

Abstract
Breast cancer treatment using a single drug is associated with a high failure rate due, in part, to the heterogeneity of drug response within individuals, nonspecific target action, drug toxicity, and/or development of resistance. Use of dual‐drug therapies, including drug conjugates, may help overcome some of these roadblocks by more selective targeting of the cancer cell and by acting at multiple drug targets rather than one. Drug‐conjugate approaches include linking drugs to antibodies (antibody‐drug conjugates), radionuclides (radioimmunoconjugates), nanoparticles (nanoparticle‐drug conjugates), or to other drugs (drug‐drug conjugates). Although all of these conjugates might be designed as effective treatments against breast cancer, the focus of this review will be on drug‐drug conjugates because of the increase in versatility of these types of drugs with respect to mode of action at the level of the cancer cell either by creating a novel pharmacophore or by increasing the potency and/or efficacy of the drugs’ effects at their respective molecular targets. The development, synthesis, and pharmacological characteristics of drug‐drug conjugates will be discussed in the context of breast cancer with the hope of enhancing drug efficacy and reducing toxicities to improve patient quality of life.
Keywords: breast cancer, drug conjugates, hybrid ligands, nanoparticles
1. INTRODUCTION
Every year, the FDA approves a considerable number of new drugs, with 30%‐50% being first in a therapeutic class (Figure 1). The average cost for a new drug—from the synthesis to marketing—is approximately $2.5 billion, including drugs that meet with failure.22 During the clinical phases of drug development, failure often arises due to nonspecific target actions and poor bioavailability.55, 60 Many anticancer drugs target rapidly dividing cells, and drug toxicity may therefore occur in those tissues with high rates of proliferation (eg, bone marrow, oral cavity, skin, nails). More severe signs of toxicity include bone marrow depression and gastrointestinal, nervous system, hepatic, urinary tract, cardiac, or pulmonary toxicity. Metabolic abnormalities may also arise due to the lysis of dead cancer cells and the release of their intracellular contents.91 In addition to these safety challenges, there is no guarantee that a new preclinically validated drug will be efficacious when tested in the patient population.
Figure 1.
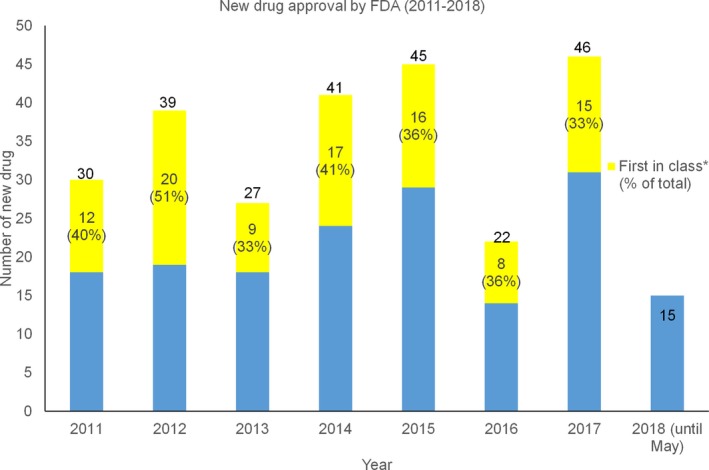
Number of new drugs approved by FDA from 2011 to 2018 (data collected from https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm)
Breast cancer is a complex disease and, unlike many other diseases, can readily develop drug resistance; this could be due to a set of gene mutations resulting in dysregulation of the balance between prosurvival and proapoptotic signaling cascades.80, 105 For example, patients with BRCA1 and/or BRCA2 gene mutations exhibit an increased risk of developing breast cancer.36 Mutation(s) of a specific signaling protein may also lead to drug resistance. For example, estrogen receptor‐positive breast cancer cells (MCF‐7) are known to display drug resistance by modulation of the antiapoptotic protein, Bcl2, in the presence of estrogenic stimulation.108 Resistance may also arise through activation of drug transporters that prevent the accumulation of the anticancer drug within the cell.46 For example, anthracycline drugs are readily effluxed from cancer cells overexpressing P‐glycoprotein or multidrug resistance protein.27 In addition, cancer phenotypes in different human populations vary, and a response to a single drug may not be equally efficacious across all individuals.84
Drug conjugates represent a growing class of anticancer agents designed to (1) increase target selectivity and reduce the bystander effect (eg, drug‐antibody conjugates; Figure 2A; Table 1), (2) enhance cytotoxicity and tumor targeting (eg, radionuclide‐drug conjugates, nanoparticle‐drug conjugates; Figure 2B; Tables 2 and 3) or (3) enhance the potency and/or efficacy of anticancer therapy (eg, drug‐drug conjugates; Figure 2C; Table 4). To overcome low efficacy, drug resistance, and/or toxicity associated with single drug use or monotherapy, dual‐drug, and even triple‐drug therapies are being developed or are already in clinical use.1 For example, paclitaxel and trastuzumab combination drugs, or capecitabine and docetaxel combination drugs have been shown to reduce mortality and improve safety compared to their administration as single drugs.78 However, most of these therapies deliver drug combinations as separate entities.
Figure 2.
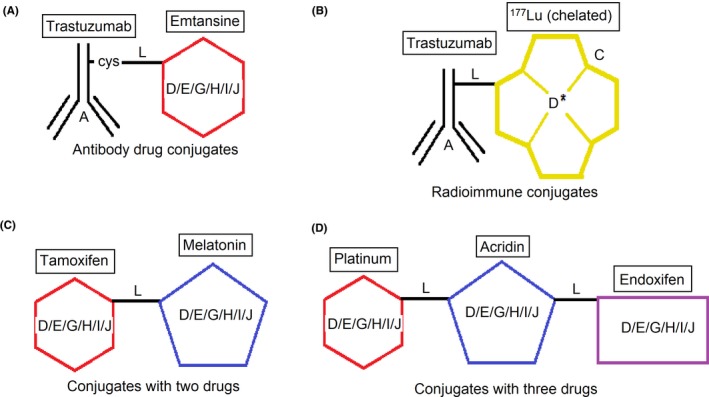
Possibilities of drug conjugates. A = antibody targets‐specific receptor (mentioned in table 1), B = radioactive isotope (mentioned in Table 2), C = chelating agent (DOTA, EDTA) D = cytotoxic drug (mentioned in Table 1), E = binds with a receptor (tamoxifen, endoxifen), F = regulates a signal pathway (anti‐NF‐КB, DNA intercalator), G = regulates an enzyme (kinase inhibitors, HDAC inhibitors), I = endogenous compound (melatonin), J = nanoparticle (gold). L = linker for conjugation (valine‐citrulline, hydrazine)
Table 1.
Antibody‐drug conjugates for breast cancer
| Drug name | Status | Antibody, target | Cytotoxic drug, target | Target patients | Potency/Efficacy |
|---|---|---|---|---|---|
| Kadcyla® | Approved | Trastuzumab, HER2 | Emtansine, antimicrotubule | HER2‐positive metastatic breast cancer | Improved overall survival compared to lapatinib plus capecitabine.112 |
| Glembatumumab vedotin | Phase 2b | Glembatumumab, Glycoprotein NMB | Monomethyl auristatin E (MMAE), tubulin inhibitor | gpNMB overexpressing metastatic triple‐negative breast cancer | Well‐tolerated in pretreated patients.122 |
| BMS‐182248‐1 (discontinued) | Phase 2 | BR96, Lewis‐Y antigen | Doxorubicin, topoisomerase II inhibitor | Metastatic breast cancer | Limited clinical activity.110 |
| IMMU‐132 | Phase I | Sacituzumab govitecan, antitrop‐2 | SN‐38, topoisomerase I inhibitor | Metastatic triple‐negative breast cancer | Well‐tolerated and robust response.6 |
| BAY 1187982 | Phase I | FGFR2, FGFR2 receptor | Auristatin, antimicrotubule | Cancer cells overexpressing FGFR | Effective compared to unconjugated antibodies in vitro102 |
| SYD985 | Phase I | Trastuzumab, HER2 receptor | Seco‐duocarmycin, DNA‐alkylating agent | BT‐474 cells and BT‐474 xenografted mice | Effective in BT‐474 xenografted in vivo model.33 |
| Anti‐PTK7‐Aur0101 | In vivo | Anti‐PTK7, antiprotein tyrosine kinase 7 antibody | Aur0101, microtubule inhibitor | Triple‐negative breast cancer | Induced sustained tumor regression.17 |
| REGN2878‐DM1 | In vivo | REGN2878, prolactin receptor | DM1, maytansine derivative | Prolactin receptor‐positive breast cancer | Significant antigen‐specific antitumor activity.57 |
| DS‐8201a | In vivo | Trastuzumab, HER2 receptor | Exatecan derivatives, topoisomerase I inhibitor | Both HER2 + and HER2‐ breast cancer cells | Showed bystander toxicity.83 |
| BT‐2111 | In vivo | Trastuzumab, HER2 receptor | Melanotransferrin, cross blood‐brain barrier | Breast cancer metastasis to the brain in NuNu mice | 68% reduction of metastasis in the brain compared to trastuzumab alone.82 |
| FGF1V‐MMAE | In vitro | FGF1 receptor ligand variant (FGF1v), FGFR | MMAE, tubulin inhibitor | Cancer cells overexpressing FGFR | Conjugate showed higher potency than MMAE.106 |
Antibody‐cytotoxic drug conjugates (ADCs) are the most widely investigated drug conjugates to treat breast cancer.89 In general, a cytotoxic drug is attached to a monoclonal antibody that is specific for the target receptor (Figure 2A). The antibody binds to the receptor of the cancer cell where the cytotoxic drug is intended to exert its actions. Therefore, the cancer cells should ideally densely express the receptor for antibody binding. Although over 55 ADCs are currently in clinical trials,13 only 3 ADCs have been approved by the FDA.77 However, gemtuzumab ozogamicin (marketed as Mylotarg® by Wyeth‐Ayerst) was withdrawn in 2010 due to increased patient mortality and demonstrating no clinical benefit over conventional therapy, which leaves only 2 ADCs available for clinical use. One of them is for HER2‐positive metastatic breast cancer—the trastuzumab‐emtansine conjugate marketed as Kadcyla® by Genentech and Roche. Another one is brentuximab‐vedotin (Adcetris® marketed by Seattle Genetics) for Hodgkin lymphoma or anaplastic large cell lymphoma. In many ways, ADCs may exert potential benefits over conventional treatment. For example, highly cytotoxic drugs might become safer for normal cells when they are bound to cancer cell‐specific antibodies.115
Table 2.
