn1a+/+; Pdgfb-icreERT2 control mice, both heterozygous and homozygous conditional deletion mutants showed hyaloid persistence (Fig. 6B-E). The degree of persistence depended on gene dose, with heterozygotes showing a phenotype intermediate between the control and the homozygote (Fig. 6E). This is very similar to the hyaloid vessel response observed when Myc is conditionally deleted from VECs (Fig. 5).
CDKN1A-deficient hyaloid VECs over-proliferate and show reduced levels of apoptosis
To assess the cellular cause of the hyaloid persistence in the Cdkn1a conditional deletion mice, we quantified proliferation by BrdU labeling and rates of cell death with TUNEL. This showed that, in contrast to control mice, Pdgfb-icreERT2; Cdkn1afl/fl mice showed a dramatically elevated BrdU labeling index (Fig. 6F-H), indicating that VECs were overproliferating. These data indicate that as might be expected, CDKN1A is required to suppress cell cycle entry in hyaloid VECs. The level of apoptotic segments in Pdgfb-icreERT2; Cdkn1afl/fl mice is significantly lower than in control (Fig. 6I) but higher than the very low level observed in the Myc conditional mutant (compare with Fig. 5J).
It has been shown that MYC can repress Cdkn1a at the transcriptional level (Amati et al., 1998; Claassen and Hann, 2000; Gartel and Radhakrishnan, 2005; Gartel et al., 2001). To determine whether this type of regulation might occur in hyaloid VECs, we performed immunoblotting for CDKN1A in hyaloid vessels from conditional Myc and Lrp5/6 CKO mice (Fig. 6J,K). In both control genotypes, levels of CDKN1A were low (Fig. 6J,K, left-most track). When Myc or the Lrp5, Lrp6 combination was conditionally deleted, CDKN1A levels became elevated. Although this interpretation is somewhat complicated by the smaller number of myeloid cells and pericytes that are hyaloid associated, these findings are consistent with MYC-dependent suppression of Cdkn1a expression, with the anticipated CDKN1A suppression of cell cycle entry, and with the very low rate of cell cycle entry observed in the VEC conditional Myc mutants (Fig. 5).
To eliminate the potential confounding effects of pericytes and myeloid cells, we assessed the interaction between the Wnt/β-catenin pathway and Cdkn1a in cultured BMVECs. BMVECs were isolated from P6 control (Lrp5fl/fl; Lrp6fl/fl) and Lrp5/6 CKO mice (after in vivo tamoxifen injection). Cadherin 5 (CDH5) labeling showed that Wnt co-receptor deletion did not significantly impair the isolation and culture of BMVECs (Fig. 7A,B). EdU labeling of control and Lrp5/6 CKO BMVECs was assessed either with or without lentivirus-delivered shRNA for Cdkn1a (Cdkn1a knockdown, KD). Quantification of EdU incorporation revealed the expected reduction in proliferation in the Lrp5/6 CKO (Fig. 7C-G). Interestingly, when CDKN1A was knocked down in the Lrp5/6 CKO, this produced a significantly higher rate of proliferation, a difference that was absent in the CDKN1A KD of Lrp5/6 control cells. This interaction between Lrp5/6 CKO and Cdkn1a KD shows that there is a role for CDKN1A in regulating Wnt pathway-dependent proliferation. It also shows that the Wnt pathway and CDKN1A have opposing influences on proliferation, an outcome consistent with the changes observed in vivo.
Fig. 7.
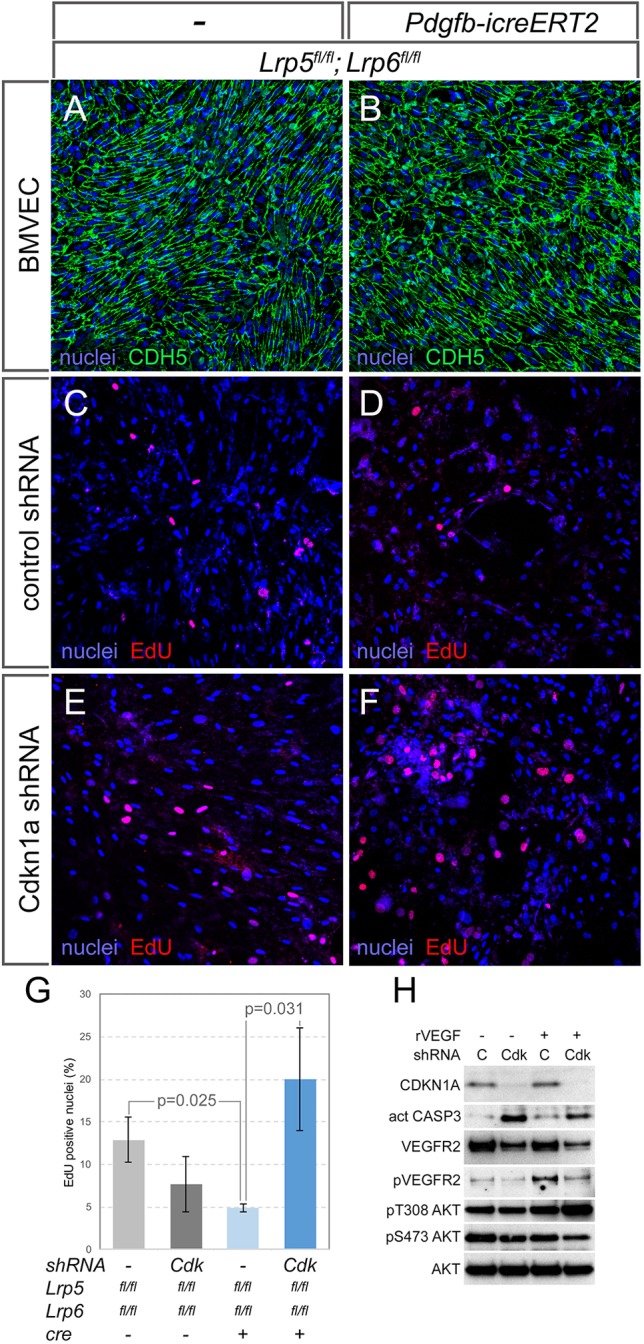
Wnt/β-catenin pathway interaction with CDKN1A and the apoptotic response. (A,B) BMVECs isolated from control (Lrp5fl/fl; Lrp6fl/fl) and conditional knockout (Pdgfb-icreERT2; Lrp5fl/fl; Lrp6fl/fl) mice labeled for nuclei (blue) and cadherin 5 (CDH5, green). (C-F) BMVECs from the indicated genotypes and shRNA treatments labeled for nuclei (blue) and EdU incorporation (red). (G) Quantification of EdU incorporation in BMVECs from the indicated genotypes and shRNA treatments (n≥3). Statistical significance was determined using two-way ANOVA with Bonferroni post-hoc test. Data are mean±s.e.m. (H) Immunoblots from lysates of BMVECs treated with combinations of recombinant VEGFA (rVEGFA) and an shRNA to Cdkn1a (C, control shRNA; Cdk, shRNA to Cdkn1a). In descending order, the immunoblots show detection of CDKN1A, activated caspase 3, VEGFR2, phospho-VEGFR2, phospho-T308-Akt, phospho-S473-Akt and total Akt.
