

Abstract
Parkinson's disease is the most common neurodegenerative movement disorder. It arises as a result of neuronal cell death in specific brain regions, notably the substantia nigra pars compacta, and is characterized by the accumulation of α‐synuclein in these brain regions. Current pharmacological therapies alleviate the motor symptoms of the disease and are particularly effective in the early stages of the disease. Ongoing drug development efforts focus on disease‐modifying strategies that aim to halt or slow disease progression. In this review, we explore a number of emerging disease‐modifying strategies with a focus on direct and indirect targeting of α‐synuclein dysfunction. We summarize newer classes of small molecules and biological agents intended to attenuate protein aggregation or to target enzymes that may increase the degradation of the pathogenic forms of α‐synuclein. Finally, we discuss emerging strategies that are demonstrating the potential for disease modification at the preclinical stage.
Abbreviations
- NAC
non‐Aβ component
- O‐GlcNAc
O‐linked β‐N‐acetyl glucosamine
- AD
Alzheimer's disease
- COMT
catechol‐O‐methyltransferase
- DLB
dementia with Lewy bodies
- ER
endoplasmic reticulum
- GCase
glucocerebrosidase
- LBs
Lewy bodies
- L‐Dopa
levodopa
- LRRK2
leucine‐rich repeat kinase 2
- MAO‐B
monoamine oxidase B
- MSA
multiple system atrophy
- PD
Parkinson's disease
- PINK1
PTEN‐induced putative kinase 1
- SNpc
substantia nigra pars compacta
- UPDRS
Unified Parkinson's Disease Rating Scale
Introduction
Parkinson's disease (PD) is a debilitating neurodegenerative condition characterized by the prominent loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) of the midbrain. PD is the most common neurodegenerative movement disorder, and age is the greatest risk factor for developing the disease. PD is largely sporadic, although 5 to 10% of cases are due to genetic causes such as mutations in the SNCA gene that encodes the protein α‐synuclein (Kalia and Lang, 2015). The SNpc is part of the basal ganglia, a group of neuronal nuclei primarily associated with regulating motor function; thus, progressive cell death in this region predominantly affects movement resulting in the cardinal motor symptoms of the disease, including bradykinesia, muscular rigidity and tremor. As the disease progresses, other types of cells such as serotonergic, noradrenergic and cholinergic neurons in other areas of the brain also degenerate (Kalia et al., 2013). Aside from having motor symptoms, PD patients will often also experience non‐motor symptoms, such as cognitive dysfunction, sleeping difficulties, psychiatric symptoms and autonomic dysfunction, partly due to the involvement of these other neurotransmitter systems (Chaudhuri and Schapira, 2009).
The main pathological hallmarks of PD are loss of SNpc dopaminergic neurons as well as Lewy pathology that occurs in the majority of cases. Lewy pathology includes Lewy bodies (LBs) and Lewy neurites, which are found in the cytosol of neuronal soma and neurites, respectively, and consist of a dense core of aggregated protein surrounded by loose filaments. These aggregates are predominantly composed of the misfolded, insoluble and fibrillar forms of α‐synuclein, although ubiquitin and other cellular components are also contained in LBs (Kalia and Kalia, 2015). Whether Lewy pathology is neuroprotective, neurotoxic or both, depending on the context, remains an area of active investigation (Halliday and Mccann, 2010). Although SNCA mutations (missense and multiplications) account for a very small proportion of cases of genetic forms of PD, there is little doubt that aggregation of the protein has an important role in the pathogenesis of this condition and other related synucleinopathies, such as dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). The normal ageing process gives rise to increased levels of cytoplasmic α‐synuclein, and a current hypothesis is that, during the development of PD, α‐synuclein misfolds, accumulating into soluble pathogenic protein oligomers and later insoluble fibrils and LBs (Burre et al., 2017).
There is much evidence for a causative role of α‐synuclein in PD, yet a number of other gene mutations have also been linked to the development of this disease, most commonly mutations in http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2059 which is also autosomal dominant. The LRRK2 gene encodes leucine‐rich repeat kinase 2 (LRRK2), a kinase for which the exact function is unknown. It is currently postulated that progression of the disease in LRRK2‐related PD may result from an increase in kinase activity, an impairment in GTPase function and/or a change in the ability of the protein to dimerize, depending on the domain in which the mutation occurs (Schulte and Gasser, 2011). Other genes associated with autosomal recessive forms of the disease include Parkin, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2161 and DJ1. Patients with PD as a result of mutations within these genes can present with similar symptoms and pathological features except in a subset of cases with mutations in Parkin and LRRK2 in which Lewy pathology is not present (Kalia et al., 2015; Schneider and Alcalay, 2017).
