

Abstract
Proteotoxicity plays a key role in many devastating human disorders, including Alzheimer's, Huntington's and Parkinson's diseases; type 2 diabetes; systemic amyloidosis; and cardiac dysfunction, to name a few. The cellular mechanisms of proteotoxicity in these disorders have been the focus of considerable research, but their role in prevalent and morbid disorders, such as diabetes, is less appreciated. There is a large body of literature on the impact of glucotoxicity and lipotoxicity on insulin‐producing pancreatic β‐cells, and there is increasing recognition that proteotoxicty plays a key role. Pancreatic islet amyloidosis by the hormone IAPP, the production of advanced glycation endproducts (AGE), and insulin misprocessing into cytotoxic aggregates are all sources of β‐cell proteotoxicity in diabetes. AGE, produced by the reaction of reducing sugars with proteins and lipids are ligands for the receptor for AGE (RAGE), as are the toxic pre‐fibrillar aggregates of IAPP produced during amyloid formation. The mechanisms of amyloid formation by IAPP in vivo or in vitro are not well understood, and the cellular mechanisms of IAPP‐induced β‐cell death are not fully defined. Here, we review recent findings that illuminate the factors and mechanisms involved in β‐cell proteotoxicity in diabetes. Together, these new insights have far‐reaching implications for the establishment of unifying mechanisms by which pathological amyloidoses imbue their injurious effects in vivo.
Keywords: proteotoxicity, islet amyloid polypeptide, RAGE, pancreatic β‐cells, diabetes, amyloidosis, aggregation
Introduction
Proteotoxicity plays a role in many prevalent human disorders, including amyloidosis diseases. A range of human diseases is associated with amyloid formation and additional disorders are associated with proteotoxic aggregates that are not classified as classical amyloids. Amyloid formation refers to the aggregation of normally soluble, functional proteins into insoluble fibrils that are partially ordered and β‐sheet rich in structure. This process can occur in many different organs and tissues in the body, and plays a role in a broad range of over 40 different diseases, including Alzheimer's disease (AD), Parkinson's disease, and pancreatic islet amyloidosis; the latter being a major source of proteotoxicity to insulin‐producing pancreatic β‐cells in type 2 diabetes (T2D). An increasing number of studies on a variety of amyloidogenic peptides and proteins suggest that there are underlying commonalities in the mechanism(s) of amyloid formation and proteotoxicity, which are independent of the details of the polypeptide sequence. Consistent with the hypothesis that many amyloidogenic proteins invoke common mechanisms of cytotoxicity,1 the receptor for advanced glycation endproducts (RAGE) engages multiple amyloidogenic proteins, such as islet amyloid polypeptide (IAPP or amylin), amyloid β (Aβ) peptide, serum amyloid A and prion‐derived peptides.2, 3, 4, 5, 6 However, of the amyloidogenic proteins that bind RAGE and activate RAGE‐mediated pathological cellular signaling, only the specific kinetic species of IAPP that engage RAGE during amyloid formation have been meticulously characterized.2 This provides a foundation and blue print for defining distinct toxic RAGE‐binding entities in amyloidosis diseases, besides T2D, for which RAGE has been shown to impart pathogenic consequences.
T2D is characterized by the loss of β‐cell function and mass, resulting in uncontrolled hyperglycemia and downstream complications. In T2D, β‐cells are exposed to multiple diabetes‐promoting factors, such as hyperglycemia (gluco‐ and glycotoxicity), hyperlipidemia (lipotoxicity), islet amyloidosis (proteotoxicity), inflammatory cytokines, and other factors for prolonged periods in subjects susceptible to metabolic disease. Each of these conditions triggers inflammatory perturbation in islet cells and other organs and, collectively, these considerations underscore the need for investigations focused on examining the impact of islet inflammation on β‐cell fate and function. Pancreatic β‐cells co‐produce and co‐secrete insulin and islet amyloid polypeptide (IAPP or amylin) in response to changes in blood glucose concentration. In the homeostatic state, IAPP acts both in the periphery and in the brain to regulate satiety, adiposity and metabolism. However, in T2D, IAPP undergoes amyloidosis in pancreatic islets by an unknown mechanism and self‐assembles into proteotoxic oligomers that directly activate multiple cellular pathways leading to islet β‐cell stress, dysfunction and death.7, 8, 9 Recent work also demonstrates a role for human (h)IAPP in the cardiovascular complications of T2D,10 and implicates a potential role for hIAPP in the pathogenesis of type 1 diabetes (T1D).11 However, a lack of molecular understanding about the in vivo factors that promote pathological aggregation, and the upstream cellular mediators that regulate the downstream deleterious events in hIAPP‐induced cytotoxicity has prevented identification of therapies to treat or prevent its pathologies. Here, we summarize the key biophysical and biochemical features, and cellular mechanisms by which pancreatic islet amyloidosis and other sources of β‐cell proteotoxicity exacerbate diabetes; factors that are likely to play a role in the proteotoxicity of other amyloidogenic proteins.