CDKN1A has a complex biology that includes a role in suppressing apoptosis (Abbas and Dutta, 2009; Jänicke et al., 2007; Piccolo and Crispi, 2012). To determine whether BMVECs could illustrate this activity, we treated them with lentivirus-delivered shRNA and VEGFA, a potent survival stimulus for VECs. We found that CDKN1A knockdown (Fig. 7H, immunoblot row 1) resulted in elevated levels of activated caspase 3 (Fig. 7H, row 2) even in the presence of VEGFA stimulation. As might be expected, VEGFA stimulation activated VEGFR2 through phosphorylation at Y1175 (Fig. 7H, row 4). CDKN1A knockdown has the effect of diminishing levels of VEGFR2 (Fig. 7H, row 3) and of its activated form both in the absence and presence of VEGFA (Fig. 7H, row 4). Furthermore, CDKN1A knockdown elevates Akt phosphorylation of T308 and diminishes phosphorylation of S473. These data show that CDKN1A can suppress apoptosis pathways in cultured BMVECs. However, this is an activity not reflected in the response of hyaloid vessels to deletion of Cdkn1a, and suggests that CDKN1A activity cannot fully explain the influence of the Wnt/β-catenin pathway and MYC on the VEC apoptosis and hyaloid vessel regression.
DISCUSSION
The current study investigates the role of the Wnt signaling pathway in regression of the hyaloid vascular system. Deletion of the canonical Wnt co-receptors LRP5 and LRP6 in an allelic series in hyaloid VECs, resulted in gene dose-dependent hyaloid persistence. Similarly, VEC deletion of Myc, a target of canonical Wnt signaling (He et al., 1998; Yochum et al., 2008), also resulted in hyaloid persistence. Consistent with a previous model suggesting that hyaloid VEC cell death is coupled to cell cycle progression (Rao et al., 2007), both types of mutant displayed a reduction in proliferation and apoptosis. The observation that loss of function of the cyclin-dependent kinase inhibitor CDKN1A (Claassen and Hann, 2000; Coller et al., 2000; Gartel et al., 2000; Gartel and Shchors, 2003) in VECs results in hyaloid persistence adds weight to the idea that cell cycle regulators are mediators of regression in this system.
Wnt signaling in vascular growth and regression
The involvement of Wnt signaling in vascular regression has now been shown in two separate vascular beds: the hyaloid vasculature (Lobov et al., 2005; Rao et al., 2007) and the CNS vasculature (Ma et al., 2013). In the latter, ectopic Wnt signaling in cortical endothelial cells resulted in vascular regression because of an increase in endothelial expression of matrix metalloproteinases (MMPs). Thus, vessel stabilization required switching off Wnt signaling in endothelial cells. Although we cannot rule out the involvement of MMPs in the context of the hyaloid vasculature, it is clear that the canonical Wnt signaling pathway regulates vascular regression in different vascular beds via multiple mechanisms. This is in contrast to the developing retina, where endothelial Wnt signaling is involved in maintaining vessel stability (Birdsey et al., 2015; Phng et al., 2009).
Canonical Wnt signaling is also crucial for CNS angiogenesis but previous analysis suggests that not all CNS regions require the same set of Wnt ligands, receptors and co-receptors (Daneman et al., 2009; Kato et al., 2002; Stenman et al., 2008; Wang et al., 2012; Zhou et al., 2014). In the retina, Norrin, a member of TGFβ family, binds with Wnt-like high affinity to FZD4 and to its co-receptor LRP5 to initiate canonical Wnt signaling, with TSPAN12 potentially involved in multimerization of the receptor (Junge et al., 2009). Recently, a comprehensive analysis of an allelic series of germline Fzd4 and conditional endothelial cell deletion of Lrp5 and Lrp6 showed that FZD4 is required for retinal and brain vascularization, whereas angiogenesis in these organs differs in terms of their requirement for co-receptors (Zhou et al., 2014). Embryonic brain vascularization and blood-brain barrier formation requires at least one allele of Lrp5 or Lrp6, whereas the retina requires at least one copy of Lrp5 for normal vascularization (Zhou et al., 2014). The authors did not address the hyaloid phenotype in these mutants, as the conditional deletions were introduced several days after birth, well beyond the period when hyaloid regression begins.