Interestingly, many of the genes associated with PD encode for proteins involved in pathways regulating mitochondrial and/or proteasomal and lysosomal dysfunction. In healthy individuals, PTEN‐induced putative kinase 1 (PINK1) and Parkin play important roles in regulating mitochondrial quality control by inducing clearance of dysfunctional mitochondria via mitophagy. Under basal conditions, low levels of the kinase PINK1 span both the mitochondrial inner and outer membranes, where it acts in respirasomes and influences complex I activity (Morais et al., 2009). Parkin is an E3 ubiquitin ligase normally present in the cytosol, but it is rapidly recruited to mitochondria in the context of mitochondrial stress. Parkin mutations are associated with reduced levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713, which is possibly related to the E3 ubiquitin ligase activity of the protein as it regulates the expression levels of many of the electron transport chain subunits (Sarraf et al., 2013), while defective mitophagy will also induce increases in ROS production and overall mitochondrial dysfunction. Additionally, α‐synuclein has been shown to have a mitochondrial targeting sequence in its N‐terminal region, and accumulation of the protein at the mitochondria reduces complex I activity and increases oxidative damage (Devi et al., 2008). Mutant α‐synuclein blocks lysosomal translocation, which in turn impairs its own degradation, enhancing accumulation of the misfolded protein (Maria Cuervo et al., 2004, Martinez‐Vicente et al., 2008). Overexpression of α‐synuclein also causes the autophagosome formation protein Atg9 to mislocalize and thus impairs macroautophagy (Winslow et al., 2010). Mutations in LRRK2 have been shown to accelerate age‐related autophagy dysfunction, through reductions in chaperone‐mediated autophagy of mutant LRRK2 (Orenstein et al., 2013), and expression of wild‐type LRRK2 improves autophagic clearance (Saha et al., 2015). The most common genetic risk factor for PD development is a mutation of glucocerebrosidase (GBA), which codes for the lysosomal enzyme http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2978 (GCase) that is important in glycolipid metabolism. GBA mutations can cause Gaucher disease, an autosomal recessive lysosomal storage disorder. Interestingly, patients with Gaucher disease and GBA mutation carriers have an increased risk of developing PD, DLB and possibly MSA. The pathogenic mechanism has yet to be elucidated but it is hypothesized to be a loss of GCase enzyme activity resulting in a deterioration of lysosomal function and endoplasmic reticulum (ER) stress (O'Regan et al., 2017). There is also evidence of an interaction between GCase and α‐synuclein where a reciprocal relationship exists with decreases in GCase enzyme activity resulting in increased intracellular α‐synuclein and intracellular α‐synuclein inhibiting the lysosomal activity of normal GCase in neurons (Manning‐Bog et al., 2009, Mazzulli et al., 2011). Taken together, research to date suggests that multiple molecular pathways are involved in the pathobiology of PD, many of which act upstream of α‐synuclein aggregation.
Current treatments for PD focus on increasing dopaminergic tone through the use of the dopamine precursor http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3639 (L‐Dopa), dopamine agonists and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2490 (MAO‐B) or http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2472 (COMT) inhibitors (Figure 1). L‐Dopa has been regarded as the most effective treatment for PD since its introduction in 1967 and it is included in most treatment regimens, but treatment complications occur over time (Fahn et al., 2004). Small molecules such as pramipexole, ropinirole and apomorphine act as agonists of dopamine receptors, with particular affinity for D2‐like receptors, while antagonizing serotonergic and β‐adrenergic neurotransmission (Nutt et al., 2000). MAO‐B and COMT inhibitors such as selegiline and entacapone, respectively, act to slow down the breakdown of dopamine in the synapse and increase overall levels in the brain. Over the course of the disease, these treatments are effective at managing many of the motor symptoms of PD, especially when used in combination, but do not halt or slow disease progression. An important focus remains on the development of therapeutics that will halt the neurodegeneration that occurs in the disease, rather than simply provide temporary symptomatic relief. This review will focus on some of the disease‐modifying therapeutics in clinical development that directly or indirectly target α‐synuclein as well as examine other potential disease‐modifying therapies that have not yet entered clinical trials (Figure 2).
Figure 1.
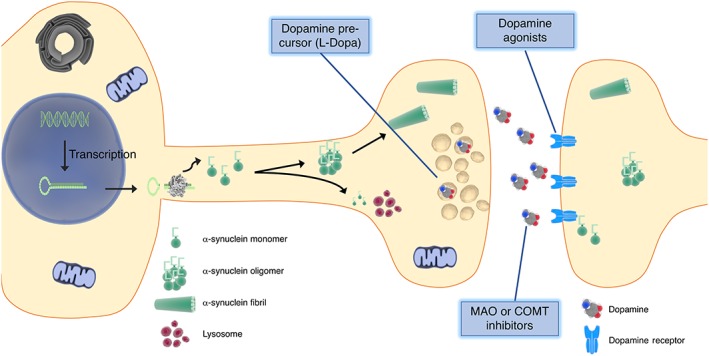
Current symptomatic pharmacological therapies for Parkinson Disease. Current treatments involve the use of the dopamine precursor L‐Dopa to increase dopaminergic neurotransmission, dopamine agonists to mimic dopaminergic neurotransmission and MAO‐B or COMT inhibitors to slow dopamine metabolism and re‐uptake by neighbouring glial cells.
Figure 2.
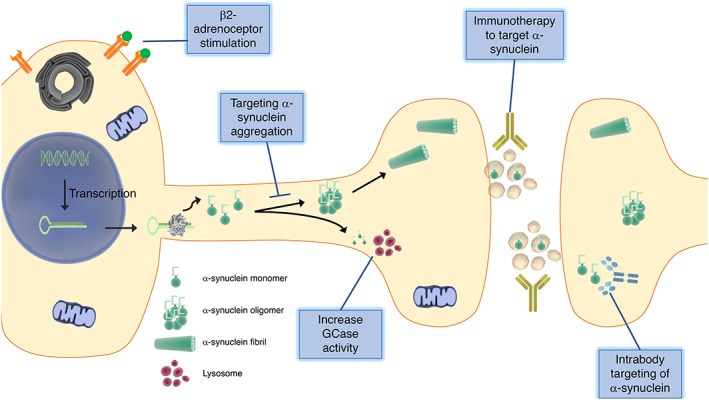
Potential disease‐modifying strategies for Parkinson disease. Development of novel treatments for PD focus on targeting α‐synuclein by decreasing its accumulation into oligomers, increasing its degradation by lysosomes, removing it from the extracellular space using immunotherapy or reducing transcription of the protein by activating β2‐adrenoceptors.