Potential radionuclide conjugates for breast cancer
| Isotope | Status | Antibody | Model | Potency/Efficacy |
|---|---|---|---|---|
| 177Lu (chelated) | Pilot clinical study | Trastuzumab | HER2‐positive breast cancer patients | No drug uptake for HER2‐negative patients, whereas the radioimmunoconjugate retains its integrity up to 7 days in vivo.8 |
| 213Bi (chelated) | In vivo | Antibody to human aspartyl β‐hydroxylase | 4T1 tumor mice | Significant effect on the tumor.92 |
| 213Bi (chelated) | In vivo | Antibody to chondroitin sulfate proteoglycan 4 | TNBC xenograft and in vitro | Significantly inhibited tumor and cell growth.53 |
| 225Ac (chelated) | Anti‐rat HER2′/In vivo | Anti‐rat HER2 | Metastatic breast cancer mouse model | Complete eradication of lung metastasis and more efficacious than213Bi.103 |
| 111In/90Y (peptide linked) | Phase I | M170 | Advanced breast cancer patients | Patients had grade 3 or 4 myelosuppression93 |
| 213Bi (chelated) | In vivo | Plasminogen activator inhibitor‐2 | MDA‐MB‐231 cell inoculation in mice | Inhibit growth at 2 days after inoculation.2 |
| 90Y (chelated) | In vivo | Cilengitide | HBT 3477 cell xenografted mice | Increased efficacy compared to radiotherapy/cilengitide alone9 |
| 131I | In vivo | Humanized anti‐Lewis Y | MCF‐7 xenografted mouse | Significant tumor growth inhibition compared to control radiolabeled antibody15 |
| 131I | In vivo | 131I‐IgG2a (rat) | MDA‐MB‐361 xenograft | Tumor growth inhibition for more than 24 days 101 |
| 111In/90Y | Phase I | IgG1 (BrE3) | Metastatic breast cancer patients | Risk of developing HAMA response 19 |
| 131I | Pilot clinical study | 131I‐IgG1 (ChL6) | Metastatic breast cancer patients | Partial response achieved with the development of thrombocytopenia and granulocytopenia20, 21 |
Radiation emitted from a radionuclide can be used to kill cells. Radioactive compounds can also attack noncancerous cells; therefore, targeted delivery of the radionuclide with the help of a monoclonal antibody is desirable. The radionuclide can be linked to a monoclonal antibody by a linker, or the antibody could be labeled with radioisotope by a chelation method (Figure 2B).
Table 3.
Drug‐delivery system conjugates
| Drug | Status | Delivery system | Model | Efficacy/Potency |
|---|---|---|---|---|
| Tamoxifen | In vivo | Naringen (P‐gp efflux inhibitor) | MCF‐7 cells and female Wistar rats | The conjugate showed 22‐fold increased cytotoxicity compared to tamoxifen or the combination.95 |
| Tamoxifen | In vivo | Chitosan‐stearic acid‐based polymeric micelles | MCF‐7 cells | Enhanced cytotoxicity and modified pharmacokinetic profiles.109 |
| Tamoxifen | In vitro | Trans‐2‐phenylcyclopropylamine | Lysine‐specific demethylase 1‐triggered controlled release | No toxic effect on normal cells.85 |
| Tamoxifen | In vitro | Glucosamine‐porphyrin | MCF‐7 cells | Works through necrosis/apoptosis pathways.4 |
| Tamoxifen | In vivo | Bile (cholic) acid | 4T1 in vivo model | More potent than tamoxifen.104 |
| Tamoxifen | In vitro | Thiol‐polyethylene glycol gold nanoparticle | MCF‐7 cells | The conjugate showed 2.7 folds higher potency than tamoxifen with less cytotoxicity to cancer cells.28 |
| Tamoxifen | In vitro | Pyropheophorbide | MCF‐7 cells | Showed light‐specific cytotoxicity35 |
| Gefitinib | In vitro | Polyarginine peptoids | MDA‐MB‐468, NME, and LM1 cell lines | NArg‐based conjugate blocked STAT3 phosphorylation without affecting ERK1/27 |
| Mitoxantrone | In vivo | Folic acid‐tocopheryl polyethylene glycol | MCF‐7 xenografted mice | MTO‐FMCT showed improved cellular uptake with higher MCF‐7 cytotoxicity. MTO‐FMCT showed higher potency to reduce MCF‐7 cell viability compared to MTO alone43 |
| Polymeric doxorubicin | In vivo | Aminopropyltriethoxysilane‐modified porous silicon particles | MDA‐MB‐231 and 4T1 mouse models of metastatic breast cancer | Nanoparticles showed enhanced efficacy with functional cures in 40%‐50% of treated mice120 |
| Doxorubicin | Retrospective Clinical Study | Pegylated liposomal nanoparticles | Stage I‐III triple‐negative breast cancer patients | Adjuvant chemotherapy was as effective as conventional chemotherapy with reduced toxicity66 |
| Paclitaxel (Abraxane®) | FDA approved | Albumin‐bound nanoparticles | Clinical trials on metastatic breast cancer | Abraxane® showed superior efficacy and reduced toxicity compared with paclitaxel39 |
The uptake of nanoparticles by a tissue depends on the hydrophobicity of that nanoparticle. For example, nanoparticles deposited in certain organs such as the liver, spleen, and reticuloendothelial system correlate positively with the increasing hydrophobicity of the polymer.37 Although several nanoparticle‐based drug delivery systems have been developed, only albumin‐bound paclitaxel nanoparticle (Abraxane®) was approved by FDA for metastatic breast cancer and nonsmall cell lung carcinoma.69 Nanotechnology can also be effectively used in breast cancer treatment. Nanoparticle conjugates may show increased potency by penetrating the cells by endocytosis instead of the diffusion method used for a single drug.28 This could be a mechanism to avoid efflux by drug transporters such as P‐glycoprotein.14 In a phase III clinical trial, paclitaxel nanoparticles bonded with albumin showed superior efficacy and safety compared to paclitaxel dissolved in castor oil.39 Furthermore, doxorubicin linked with poly(L‐glutamic acid) by a pH‐sensitive cleavable linker showed enhanced efficacy in MDA‐MB‐231 and 4T1 metastatic breast cancer mouse model.120
Table 4.
Hybrid drug conjugates targeting breast cancer
| Drug conjugate/Drug class | Status/model | Potency/efficacy |
|---|---|---|
Ribociclib‐vorinostat/cyclic‐dependent kinase CDK‐4–HDAC inhibitor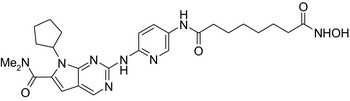
|
In vitro MDA‐MB‐231 cellsIn vivo 4T1 cells of rat breast cancer | Conjugate showed higher cytotoxicity on MDA‐MB‐231 cells (IC50 = 1.86 μmol/L) than vorinostat (IC50 = 2.59 μmol/L) and ribociclib (IC50 > 10 μmol/L) and stronger tumor growth inhibition in 4T1 cells (79%) than vorinostat (75.6%) and ribociclib (38.9%)65 |
Fibroblast growth factor 1 inhibitor‐nexturastat/FGFR 1‐HDAC‐6 inhibitor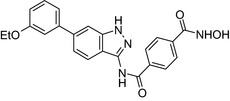
|
In vitro MCF‐7 cells | Conjugate showed cytotoxic activity on MCF‐7 cells (IC50 = 9 μmol/L)67 |
Raloxifen‐dimethyl fumarate/SERM–anti‐NF‐κB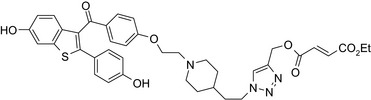
|
In vitro MCF‐7 cells | Higher inhibition of NF‐κB than fumarate alone54 |
Olaparib‐vorinostat/PARP inhibitor–HDAC inhibitor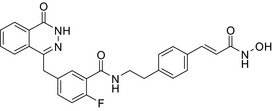
|
In vitro MDA‐MB‐231 and HCC1937 cells | Conjugate showed more potent activity than olaparib and vorinostat with 4.1‐fold less cytotoxicity to MCF‐10A123 |