The current study reinforces the importance of endothelial cell-specific Wnt signaling in hyaloid regression and reveals several novel observations. First, in addition to LRP5, canonical Wnt signaling mediated by LRP6 is also necessary for hyaloid regression, as the phenotype exhibited by the double receptor mutants was more severe than that of the individual mutants. Until now, only the FZD4 receptor and the LRP5 co-receptor were thought to be necessary for canonical Wnt signal-mediated hyaloid regression (Gong et al., 2001; Jiao et al., 2004; Robitaille et al., 2002; Toomes et al., 2004). Second, when compared with the Lrp5 germline null (Kato et al., 2002), the phenotype exhibited by the endothelial-specific LRP5 mutants in our study is less severe. This could potentially be because of inefficient cre-mediated deletion of Lrp5fl and Lrp6fl, but could also be attributed to LRP5/6 activity in multiple cell types, including the hyaloid macrophages and pericytes, which are also involved in the regression process (Lobov et al., 2005; Rao et al., 2007). Finally, the elevated number of macrophages in the Lrp5/6 CKO suggests that there is a Wnt pathway-dependent VEC activity that normally suppresses macrophage recruitment and proliferation. It will be interesting to try and understand the nature of this VEC-to-macrophage communication.
Coupling of cell cycle progression and cell death
The ability of canonical Wnt signaling to induce endothelial cell proliferation has been shown in several instances (Chen et al., 2003; Cheng et al., 2008; Goodwin et al., 2007; Masckauchán et al., 2006, 2005). Although Wnt signaling is known to enhance cyclin D1 levels, it also induces the expression of MYC, which has a pivotal role in stimulating cell growth and proliferation (Dang, 2013; He et al., 1998; Phng et al., 2009; Yochum et al., 2008). It has also been shown that MYC couples cell cycle progression to apoptotic cell death in some settings (Amati et al., 1998; Diez-Roux et al., 1999; Evan et al., 1992; Muthalagu et al., 2014; Shi et al., 1992).
MYC-binding sites are thought to be in the order of 10,000 or more, with target genes involved in energy metabolism, cell growth, proliferation and apoptosis (Dang, 2013; Hoffman and Liebermann, 2008; McMahon, 2014; Zeller et al., 2006). In recent years however, the mechanism by which MYC activates transcription has been examined by two studies which suggest that MYC is a global amplifier of gene expression (Lin et al., 2012; Nie et al., 2012). While this is still being debated (Kress et al., 2015), the results of our study, in which CDKN1A was upregulated on upon Myc deletion, can be explained by viewing MYC as a transcription factor that differentially regulates its targets. MYC regulation of angiogenesis in the retina was recently reported in a study that investigated the relationship between metabolic activity and vascular growth (Wilhelm et al., 2016). Here, Myc deletion resulted in hypovascularization of the retina. It was shown that MYC suppression by FOXO1 monitors cell proliferation and metabolic activity that in turn regulates retinal angiogenesis. The role of MYC therefore, will depend on the cellular and environmental context.
It is known that Wnt signaling can serve as the stimulus for cell cycle entry in VECs during hyaloid regression (Lobov et al., 2005; Rao et al., 2007). The Wnt co-receptor and Myc mutant hyaloid phenotypes are therefore consistent with deficient cell cycle entry stimuli that consequently reduce the rate of apoptosis. However, deletion of vascular endothelial-specific CDKN1A most likely leads to hyaloid persistence because of the dramatic oversupply of VECs. In addition, CDKN1A is known to regulate apoptosis (Abbas and Dutta, 2009; Jänicke et al., 2007; Piccolo and Crispi, 2012) but depending on the setting this can result in either a positive or a negative effect. The modest reduction in the rate of apoptosis in the CDKN1A hyaloid VEC mutants may reflect a balance of the pro- and anti-apoptotic activities of CDKN1A.
Signaling patterns that underlie hyaloid regression
The current data strengthen the prevailing model for the cellular mechanism of hyaloid regression and add more detail (Fig. 8A,B). Previously, it was shown that regression begins around P5, when VEGFA levels in the vitreous are held in check by a light- and melanopsin-dependent pathway (Rao et al., 2013). Pericytes in the hyaloid vessels express Ang2, which promotes endothelial cell death and capillary regression (Lobov et al., 2002; Rao et al., 2007; Yancopoulos et al., 2000). Ang2 antagonizes Ang1 function and suppresses Akt signaling, further reducing cell survival (Datta et al., 1999) (Fig. 8A,B). However, inhibition of Ang1 signaling also leads to destabilization of β-catenin and loss of the cell cycle entry stimulus. This is compensated for by release of WNT7b by macrophages, which activates Wnt signaling in hyaloid endothelial cells (Lobov et al., 2005). The Wnt-mediated stimulus for cell cycle entry is central to initiating cell death, as shown previously and by the current study, where disruption of endothelial Wnt signaling leads to a reduction in apoptosis (Lobov et al., 2005; Rao et al., 2007). The updated model includes a role for MYC as an essential promotor of cell cycle entry and cell death.
Fig. 8.

Schematic describing the proposed role of MYC and CDKN1A in Wnt/β-catenin (CTNNB1) and angiopoietin pathway-dependent hyaloid vessel regression. This model describes the role of three cell types, macrophages, pericytes and VECs, in hyaloid vessel regression. Prior to the initiation of regression (A, maintenance phase) macrophages do not express Wnt7b. Through mechanisms not currently understood, Akt activity is sufficient to maintain cell survival during this phase. Regression phase (B) is initiated by the suppression of VEGFA expression by a light-response pathway (not shown) and by the upregulation of Wnt7b expression in macrophages. Expression of Wnt7b is dependent on expression of Ang2 in pericytes. Ang2 has the dual role of suppressing the activity of Akt to promote cell death but at the same time providing an alternative means with which to activate β-catenin and thus promote cell cycle entry. Myc is known to be positively regulated by β-catenin to promote cell cycle entry and cell death. The loss of both of these cellular responses in the hyaloid vessels of Myc conditional mutant VECs is consistent with these observations. In the hyaloid vessels, CDKN1A is crucial for regulating cell cycle progression and thus influences the balance between maintenance and regression. CDKN1A levels are suppressed by MYC but are also likely to be regulated directly via Akt-dependent phosphorylation.