Strategies at the clinical trial stage
Direct targeting of α‐synuclein
α‐Synuclein as a therapeutic target
α‐Synuclein is a pre‐synaptic protein of 140 amino acids whose exact physiological function remains unknown. It is known to associate with membranes where it forms α‐helical structures when bound to negatively charged lipids and, after longer incubation periods, can form β‐sheet structures. There are three distinct regions within the protein: an N‐terminus that is an amphipathic region, a non‐Aβ component (NAC) domain that is important for fibril formation and a C‐terminus that is negatively charged and contains an aggregation inhibition region (Rosborough et al., 2017). Immunohistochemical staining reveals that α‐synuclein follows a pattern consistent with the pre‐synaptic terminals and associates primarily with, but not within, synaptic vesicles (Kahle, 2008). Some evidence points to α‐synuclein acting to reduce neurotransmission after sustained periods of firing as it disperses from vesicles after fusion with the outer neuronal membrane but returns to inhibit refilling of the readily releasable synaptic vesicle pool (Fortin et al., 2005). Both wild‐type and mutant forms of the protein form β‐sheet structures (similar to the β‐sheet structure of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4865) and subsequently fibrils upon long‐term incubation in solution, and these fibrils are widely acknowledged to be the main component of Lewy pathology in PD. Prior to the formation of mature fibrils, there are a number of intermediate forms of α‐synuclein that assume ring, string and spherical‐like characteristics, and it is these forms that are believed to be the pathogenic oligomeric forms of the protein (Kalia et al., 2013). Direct targeting of α‐synuclein aims to prevent this type of pathogenic aggregation and thus halt disease progression by inhibiting α‐synuclein toxicity.
Small molecules to target α‐synuclein
Several α‐synuclein inhibitors are being investigated for efficacy at reducing aggregation and toxicity. NPT200‐11 is a small molecule that works by targeting α‐synuclein and stabilizing its physiological structure, leaving it incapable of assembling into toxic oligomers. It has been shown to prevent the formation of pathogenic α‐synuclein oligomeric aggregates and to lead to improvements in neuropathological and biochemical outcomes in α‐synuclein transgenic murine models (Koike et al., 2014, Price et al., 2014). Furthermore, NPT200‐11 has been tested for safety in humans in a phase I clinical trial (NCT02606682). Another small molecule known as ANLE138b has been shown to cross the blood–brain barrier and reduce aggregation of α‐synuclein as well as block the aggregation of pathogenic forms of prion protein in cell culture and mouse models (Levin et al., 2014, Wagner et al., 2013).
NPT088 is a compound that binds to a putative amyloid fold common to several toxic misfolded proteins including α‐synuclein, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9275 and β‐amyloid. It is a fusion protein that combines a human immunoglobulin backbone with a general amyloid interaction motif (GAIM) and is the most promising candidate in this class of drugs. Gene 3 protein (g3p) of filamentous bacteriophage (the active protein fused to immunoglobulin) has shown promise in a mouse model of PD where it binds α‐synuclein aggregates (Krishnan et al., 2014). This compound has also shown promise for the treatment of Alzheimer's disease (AD) where it reduced β‐amyloid plaques, improved cognitive performance and reduced phosphorylated http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9275 pathology in the brains of transgenic mouse models of the disease (Levenson et al., 2016).
An intrabody is an antibody fragment that works within the cell to bind an intracellular protein. The single chain variable fragment (scFv) can be expressed without the full‐length immunoglobulin, thus retaining conventional antibody specificity but allowing increased stability in the intracellular environment. Different intrabodies have been developed, which can bind monomeric, oligomeric and fibrillary forms of α‐synuclein and appear to be neuroprotective by modulating aggregated forms of the protein (Bhatt et al., 2013). VH14 is a single domain intrabody against the NAC hydrophobic interaction domain which, when fused to a proteasome targeting signal (VH14PEST), can protect against α‐synuclein toxicity in cell culture systems. NbSyn87PEST, a similar nanobody targeting the C‐terminus, was similarly effective at degrading α‐synuclein (Butler et al., 2016). Further research has shown that these two intrabodies have the ability to eliminate aggregated α‐synuclein, replenish striatal dopamine and improve motor function in rats overexpressing wild‐type α‐synuclein (Brundin et al., 2017). While these therapeutics show promise, a significant challenge is maintaining high levels of the intrabodies in specific brain regions for prolonged periods of time, and thus, current drug delivery technology would require direct delivery to affected regions using viral vectors.
Immunotherapy to target α‐synuclein
The current ‘prion‐like spreading’ hypothesis for PD proposes cell‐to‐cell transfer of α‐synuclein as a leading cause of propagation of PD pathology throughout the striatum into other brain regions as the disease progresses. This phenomenon would likely require α‐synuclein to traverse the synapse or alternate extracellular spaces, and so reducing the extracellular pool of the misfolded or aggregated protein may reduce the propagation or act as a sink. Thus, a number of companies have begun to target this property of α‐synuclein by using antibodies generated by active immunization or passive transfer. Active immunization stimulates the body's own immune system to produce antibodies against the toxic protein while passive transfer requires direct administration of antibodies. There is a possibility of off‐target responses and non‐specific inflammatory responses with these types of treatment, but a number of therapeutics have advanced to the clinical trial stage and shown promise to date.
C‐terminal cleavage of α‐synuclein by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2336 is thought to contribute to the formation of toxic oligomers (Dufty et al., 2007), and many immunization approaches are designed to directly target this domain of the protein. Using the active immunotherapy approach, Affris is currently testing two short immunogenic peptides that mimic the C‐terminus region of α‐synuclein, known as PD01A and PD03A. These AFFITOPEs® illicit an immune response in the absence of a T‐cell response, thus reducing the potential for a damaging autoimmune response. Initial vaccination in mouse models of PD resulted in high antibody titres in both the CSF and plasma. These antibodies were also shown to cross the blood–brain barrier and target α‐synuclein directly. The results indicated a decreased accumulation of α‐synuclein in axons and synapses and a reduction in the degeneration of dopaminergic neuron fibres in the striatum as well as an improvement in motor symptoms (Mandler et al., 2014). Both of these vaccines are currently in phase I development while early results have indicated that 55% (12 of 22) of subjects administered PD01A generated serum antibodies against α‐synuclein. Results from a follow‐up ‘booster’ study have shown that 86% of subjects generated an immune response, 63% of whom generated α‐synuclein specific antibodies, and a cohort of these subjects did not require any increase in symptomatic dopaminergic medications during the study period (NCT02216188). It was reported that, in a phase I safety trial of PD03A, both high and low dose groups showed no serious adverse events or reactions and both doses were well tolerated (NCT02267434). The company has also reported that vaccination with PD03A exhibited a dose‐dependent immune response against the peptide itself and cross reactivity against the α‐synuclein targeted epitope over time.