Diabetes: A Growing Worldwide Crisis
Diabetes mellitus, commonly referred to as diabetes, describes a group of life‐threatening metabolic disorders characterized by elevated blood glucose levels (hyperglycemia), resulting from the inability of the body to produce and/or utilize the essential hormone insulin, which is produced by pancreatic β‐cells and regulates the transport of dietary glucose from the bloodstream into cells where the glucose is converted into energy. Individuals with diabetes have a higher risk of morbidity and mortality than the general population. Chronic hyperglycemia leads to the damage of various body organs and is the leading cause of disabling health complications, such as retinopathy, neuropathy, nephropathy, and cardiovascular disease. The worldwide prevalence of diabetes in adults has been steadily increasing over recent decades. According to the International Diabetes Federation (IDF), over 425 million people (one in eleven adults) worldwide are currently estimated to suffer from diabetes (not including 212 million who are undiagnosed), and the number is expected to grow to more than 640 million by 2045, representing slightly more than 10% of the world's adult population if current trends continue. The largest increases are expected to take place in the regions where society is transitioning from low‐income to middle‐income economies, where three quarters of people with diabetes live currently.12, 13 The human cost of diabetes is enormous. Diabetes is estimated to cause 5 million deaths per year among the 20 to 79‐year‐old age group, a number which exceeds the combined mortality from HIV/AIDS, tuberculosis and malaria.12 The life expectancy of a diabetic patient is half that of an age‐matched control subject in the United States.2 The financial cost of diabetes is also enormous: 12% of global health expenditure is currently spent on diabetes ($727 billion), with over $673 billion worldwide in 2015 for T2D.12, 14
The diagnosis and classification of diabetes are complex and have been debated and redefined over many decades. Diabetes can be diagnosed by measures such as blood tests that detect glycated hemoglobin (A1C), which indicate the average blood sugar level over the past two or three months, and/or by two or more separate blood sugar tests after overnight fasting.15 It is currently well accepted that there are many types of diabetes; three of these constitute the main forms: T1D, an autoimmune disorder whereby the body's immune system attacks insulin‐producing pancreatic islet β‐cells resulting in the inability to produce insulin and islet amyloid polypeptide (also known as IAPP or amylin); T2D, which develops as a result of inadequate insulin production and/or utilization (insulin resistance) over time due to various factors; and gestational diabetes mellitus, which is first detected during pregnancy.12 Of the different forms of diabetes, T2D is the most common, accounting for 90% of diabetes cases.12
The Receptor for Advanced Glycation Endproducts (RAGE) and Its Ligands
Advanced glycation endproducts
The accumulation of advanced glycation endproducts (AGEs) in pancreatic islets, kidney, retina, nerves, and atherosclerotic plaques has been linked to diabetic complications.2, 16 AGEs are a heterogeneous repertoire of compounds formed by reducing sugars, such as glucose, that non‐enzymatically modify amino acids, lipids, and nucleic acids, particularly during diabetes. In theory, every protein can be modified by glycation. A multitude of pathways lead to the formation of AGEs, which yield a large variety of products, including high molecular weight compounds and aggregated endproducts. First, a reversible Schiff base is formed by the reaction of the aldehyde group in the sugar with the ɛ‐amino group of a lysine residue generating a double bond between the carbon atom of the sugar and the nitrogen in the lysine sidechain.17 Subsequent Amadori rearrangement then leads to maturation of an AGE, or upon oxidation, may form glycoxidation products that facilitate the production of other distinct families of AGEs.18 While AGEs come in many arrangements, there are three common outcomes following their formation: (1) Covalent cross‐linking (particularly at basement membranes and on collagen), (2) disrupted osmotic balance of the system, and (3) receptor engagement by the best‐characterized AGE receptor (RAGE) and other cell surface binding sites.19 All of these may result in pathological consequences.17