The interpretation of CDKN1A activity during hyaloid vessel regression is more complex. In other settings, CDKN1A functions downstream of the Akt pathway to inhibit both cell cycle entry and apoptosis (Brühl et al., 2004; Gartel and Tyner, 2002; Li et al., 2002; Rossig et al., 2001, 2002). In the context of the hyaloid vessels, consistent with expectation, CDKN1A normally suppresses cell cycle entry and, in isolated BMVECs, can suppress apoptotic pathways (Fig. 8A,B). However, the deletion of Cdkn1a in vivo results in an elevated level of cell proliferation but not cell death. This is probably explained by the previous observation that apoptotic segments arise because of flow stasis after the vessel lumen narrows as a result of accumulated loss of apoptotic VECs (Meeson et al., 1996). In the Cdkn1a conditional mutant it is likely that the oversupply of VECs can compensate and maintain lumen dimensions. In the future, it will be interesting to address more closely the many activities of CDKN1A in vascular development.
MATERIALS AND METHODS
Mice
Animals were housed in a pathogen-free vivarium in accordance with institutional policies. All animal experiments were approved by the Institutional Animal Care and Use Committee at Cincinnati Children's Hospital Medical Center. Pdgfb-icreERT2 (Claxton et al., 2008), Lrp5flox and Lrp6flox (Joeng et al., 2011), TCF/Lef:H2B-GFP (Ferrer-Vaquer et al., 2010) (JAX #013752), Ai14 (Madisen et al., 2010) (JAX #007914), and c-Mycflox (Trumpp et al., 2001), lines have been described. To induce endothelial cell gene deletion in Pdgfb-icreERT2 pups, peanut oil-dissolved tamoxifen (Sigma) was introduced intraperitoneally daily from P1 to P3 at 20 μg/g body weight for Mycflox and CDKN1Aflox, or intragastrically at P2, P4 and P6 for Lrp5flox and Lrp6flox. Tamoxifen-injected Pdgfb-icreERT2 negative littermates were used as controls. Birthdate was defined as P1.
Generation of the Cdkn1a conditional allele
A conditional loss-of-function allele, Cdkn1aflox, was generated by conventional gene targeting (Fig. 6A). With the intent of generating a recombination reporter, this allele incorporated an artificial dsRed exon. This feature of the allele did not function successfully. An FRT-flanked neo followed by an inverted dsRed cassette was inserted in the intron upstream of exon 2. The dsRed gene is preceded by an internal ribosome entry site and flanked with splice acceptor and poly-A signal, all of which are in the reverse orientation. In addition, wild-type and variant LoxP (Lee and Saito, 1998) sites are inserted to allow cre recombinase-dependent conditional knockout of the gene (LoxP-LoxP recombination) and reorientation of dsRed (LoxP5171-LoxP5171 recombination). In this allele, flip recombinase action at frt sites excised the neo cassette. The following primer set was used for genotyping the Cdkn1a floxed allele: forward primer, TGCCAGCGTCCTTTGGAAAGG; reverse primer, TCCATCAGGCCAATCAAAAGTACC.
Dissections, immunostaining and imaging
Hyaloid vessel preparations were generated and labeled as previously described (Diez-Roux et al., 1999; Lobov et al., 2005; Stefater et al., 2011). Labeling reagents were as follows: Alexa 488-Isolectin IB4 (Life Technologies, I21411, 1:500), Iba1 (Wako, 019-19741, 1:100), cleaved caspase 3 (R&D Systems, AF835, 1:100), CDH5 (BD Pharmingen, #550548, 1:1000), anti-BrdU (Dako, M0744, 1:100), and EdU (ThermoFisher Scientific, C10640; for mice, EdU was injected intraperitoneally at 25 μg/g and samples harvested after 24 h; for BMVECs, EdU was used at 10 μM and chased for 24 h). Secondary antibodies labeled with Alexa fluorochromes (ThermoFisher Scientific) were used at a 1:500 dilution. TUNEL labeling of apoptotic cells was performed using the In Situ Cell Death Detection Kit (Roche Applied Science). Images were captured using a Zeiss ApoTome AX10 or Zeiss LSM700 confocal microscope, and processed using ImageJ and Adobe Photoshop.
Brain microvascular endothelial cell isolation and lentivirus use
Isolation and culture of BMVECs has been described elsewhere (Lopez-Ramirez et al., 2017). Lentivirus shRNA-Cdkn1a (Clone ID: TRCN0000042583, MilliporeSigma) or non-targeting shRNA control (SHC002) was prepared by transfecting MISSION shRNA (MilliporeSigma) and packaging plasmids to HEK293T packaging cells. Filtered supernatant containing lentivirus was introduced to the primary culture of BMVECs. The following day medium was replaced and cells were analyzed at day 4. For induction of Wnt or the VEGF signaling pathway, Wnt3a (R&D Systems, 1324-WNP-010, 100 ng/ml) and hrVEGFA (Goldbio, #1350, 100 ng/ml) were used, respectively.
Western blotting
Western blots were performed using standard protocols. Hyaloids were harvested at P5 and lyzed in Laemmli sample buffer [4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromphenol blue and 0.125 M Tris HCl, (pH 6.8)] with sonication. Blots were incubated with the following antibodies: CDKN1A (Abcam, ab7960, 1:100, or Abcam, ab109199 1:100), active-β-catenin (MilliporeSigma, 05-665, 1:1000), Myc (Cell Signaling Technology, #5605, 1:1000), active caspase 3 (Abcam, ab3623, 1:1000), VEGFR2 (Cell Signaling Technology, #9698, 1:1000), phosphor-VEGFR2 (Cell Signaling Technology, #2478, 1:1000), Akt (Cell Signaling Technology, #4691, 1:1000), phospho-Akt T308 (Cell Signaling Technology, #13038, 1:1000), phospho-Akt S473 (Cell Signaling Technology, #4060, 1:1000), GFP (Abcam, ab13970, 1:300), and total actin (Seven Hills Bioreagents, LMAB-C4, 1:500) or β-tubulin (Abcam, ab6046, 1:5000) as loading control. HRP-conjugated secondary antibodies were used at 1:3000 dilution and detected by enhanced chemiluminescence (ThermoFisher Scientific, #34577).