PRX002 (Prothena/Roche) is a humanized monoclonal antibody, which specifically binds to the C‐terminus of α‐synuclein, developed for passive immunization. Results from PD mouse models showed antibody treatment attenuated synaptic and axonal pathology, reduced the accumulation of C‐terminal truncated α‐synuclein and improved motor and memory deficits (Games et al., 2014). It has been shown to be well tolerated in a pilot phase I trial while reducing α‐synuclein serum levels up to 96.5% in a dose‐dependent manner (NCT02157714) (Schenk et al., 2017). A phase II trial is currently recruiting and will test the effects of the antibody versus placebo for 52 weeks in 300 patients with early stage PD who have not received L‐Dopa treatment. BIIB054 is a monoclonal antibody developed by Biogen that targets forms of α‐synuclein thought to be pathogenic. A randomized double‐blind, placebo‐controlled phase I trial in 48 healthy subjects found BIIB054 was well tolerated, with the exception of the highest dose (135 mg·kg−1), and Cmax values were dose proportional, with a 28‐day serum half‐life. BIIB054 was detectable in CSF in the range expected for monoclonal antibodies (Alzforum, 2017). Phase II trials were planned to start in November 2017 (NCT03318523). MEDI1341 is a combined Astra Zeneca/Takeda effort aimed at targeting extracellular α‐synuclein through passive immunization. A press release highlighted the fact that the antibody has a high affinity for α‐synuclein and reduced effector function, making this therapeutic more efficacious and safer by decreasing immune system interaction. A phase I safety trial involving treatment with a single ascending dose began in September 2017 (NCT03272165). Finally, BioArctic Neuroscience and AbbVie have combined to produce a clinical phase antibody called BAN0805, which targets oligomeric forms of pathogenic α‐synuclein. This therapeutic has shown promise in mouse models but has not yet entered the clinical trial stage (Fagerqvist et al., 2013).
The success of these approaches likely depends on validation of the ‘prion‐like spreading’ hypothesis of α‐synuclein propagation throughout the brain and how much of the pathogenic protein is maintained in the extracellular space for a limited amount of antibody to target it. The potential off‐target effects of these antibodies (e.g. due to affecting α‐synuclein outside of the brain) will need to be examined.
Indirect targeting of α‐synuclein
Increasing GCase activity
As mentioned above, mutations in GBA are a strong risk factor for the development of PD as well as other synucleinopathies. It is estimated that 7 to 10% of PD patients carry a GBA mutation, which confers up to a 30‐fold increased risk of developing PD (Migdalska‐Richards and Schapira, 2016). Most potential GBA‐related therapeutics focus on enhancing enzyme activity since experimental evidence demonstrates that decreased GCase activity leads to an increase in α‐synuclein misfolding. Additional evidence suggests that mutations in GBA interfere with the correct function of the autophagy‐lysosomal system as decreased GCase activity may result in a build‐up of its substrate in the lysosome, while there is also evidence of ER stress (O'regan et al., 2017). Such evidence suggests that any enhancement of GCase activity pharmacologically could decrease α‐synuclein accumulation but also enhance the function of the lysosome and ER, both of which have been shown to be dysfunctional in PD, and thus enhance degradation of the misfolded protein (Kilpatrick et al., 2016).
At present, it is not possible to directly supplement the enzyme using enzyme replacement therapy as those currently available do not penetrate the blood–brain barrier. Research is focusing on other upstream targets, which limit substrates, or on molecular chaperones (Figure 3). Glucosylceramide, a glycosphingolipid, is a substrate of GCase that is thought to stabilize α‐synuclein oligomers. Sanofi/Genzyme have started a phase II clinical trial (NCT02906020), which will test the efficacy of a glucosylceramide inhibitor called ibiglustat (SAR402671) in patients with early stage PD carrying a GBA mutation. An alternative target within this pathway is to use molecular chaperones to alleviate misfolding of mutated forms of GCase in the ER and thus reduce build‐up of the enzyme in the lysosome and increase its activity. Ambroxol is an anti‐mucolytic drug, used in the treatment of respiratory diseases, which has recently been shown to enhance the activity of GCase in fibroblasts from control and GBA mutation carriers (Sanchez‐Martinez et al., 2016). A phase I proof‐of‐concept trial has now begun examining the effect of ambroxol on brain GCase activity and α‐synuclein accumulation in PD patients with and without GBA mutations (NCT02914366). Lysosomal Therapeutics has developed a brain‐penetrant, small molecule that targets GCase, increasing its activity, and thus reducing levels of glucosylceramide. Animal studies have demonstrated that LTI‐291 reduces glucosylceramide levels in the brain, and GCase activation effects are exacerbated when GCase enzyme activity is more impaired and glucosylceramide levels are higher (Lysosomal‐Therapeutics‐Website, 2017).
Figure 3.
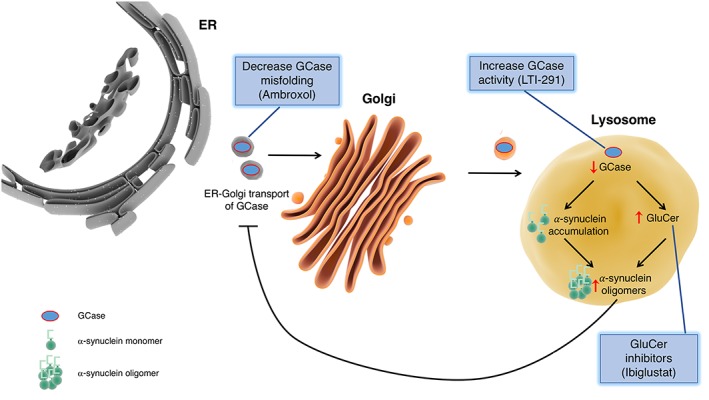
Targeting GCase as a potential disease‐modifying strategy for Parkinson disease. Development of therapeutics targeting GCase for treatment of PD focus on increasing enzyme activity, decreasing GCase misfolding and inhibiting glucosylceramide (GluCer), a substrate of GCase. The net result of these therapeutics is anticipated to be decreased α‐synuclein accumulation and enhanced lysosomal function.