Ruxolitinib‐vorinostat/Janus kinase‐HDAC inhibitor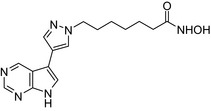
|
In vitro MCF‐7 cells | Conjugate was equipotent on MCF‐7 cells (IC50 = 0.84 μmol/L) to vorinosta (IC50 = 0.84 μmol/L) and more potent than ruxolitinib (IC50 = 10 μmol/L)121 |
Combretastatin‐cyclofenil/Antimitotic‐SERM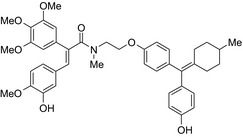
|
In vitro MCF‐7 cells | Cyclofenil‐combretastatin conjugate (IC50 = 187 nmol/L) showed potent antiproliferative activity to MCF‐7 cells56 |
Combretastatin endoxifen/Antimitotic‐SERM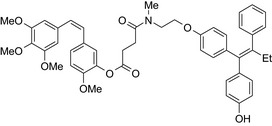
|
In vitro MCF‐7 cells | Endoxifen‐combretastatin conjugate (IC50 = 5.7 nmol/L) showed potent antiproliferative activity to MCF‐7 cells56 |
Endoxifen‐combretastatin/Antimitotic‐SERM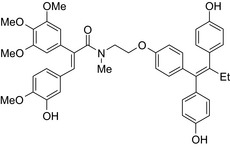
|
In vitro MCF‐7 and MDA‐MB‐231 cells | The conjugate showed potent antiproliferative activity (IC50 = 5 nmol/L) to MCF‐7 cells56 |
Vandetanib‐vorinostat/VEGFR‐HDAC inhibitor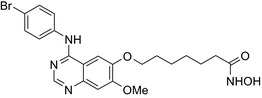
|
In vitro MCF‐7 cells | Conjugate was more potent on MCF‐7 cells (IC50 = 0.85 μmol/L) than vandetanib (IC50 = 18.5 μmol/L) and vorinostat (IC50 = 4.5 μmol/L)87 |
TBB‐triazole hydroxamic acid/Casein kinase 2–HDAC inhibitor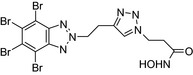
|
In vitro MCF‐7 cells | The conjugate showed cytotoxic activity (IC50 = 4.26 μmol/L) on MCF‐7 cells90 |
Oxabicycloheptene sulfonate‐vorinostat/ERα antagonist–HDAC inhibitor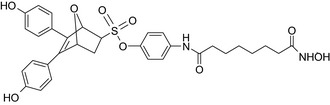
|
In vitro MCF‐7 cells | The conjugate showed higher potency than tamoxifen.107 |
ICI‐164,384‐N‐butylvorinostat/ER antagonist‐HDAC inhibitor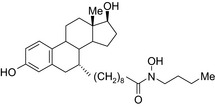
|
In vitro MCF‐7 and MDA‐MB‐237 cells | Conjugate was more potent on MCF‐7 cells (IC50 = 0.34 μmol/L) than ICI‐164,384 (IC50 = 0.93 μmol/L) and vorinostat (IC50 = 0.32 μmol/L)76 |
Semaxanib‐vorinostat / VEGFR‐HDAC inhibitor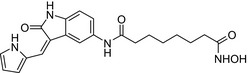
|
In vitro MDA‐MB‐237 cells | Conjugate was equipotent on MDA‐MB‐237 cells (IC50 = 117 nmol/L) to vorinostat (IC50 = 118 nmol/L)86 |
Melatonin‐tamoxifen/SERM–melatonin receptor agonist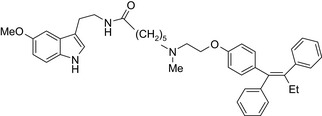
|
In vitro BC cells In vivo ovariectomized FVB/n mice | Hybrid conjugate did not increase uterus weight compared to tamoxifen, and showed efficacy against different BC cells including tamoxifen‐resistant MCF‐7 cells, to be published,116 US patent no. ‐ 08785501) |
Colchicin‐pironetin/Β‐tubulin inhibitor–α‐tubulin inhibitor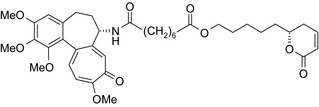
|
In vitro MCF‐7 cells | All conjugates showed lower cytotoxicity values than the parental molecules, whereas the binding of the conjugates to tubulin depends on the length of the linkers113 |
c‐Src kinase inhibitor vorinostat/c‐Src‐HDAC inhibitor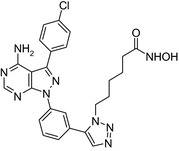
|
In vitro SK‐BR‐3 cells | Conjugate was more potent on SK‐BR‐3 (IC50 = 0.2 μmol/L) than vorinostat (IC50 = 1.2 μmol/L)59 |
Platinum‐acridin‐endoxifen/DNA intercalation & platination–SERM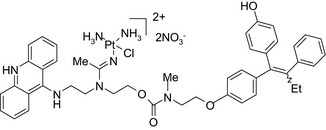
|
In vitro MCF‐7 and MDA‐MB‐231 cells | One conjugate showed higher potency on MCF‐7 cells compared to cisplatin or tamoxifen23 |
Endoxifen‐endoxifen/Bivalent SERM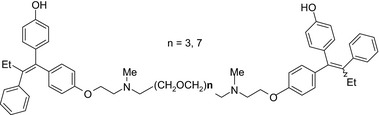
|
In vitro MCF‐7 and MDA‐MB‐231 cells | Bivalent ligands showed higher potency than 4OH tamoxifen98 |
Tamoxifen‐vorinostat/SERM‐HDAC inhibitor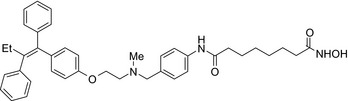
|
In vitroMCF‐7 and MDA‐MB‐231 cells | The conjugate showed higher cytotoxicity on MCF‐7 (IC50 = 3.8 μmol/L) and on MDA‐MB‐231 cells (IC50 = 8.1 μmol/L) than tamoxifen and vorinostat40 |
Doxorubicin‐RU 39411/Topoisomerase inhibitor–ER antagonist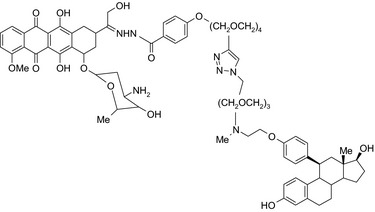
|
In vitro MCF‐7 and MDA‐MB‐231 cells | The conjugate was about 70‐fold more potent than doxorubicin to inhibit MCF‐7 cell proliferation18 |
Erlotinib‐vorinostat CUDC‐101/EGFR‐HER2‐HDAC inhibitor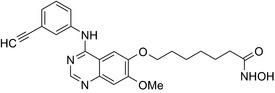
|
In vitro MCF‐7 and MDA‐MB‐231 cellsIn vivo xenograft mice | Conjugate was more potent on MCF‐7 cells (IC50 = 0.55 μmol/L) than erlotinib (IC50 = 20 μmol/L) and vorinostat (IC50 = 2.8 μmol/L) and the combination of the parent drugs (IC50 = 2.7 μmol/L)64 |
Lapitanib‐panobinostat/EGFR‐HER2‐HDAC inhibitor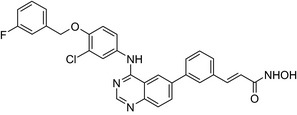
|
In vitroSKBR3 cells | Conjugate is more potent on SMBR3 cells than lapitanib and vorinostat70 |
Estradiol‐cisplatin/ER agonist–antineoplastic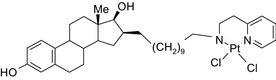
|
In vivoMCF‐7 and MDA‐MB‐468 mouse xenografts | The conjugates decreased tumor volume compared to cisplatin in ER‐positive mice111 |
Retinoic acid‐butyric acid/RAR & RXR agonist–HDAC inhibitor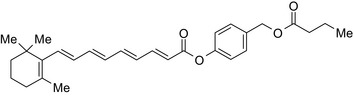
|
In vitroMCF‐7 andMDA‐MB‐231 cells | The conjugate showed 1085‐fold higher potency than parent retinoic acid and 100000‐fold higher potency than butyric acid38 |
Tamoxifen‐ferrocene/SERM–organometallic complex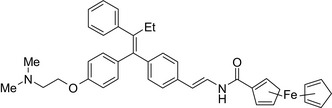
|
In vitroMCF‐7 cells | Increased apoptotic events compared to tamoxifen/ferrocene117 |
Doxorubicin‐4OH tamoxifen/Topoisomerase inhibitor–SERM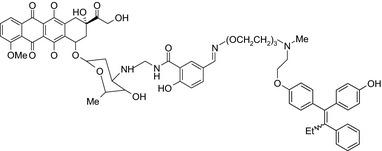
|
In vitroMCF‐7, MCF‐7 resistant,MDA‐MB‐231, MDA‐MB‐435 cells | The conjugates showed 4‐ to 140‐fold higher potency than doxorubicin10 |
Aniline mustard‐estradiol/DNA‐alkylating agent–ER agonist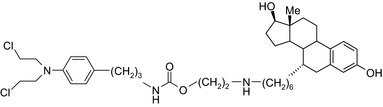
|
In vitroMCF‐7 and MDA‐MB‐231 cells | The conjugate showed higherpotency compared to chlorambucil79 |
Aniline mustard‐phenylindole/DNA‐alkylating agent–SERM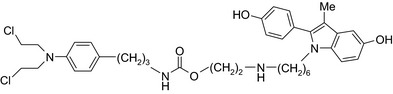
|
In vitroMCF‐7 and MDA‐MB‐231 cells | Two conjugates showed higher toxicity to MCF‐7 than to MDA‐MB‐231 cells94 |
SERM, Selective estrogen receptor modulator, GPCR, G protein‐coupled receptor.
Drug conjugates offer an alternative approach in situations where a single drug or monotherapy fails. A drug‐drug conjugate comprises 2 or more drugs connected by a chemical linker (Figure 2C and D). These novel types of drugs may be useful for targeted drug delivery and modulation of pharmacokinetic parameters, rendering the drugs more effective and less toxic than in isolation. Furthermore, conjugation of 2 drugs (eg, melatonin‐tamoxifen) with a noncleavable linker may produce a novel molecule with unique pharmacological characteristics,116 (US Patent No. 08785501). Although antibody‐drug conjugates comprise a large segment of conjugated drugs (see Table 1), this class of drugs will not be discussed here, and the reader is referred to recent excellent papers.51, 89 The focus of this review will be on the emerging field of drug‐drug conjugates reported in the literature, their design and their pharmacological characteristics with respect to drug targets (receptors, intracellular signaling proteins) and efficacy as antibreast cancer agents.