Next generation RNAseq and data availability
Total RNA was prepared from lysate of two hyaloid tissues at P5 in Tri Reagent (ThermoFisher Scientific) and purified using the RNeasy Micro column (Qiagen). After RNA quality was verified using an Agilent Bioanalyzer (Agilent Technologies), the Ovation RNA-Seq System v2 (NuGEN) was used for RNA amplification and double-stranded cDNA construction. DNA concentration was measured by Qubit (ThermoFisher Scientific), and the DNA length was determined by the DNA 1000 Chip (Agilent Technologies). DNA library templates were then created using the Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced for the paired-end sequencing with 25-30 million reads by Illumina HiSeq2500 (Illumina). The data are available in GEO under accession number GSE113294.
Statistical analysis
Vessel number was quantified using established methods (Ito and Yoshioka, 1999). Sample sizes were determined using Power and Sample size calculators (statisticalsolutions.net/; www.anzmtg.org/stats/PowerCalculator/PowerANOVA), and tissues were collected from animals across different litters. All data are presented as mean±s.e.m. Student's t-test and one-way ANOVA with Tukey's post-test (GraphPad Prism 6) were used to assess statistical significance.
Supplementary Material
Acknowledgements
We thank Paul Speeg for excellent technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.R., R.A.L.; Methodology: G.N., Y.O., V.P., A.F.S., S.V., J.D.M., A.T., B.W., S.R., R.A.L.; Validation: G.N., Y.O., E.Y.; Formal analysis: G.N., Y.O., S.R., R.A.L.; Investigation: G.N., Y.O., E.Y., S.V., S.R.; Resources: V.P., J.D.M., A.T., B.W.; Data curation: V.P., A.F.S., S.V.; Writing - original draft: G.N., Y.O., R.A.L.; Writing - review & editing: G.N., Y.O., S.V., R.A.L.; Visualization: G.N., Y.O., S.R., R.A.L.; Supervision: J.D.M., R.A.L.; Project administration: R.A.L.; Funding acquisition: R.A.L.
Funding
We acknowledge support from the National Institutes of Health (R01 EY021636, EY027711 and EY027077) and from the National Eye Institute and the Abrahamson Pediatric Eye Institute. Deposited in PMC for release after 12 months.
Data availability
The data are available in GEO under accession number GSE113294.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.154898.supplemental
References
- Abbas T. and Dutta A. (2009). p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400-414. 10.1038/nrc2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y., Cederlund M. L., Cottell D. C., Bill B. R., Ekker S. C., Torres-Vazquez J., Weinstein B. M., Hyde D. R., Vihtelic T. S. and Kennedy B. N. (2007). Genetic determinants of hyaloid and retinal vasculature in zebrafish. BMC Dev. Biol. 7, 114 10.1186/1471-213X-7-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B., Alevizopoulos K. and Vlach J. (1998). Myc and the cell cycle. Front. Biosci. 3, d250-d268. 10.2741/A239 [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T. et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew D. S., Ashmun R. A., Simmons B. C. and Cleveland J. L. (1991). Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6, 1915-1922. [PubMed] [Google Scholar]
- Benedito R., Rocha S. F., Woeste M., Zamykal M., Radtke F., Casanovas O., Duarte A., Pytowski B. and Adams R. H. (2012). Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484, 110-114. 10.1038/nature10908 [DOI] [PubMed] [Google Scholar]
- Birdsey G. M., Shah A. V., Dufton N., Reynolds L. E., Osuna Almagro L., Yang Y., Aspalter I. M., Khan S. T., Mason J. C., Dejana E. et al. (2015). The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/beta-catenin signaling. Dev. Cell 32, 82-96. 10.1016/j.devcel.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brühl T., Heeschen C., Aicher A., Jadidi A. S., Haendeler J., Hoffmann J., Schneider M. D., Zeiher A. M., Dimmeler S. and Rössig L. (2004). p21Cip1 levels differentially regulate turnover of mature endothelial cells, endothelial progenitor cells, and in vivo neovascularization. Circ. Res. 94, 686-692. 10.1161/01.RES.0000119922.71855.56 [DOI] [PubMed] [Google Scholar]
- Chappell J. C., Mouillesseaux K. P. and Bautch V. L. (2013). Flt-1 (vascular endothelial growth factor receptor-1) is essential for the vascular endothelial growth factor-Notch feedback loop during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 33, 1952-1959. 10.1161/ATVBAHA.113.301805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., ten Berge D., Brown J., Ahn S., Hu L. A., Miller W. E., Caron M. G., Barak L. S., Nusse R. and Lefkowitz R. J. (2003). Dishevelled 2 recruits ß-arrestin 2 to mediate Wnt5A-stimulated endocytosis of frizzled 4. Science 301, 1391-1394. 10.1126/science.1082808 [DOI] [PubMed] [Google Scholar]
- Cheng C.-W., Yeh J.-C., Fan T.-P., Smith S. K. and Charnock-Jones D. S. (2008). Wnt5a-mediated non-canonical Wnt signalling regulates human endothelial cell proliferation and migration. Biochem. Biophys. Res. Commun. 365, 285-290. 10.1016/j.bbrc.2007.10.166 [DOI] [PubMed] [Google Scholar]
- Claassen G. F. and Hann S. R. (2000). A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc. Natl. Acad. Sci. USA 97, 9498-9503. 10.1073/pnas.150006697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K. and Fruttiger M. (2008). Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis 46, 74-80. 10.1002/dvg.20367 [DOI] [PubMed] [Google Scholar]