AGE formation can induce aggregation of proteins into cytotoxic species.20, 21, 22, 23, 24, 25, 26, 27, 28 The toxicity of glycated polypeptides may be due to the AGE modification or due to the conformational properties of the aggregated state. AGEs have also been demonstrated to play a mechanistic role in protein aggregation related to amyloidosis.29, 30, 31 Glycation of albumin was first shown to promote the refolding of this globular protein from a largely α‐helical to a cross‐β structure commonly shared by all amyloids, demonstrating that glycation may be a predisposing factor for amyloid formation.32 AGE‐modified amyloid deposits have been found in the brain tissue of AD33 and transmissible spongiform encephalopathy patients,34 and in the pancreatic islets of diabetic patients.35 High levels of argpyrimidine have been observed in protein aggregates derived from cases of familial amyloidosis but not in controls lacking disease, indicating an important potential role for methylglyoxal, an AGE precursor, in pathological protein aggregation.36 Similar mechanisms have been reported in neurodegenerative disorders.37, 38, 39 Studies have shown a correlation between AGEs and both the induction and progression of AD pathogenesis.40, 41, 42, 43, 44 These data indicate that glycation of polypeptides, which have the propensity to aggregate into amyloid fibrils, can accelerate the formation of cross β‐sheet structure.
AGE adducts and RAGE‐mediated proteotoxicity
One of the main mechanisms by which both protein‐AGE adducts and amyloidogenic proteins exert their action in vivo is via binding to multi‐ligand cell surface receptors, such as RAGE, CD36, scavenger receptor class A and B type I, the serine protease tissue‐type plasminogen activator receptor, and other pattern recognition receptors, which are expressed on a wide range of cell types.45, 46, 47, 48, 49, 50, 51, 52 Among the AGE binding receptors, RAGE is particularly important and well‐characterized.19, 46, 53, 54, 55, 56 RAGE is an immunoglobulin‐like, multimeric cell surface receptor with three distinct extracellular domains, including the variable (V)‐type immunoglobulin (Ig) domain, which is primarily implicated in ligand engagement19, 57 [Fig. 1(A)]. RAGE is ubiquitously expressed on a variety of cell types including monocytes/macrophages, smooth muscle cells, endothelial cells, and pancreatic β‐cells. Upon binding AGEs or one of its other ligands, including: IAPP, Aβ, prion peptides, transthyretin, S100/calgranulins, high mobility group box (HMGB) proteins, phosphatidylserine, and others, RAGE transduces signals through the intracellular formin molecule, Diaphanous 1 (DIAPH1).2, 58, 59, 60, 61 A growing body of work in other cell types, particularly smooth muscle cells, cardiomyocytes and peripheral monocytes/macrophages, has revealed that DIAPH1 is required for RAGE‐dependent upregulation of cytokines, oxidative stress, alterations in cellular migration and retention, proliferation and survivability, and other canonical facets of RAGE signaling58, 62, 63 [Fig. 1(B)]. DIAPH1 is expressed in pancreatic β‐cells. However, very little is known about the impact of DIAPH1 in these cells. Thus, further investigation of this pathway could potentially lead to a better understanding of the pathological consequences of RAGE engagement with its ligands in β‐cells, particularly in diabetes. Of note, there are also other isoforms of RAGE, including soluble RAGE (sRAGE), which lacks the transmembrane and intracellular domains, and therefore fails to signal through DIAPH1 upon ligand engagement. sRAGE has been demonstrated to be protective as it acts as a decoy receptor that binds AGEs and other RAGE ligands, including some forms of amyloidogenic proteins, preventing their interaction with cell membrane‐bound RAGE and the activation of pathological cellular signaling.64 Together, the findings suggest that glycated proteins may constitute a family of amyloidogenic ligands with shared biophysical and biochemical properties that facilitate engagement and activation of pathological pattern recognition receptors. This hypothesis rationalizes how the V‐domain of RAGE is capable of interacting with a diverse family of ligands. More work is needed to fully determine the specific protein structures and posttranslational modifications involved in the generation of toxic aggregates by AGEs.