Inhibiting c‐Abl activity
Another approach is to inhibit a tyrosine kinase, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1923, which has been shown to have enhanced activity in the brain of PD patients, leading to a downstream increase in phosphorylation, and subsequently, aggregation of α‐synuclein. Interestingly, enhanced c‐Abl activity inhibits the ubiquitination of Parkin, reducing its activity (Mahul‐Mellier et al., 2014). Mutations in c‐Abl enhance its kinase activity, leading to an increase in cell proliferation, so inhibitors of c‐Abl have primarily been developed as an anti‐cancer treatment. Nilotinib is a drug approved for the treatment of imatinib‐resistant chronic myelogenous leukaemia and has previously been shown to penetrate into the brain and protect dopaminergic neurons from toxicity in a viral‐vector mediated α‐synuclein overexpression mouse model (Hebron et al., 2013). Phase I safety trials have indicated two doses (150 and 300 mg) are safe and tolerable in PD and DLB patients and suggested possible target engagement, although this was an open label trial (Pagan et al., 2016). A phase II, randomized, double‐blind, placebo‐controlled clinical trial to measure the effects of nilotinib treatment on clinical outcomes and biomarkers in mid‐stage PD patients is currently underway (NCT03205488).
Altering levels of endogenous metals
Metals are a vital component of the earth's ecosystem and also play integral roles in the human body where they act as co‐factors in a variety of enzymatic reactions. Essential metals include iron, copper (Cu), zinc and manganese, and these are important in regulating gene expression, neurotransmission and the immune response. There is accumulating evidence from post‐mortem studies, MRI and ultrasound studies that PD patients have high levels of iron deposition in the neurons of the SNpc (Gotz et al., 2004). The presence of excess amounts of low molecular weight iron can cause oxidative damage to lipids, proteins and DNA through the generation of increased amounts of ROS, which cannot be scavenged by the usual enzymes. Cellular damage due to ROS is believed to be an important contributor to neurodegeneration in the PD brain (Manoharan et al., 2016). Furthermore, iron can translationally increase protein levels of α‐synuclein through its promoter region, and iron markedly induces aggregation of α‐synuclein into intracellular inclusions. Accordingly, iron chelation has been shown to reduce the amount of insoluble α‐synuclein aggregates and rescue behavioural deficits in transgenic murine models. A number of iron chelators have shown favourable results in preclinical testing for treatment of PD including desferrioxamine, deferasirox, VK28 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7456. Deferiprone is of particular interest as it is already approved for use in the treatment of thalassaemia and has been identified as a potential therapeutic for Freidreich's ataxia, another neurodegenerative condition. Deferiprone works by translocating iron from cells with excessive levels of iron to those with deficient levels, keeping overall iron content relatively stable (Cabantchik et al., 2013). Two trials have demonstrated that deferiprone treatment reduced iron content in the SNpc in some PD patients while all patients showed a trend towards improved Unified Parkinson's Disease Rating Scale (UPDRS) motor scores (Devos et al., 2014; Martin‐Bastida et al., 2017). Based on these results, larger phase II trials will assess the ability of deferiprone to attenuate the progression of PD (NCT02655315) and examine the effects of an extended‐release tablet formulation of the drug on motor features (measured using UPDRS motor scores) over a 9‐month treatment period (NCT02728843).
Cu is another essential trace element of which elevated brain levels have been associated with increased ROS generation, DNA damage and mitochondrial dysfunction (Desai and Kaler, 2008). Rasia et al. (2005) demonstrated that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4164 ions are effective in accelerating α‐synuclein aggregation at physiologically relevant concentrations by binding to specific sites on the N‐ and C‐termini. Yet this effect is abolished in the acetylated form of α‐synuclein, which is more commonly found in the brain (Moriarty et al., 2014). Interestingly, copper concentrations in the SNpc of PD patients are not elevated but are 34 to 51% lower than age‐matched controls; this decrease is even greater when examined at the single cell level (Davies et al., 2016). Cell death seen with decreased copper levels in later disease stages may be mediated by decreased function of copper‐binding proteins, such as ceruloplasmin and SOD1, both of which have decreased activity in PD patients (Trist et al., 2017). These findings suggest that augmenting or redistributing copper in affected brain regions may hold therapeutic potential for the treatment of PD. Indeed, increasing copper delivery using the compound Cu2+ complex of diacetylbis(4‐methylthiosemicarbazone) (Cu2+(atsm)) was shown to have neuroprotective properties in four different animal models of PD (Hung et al., 2012). Treatment with this drug resulted in a decrease in α‐synuclein dimers in MPTP‐lesioned mice as well as in MPTP‐lesioned A53T α‐synuclein transgenic mice, possibly due to peroxynitrite inhibition, which decreases α‐synuclein nitration. A phase I dose escalation clinical trial of Cu2+(atsm) for early idiopathic PD has started recruiting subjects (NCT03204929). In addition, a metal protein attenuating compound called PBT2 demonstrated safety in a phase I trial for AD and significantly lowered levels of Aβ42 in CSF (Faux et al., 2010). As of yet, no clinical trials examining the safety or efficacy of PBT2 in PD have begun, yet preclinical results with related compounds have suggested it may hold promise.