- Coller H. A., Grandori C., Tamayo P., Colbert T., Lander E. S., Eisenman R. N. and Golub T. R. (2000). Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 97, 3260-3265. 10.1073/pnas.97.7.3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M., Orsenigo F., Morini M. F., Pitulescu M. E., Bhat G., Nyqvist D., Breviario F., Conti V., Briot A., Iruela-Arispe M. L. et al. (2013). Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 4, 2609 10.1038/ncomms3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R., Agalliu D., Zhou L., Kuhnert F., Kuo C. J. and Barres B. A. (2009). Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 106, 641-646. 10.1073/pnas.0805165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V. (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 3, a014217 10.1101/cshperspect.a014217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. R., Brunet A. and Greenberg M. E. (1999). Cellular survival: a play in three Akts. Genes Dev. 13, 2905-2927. 10.1101/gad.13.22.2905 [DOI] [PubMed] [Google Scholar]
- Deng C., Zhang P., Harper J. W., Elledge S. J. and Leder P. (1995). Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675-684. 10.1016/0092-8674(95)90039-X [DOI] [PubMed] [Google Scholar]
- Diez-Roux G., Argilla M., Makarenkova H., Ko K. and Lang R. A. (1999). Macrophages kill capillary cells in G1 phase of the cell cycle during programmed vascular regression. Development 126, 2141-2147. [DOI] [PubMed] [Google Scholar]
- Evan G. I., Wyllie A. H., Gilbert C. S., Littlewood T. D., Land H., Brooks M., Waters C. M., Penn L. Z. and Hancock D. C. (1992). Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, 119-128. 10.1016/0092-8674(92)90123-T [DOI] [PubMed] [Google Scholar]
- Ferrer-Vaquer A., Piliszek A., Tian G., Aho R. J., Dufort D. and Hadjantonakis A.-K. (2010). A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev. Biol. 10, 121 10.1186/1471-213X-10-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco C. A., Jones M. L., Bernabeu M. O., Vion A. C., Barbacena P., Fan J., Mathivet T., Fonseca C. G., Ragab A., Yamaguchi T. P. et al. (2016). Non-canonical Wnt signalling modulates the endothelial shear stress flow sensor in vascular remodelling. Elife 5, e07727 10.7554/eLife.07727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale N. W., Thurston G., Hackett S. F., Renard R., Wang Q., McClain J., Martin C., Witte L., Witte M. H., Jackson D. et al. (2002). Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter is rescued by angiopoietin-1. Dev. Cell 3, 411-423. 10.1016/S1534-5807(02)00217-4 [DOI] [PubMed] [Google Scholar]
- Gartel A. L. and Radhakrishnan S. K. (2005). Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 65, 3980-3985. 10.1158/0008-5472.CAN-04-3995 [DOI] [PubMed] [Google Scholar]
- Gartel A. L. and Shchors K. (2003). Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 283, 17-21. 10.1016/S0014-4827(02)00020-4 [DOI] [PubMed] [Google Scholar]
- Gartel A. L. and Tyner A. L. (2002). The role of the cyclin-dependent kinase inhibitor p21 in apoptosis 1 supported in part by NIH grant R01 DK56283 (to A. L. T.) for the p21 research and campus research board and illinois department of public health penny severns breast and cervical cancer grants (to A. L. G.).1. Mol. Cancer Ther. 1, 639-649. [PubMed] [Google Scholar]
- Gartel A. L., Goufman E., Najmabadi F. and Tyner A. L. (2000). Sp1 and Sp3 activate p21 (WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 19, 5182-5188. 10.1038/sj.onc.1203900 [DOI] [PubMed] [Google Scholar]
- Gartel A. L., Ye X., Goufman E., Shianov P., Hay N., Najmabadi F. and Tyner A. L. (2001). Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl Acad. Sci. USA 98, 4510-4515. 10.1073/pnas.081074898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D. A. II, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A. et al. (2005). Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751-764. 10.1016/j.devcel.2005.02.017 [DOI] [PubMed] [Google Scholar]
- Gong Y., Slee R. B., Fukai N., Rawadi G., Roman-Roman S., Reginato A. M., Wang H., Cundy T., Glorieux F. H., Lev D. et al. (2001). LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513-523. 10.1016/S0092-8674(01)00571-2 [DOI] [PubMed] [Google Scholar]
- Goodwin A. M., Kitajewski J. and D'Amore P. A. (2007). Wnt1 and Wnt5a affect endothelial proliferation and capillary length; Wnt2 does not. Growth Factors 25, 25-32. 10.1080/08977190701272933 [DOI] [PubMed] [Google Scholar]
- He T.-C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B. and Kinzler K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509-1512. 10.1126/science.281.5382.1509 [DOI] [PubMed] [Google Scholar]
- Hoffman B. and Liebermann D. A. (2008). Apoptotic signaling by c-MYC. Oncogene 27, 6462-6472. 10.1038/onc.2008.312 [DOI] [PubMed] [Google Scholar]
- Ito M. and Yoshioka M. (1999). Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat. Embryol. 200, 403-411. 10.1007/s004290050289 [DOI] [PubMed] [Google Scholar]
- Jänicke R. U., Sohn D., Essmann F. and Schulze-Osthoff K. (2007). The multiple battles fought by anti-apoptotic p21. Cell Cycle 6, 407-413. 10.4161/cc.6.4.3855 [DOI] [PubMed] [Google Scholar]
- Jiao X., Ventruto V., Trese M. T., Shastry B. S. and Hejtmancik J. F. (2004). Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 75, 878-884. 10.1086/425080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joeng K. S., Schumacher C. A., Zylstra-Diegel C. R., Long F. and Williams B. O. (2011). Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev. Biol. 359, 222-229. 10.1016/j.ydbio.2011.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner D. M., Ke J., Zhong Z., Xu H. E. and Williams B. O. (2013). LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 24, 31-39. 10.1016/j.tem.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge H. J., Yang S., Burton J. B., Paes K., Shu X., French D. M., Costa M., Rice D. S. and Ye W. (2009). TSPAN12 regulates retinal vascular development by promoting Norrin- but not wnt-induced FZD4/β-catenin signaling. Cell 139, 299-311. 10.1016/j.cell.2009.07.048 [DOI] [PubMed] [Google Scholar]