2. DRUG CONJUGATES/HYBRID LIGANDS
Ideally, a drug‐drug conjugate should be stable in the systemic circulation before arriving at the target site, which is not only valid for drug conjugates with a noncleavable linker, but also for those formed by a cleavable spacer such that drug release will occur only at the intended site of action.26 A noncleavable linker may also be designed to maintain its chemical integrity, allowing it to act as a single moiety/pharmacophore at its receptor or receptors.72 Unlike combination drug therapy, in which 2 individual unlinked drugs are administered simultaneously, conjugated drug‐drug therapies may offer distinct advantages therapeutically, aesthetically, or both.63 With respect to esthetics, it may be more convenient for a patient to take a single drug conjugate instead of 2 drugs, thus improving compliance.63 Concerning therapeutic advantages, conjugated drug therapies may improve the therapeutic index by lowering the minimum effective dose and by increasing the maximum tolerated dose compared to traditional combination therapies.73 Drug‐drug conjugates may also prevent drug resistance, as tumors are less likely to become resistant to drugs with distinct targets (ie, receptors, and/or intracellular signaling proteins).1, 16 A drug conjugate may display superior activity (potency, and/or efficacy) compared to a single drug; this may be a result of improved bioavailability119 or through additive or synergistic action of the 2 drugs working in concert.97 Moreover, 2 conjugated pharmacophores targeted for 2 different receptors may show higher affinity by coactivation in the same cells.48 For example, paclitaxel and doxorubicin drug conjugates showed superior efficacy and safety compared to the combination by acting synergistically because it delivers both drugs to the same target site at a synergistic ratio.71 The increase in potency of a drug‐drug conjugate may also produce less toxicity as the recommended dose could be lowered.5, 24, 61, 62 For example, chimeric c‐Src kinase and histone deacetylase inhibitors59 and the multiacting EGFR/HER2 and histone deacetylase inhibitor CUDC‐10112 were reported to be not only more active than the combination of the 2 constituting parent compounds, but also less toxic with a higher improved therapeutic index. These findings indicate that molecular hybridization does not necessarily lead to the additive toxicity often observed for the combination therapy.
3. DESIGNING DRUG CONJUGATES
3.1. Drug targets
Stability and efficacy of drug conjugates need to be carefully considered during their design. Concerning stability, conjugates containing a cleavable linker should be designed to release drugs only at the target site.24 With respect to improved efficacy, conjugates can be designed to target breast tumors with unique phenotypes, such as luminal A (ER+/HER2+; PR+/HER2−), luminal B (ER+/HER2+; PR+/HER2+), HER2 (ER‐/PR‐/HER2+), or to target specific intracellular signaling proteins in triple‐negative breast cancer, (eg, mTOR, PI3K, PARP, TROP‐2, VEGFR, EGFR, FGFR, CYP17A1, MEK, AKT, anti‐CD27, anti‐CD52, hedgehog).124 In such cases, combining 2 drugs with different pharmacological targets ensures their equivalent delivery to the cancer tissue, which may not be the case if the 2 combination drugs differ in pharmacokinetic properties of tissue distribution and elimination.
3.2. Linker design
The following questions need to be addressed when designing chemical linkers: Should it be stable in the circulation? Should it release the 2 active drugs at the target site? Should it be stable at the target site? Should it influence drug activity? Stable linkers can be simple (CH2)n‐chains (eg, tamoxifen‐melatonin drug conjugate or more hydrophilic amides (eg, valine‐citrulline linker), and ether‐containing spacers (eg, maleimidomethyl cyclohexane‐1‐carboxylate linker); the design depends on the drugs themselves and sites of attachment; and on the desired action on the tumor.24, 74, 116 Furthermore, if the efficacy of the drug conjugate is dependent upon linker stability, then noncleavable linkers may be superior because they can only be degraded once they are internalized by lysosomes within cells.74 However, it is also possible that the conjugate will exert its action by maintaining the integrity of the linker until it reaches its target site(s) where it will then be broken down chemically or enzymatically depending on the structure of the linker; this type of linker is referred to as a cleavable linker.74 One drawback to the type of drug conjugate is that the local release of drugs at their target site(s) may also impact neighboring “nontarget” sites, which may cause toxicity depending upon the cellular milieu.47
Drugs conjugated with a chemically cleavable linker are released under specific biochemical environments. A hydrazone linker is usually stable in standard blood pH conditions (pH 7.4) and hydrolyzed under acidic conditions, such as in lysosomes or in the more acidic tumor microenvironment.30 For example, adriamycin conjugated to PEO‐b‐PAsp copolymers with a hydrazone linker exhibited release of the active drug at pH levels below 5.44 However, one drawback to this type of linker may be its instability in acidic buffers and excipients as observed for hydrazone linkers.52 In a recent study, a drug‐antibody conjugate using this type of linker [ie, gemtuzumab ozogamicin (Mylotarg®)] was withdrawn due to its narrow therapeutic index, poor plasma stability, and lack of benefit over conventional chemotherapy.30, 74, 88 Even though this was with a drug‐antibody conjugate, these findings need to be taken into consideration when designing a drug‐drug conjugate.
Chemical linkers containing disulfide bonds are also chemically cleavable. For example, disulfide bonds may be reduced in vivo by glutathione. As glutathione levels are relatively low in plasma (2‐20 μmol/L) compared to the cytoplasm (0.5‐10 mmol/L)75, 115, 118 disulfide bond‐containing linkers remain relatively stable in blood. This property improves the pharmacokinetic profiles of the conjugates compared to their unconjugated antibody component. For example, the disulfide linker containing a doxorubicin‐gold drug conjugate exhibited greater intracellular uptake than free doxorubicin in multidrug‐resistant cancer cells.41 One point to consider when creating drug conjugates with a disulfide linker is its stability during the sterilization process. Heat generated during the sterilization process can break down disulfide bonds.68 This obstacle could be overcome through filter sterilization.31
Linkers may be designed to be degraded by enzymes within the breast cancer cell to control drug delivery at the target site; this may avoid premature release into the systemic circulation due to factors such as low pH.30 For example, lysosomal enzymes located in breast cancer cells can be leveraged to design conjugates that will only release the drug intracellularly. A commonly used peptide linker, valine‐citrulline, is cleaved by cathepsin B and exhibits superior stability compared to the hydrazone linker.25 For example, a drug‐antibody conjugate (ie, a Lewis Y‐specific antibody conjugated to monomethyl auristatin E) connected by a valine‐citrulline linker showed superior stability in buffer and plasma, suggesting that the conjugate will release the drug only in the presence of cathepsin.25 In another study, it was shown that the valine‐citrulline linker exhibits greater stability compared to the Phe‐Lys peptide linker in the presence of cathepsin B alone, as doxorubicin release from the conjugate was 30‐fold faster for the Phe‐Lys linker than the valine‐citrulline linker.29 Glucuronide‐based linkers can also be used as nonpeptide linkers that release drugs within the interior of the cell through the action of lysosomal β‐glucuronidases.11, 50, 74 For example, a camptothecin analog conjugated to a CD30 or Lewis Y antibody with a glucuronide linker demonstrated greater potency to inhibit breast cancer cell viability compared to these same drugs connected using a dipeptide linker.11 Even though these examples were based on drug‐antibody conjugates, these approaches may also be useful in designing drug‐drug conjugates.
Attention should also be drawn to the attachment points of the linker to the parent drugs. In particular, the chemical nature and attachment positions of the functional groups connecting the 2 pharmacophores should be chosen in accordance with the structure‐activity relationships with the given target to maintain the pharmacological actions of the 2 constituents in the dually acting molecular hybrid.81
4. PHARMACOLOGY OF DRUG‐DRUG CONJUGATES
Drug conjugates (molecular hybrids, chimeric drugs) are compounds that incorporate 2 drugs into a single molecule (Figure 2C). The 2 pharmacophores could be either directly linked or connected by a long cleavable/noncleavable linker/spacer described previously. Drug conjugates are expected to exert simultaneous action at the biological targets specific to each drug, which may enhance their potency, and/or efficacy compared to their administration as 2 individual agents. The efficacy can be improved due to either an increased potency of the conjugate or by improving pharmacokinetic parameters. For example, ribociclib‐vorinostat drug conjugates demonstrated higher potency (IC50 = 1.86 μmol/L) than vorinostat (IC50 = 2.59 μmol/L) and ribociclib (IC50 > 10 μmol/L) in the triple‐negative breast cancer cells, MDA‐MB‐231.65 Although this class of conjugates is not yet available on the market, some show promise as breast cancer drugs both in vivo and in vitro, as described in Table 4.
Antiestrogen/cytotoxic drug conjugates are commonly researched against ER+ type breast cancer, in which estrogen plays an indispensable role in tumor progression. Tamoxifen is the most commonly used antiestrogen drug for the synthesis of the anticancer drug hybrids (Tables 3 and 4). For example, a tamoxifen‐melatonin drug conjugate with similar affinity to estrogen receptors and MT1 melatonin receptors as tamoxifen or melatonin, respectively, demonstrated unique pharmacological characteristics at higher (>10 μmol/L) concentrations where the total number of estrogen receptor and MT1 melatonin receptor ligand‐binding sites increased compared to the decreases observed in the presence of tamoxifen or melatonin alone. Because this was not observed for tamoxifen alone or melatonin alone, this suggests that the melatonin‐tamoxifen conjugate may be creating a unique pharmacophore between estrogen receptors and MT1 melatonin receptors. This is underscored in an in vivo study in female mice where tamoxifen alone and tamoxifen plus melatonin (coadministered but unlinked) increased uterine weight while the melatonin‐tamoxifen conjugate was without effect and similar to control mice,116 (US Patent No. 08785501). The melatonin‐tamoxifen conjugates may be creating a unique pharmacophore in mouse uterus preventing it from excessive stimulation by tamoxifen.
Histone deacetylase (HDAC) inhibitors were reported to show promising activity in triple‐negative breast cancer.34 Indeed, linking of tamoxifen to the histone deacetylase inhibitor vorinostat resulted in a conjugate with higher cytotoxicity in MCF‐7 and MDA‐MB‐231 cells than the parent drugs.40 The RU39411‐like antiestrogen compound conjugated with doxorubicin exhibited lower IC50 values (ie, greater potency) compared to doxorubicin only or doxorubicin plus the linker when tested in MCF‐7 cells, but not MDA‐MB‐231 cells.18 The triple drug conjugate platinum‐acridine‐endoxifen exhibited higher cytotoxicity compared to a tamoxifen/cisplatin combination therapy, which warrants further investigation.23 Another triple conjugate, 4‐hydroxytamoxifen‐doxorubicin‐formaldehyde showed increased drug uptake in estrogen receptor‐positive breast cancer cell lines compared to untargeted doxorubicin−formaldehyde conjugates.10 These studies further support the versatility and therapeutic efficacy of drug conjugates, which can be expanded from a dual‐drug approach to a triple‐drug conjugate approach.