Figure 1.

Schematic diagrams of extracellular RAGE structure and activation. RAGE is a cell surface receptor that is comprised of three extracellular immunoglobulin‐like domains (a V‐type domain and two C‐type domains), a transmembrane domain (TM) and an intracellular domain (ID) that is required for signaling. (A) A ribbon diagram of the extracellular V‐C1 domains of RAGE, which form an integrated structural unit. β‐sheets are yellow, helices are purple and loops are blue and white (PDB code 303U).54 (B) Activation of RAGE by its ligands leads to recruitment and binding of the intracellular formin molecule, DIAPH1, resulting in pathological intracellular signaling that can induce oxidative stress, inflammation, cellular dysfunction and apoptosis.
Diabetes is among the diverse range of inflammatory diseases associated with RAGE activation
Increasing evidence implicates RAGE as an important mediating factor in diabetes and pancreatic β‐cell dysfunction. A Genome Wide Association Study (GWAS) conducted on 3624 Finnish individuals revealed that the rs2070600 and rs17493811 AGER (AGER is the gene encoding RAGE) polymorphisms predicted increased risk of developing T1D.65 rs2070600 has previously been shown to cause a decrease in sRAGE levels, and an increase in ligand affinity.66, 67, 68 Other studies have also shown that RAGE induction and increased levels of RAGE ligands (S100B and other members of the S100/calgranulin family, HMGB1, and AGEs) drive dysfunctional glucose stimulated insulin secretion (GSIS),69 amplify oxidative stress and impairment of adenosine triphosphate (ATP) synthesis, and increase apoptotic cell death in pancreatic β‐cells.70, 71 In addition, RAGE inhibition in vitro, mediated by anti‐RAGE antibodies or RNA silencing techniques, protected rodent and human β‐cells from GSIS dysfunction and apoptosis.72 Long‐term treatment of cultured rat pancreatic islets with aminoguanidine, which inhibits AGE formation, conferred a benefit to β‐cell insulin secretion and biosynthesis.73 Subsequent studies have indicated that short term AGE exposure to rat pancreatic islets protected against apoptosis, whereas prolonged exposure promoted death. These findings suggest that the effect of RAGE activation may be dependent on the temporal dynamics of RAGE/ligand engagement and on ligand concentration.74 AGEs have also been implicated in destabilizing PDX‐1 protein, a critical factor for β‐cell survival, thereby impairing insulin synthesis.75 Recent breakthroughs in the transplantation of human fetal pancreatic progenitor cells have shown promise in preventing RAGE upregulation in the kidney, protecting against diabetic nephropathy.76 While these advancements are encouraging, more work is needed to fully determine how RAGE and its ligands influence pancreatic β‐cell fate, and how modulation of ligand production, engagement of the ligand with the RAGE receptor, RAGE receptor, and DIAPH1‐dependent intracellular processes may be harnessed to prevent β‐cell dysfunction, diabetes induction, and subsequent pathological consequences.