- Kato M., Patel M. S., Levasseur R., Lobov I., Chang B. H.-J., Glass D. A., Hartmann C., Li L., Hwang T.-H., Brayton C. F. et al. (2002). Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303-314. 10.1083/jcb.200201089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn C., Scholz B., Hu J., Srivastava K., Wojtarowicz J., Arnsperger T., Adams R. H., Boutros M., Augustin H. G. and Augustin I. (2014). Endothelial cell-derived non-canonical Wnt ligands control vascular pruning in angiogenesis. Development 141, 1757-1766. 10.1242/dev.104422 [DOI] [PubMed] [Google Scholar]
- Kress T. R., Sabò A. and Amati B. (2015). MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593-607. 10.1038/nrc3984 [DOI] [PubMed] [Google Scholar]
- Lang R. A. and Bishop M. J. (1993). Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell 74, 453-462. 10.1016/0092-8674(93)80047-I [DOI] [PubMed] [Google Scholar]
- Lang R., Lustig M., Francois F., Sellinger M. and Plesken H. (1994). Apoptosis during macrophage-dependent ocular tissue remodelling. Development 120, 3395-3403. [DOI] [PubMed] [Google Scholar]
- Lee G. and Saito I. (1998). Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene 216, 55-65. 10.1016/S0378-1119(98)00325-4 [DOI] [PubMed] [Google Scholar]
- Li Y., Dowbenko D. and Lasky L. A. (2002). AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 277, 11352-11361. 10.1074/jbc.M109062200 [DOI] [PubMed] [Google Scholar]
- Lin C. Y., Lovén J., Rahl P. B., Paranal R. M., Burge C. B., Bradner J. E., Lee T. I. and Young R. A. (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56-67. 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov I. B., Brooks P. C. and Lang R. A. (2002). Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl Acad. Sci. USA 99, 11205-11210. 10.1073/pnas.172161899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov I. B., Rao S., Carroll T. J., Vallance J. E., Ito M., Ondr J. K., Kurup S., Glass D. A., Patel M. S., Shu W. et al. (2005). WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature 437, 417-421. 10.1038/nature03928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ramirez M. A., Fonseca G., Zeineddine H. A., Girard R., Moore T., Pham A., Cao Y., Shenkar R., de Kreuk B.-J., Lagarrigue F. et al. (2017). Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J. Exp. Med. 214, 3331-3346. 10.1084/jem.20171178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe S. W., Cepero E. and Evan G. (2004). Intrinsic tumour suppression. Nature 432, 307-315. 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- Ma S., Kwon H. J., Johng H., Zang K. and Huang Z. (2013). Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 11, e1001469 10.1371/journal.pbio.1001469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R. et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133-140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masckauchán T. N., Shawber C. J., Funahashi Y., Li C.-M. and Kitajewski J. (2005). Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 8, 43-51. 10.1007/s10456-005-5612-9 [DOI] [PubMed] [Google Scholar]
- Masckauchán T. N. H., Agalliu D., Vorontchikhina M., Ahn A., Parmalee N. L., Li C.-M., Khoo A., Tycko B., Brown A. M. C. and Kitajewski J. (2006). Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol. Biol. Cell 17, 5163-5172. 10.1091/mbc.e06-04-0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon S. B. (2014). MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 4, a014407 10.1101/cshperspect.a014407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeson A., Palmer M., Calfon M. and Lang R. A. (1996). A relationship between apoptosis and flow during programmed capillary regression is revealed by vital analysis. Development 12, 3929-3938. [DOI] [PubMed] [Google Scholar]
- Muthalagu N., Junttila M. R., Wiese K. E., Wolf E., Morton J., Bauer B., Evan G. I., Eilers M. and Murphy D. J. (2014). BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 8, 1347-1353. 10.1016/j.celrep.2014.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namura S., Zhu J., Fink K., Endres M., Srinivasan A., Tomaselli K. J., Yuan J. and Moskowitz M. A. (1998). Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 18, 3659-3668. 10.1523/JNEUROSCI.18-10-03659.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D. R., Tessarollo L., Casellas R. et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68-79. 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K. G., Kontos C. D., Lin P. C., Wong A. L., Rao P., Huang L., Dewhirst M. W. and Sankar S. (2004). Functional significance of Tie2 signaling in the adult vasculature. Recent Prog. Horm. Res. 59, 51-71. 10.1210/rp.59.1.51 [DOI] [PubMed] [Google Scholar]
- Phng L.-K., Potente M., Leslie J. D., Babbage J., Nyqvist D., Lobov I., Ondr J. K., Rao S., Lang R. A., Thurston G. et al. (2009). Nrarp coordinates endothelial notch and Wnt signaling to control vessel density in angiogenesis. Dev. Cell 16, 70-82. 10.1016/j.devcel.2008.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo M. T. and Crispi S. (2012). The dual riole played by p21 may influencethe aopototic or anti-apoptotic fate in cancer. J. Cancer Research Updates 1, 189-202. [Google Scholar]
- Poché R. A., Hsu C.-W., McElwee M. L., Burns A. R. and Dickinson M. E. (2015). Macrophages engulf endothelial cell membrane particles preceding pupillary membrane capillary regression. Dev. Biol. 403, 30-42. 10.1016/j.ydbio.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter J. A., Ali M., Gilmour D. F., Rice A., Kondo H., Hayashi K., Mackey D. A., Kearns L. S., Ruddle J. B., Craig J. E. et al. (2010). Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am. J. Hum. Genet. 86, 248-253. 10.1016/j.ajhg.2010.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Lobov I. B., Vallance J. E., Tsujikawa K., Shiojima I., Akunuru S., Walsh K., Benjamin L. E. and Lang R. A. (2007). Obligatory participation of macrophages in an angiopoietin 2-mediated cell death switch. Development 134, 4449-4458. 10.1242/dev.012187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Chun C., Fan J., Kofron J. M., Yang M. B., Hegde R. S., Ferrara N., Copenhagen D. R. and Lang R. A. (2013). A direct and melanopsin-dependent fetal light response regulates mouse eye development. Nature 494, 243-246. 10.1038/nature11823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle R. C., Diegel C. R., Leslie J. M., Van Koevering K. K., Faugere M.-C., Clemens T. L. and Williams B. O. (2013). Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE 8, e63323 10.1371/journal.pone.0063323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille J., MacDonald M. L., Kaykas A., Sheldahl L. C., Zeisler J., Dubé M.-P., Zhang L.-H., Singaraja R. R., Guernsey D. L., Zheng B. et al. (2002). Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 32, 326-330. 10.1038/ng957 [DOI] [PubMed] [Google Scholar]