Bivalent ligands (2 identical pharmacophores connected by a spacer) can also serve as drug conjugates. For example, bivalent ligands of hydroxytamoxifen—an estrogen receptor antagonist in breast tissue—exhibited more potent (IC50 ≤ 0.11 μmol/L) growth inhibitory effects in MCF‐7 cells compared to monomeric hydroxytamoxifen (IC50 = 0.15 μmol/L); in the same study, bivalent ligation with an estrogen receptor agonist (diethylstilbestrol) did not show any effect.98 The activity of the bivalent conjugate depends on the length of the spacer with a maximal estrogen receptor binding achieved for the (CH2OCH2)n‐linked endoxifen‐endoxifen bivalent ligands with an n of 3 or 798; Table 4).
An estradiol‐cisplatin hybrid conjugate showed antiproliferative activity in the low micromolar range, which was more potent than cisplatin alone in estrogen‐dependent and estrogen‐independent breast cancer cell lines MCF‐7 and MDA‐MB‐231, respectively.111 Additionally, this conjugate displayed high affinity toward the estrogen receptor α and was superior to cisplatin in inducing tumor regression in the MCF‐7 human breast cancer tumors in a mouse model. Recently, conjugates of selective estrogen receptor modulators (ie, endoxifen or cyclofenil) linked to combretastatin, an antimitotic agent that binds to the beta subunit of tubulin, demonstrated potent (nmol/L) antiproliferative activity against MCF‐7 cells (Table 4).56, 58
An estrogen receptor antagonist, oxabicycloheptene sulfonate, linked to the histone deacetylase (HDAC) inhibitor vorinostat (suberanilohydroxamic acid) showed higher antitumor potency in MCF‐7 cells compared to the oxabicycloheptene sulfonate alone, indicating that parallel inhibition of HDAC and the estrogen receptor is beneficial for anticancer action.107
DNA‐alkylating agents linked to estrogen receptor ligands is yet another approach to developing novel hybrid drugs targeting specific phenotypes of breast cancer.94 For example, a hybrid drug conjugate consisting of an aniline nitrogen mustard with an estrogen receptor ligand belonging to the class of 2‐phenylindoles was reported to display selective toxicity toward estrogen receptor‐positive MCF‐7 cells compared with the estrogen receptor‐negative line, MDA‐MB‐231.94 Drug conjugates created by replacing the phenylindole group with estradiol and combining this to bis‐chloroethyl aniline mustard demonstrated selective toxicity toward estrogen receptor‐positive cancer cells.79, 99
The HDAC inhibitors, butyric acid and MS‐275 (entinostat), were each conjugated to trans‐retinoic acid via an ester linkage.3 Trans‐retinoic acid induces differentiation and arrests proliferation in a wide range of cancer cells.96 The 2 retinoic acid conjugates displayed potent antiproliferative activity in a hormone‐insensitive breast cancer cell line, MDA‐MB‐231 and some drug‐resistant breast cancer cell lines, MCF‐7TAMR, MCF‐7 HOX‐B7, LTLC and LTLT‐Ca.38
Another drug conjugate linked a β‐tubulin inhibitor, colchicine, to an analog of the α‐tubulin‐binding agent, pironetin, through an ester‐amide spacer. Although colchicine and the pironetin analog administered individually displayed similar toxicity toward the breast adenocarcinoma (MCF‐7) and the nontumoral HEK‐293 cell lines, the colchicine‐pironetin drug conjugate was 100‐fold more toxic to the MCF‐7 cell lines than either parent drug113 perhaps due to enhanced antimitotic actions in the more rapidly dividing cancer cell.
A conjugate consisting of vorinostat (an HDAC type I and II inhibitor) with olaparib [poly (ADP‐ribose) polymerase (PARP) inhibitor] demonstrated higher antiproliferative activity than olaparib or vorinostat alone in a variety of cancer cells, including breast cancer (MDA‐MB‐231, HCC1937) and Burkitt's lymphoma cells. This vorinostat‐olaparib drug conjugate also demonstrated lower cytotoxicity in normal mammary (MCF‐10A) cells compared to vorinostat, indicating that simultaneous targeting of PARP and HDAC might be beneficial for breast cancer therapy with a better safety profile.123
Many molecular hybrids have been readily designed for cytotoxic activity against a variety of cancer cells, such as the dual protein tyrosine kinase—HDAC inhibitors. For a more detailed report on chimeric HDAC inhibitors, the reader is referred to a very recent comprehensive review.45 The clinically most advanced agent is a multitarget EGFR/HER2‐HDAC inhibitor CUDC‐101,12, 114) that reached phase‐1 clinical trial in patients with advanced solid tumors.100 CUDC‐101 was more potent (IC50 = 0.55 μmol/L) than the combination of the parent drugs erlotinib + vorinostat (2.7 μmol/L) in MCF‐7 cells and promoted tumor inhibition in various cancer xenograph models including breast cancer.64 Other dual protein tyrosine kinase—HDAC inhibitors (Table 4) that were reported to display higher or equal cytotoxicity compared to the parent agents in various breast cancer cell lines include hybrids of ribociclib (CDK‐4 inhibitor) and vorinostat, FGFR‐1‐inhibitor and nexturastat,67 ruxolitinib (Janus kinase inhibitor)‐vorinostat,121 vandetanib (VEGFR inhibitor)–vorinostat,87 casein kinase 2 inhibitor and triazole hydroxamic acid (HDAC inhibitor),90 semaxanib (VEGFR inhibitor) and vorinostat,86 c‐Src kinase inhibitor and vorinostat,59 and lapitanib (EGFR inhibitor) and panabinostat.70
The concept of drug conjugates can also be used in the development of targeted drug delivery systems. Targeted drug delivery systems are generally stable in the systemic circulation, and they might be designed to release the cytotoxic drugs only after internalization into cancer cells.49 Some drug conjugates were specifically designed to deliver the cytotoxic drug at the target site using a pharmacologically inactive drug. Examples of tamoxifen drug conjugates targeted for specific drug delivery are described in Table 3.
5. CONCLUSION
Hundreds of receptors and signaling pathways are dysregulated in breast cancer, a highly heterogeneous disease that exploits multiple targets, such as receptors from different classes and myriad signal transduction cascades.42 Because of these factors, breast cancer drugs need to be selective and personalized based on the patient's genotype or phenotype. Drug‐drug conjugates offer a partial solution to these issues because conceptually they target multiple proteins and signaling pathways at the same time within a cancer cell. For 2 drugs with very different pharmacokinetic properties of distribution and elimination, it may not be practical to achieve therapeutic concentrations in a tumor at the same time, unless the drugs are chemically linked. Although only 32 unique generic medications are currently approved to prevent and treat breast cancer (data collected from https://www.cancer.gov/about-cancer/treatment/drugs/breast), there are 496 drug conjugate possibilities if 2 of the 32 drugs are linked. As summarized in Tables 1, 2, 3, 4, several in vitro and preclinical examples suggest that conjugates may harbor a therapeutic advantage over a single drug or standard drug combination treatment regimens.
As almost 60% of breast cancer patients are hormone receptor‐positive, antiestrogen drug conjugates hold promise as effective therapeutics.32 Although some tamoxifen drug conjugates display higher potency compared to tamoxifen alone, there remains the risk of tamoxifen drug resistance. Therefore, testing these conjugates in tamoxifen‐resistant breast cancer lines is critical. A recently reported series of tamoxifen‐melatonin conjugates (US Patent No. 08785501) hold promise against ER, HER2, triple‐negative, and tamoxifen‐resistant breast cancer cells (to be published). Clinical studies need to be performed to determine if drug conjugates demonstrate greater convenience for patients who are prescribed multiple medications resulting in better patient management by the physician. With safety and efficacy always being major concerns in anticancer treatments, clinical studies need to determine if drug‐drug conjugates offer an advantage over cancer therapies utilizing 2 unlinked drugs by improving pharmacokinetic and pharmacodynamic drug profiles; all of these may mitigate the toxic bystander effect and spare healthy neighboring cells. These collective features need to be further studied, both preclinically and clinically to determine if they result in superior safety profiles and patient outcomes.
All drug conjugate classes hold promise, whether they are linked to antibodies, radionuclides, or nanoparticles. Indeed, 2 clinical trials are currently assessing the efficacy of CDX‐011, a glembatumumab vedotin antibody‐drug conjugate against triple‐negative breast cancer (NCT01997333), and SYD985, a trastuzumab‐duocarmazine antibody‐drug conjugate against HER2 breast cancer (NCT03262935). Thus, drug conjugates are emerging as a novel and effective treatment approach for breast cancer.
AUTHORSHIP CONTRIBUTIONS
Conception of review (MH; PWE); Writing of manuscript (MH; DPZ; PWE); Reviewed/Edited/Revised (PWE; RKL; RES).
DISCLOSURE
US Patent 08785501.