Islet Amyloid Formation Is a Significant Source of β‐Cell Proteotoxicity in T2D
The biosynthesis and processing of IAPP
IAPP belongs to the calcitonin peptide family, which is comprised of α‐ and β‐calcitonin gene‐related peptide (CGRP), calcitonin, intermedin, and adrenomedullin. These peptides all include an intramolecular disulfide bridge near the N‐terminus and an amidated aromatic residue at the C‐terminus.77 IAPP has been found in all mammals studied and, like many polypeptide hormones, is synthesized as a pre‐pro‐form. In the case of the human polypeptide, the pre‐pro‐hormone is 89 residues in length and includes a 22 amino acid residue signal sequence at the N‐terminus. The remaining 67 residues make up the pro‐form (proIAPP), which includes N‐ and C‐terminal extensions relative to mature IAPP (Fig. 2). ProIAPP is processed in the Golgi and in the insulin β‐cell secretory granule to yield the mature hormone, which is secreted in tandem with insulin.78, 79, 80 The N‐ and C‐terminal flanking sequences of the pro‐form are cleaved by the prohormone convertases, PC2 and PC1/3.80 PC2 is responsible for cleavage of the N‐terminal extension and PC1/3 is primarily responsible for the initial step in the C‐terminal processing, although PC2 can cleave here as well. Processing at the C‐terminus, which leads to an amidated C‐terminal Tyr, is a multi‐step process. The initial C‐terminal cleavage leaves a Gly‐Lys‐Arg sequence as the new C‐terminus. The Lys‐Arg dipeptide is trimmed by carboxypeptidase, leaving the Gly, which donates the nitrogen during amidation by the peptidyl amidating monooxygenase complex (PAM).79, 80
Figure 2.
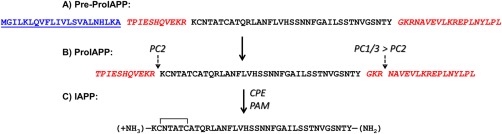
Processing of human pre‐proIAPP into the mature IAPP sequence. (A) The 89 amino acid residues of pre‐pro‐peptide, including the 22‐residue signal sequence which is shown in blue underlined font, and the flanking peptide regions of the pro‐hormone, which are illustrated in red italicized font. (B) The sequence of proIAPP showing the cleavage sites for PC2 at the N‐terminus of mature IAPP and PC1/3 after three residues (GKR) in the C‐terminal flanking region. (C) Amidation of the C‐terminal Tyr is a multi‐step process. CPE removes the KR pair of dibasic residues that remain after PC1/3 cleavage, and the glycine is the nitrogen donor for amidation by PAM
Defective post‐translational processing of proIAPP may be another factor that promotes islet amyloid formation and proteotoxicity in T2D. There is indirect evidence that normal processing of proIAPP, at the N‐terminal cleavage site, may be incorrect/incomplete in T2D, resulting in secretion of an N‐terminal extended proIAPP intermediate.77, 81, 82, 83 Comparatively little work has been done on amyloid formation by partially processed proIAPP, but it has been proposed that retention of the N‐terminal pro‐sequence leads to binding to sulfated proteoglycans in the extracellular matrix, which in turn, could generate a high local concentration of the polypeptide and facilitate the formation of active amyloidogenic “seeds.”82, 84, 85 In vitro biophysical studies demonstrate that this partially processed form can bind to the glucosaminoglycan portion of proteoglycans, and that this interaction promotes amyloid formation.82, 84 Furthermore, the deposits formed can seed amyloid formation by mature fully processed IAPP.84 Fully processed IAPP is stored in the halo region of the insulin secretory granule, while insulin is found in the dense core of the granule.77, 78 The concentration of IAPP in the granule is only about 1–2% that of insulin, but this is a much higher concentration than required to promote aggressive amyloid formation in vitro.77, 86, 87, 88, 89 This suggests that there are factors that inhibit irreversible aggregation of IAPP in the granule.