- Rossig L., Jadidi A. S., Urbich C., Badorff C., Zeiher A. M. and Dimmeler S. Z. (2001). Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol. Cell. Biol. 21, 5644-5657. 10.1128/MCB.21.16.5644-5657.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössig L., Badorff C., Holzmann Y., Zeiher A. M. and Dimmeler S. (2002). Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem. 277, 9684-9689. 10.1074/jbc.M106157200 [DOI] [PubMed] [Google Scholar]
- Salic A. and Mitchison T. J. (2008). A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl Acad. Sci. USA 105, 2415-2420. 10.1073/pnas.0712168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Glynn J., Guilbert L., Cotter T., Bissonnette R. and Green D. (1992). Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 257, 212-214. 10.1126/science.1378649 [DOI] [PubMed] [Google Scholar]
- Stalmans I., Ng Y.-S., Rohan R., Fruttiger M., Bouché A., Ÿuce A., Fujisawa H., Hermans B., Shani M., Jansen S. et al. (2002). Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109, 327-336. 10.1172/JCI0214362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefater J. A. III, Lewkowich I., Rao S., Mariggi G., Carpenter A. C., Burr A. R., Fan J., Ajima R., Molkentin J. D., Williams B. O. et al. (2011). Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 474, 511-515. 10.1038/nature10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J. M., Rajagopal J., Carroll T. J., Ishibashi M., McMahon J. and McMahon A. P. (2008). Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247-1250. 10.1126/science.1164594 [DOI] [PubMed] [Google Scholar]
- Toomes C., Bottomley H. M., Jackson R. M., Towns K. V., Scott S., Mackey D. A., Craig J. E., Jiang L., Yang Z., Trembath R. et al. (2004). Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 74, 721-730. 10.1086/383202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomes C., Downey L. M., Bottomley H. M., Mintz-Hittner H. A. and Inglehearn C. F. (2005). Further evidence of genetic heterogeneity in familial exudative vitreoretinopathy; exclusion of EVR1, EVR3, and EVR4 in a large autosomal dominant pedigree. Br. J. Ophthalmol. 89, 194-197. 10.1136/bjo.2004.042507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A., Refaeli Y., Oskarsson T., Gasser S., Murphy M., Martin G. R. and Bishop J. M. (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768-773. 10.1038/414768a [DOI] [PubMed] [Google Scholar]
- Udan R. S., Culver J. C. and Dickinson M. E. (2013). Understanding vascular development. Wiley Interdiscip Rev. Dev. Biol. 2, 327-346. 10.1002/wdev.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S., Lorenzin F., Morton J., Wiese K. E., von Eyss B., Herold S., Rycak L., Dumay-Odelot H., Karim S., Bartkuhn M. et al. (2014). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483-487. 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Rattner A., Zhou Y., Williams J., Smallwood P. M. and Nathans J. (2012). Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151, 1332-1344. 10.1016/j.cell.2012.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M. F., Lim R., Zimmermann B., Aspalter I. M., Franco C. A., Boettger T. et al. (2016). FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Murphy M. J., Oskarsson T., Kaloulis K., Bettess M. D., Oser G. M., Pasche A. C., Knabenhans C., Macdonald H. R. and Trumpp A. (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747-2763. 10.1101/gad.313104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E., Gebhardt A., Kawauchi D., Walz S., von Eyss B., Wagner N., Renninger C., Krohne G., Asan E., Roussel M. F. et al. (2013). Miz1 is required to maintain autophagic flux. Nat. Commun. 4, 2535 10.1038/ncomms3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K. et al. (2004). Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883-895. 10.1016/S0092-8674(04)00216-8 [DOI] [PubMed] [Google Scholar]
- Yancopoulos G. D., Davis S., Gale N. W., Rudge J. S., Wiegand S. J. and Holash J. (2000). Vascular-specific growth factors and blood vessel formation. Nature 407, 242-248. 10.1038/35025215 [DOI] [PubMed] [Google Scholar]
- Yang J.-H., Wylie-Sears J. and Bischoff J. (2008). Opposing actions of Notch1 and VEGF in post-natal cardiac valve endothelial cells. Biochem. Biophys. Res. Commun. 374, 512-516. 10.1016/j.bbrc.2008.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum G. S., Cleland R. and Goodman R. H. (2008). A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 28, 7368-7379. 10.1128/MCB.00744-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller K. I., Zhao X., Lee C. W. H., Chiu K. P., Yao F., Yustein J. T., Ooi H. S., Orlov Y. L., Shahab A., Yong H. C. et al. (2006). Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl Acad. Sci. USA 103, 17834-17839. 10.1073/pnas.0604129103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Wang Y., Tischfield M., Williams J., Smallwood P. M., Rattner A., Taketo M. M. and Nathans J. (2014). Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 124, 3825-3846. 10.1172/JCI76431 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.