Hasan M, Leak RK, Stratford RE, Zlotos DP, Witt‐Enderby PA. Drug conjugates—an emerging approach to treat breast cancer. Pharmacol Res Perspect. 2018;e00417 https://doi.org/10.1002/prp2.417
REFERENCES
- 1. Al‐Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post‐genomic era. Nat Biotechnol. 2012;30:679‐692. https://doi.org/10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 2. Allen BJ, Tian Z, Rizvi SMA, Li Y, Ranson M. Preclinical studies of targeted [alpha] therapy for breast cancer using 213Bi‐labelled‐plasminogen activator inhibitor type 2. Br J Cancer. 2003;88:944‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181.https://doi.org/10.1038/35106036. https://www.nature.com/articles/35106036#supplementary-information [DOI] [PubMed] [Google Scholar]
- 4. Amanlou M, Heidari Z, Siadat SD, et al. Nanosized tamoxifen‐porphyrin‐glucose [TPG] conjugate: Novel selective anti‐breast‐cancer agent, synthesis and in vitro evaluations. Med Chem. 2013;9:526‐538. [DOI] [PubMed] [Google Scholar]
- 5. Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377‐380. [DOI] [PubMed] [Google Scholar]
- 6. Bardia A, Mayer IA, Diamond JR. Efficacy and safety of anti‐trop‐2 antibody drug conjugate sacituzumab govitecan (IMMU‐132) in heavily pretreated patients with metastatic triple‐negative breast cancer. J Clin Oncol. 2017;JCO2016708297. https://doi.org/10.1200/jco.2016.70.8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartolowits MD, Brown W, Ali R, et al. Selective inhibition of STAT3 phosphorylation using a nuclear‐targeted kinase inhibitor. ACS Chem Biol. 2017;12:2371‐2378. https://doi.org/10.1021/acschembio.7b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhusari P, Vatsa R, Singh G, et al. Development of Lu‐177‐trastuzumab for radioimmunotherapy of HER2 expressing breast cancer and its feasibility assessment in breast cancer patients. Int J Cancer. 2017;140:938‐947. https://doi.org/10.1002/ijc.30500. [DOI] [PubMed] [Google Scholar]
- 9. Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of αvβ3 integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Can Res. 2002;62:4263. [PubMed] [Google Scholar]
- 10. Burke PJ, Kalet BT, Koch TH. Antiestrogen binding site and estrogen receptor mediate uptake and distribution of 4‐hydroxytamoxifen‐targeteddoxorubicin‐formaldehyde conjugate in breast cancer cells. J Med Chem. 2004;47:6509‐6518. https://doi.org/10.1021/jm049496b. [DOI] [PubMed] [Google Scholar]
- 11. Burke PJ, Senter PD, Meyer DW, et al. Design, synthesis, and biological evaluation of antibody‐drug conjugates comprised of potent camptothecin analogues. Bioconjug Chem. 2009;20:1242‐1250. https://doi.org/10.1021/bc9001097. [DOI] [PubMed] [Google Scholar]
- 12. Cai X, Zhai H, Wang J, et al. Discovery of 7‐(4‐(3‐ethynylphenylamino)‐7‐methoxyquinazolin‐6‐yloxy)‐Nhydroxyheptanamide (CUDC‐101) as a potent multi‐acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53:2000‐2009. [DOI] [PubMed] [Google Scholar]
- 13. Chari RV. Expanding the reach of antibody‐drug conjugates. ACS Med Chem Lett. 2016;7:974‐976. https://doi.org/10.1021/acsmedchemlett.6b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310. [DOI] [PubMed] [Google Scholar]
- 15. Clarke K, Lee F‐T, Brechbiel MW, Smyth FE, Old LJ, Scott AM. In vivo biodistribution of a humanized anti‐Lewis Y monoclonal antibody (hu3S193) in MCF‐7 xenografted BALB/c nude mice. Cancer Res. 2000;60:4804‐4811. [PubMed] [Google Scholar]
- 16. Crystal AS, Shaw AT, Sequist LV, et al. Patient‐derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480‐1486. https://doi.org/10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Damelin M, Bankovich A, Bernstein J, et al. A PTK7‐targeted antibody‐drug conjugate reduces tumor‐initiating cells and induces sustained tumor regressions. Sci Transl Med. 2017;9: https://doi.org/10.1126/scitranslmed.aag2611. [DOI] [PubMed] [Google Scholar]
- 18. Dao KL, Sawant RR, Hendricks JA, Ronga V, Torchilin VP, Hanson RN. Design, synthesis, and initial biological evaluation of a steroidal anti‐estrogen‐doxorubicin bioconjugate for targeting estrogen receptor‐positive breast cancer cells. Bioconjug Chem. 2012;23:785‐795. https://doi.org/10.1021/bc200645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeNardo SJ, Kramer EL, O'Donnell RT, et al. Radioimmunotherapy for breast cancer using indium‐111/yttrium‐90 BrE‐3: results of a phase I clinical trial. J Nucl Med. 1997a;38:1180‐1185. [PubMed] [Google Scholar]
- 20. Denardo SJ, O'Grady LF, Richman CM, et al. Radioimmunotherapy for advanced breast cancer using I‐131‐ChL6 antibody. Anticancer Res. 1997b;17:1745‐1751. [PubMed] [Google Scholar]
- 21. DeNardo SJ, Warhoe KA, O'Grady LF, et al. Radioimmunotherapy for breast cancer: treatment of a patient with I‐131 L6 chimeric monoclonal antibody. Int J Biol Markers. 1991;6:221‐230. [DOI] [PubMed] [Google Scholar]
- 22. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. 2014; Retrieved from http://csdd.tufts.edu/files/uploads/Tufts_CSDD_briefing_on_RD_cost_study_-_Nov_18,_2014..pdf [DOI] [PubMed]
- 23. Ding S, Qiao X, Kucera GL, Bierbach U. (2013). Design of a platinum–acridine–endoxifen conjugate targeted at hormone‐dependent breast cancer. Chem Commun (Camb), 49, 2415‐2417. https://doi.org/10.1039/c3cc38957j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody‐drug conjugates. mAbs 2016; 8:659‐671. https://doi.org/10.1080/19420862.2016.1156829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doronina SO, Toki BE, Torgov MY, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotech. 2003;21:778‐784. [DOI] [PubMed] [Google Scholar]
- 26. Dorywalska M, Strop P, Melton‐Witt JA, et al. Site‐dependent degradation of a non‐cleavable auristatin‐based linker‐payload in rodent plasma and its effect on adc efficacy. PLoS ONE. 2015;10:e0132282 https://doi.org/10.1371/journal.pone.0132282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF‐7 breast cancer cells. Proc Natl Acad Sci. 1998;95:15665‐15670. https://doi.org/10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dreaden EC, Mwakwari SC, Sodji QH, Oyelere AK, El‐Sayed MA. Tamoxifen‐poly(ethylene glycol)‐thiol gold nanoparticle conjugates: enhanced potency and selective delivery for breast cancer treatment. Bioconjug Chem. 2009;20:2247‐2253. https://doi.org/10.1021/bc9002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dubowchik GM, Firestone RA, Padilla L, et al. Cathepsin B‐labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen‐specific in vitro anticancer activity. Bioconjug Chem. 2002;13:855‐869. [DOI] [PubMed] [Google Scholar]
- 30. Ducry L, Stump B. Antibody−drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5‐13. https://doi.org/10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 31. Duncan R. Development of HPMA copolymer‐anticancer conjugates: Clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131‐1148. https://doi.org/10.1016/j.addr.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 32. Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9:R6 https://doi.org/10.1186/bcr1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elgersma RC, Coumans RG, Huijbregts T, et al. Design, synthesis, and evaluation of linker‐duocarmycin payloads: toward selection of HER2‐targeting antibody‐drug conjugate SYD985. Mol Pharm. 2015;12:1813‐1835. https://doi.org/10.1021/mp500781a. [DOI] [PubMed] [Google Scholar]
- 34. Fedele P, Orlando L, Cinieri S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin Investig Drugs. 2017;26:1199‐1206. https://doi.org/10.1080/13543784.2017.1386172. [DOI] [PubMed] [Google Scholar]
- 35. Fernandez Gacio A, Fernandez‐Marcos C, Swamy N, Dunn D, Ray R. Photodynamic cell‐kill analysis of breast tumor cells with a tamoxifen‐pyropheophorbide conjugate. J Cell Biochem. 2006;99:665‐670. https://doi.org/10.1002/jcb.20932. [DOI] [PubMed] [Google Scholar]
- 36. Gabai‐Kapara E, Lahad A, Kaufman B, et al. Population‐based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci USA. 2014;111:14205‐14210. https://doi.org/10.1073/pnas.1415979111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaur U, Sahoo SK, De TK, Ghosh PC, Maitra A, Ghosh PK. Biodistribution of fluoresceinated dextran using novel nanoparticles evading reticuloendothelial system. Int J Pharm. 2000;202:1‐10. https://doi.org/10.1016/S0378-5173(99)00447-0. [DOI] [PubMed] [Google Scholar]
- 38. Gediya LK, Khandelwal A, Patel J, et al. Design, synthesis, and evaluation of novel mutual prodrugs (hybrid drugs) of all‐trans‐retinoic acid and histone deacetylase inhibitors with enhanced anticancer activities in breast and prostate cancer cells in vitro. J Med Chem. 2008;51:3895‐3904. https://doi.org/10.1021/jm8001839. [DOI] [PubMed] [Google Scholar]
- 39. Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin‐bound paclitaxel compared with polyethylated castor oil‐based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794‐7803. https://doi.org/10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 40. Gryder BE, Rood MK, Johnson KA, et al. Histone deacetylase inhibitors equipped with estrogen receptor modulation activity. J Med Chem. 2013;56:5782‐5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gu YJ, Cheng J, Man CW, Wong WT, Cheng SH. Gold‐doxorubicin nanoconjugates for overcoming multidrug resistance. Nanomedicine. 2012;8:204‐211. https://doi.org/10.1016/j.nano.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 42. Guille A, Chaffanet M, Birnbaum D. Signaling pathway switch in breast cancer. Cancer Cell Int. 2013;13:66 https://doi.org/10.1186/1475-2867-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guissi NEI, Li H, Xu Y, et al. Mitoxantrone‐ and folate‐TPGS2k conjugate hybrid micellar aggregates to circumvent toxicity and enhance efficiency for breast cancer therapy. Mol Pharm. 2017;14:1082‐1094. https://doi.org/10.1021/acs.molpharmaceut.6b01009. [DOI] [PubMed] [Google Scholar]