Strategies at the preclinical stage
β‐Adrenoceptor stimulation
Another potential strategy to reduce the risk of developing PD is to regulate the amount of α‐synuclein transcribed from the SNCA gene. Transcription is regulated by GATA transcription factor occupancy of enhancers, the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5026 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924 (FGF‐2) pathway, methylation and microRNAs. An eloquent study by Mittal et al. (2017) identified three agonists of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 that could regulate SNCA gene expression in a high‐throughput drug screen: metaproterenol, clenbuterol and salbutamol. Treatment of rat primary cortical neurons with these three candidate drugs significantly reduced endogenous SNCA mRNA and α‐synuclein protein levels while treatment with propranolol (a β‐blocker) increased endogenous SNCA mRNA and α‐synuclein protein levels in SK‐N‐MC cells. The authors identified histone 3 lysine 27 (H3K27) acetylation signals at the promotor and enhancer regions of SNCA and showed clenbuterol treatment decreased H3K27 acetylation levels and relative SNCA mRNA abundance. Thus, the β2‐adrenoceptor regulates the transcription of α‐synuclein via a mechanism that controls H3K27 acetylation levels across the promotor and enhancers in the human SNCA locus. Finally, the authors examined the risk of developing PD amongst populations prescribed salbutamol and propranolol, the most commonly used β2‐adrenoceptor agonist and antagonist, respectively, in Norway and found that salbutamol (a drug commonly used to treat asthma) was associated with a decreased risk of PD (rate ratio of 0.66) and propranolol was associated with a markedly increased risk of PD (rate ratio of 2.20). β2‐adrenoceptor agonists are not currently approved for treatment of PD, but these results demonstrate a potential therapeutic role for these drugs if further trials prove beneficial in patients with PD.
Targeting O‐linked β‐N‐acetyl glucosamine
The O‐linked β‐N‐acetyl glucosamine (O‐GlcNAc) modification is a type of glycosylation that involves the addition or removal of an uncharged acetylated hexosamine sugar to a serine or threonine residue on a nucleocytoplasmic or mitochondrial target protein. The addition or removal of O‐GlcNAc is catalysed by the intracellular enzymes O‐GlcNAc transferase and O‐GlcNAcase, respectively (Bond and Hanover, 2015). Transcripts encoding these enzymes are most highly expressed in immune cells, brain and pancreas, and the post‐translational modification has been linked to increased risk for the development of numerous human diseases including lupus, AD, autism and X‐linked dystonia‐parkinsonism (Bond and Hanover, 2013). O‐GlcNAc cycling has become an intense area of focus for research into neurodegenerative diseases as a result of the discovery that many pathogenic protein aggregates appear to have altered levels of O‐GlcNAcylation (Yuzwa et al., 2014). Much of the research thus far has focused on the role of O‐GlcNAc in the aggregation of β‐amyloid and tau, but α‐synuclein has also been shown to have a number of O‐GlcNAcylation sites (Cole and Hart, 2001). Many of the O‐GlcNAc sites of α‐synuclein identified are located within the NAC region of the protein, with O‐GlcNAc at threonine 72 (T72) appearing to be particularly important. One study has shown that the addition of a single O‐GlcNAc residue at T72 completely blocks the formation of both fibril and oligomer aggregates but does not affect the membrane binding capabilities of α‐synuclein. In addition, researchers showed that this post‐translational modification inhibits the toxicity of α‐synuclein when it is added to neurons in culture (Marotta et al., 2015). A more recent study has also shown that increasing O‐GlcNAc levels in neurons resulted in an increase in the accumulation of monomeric α‐synuclein (Wani et al., 2017). Perhaps the most important observation in relation to a role for O‐GlcNAc in PD pathogenesis is the discovery that O‐GlcNAcylation at T72 and serine 87 inhibit the cleavage of α‐synuclein by calpain, which is associated with aggregation of the protein (Levine et al., 2017). There are no toxic effects reported in mice with long‐term treatment with thiamet G, a potent O‐GlcNAcase inhibitor (Hastings et al., 2017). Further preclinical studies are required before thiamet G or a similar compound ASN120290 can be considered for disease‐modifying therapy trials for PD.
Intestinal microbiota targeted therapies
Much evidence has begun to accumulate for the existence of a gut‐brain axis and the influence of the intestinal microbiota on neurodevelopment, modulation of behaviour and neurological disorders. Research has shown that PD patients have a significantly different microbiome to that of healthy controls, with associations between abundance of particular types of bacteria and severity of gait difficulty (Scheperjans et al., 2015). These findings open the possibility of analysing an individual's microbiome as a potential biomarker for early stage PD. Furthermore, a recent paper reports that gut microbiota is required for motor deficits, microglial activation and α‐synuclein pathology in mice overexpressing α‐synuclein (Sampson et al., 2016). The authors showed colonization of these mice with microbiota from human PD patients enhanced the motor dysfunction compared to microbiota transplants from healthy human donors. These remarkable findings indicate that the microbiome may represent a risk factor for the development of PD but also outlines a potential therapeutic avenue for microbiome transplants to impact the pathobiology of PD.
Conclusions
Several lines of evidence support α‐synuclein as a promising disease‐modifying therapeutic target for PD. The therapies presented above either directly or indirectly target α‐synuclein to halt or slow the disease process. Currently, the generally accepted hypothesis is that α‐synuclein is a natively unfolded, monomeric protein and its pathogenic misfolding and aggregation are driven by specific gene mutations, toxin exposure, post‐translational modifications and/or cellular dysfunction. Challenges exist in that it has not yet been fully elucidated which form(s) of the protein (dimer, trimer, oligomer, fibril) is the true pathogen in disease progression. Other caveats exist as it is not known if mitochondrial failure, oxidative stress or lysosomal dysfunction are triggers for α‐synuclein misfolding or occur after protein misfolding. Furthermore, the physiological functions of α‐synuclein within and outside the CNS remain to be fully elucidated, and thus, targeting α‐synuclein must be pursued with caution. Despite these caveats, many of the emerging therapies have shown much promise in animal models and have entered phase I/II clinical trials with encouraging initial results. In the coming years, it is likely the development of accurate biomarkers, such as the microbiome or a positron emission tomography tracer for α‐synuclein, will become ever more important considering how pathological changes begin many years before the presentation of clinical symptoms. In addition, a more personalized medicine‐based approach is expected as genome sequencing becomes more prevalent and affordable. There has been a significant re‐evaluation into how we may treat PD in recent years with many promising early results, yet more basic research into disease pathogenesis is vital to refine the newer class of emerging therapies.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgement
Figures were prepared using Library of Science & Medical Illustrations ‐ available at http://www.somersault1824.com/science-illustrations/.