Variations in the Primary Sequence of IAPP Correlate with its Ability to Form Amyloid In Vitro and In Vivo
IAPP is produced by all species examined to date, but not all IAPP sequences form amyloid. The sequence of IAPP is strongly conserved, however examined interspecies variations correlate significantly with the ability to form amyloid in vitro and with the presence or absence of islet amyloid in vivo. In particular, rat IAPP (rIAPP), which has the same amino acid sequence as mouse IAPP, differs from the human polypeptide at six positions and does not form islet amyloid in vivo. While there are some variations in the IAPP sequences of rodents, rodent IAPP generally is non‐amyloidogengic in vitro under conditions where the human peptide rapidly forms amyloid (Fig. 3). Early comparison of the rodent and human sequences led to the hypothesis that differences in the sequence of IAPP within positions 20–29 correlated with the ability to form amyloid.14 Four of the six sequence differences between hIAPP and rIAPP are found within this region (Fig. 3). The most important of these are three proline residues in rIAPP at positions 25, 28, and 29. Proline disrupts interstrand hydrogen bonding and intermolecular β‐sheet formation, and this helps to explain the lack of islet amyloid in rodents. Another important difference between hIAPP and rIAPP is the replacement of His‐18 in hIAPP with Arg in rIAPP. This substitution ensures that the sidechain at position‐18 will be positively charged at all physiologically relevant pH values; the increased charge decreases the polypeptide's tendency to aggregate and form amyloid.90, 91 Naturally occurring sequence variations in IAPP have been reviewed recently.86 Studies with synthetic variants of hIAPP have shown that residues outside of the 20–29 segment play an important role in modulating amyloidogenicity. For example, substitution of Asn‐14 or Asn‐21 can have drastic effects, as can proline substitutions outside the 20–29 region.92, 93 Conversely, replacement of residues Arg‐18, Leu‐23, and Val‐26 in rIAPP by the corresponding amino acids of hIAPP (His‐18, Phe‐23, Ile‐26) has been reported to lead to a weakly amyloidogenic polypeptide, despite it still containing the three proline substitutions found in rIAPP.94 Taken together, these studies and other work demonstrate that additional factors other than just the sequence within residues 20–29 play an important role in dictating amyloidogenicity. Elucidating the factors that control hIAPP amyloidosis is important since it can aid in the rational design of next generation soluble analogs of hIAPP for clinical usage in the maintenance of metabolic homeostasis.95
Figure 3.
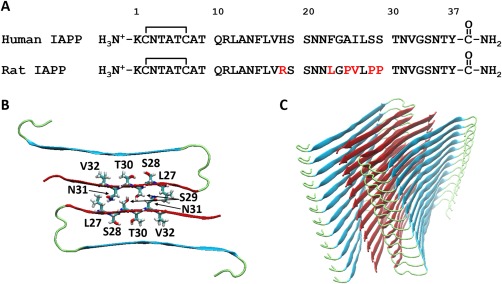
(A) The primary sequence of human and rat/mouse IAPP. (B) A cross section of the UCLA model of hIAPP amyloid fibril (104). Two symmetric related peptides are shown in a top‐down view of a stack. Several residues which have been proposed to make key inter‐stack contacts via sidechain interactions are indicated. (C) A ribbon diagram showing the arrangement of the polypeptide orientation in the amyloid fibril
The structure of monomeric hIAPP and of IAPP amyloid fibrils
Mature hIAPP is a positively charged, hydrophobic peptide that contains no negative charges at physiologically relevant pH's due to the lack of acidic residues and the amidated C‐terminus. hIAPP does not adopt a well‐defined globular conformation in its unaggregated state, but samples an ensemble of rapidly interconverting, partial and less structured conformations.96 Thus, IAPP is considered to be a so‐called “natively unfolded” or “intrinsically disordered” monomer, although residues 5 through 22 of hIAPP transiently sample helical phi psi angles in solution.96 Interactions with membranes, particularly ones with a high content of anionic lipids, can promote formation of a more ordered partial helical state.97, 98 The tendency to form helical conformations may facilitate the conversion of hIAPP to β‐sheet rich amyloid fibrils by promoting initial oligomerization.99, 100, 101, 102
Several high resolution structural models have been proposed for hIAPP amyloid fibrils formed in vitro and, although they differ in the details, they share common overall features.103, 104 The basic fibril structure is made of two U‐shaped stacks of IAPP monomers with two β‐strands per monomer connected by a less ordered loop/turn region (Fig. 3). Each monomer is hydrogen bonded to its immediate neighbors in the same stack and the polypeptide forms parallel β‐sheets. The backbone hydrogen bonds are between different molecules rather than within a single polypeptide, and are thus oriented parallel to the long axis of the fibril. The pair of U‐shaped stacks are aligned so that hIAPP molecules in adjacent stacks are oriented antiparallel to each other, and the stacks interact via networks of side chain interactions. The different models vary in the exact location of the two β‐strands and the loop/turn region. Both models place the disulfide‐bridged loop between residues 2 and 7, outside of the ordered core of the cross β‐structure (Fig. 3).