- 44. Haag R. Supramolecular drug‐delivery systems based on polymeric core‐shell architectures. Angew Chem Int Ed. 2004;43:278‐282. https://doi.org/10.1002/anie.200301694. [DOI] [PubMed] [Google Scholar]
- 45. Hesham HM, Lasheen DS, Abouzid KAM. Chimeric HDAC inhibitors: Comprehensive review on the HDAC‐based strategies developed to combat cancer. Med Res Rev. 2018;1‐52. https://doi.org/10.1002/med.21505 [DOI] [PubMed] [Google Scholar]
- 46. Hembruff SL, Laberge ML, Villeneuve DJ, et al. Role of drug transporters and drug accumulation in the temporal acquisition of drug resistance. BMC Cancer. 2008;8:318 https://doi.org/10.1186/1471-2407-8-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hinrichs MJM, Dixit R. Antibody drug conjugates: nonclinical safety considerations. AAPS J. 2015;17:1055‐1064. https://doi.org/10.1208/s12248-015-9790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jacobson KA. GPCR ligand‐dendrimer (GLiDe) conjugates: future smart drugs? Trends Pharmacol Sci. 2010;31:575‐579. https://doi.org/10.1016/j.tips.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor‐targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043‐5054.https://doi.org/10.1016/j.bmc.2005.04.084 [DOI] [PubMed] [Google Scholar]
- 50. Jeffrey SC, Andreyka JB, Bernhardt SX, et al. Development and properties of beta‐glucuronide linkers for monoclonal antibody‐drug conjugates. Bioconjug Chem. 2006;17:831‐840. https://doi.org/10.1021/bc0600214. [DOI] [PubMed] [Google Scholar]
- 51. Junutula JR, Flagella KM, Graham RA, et al. Engineered thio‐trastuzumab‐dm1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2–positive breast cancer. Clin Cancer Res. 2010;16:4769. [DOI] [PubMed] [Google Scholar]
- 52. Kale AA, Torchilin VP. Design, synthesis and characterization of pH‐sensitive PEG‐PE conjugates for stimuli‐sensitive pharmaceutical nanocarriers: the effect of substitutes at the hydrazone linkage on the pH‐stability of PEG‐PE conjugates. Bioconjug Chem. 2007;18:363‐370. https://doi.org/10.1021/bc060228x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kasten B, Fan J, Ferrone S, Zinn K, Buchsbaum D. Targeted radioimmunotherapy of triple negative breast cancer with CSPG4‐specific 212Pb‐labeled monoclonal antibody. J Nucl Med. 2016;57(Suppl 2):114. [Google Scholar]
- 54. Kastrati I, Siklos MI, Brovkovych SD, Thatcher GR, Frasor J. A novel strategy to co‐target estrogen receptor and nuclear factor κB pathways with hybrid drugs for breast cancer therapy. Horm Cancer. 2017;8:135‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaur R, Sidhu P, Singh S. What failed BIA 10–2474 phase I clinical trial? global speculations and recommendations for future phase I trials. J Pharmacol Pharmacother. 2016;7:120‐126. https://doi.org/10.4103/0976-500X.189661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Keely ON, Carr M, Yassin B, et al. Design, synthesis and biochemical evaluation of novel selective estrogen receptor ligand conjugates incorporating an endoxifen‐combretastatin hybrid scaffold. Biomedicines. 2016;4: 15. https://doi.org/10.3390/biomedicines4030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kelly MP, Hickey C, Makonnen S, et al. Preclinical activity of the novel anti‐prolactin receptor (PRLR) antibody‐drug conjugate REGN2878‐DM1 in PRLR positive breast cancers. Mol Cancer Ther. 2017a;. https://doi.org/10.1158/1535-7163.MCT-16-0839. [DOI] [PubMed] [Google Scholar]
- 58. Kelly MP, Keely ON, Bright AS, et al. Novel selective estrogen receptor ligand conjugates incorporating endoxifen‐combretastatin and cyclofenil‐combretastatin hybrid scaffolds: synthesis and biochemical evaluation. Molecules. 2017b;22: 1440 https://doi.org/10.3390/molecules22091440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ko KS, Steffey ME, Brandvold KR, Soellner MB. Development of a chimeric c‐Src kinase and HDAC inhibitor. ACS Med Chem Lett. 2013;4:779‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discovery. 2004;3:711 https://doi.org/10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 61. Lee CC, Gillies ER, Fox ME, et al. A single dose of doxorubicin‐functionalized bow‐tie dendrimer cures mice bearing C‐26 colon carcinomas. Proc Natl Acad Sci USA. 2006;103:16649‐16654. https://doi.org/10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lewis Phiiiips GD, Li G, Dugger DL, et al. Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Can Res. 2008;68:9280. [DOI] [PubMed] [Google Scholar]
- 63. Li C, Wallace S. Polymer‐drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60:886‐898. https://doi.org/10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu H. CUDC‐101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70:3647‐3656. https://doi.org/10.1158/0008-5472. [DOI] [PubMed] [Google Scholar]
- 65. Li Y, Luo X, Guo Q, et al. Discovery of N 1‐(4‐((7‐cyclopentyl‐6‐(dimethylcarbamoyl)‐7 H ‐pyrrolo[2,3‐ d]pyrimidin‐2‐ yl)amino)phenyl)‐ N 8‐hydroxyoctanediamide as a novel inhibitor targeting cyclin‐dependent kinase 4/9 (CDK4/9) and histone deacetlyase1 (HDAC1) against malignant Cancer. J Med Chem. 2018;61:3166‐3192. https://doi.org/10.1021/acs.jmedchem.8b00209. [DOI] [PubMed] [Google Scholar]
- 66. Lien MY, Liu LC, Wang HC, et al. Safety and efficacy of pegylated liposomal doxorubicin‐based adjuvant chemotherapy in patients with stage I‐III triple‐negative breast cancer. Anticancer Res. 2014;34:7319‐7326. [PubMed] [Google Scholar]
- 67. Liu J, Qian C, Zhu Y, et al. Design, synthesis and evaluate of novel dual FGFR1 and HDAC inhibitors bearing an indazole scaffold. Bioorg Med Chem. 2018;26:747‐757. [DOI] [PubMed] [Google Scholar]
- 68. Lowe EK, Anema SG, Bienvenue A, Boland MJ, Creamer LK, Jiménez‐Flores R. Heat‐induced redistribution of disulfide bonds in milk proteins. 2. Disulfide bonding patterns between bovine beta‐lactoglobulin and kappa‐casein. J Agric Food Chem. 2004;52:7669‐7680. [DOI] [PubMed] [Google Scholar]
- 69. Ma P, Mumper RJ. Paclitaxel nano‐delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;4:1000164 https://doi.org/10.4172/2157-7439.1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mahboobi S, Sellmer A, Winkler M, et al. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J Med Chem. 2010;53:8546‐8555. https://doi.org/10.1021/jm100665z. [DOI] [PubMed] [Google Scholar]
- 71. Markovsky E, Baabur‐Cohen H, Satchi‐Fainaro R. Anticancer polymeric nanomedicine bearing synergistic drug combination is superior to a mixture of individually‐conjugated drugs. J Controlled Release. 2014;187:145‐157.https://doi.org/10.1016/j.jconrel.2014.05.025 [DOI] [PubMed] [Google Scholar]
- 72. Marshall DJ, Harried SS, Murphy JL, et al. Extracellular antibody drug conjugates exploiting the proximity of two proteins. Mol Ther. 2016;24:1760‐1770. https://doi.org/10.1038/mt.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martin C, Kizlik‐Masson C, Pèlegrin A, Watier H, Viaud‐Massuard M‐C, Joubert N. Antibody‐drug conjugates: Design and development for therapy and imaging in and beyond cancer, LabEx MAbImprove industrial workshop, July 27–28, 2017, tours France. mAbs. 2018;10:210‐221. https://doi.org/10.1080/19420862.2017.1412130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J. 2015;17:339‐351. https://doi.org/10.1208/s12248-014-9710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711‐760. https://doi.org/10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 76. Mendoza‐Sanchez R, Cotnoir‐White D, Kulpa J, et al. Design, synthesis and evaluation of antiestrogen and histone deacetylase inhibitormolecular hybrids. Bioorg Med Chem. 2015;23:7597‐7606. [DOI] [PubMed] [Google Scholar]
- 77. Miksinski SP. Regulatory Considerations for Antibody Drug Conjugates. 2013; Retrieved from https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM406887.pdf
- 78. Miles D, von Minckwitz G, Seidman AD. Combination versus sequential single‐agent therapy in metastatic breast cancer. Oncologist. 2002;7(Suppl 6):13‐19. [PubMed] [Google Scholar]
- 79. Mitra K, Marquis JC, Hillier SM, et al. A rationally designed genotoxin that selectively destroys estrogen receptor‐positive breast cancer cells. J Am Chem Soc. 2002;124:1862‐1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78‐S103. https://doi.org/10.1016/j.semcancer.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nepali K, Sharma S, Sharma M, Bedi PM, Dhar KL. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur J Med Chem. 2014;77:422‐487. https://doi.org/10.1016/j.ejmech.2014.03.018 [DOI] [PubMed] [Google Scholar]
- 82. Nounou MI, Adkins CE, Rubinchik E, et al. Anti‐cancer antibody trastuzumab‐melanotransferrin conjugate (BT2111) for the treatment of metastatic HER2 + breast cancer tumors in the brain: an in vivo study. Pharm Res. 2016;33:2930‐2942. https://doi.org/10.1007/s11095-016-2015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS‐8201a, a novel anti‐human epidermal growth factor receptor 2 antibody‐drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107:1039‐1046. https://doi.org/10.1111/cas.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Olopade OI, Grushko TA, Nanda R, Huo D. Advances in breast cancer: pathways to personalized medicine. Clin Cancer Res. 2008;14:7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ota Y, Itoh Y, Kaise A, et al. Targeting cancer with PCPA‐drug conjugates: LSD1 inhibition‐triggered release of 4‐hydroxytamoxifen. Angew Chem Int Ed Engl. 2016;55:16115‐16118. https://doi.org/10.1002/anie.201608711. [DOI] [PubMed] [Google Scholar]