O'Hara, D. M. , Kalia, S. K. , and Kalia, L. V. (2018) Emerging disease‐modifying strategies targeting α‐synuclein for the treatment of Parkinson's disease. British Journal of Pharmacology, 175: 3080–3089. https://doi.org/10.1111/bph.14345.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzforum 2017. Alpha‐Synuclein Antibodies Enter Phase 2, Sans Biomarker. Https://Www.Alzforum.Org/News/Conference-Coverage/Synuclein-Antibodies-Enter-Phase-2-Sans-Biomarker.
- Bhatt MA, Messer A, Kordower JH (2013). Can intrabodies serve as neuroprotective therapies for Parkinson's disease? Beginning thoughts. J Parkinsons Dis 3: 581–591. [DOI] [PubMed] [Google Scholar]
- Bond MR, Hanover JA (2013). O‐Glcnac cycling: a link between metabolism and chronic disease. Annu Rev Nutr 33: 205–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Hanover JA (2015). A little sugar goes a long way: the cell biology of O‐Glcnac. J Cell Biol 208: 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Dave KD, Kordower JH (2017). Therapeutic approaches to target alpha‐synuclein pathology. Exp Neurol 298: 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Südhof TC (2017). Cell Biology and Pathophysiology of Alpha‐Synuclein. Cold Spring Harb Perspect Med 8: a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DC, Joshi SN, Genst ED, Baghel AS, Dobson CM, Messer A (2016). Bifunctional anti‐non‐amyloid component α‐synuclein nanobodies are protective in situ. Plos One 11: E0165964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantchik ZI, Munnich A, Youdim MB, Devos D (2013). Regional siderosis: a new challenge for iron chelation therapy. Front Pharmacol 4: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Schapira AHV (2009). Non‐motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. The Lancet Neurology 8: 464–474. [DOI] [PubMed] [Google Scholar]
- Cole RN, Hart GW (2001). Cytosolic O‐Glycosylation is abundant in nerve terminals. J Neurochem 79: 1080–1089. [DOI] [PubMed] [Google Scholar]
- Davies KM, Mercer JF, Chen N, Double KL (2016). Copper dyshomoeostasis in Parkinson's disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci (Lond) 130: 565–574. [DOI] [PubMed] [Google Scholar]
- Desai V, Kaler SG (2008). Role of copper in human neurological disorders. Am J Clin Nutr 88: 855s–858s. [DOI] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008). Mitochondrial import and accumulation of α‐synuclein impair complex i in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283: 9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C et al (2014). Targeting Chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal 21: 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez‐Isla T, Leenhouts KM et al (2007). Calpain‐cleavage of alpha‐synuclein: connecting proteolytic processing to disease‐linked aggregation. Am J Pathol 170: 1725–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerqvist T, Lindstrom V, Nordstrom E, Lord A, Tucker SM, Su X et al (2013). Monoclonal antibodies selective for alpha‐synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and alpha‐synuclein transgenic mice with the disease‐causing A30P mutation. J Neurochem 126: 131–144. [DOI] [PubMed] [Google Scholar]
- Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A et al (2004). Levodopa and the progression of Parkinson's disease. N Engl J Med 351: 2498–2508. [DOI] [PubMed] [Google Scholar]
- Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J et al (2010). Pbt2 rapidly improves cognition in Alzheimer's disease: additional phase II analyses. J Alzheimers Dis 20: 509–516. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH (2005). Neural activity controls the synaptic accumulation of alpha‐synuclein. J Neurosci 25: 10913–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A et al (2014). Reducing C‐terminal‐truncated alpha‐synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease‐like models. J Neurosci 34: 9441–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz ME, Double K, Gerlach M, Youdim MB, Riederer P (2004). The relevance of iron in the pathogenesis of Parkinson's disease. Ann N Y Acad Sci 1012: 193–208. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Mccann H (2010). The progression of pathology in Parkinson's disease. Ann N Y Acad Sci 1184: 188–195. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide To Pharmacology in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings NB, Wang X, Song L, Butts BD, Grotz D, Hargreaves R et al (2017). Inhibition of O‐Glcnacase leads to elevation of O‐Glcnac tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Molecular Neurodegeneration 12: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron ML, Lonskaya I, Moussa CE (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha‐synuclein in Parkinson's disease models. Hum Mol Genet 22: 3315–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR et al (2012). The hypoxia imaging agent Cuii(Atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson's disease. J Exp Med 209: 837–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ (2008). Alpha‐synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol 115: 87–95. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Brotchie JM, Fox SH (2013). Novel nondopaminergic targets for motor features of Parkinson's disease: review of recent trials. Mov Disord 28: 131–144. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK (2015). Alpha‐synuclein and Lewy pathology in Parkinson's disease. Curr Opin Neurol 28: 375–381. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Mclean PJ, Lozano AM, Lang AE (2013). Alpha‐synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol 73: 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Lang AE (2015). Parkinson's disease. Lancet 386: 896–912. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Lang AE, Hazrati L‐NN, Fujioka S, Wszolek ZK, Dickson DW et al (2015). Clinical correlations with Lewy body pathology in LRRK2‐related Parkinson disease. JAMA Neurol 72: 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick BS, Magalhaes J, Beavan MS, Mcneill A, Gegg ME, Cleeter MWJ et al (2016). Endoplasmic reticulum and lysosomal Ca(2+) stores are remodelled in Gba1‐linked Parkinson disease patient fibroblasts. Cell Calcium 59: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike MA, Price DL, White BM, Rockstein E, Wrasidlo W, Tsigelny I et al (2014). The novel alpha‐synuclein stabilizer Npt200‐11 improves behavior, neuropathology, and biochemistry in the murine thy1‐asyn transgenic model of Parkinson's disease. 2014 Neuroscience Meeting Planner. Washington, DC. Soc Neurosci. [Google Scholar]
- Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S et al (2014). A bacteriophage capsid protein provides a general amyloid interaction motif (Gaim) that binds and remodels misfolded protein assemblies. J Mol Biol 426: 2500–2519. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Schroeter S, Carroll JC, Cullen V, Asp E, Proschitsky M et al (2016). Npt088 reduces both amyloid‐beta and tau pathologies in transgenic mice. Alzheimers Dement (N Y) 2: 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin J, Schmidt F, Boehm C, Prix C, Bötzel K, Ryazanov S et al (2014). The oligomer modulator Anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol 127: 779–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine PM, De Leon CA, Galesic A, Balana A, Marotta NP, Lewis YE et al (2017). O‐Glcnac modification inhibits the calpain‐mediated cleavage of alpha‐synuclein. Bioorg Med Chem 25: 4977–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysosomal‐Therapeutics‐Website . 2017. Lti‐291: a targeted approach [Online]. Available: Https://Lti-Staging.Squarespace.Com/Our-Science/#Lti-291 [Accessed].