- 86. Patel H, Chuckowree I, Coxhead P, et al. Synthesis of hybrid anticancer agents based on kinase and histone deacetylase inhibitors. Med Chem Commun. 2014;5:1829‐1833. [Google Scholar]
- 87. Peng FW, Xuan J, Wu TT, et al. Design, synthesis and biological evaluation of N‐phenylquinazolin‐4‐amine hybrids as dual inhibitors of VEGFR‐2 and HDAC. Eur J Med Chem. 2016;109:1‐12. https://doi.org/10.1016/j.ejmech.2015.12.033. [DOI] [PubMed] [Google Scholar]
- 88. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854‐4860. https://doi.org/10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016;68:3‐19. https://doi.org/10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- 90. Purwin M, Hernandez‐Toribio J, Coderch C, et al. Design and synthesis of novel dual‐target agents for HDAC1 and CK2 inhibition. RSC Adv. 2016;6:66595‐66608. [Google Scholar]
- 91. Remesh A. Toxicities of anticancer drugs and its management. Int J Basic Clin Pharmacol. 2017;1:2‐12. [Google Scholar]
- 92. Revskaya E, Jiang Z, Morgenstern A, et al. A radiolabeled fully human antibody to human aspartyl (Asparaginyl) beta‐hydroxylase is a promising agent for imaging and therapy of metastatic breast cancer. Cancer Biother Radiopharm. 2017;32:57‐65. https://doi.org/10.1089/cbr.2016.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Richman CM, DeNardo SJ, Donnell RT, et al. High‐dose radioimmunotherapy combined with fixed, low‐dose paclitaxel in metastatic prostate and breast cancer by using a MUC‐1 monoclonal antibody, m170, linked to indium‐111/Yttrium‐90 via a cathepsin cleavable linker with cyclosporine to prevent human anti‐mouse antibody. Clin Cancer Res. 2005;11:5920. [DOI] [PubMed] [Google Scholar]
- 94. Rink SM, Yarema KJ, Solomon MS, et al. Synthesis and biological activity of DNA damaging agents that. Proc Natl Acad Sci USA. 1996;93:15063‐15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sandhu PS, Kumar R, Beg S, et al. Natural lipids enriched self‐nano‐emulsifying systems for effective co‐delivery of tamoxifen and naringenin: Systematic approach for improved breast cancer therapeutics. Nanomedicine. 2017;13:1703‐1713. https://doi.org/10.1016/j.nano.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 96. Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer therapy. Br J Cancer. 2014;111:2039‐2045.https://doi.org/10.1038/bjc.2014.412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Senanayake TH, Lu Y, Bohling A, Raja S, Band H, Vinogradov SV. Encapsulation of poorly soluble drugs in polymer‐drug conjugates: effect of dual‐drug nanoformulations on cancer therapy. Pharm Res. 2014;31:1605‐1615. https://doi.org/10.1007/s11095-013-1265-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shan M, Carlson KE, Bujotzek A, et al. Nonsteroidal bivalent estrogen ligands: an application of the bivalent concept to the estrogen receptor. ACS Chem Biol. 2013;8:707‐715. https://doi.org/10.1021/cb3006243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sharma U, Marquis JC, Dinaut AN, et al. Design, synthesis and evaluation of estradiol‐linked genotoxicants as anticancer agents. Bioorg Med Chem Lett. 2004;14:3829‐3833. https://doi.org/10.1016/j.bmcl.2004.04.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shimizu T, LoRusso PM, Papadopoulos KP, et al. Phase I first‐in‐human study of CUDC‐101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. 2014;20:5032‐5040. https://doi.org/10.1158/1078-0432.CCR-14-0570. [DOI] [PubMed] [Google Scholar]
- 101. Smellie WJB, Dean CJ, Sacks NPM, et al. Radioimmunotherapy of breast cancer xenografts with monoclonal antibody ICR12 against c‐erbB2 p185: comparison of iodogen and N‐Succinimidyl 4‐Methyl‐3‐(tri‐n‐butylstannyl)benzoate radioiodination methods. Can Res. 1995;55(23 Suppl):5842s‐5846s. [PubMed] [Google Scholar]
- 102. Sommer A, Kopitz C, Schatz CA, et al. Preclinical efficacy of the auristatin‐based antibody‐drug conjugate BAY 1187982 for the treatment of FGFR2‐positive solid tumors. Can Res. 2016;76:6331‐6339. https://doi.org/10.1158/0008-5472.can-16-0180. [DOI] [PubMed] [Google Scholar]
- 103. Song H, Hobbs RF, Vajravelu R, et al. Radioimmunotherapy of breast cancer metastases with α‐particle emitter 225Ac: Comparing efficacy with 213Bi and 90Y. Can Res. 2009;69:8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sreekanth V, Bansal S, Motiani RK, et al. Design, synthesis, and mechanistic investigations of bile acid‐tamoxifen conjugates for breast cancer therapy. Bioconjug Chem. 2013;24:1468‐1484. https://doi.org/10.1021/bc300664k. [DOI] [PubMed] [Google Scholar]
- 105. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719‐724. https://doi.org/10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Szlachcic A, Zakrzewska M, Lobocki M, Jakimowicz P, Otlewski J. Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor‐targeted cancer therapy. Drug Des Devel Ther. 2016;10:2547‐2560. https://doi.org/10.2147/dddt.s105896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tang C, Li C, Zhang S, et al. Novel bioactive hybrid compound dual targeting estrogen receptor and histone deacetylase for the treatment of breast cancer. J Med Chem. 2015;58:4550‐4572. https://doi.org/10.1021/acs.jmedchem.5b00099. [DOI] [PubMed] [Google Scholar]
- 108. Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl‐2 proto‐oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902‐3907. [PubMed] [Google Scholar]
- 109. Thotakura N, Dadarwal M, Kumar P, et al. Chitosan‐stearic acid based polymeric micelles for the effectivedelivery of tamoxifen: cytotoxic and pharmacokinetic evaluation. AAPS PharmSciTech. 2017;18:759‐768. https://doi.org/10.1208/s12249-016-0563-6. [DOI] [PubMed] [Google Scholar]
- 110. Tolcher AW, Sugarman S, Gelmon KA, et al. Randomized phase II study of BR96‐doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol. 1999;17:478‐484. https://doi.org/10.1200/JCO.1999.17.2.478. [DOI] [PubMed] [Google Scholar]
- 111. Van Themsche C, Parent S, Leblanc V, et al. VP‐128, a novel oestradiol‐platinum(II) hybrid with selective anti‐tumour activity towards hormone‐dependent breast cancer cells in vivo. Endocr Relat Cancer. 2009;16:1185‐1195. https://doi.org/10.1677/erc-09-0113. [DOI] [PubMed] [Google Scholar]
- 112. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med. 2012; 367, 1783‐1791.https://doi.org/10.1056/nejmoa1209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vilanova C, Diaz‐Oltra S, Murga J, et al. Design and synthesis of pironetin analogue/colchicine hybrids and study of their cytotoxic activity and mechanisms of interaction with tubulin. J Med Chem. 2014;57:10391‐10403. https://doi.org/10.1021/jm501112q. [DOI] [PubMed] [Google Scholar]
- 114. Wang J, Pursell NW, Samson ME, et al. Potential advantages of CUDC‐101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther. 2013;12:925‐936. https://doi.org/10.1158/1535-7163.MCT-12-1045. [DOI] [PubMed] [Google Scholar]
- 115. Widdison WC, Wilhelm SD, Cavanagh EE, et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem. 2006;49:4392‐4408. https://doi.org/10.1021/jm060319f. [DOI] [PubMed] [Google Scholar]
- 116. Witt‐Enderby PA, Davis VL, Lapinsky D. Anti‐cancer tamoxifen‐melatonin hybrid ligand: Google Patents. (2014).
- 117. Wlassoff WA, Albright CD, Sivashinski MS, Ivanova A, Appelbaum JG, Salganik RI. Hydrogen peroxide overproduced in breast cancer cells can serve as an anticancer prodrug generating apoptosis‐stimulating hydroxyl radicals under the effect of tamoxifen‐ferrocene conjugate. J Pharm Pharmacol. 2007;59:1549‐1553. https://doi.org/10.1211/jpp.59.11.0013. [DOI] [PubMed] [Google Scholar]
- 118. Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489‐492. [DOI] [PubMed] [Google Scholar]
- 119. Xie H, Audette C, Hoffee M, Lambert JM, Blättler WA. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242‐DM1), and its two components in mice. J Pharmacol Exp Ther. 2004;308:1073. [DOI] [PubMed] [Google Scholar]
- 120. Xu R, Zhang G, Mai J, et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016;34:414‐418.https://doi.org/10.1038/nbt.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yao L, Mustafa N, Tan EC, et al. Design and synthesis of ligand efficient dual inhibitors of Janus kinase (JAK) and histone deacetylase (HDAC) based on ruxolitinib and vorinostat design and synthesis of ligand efficient dual inhibitors of Janus kinase (JAK) and histone deacetylase. J Med Chem. 2017a;60:8336‐8357. [DOI] [PubMed] [Google Scholar]
- 122. Yardley DA, Weaver R, Melisko ME, et al. EMERGE: a randomized phase II study of the antibody‐drug conjugate glembatumumab vedotin in advanced glycoprotein NMB‐Expressing breast cancer. J Clin Oncol. 2015;33:1609‐1619. https://doi.org/10.1200/jco.2014.56.2959. [DOI] [PubMed] [Google Scholar]
- 123. Yuan Z, Chen S, Sun Q, et al. Olaparib hydroxamic acid derivatives as dual PARP and HDAC inhibitors for cancer therapy. Bioorg Med Chem. 2017;25:4100‐4109. https://doi.org/10.1016/j.bmc.2017.05.058. [DOI] [PubMed] [Google Scholar]
- 124. Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple‐negative breast cancer. Breast Cancer: Basic Clin Res. 2016;10:25‐36. https://doi.org/10.4137/BCBCR.S32783. [DOI] [PMC free article] [PubMed] [Google Scholar]