- Mahul‐Mellier A‐L, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S et al (2014). C‐Abl phosphorylates α‐synuclein and regulates its degradation: implication for α‐synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum Mol Genet 23: 2858–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A et al (2014). Next‐generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol 127: 861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning‐Bog AB, Schule B, Langston JW (2009). Alpha‐synuclein‐glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30: 1127–1132. [DOI] [PubMed] [Google Scholar]
- Manoharan S, Guillemin GJ, Abiramasundari RS, Essa MM, Akbar M, Akbar MD (2016). The role of reactive oxygen species in the pathogenesis of Alzheimer's Disease, Parkinson's disease, and Huntington's disease: a mini review. Oxid Med Cell Longev 2016: 8590578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria Cuervo A, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004). Impaired Degradation of mutant alpha‐synuclein by chaperone‐mediated autophagy. Science. (New York, N.Y.). 305: 1292–1295. [DOI] [PubMed] [Google Scholar]
- Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT et al (2015). O‐Glcnac modification blocks the aggregation and toxicity of the protein α‐synuclein associated with Parkinson's disease. Nat Chem 7: 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C et al (2017). Brain iron chelation by deferiprone in a phase 2 randomised double‐blinded placebo controlled clinical trial in Parkinson's disease. Sci Rep 7: 1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV et al (2008). Dopamine‐modified alpha‐synuclein blocks chaperone‐mediated autophagy. J Clin Invest 118: 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, Mclean PJ, Caldwell GA et al (2011). Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska‐Richards A, Schapira AH (2016). The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 139 Suppl 1: 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ et al (2017). Beta2‐adrenoreceptor is a regulator of the alpha‐synuclein gene driving risk of Parkinson's disease. Science 357: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M et al (2009). Parkinson's disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol Med 1: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty GM, Minetti CA, Remeta DP, Baum J (2014). A revised picture of the Cu(Ii)‐alpha‐synuclein complex: the role of N‐terminal acetylation. Biochemistry 53: 2815–2817. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Obeso JA, Stocchi F (2000). Continuous dopamine‐receptor stimulation in advanced Parkinson's disease. Trends Neurosci 23: S109–S115. [DOI] [PubMed] [Google Scholar]
- O'regan G, Desouza RM, Balestrino R, Schapira AH (2017). Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis 7: 411–422. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez‐Carasa I et al (2013). Interplay of Lrrk2 with chaperone‐mediated autophagy. Nat Neurosci 16: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan F, Hebron M, Valadez EH, Torres‐Yaghi Y, Huang X, Mills RR et al (2016). Nilotinib effects in Parkinson's disease and dementia with Lewy bodies. J Parkinsons Dis 6: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Rockenstein EM, Mante M, Wrasidlo E, Masliah D, Bonhaus DW et al (2014). The novel alpha‐synuclein stabilizer Npt200‐11 reduces retinal deposits of asyn‐Egfp in a transgenic mouse model of Parkinson's disease/Lewy body disease. 2014 Neuroscience Meeting Planner. Washington, DC. Soc Neurosci. [Google Scholar]
- Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M et al (2005). Structural characterization of copper(Ii) binding to alpha‐synuclein: insights into the bioinorganic chemistry of Parkinson's disease. Proc Natl Acad Sci U S A 102: 4294–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosborough K, Patel N, Kalia LV (2017). Alpha‐synuclein and parkinsonism: updates and future perspectives. Curr Neurol Neurosci Rep 17: 31. [DOI] [PubMed] [Google Scholar]
- Saha S, Ash PE, Gowda V, Liu L, Shirihai O, Wolozin B (2015). Mutations in LRRK2 potentiate age‐related impairment of autophagic flux. Mol Neurodegener 10: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE et al (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167: 1469–1480 E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH (2016). Parkinson disease‐linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep 6: 31380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME (2013). Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W et al (2017). First‐in‐human assessment of Prx002, an anti‐alpha‐synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E et al (2015). Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord 30: 350–358. [DOI] [PubMed] [Google Scholar]
- Schneider SA, Alcalay RN (2017). Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 32: 1504–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte C, Gasser T (2011). Genetic basis of Parkinson's disease: inheritance, penetrance, and expression. The Application Of Clinical Genetics 4: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trist BG, Davies KM, Cottam V, Genoud S, Ortega R, Roudeau S et al (2017). Amyotrophic lateral sclerosis‐like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson's disease brain. Acta Neuropathol 134: 113–127. [DOI] [PubMed] [Google Scholar]
- Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F et al (2013). Anle138b: a novel oligomer modulator for disease‐modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol 125: 795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Ouyang X, Benavides GA, Redmann M, Cofield SS, Shacka JJ et al (2017). O‐Glcnac regulation of autophagy and α‐synuclein homeostasis; implications for Parkinson's disease. Mol Brain 10: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo‐Arozena A, Gordon DE, Peden AA et al (2010). Alpha‐synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 190: 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Jones BA, Zhao G, Woodward ML, Li X et al (2014). Pharmacological inhibition of O‐Glcnacase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/app mutant mice. Mol Neurodegener 9: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]