The physiological role of IAPP
The circulating concentration of IAPP is reported to be on the order of three to five picomolar in rats, and to rise to 15 to 20 picomolar with elevation of blood glucose levels.77 The local concentration at the site of release from the granule is significantly higher and this is likely the more relevant value for amyloid formation. hIAPP is believed to play a role in controlling gastric emptying, maintaining glucose homeostasis, and suppressing glucagon release. The hormone is also involved in controlling satiety and is proposed to act as an adiposity signal.105, 106 A reduction in weight induced by IAPP has been reported for obese rats and humans, and animal studies have led to the hypothesis that weight loss occurs through a mode of action that is similar to that found in cases of enhanced leptin sensitivity.107, 108 As the focus of this review is on proteotoxicity rather than normal function of IAPP, the interested reader is referred to several recent reviews that discuss the proposed function(s) of IAPP in more depth.77, 86, 89
Does islet amyloid have an extracellular or intracellular origin?
In human T2D and in all cell and animal models, IAPP fibrils are found in invaginations of the β‐cell membrane, but the question of where islet amyloid originates is still open to debate and there are conflicting reports in the literature concerning the initiation site for islet amyloid deposition in vivo. As noted, amyloid deposits found in human T2D appear to be extracellular and early studies with transgenic (Tg) mice were consistent with an extracellular origin for islet amyloid; but other studies with rodent models that overexpress IAPP are consistent with an intracellular origin.77, 109 It should be noted that some Tg mouse models of islet amyloidosis contain high copy numbers of the human IAPP gene and produce high levels of hIAPP. This could play a role in the reported intracellular aggregation. Defining whether islet amyloid has an intracellular or extracellular origin might affect drug design, and is thus of practical, as well as academic interest. The interested reader is referred to the recent review by Clark and co‐workers for a more in‐depth discussion of the issue of intra‐ vs. extracellular islet amyloid formation.89
Mechanisms of hIAPP‐induced toxicity
Irrespective of the intra‐ and/or extracellular origin of IAPP aggregation, it is certain that discrete toxic forms of this polypeptide cause pathological consequences in key metabolic tissues. Indeed, identification of the toxic species in amyloidosis is a major challenge and the nature of the toxic species is controversial, due to the difficulty of isolating pure fractions of transient kinetic species that form over the course of amyloid formation. The preponderance of evidence indicates that soluble oligomeric species populated during amyloid formation are responsible for toxicity in islet amyloidosis and recent in vitro time‐resolved biophysical and biological measurements have demonstrated that pre‐amyloid oligomers are more toxic than hIAPP amyloid fibrils or monomeric hIAPP.89, 109, 110 The biophysical properties of toxic hIAPP oligomers have been shown to be distinct from those produced in other amyloidosis diseases,110 providing a target for rational drug development. However, the origin of the events that lead to β‐cell death in vivo are still debated. Does induction of stress and dysfunction arise from within the cell or is it triggered from events that occur after secretion of the polypeptide from the β‐cell? It has been proposed that hIAPP toxic oligomers are present in the cytoplasm of cells which overexpress hIAPP, and reactivity to an anti‐oligomer antibody has been reported for β‐cells in Tg hIAPP mice.111, 112 However, some of these studies made use of a conformational antibody, which was not raised against hIAPP. These antibodies are clearly useful in vitro, but their applicability to IAPP detection in vivo has been questioned.113, 114 Conversely, work with a cultured islet model that produces physiologically relevant levels of hIAPP show that, in this system, the secretion of IAPP is an important factor for islet amyloid formation and β‐cell toxicity. In that study, inhibiting IAPP secretion, while maintaining the level of IAPP production, was found to reduce amyloid formation. In contrast, promoting increased secretion in this model without increasing hIAPP production led to increased toxicity and amyloid formation.115 The differences observed between the various models may be related to the level of production of hIAPP.89, 114
The pathways that lead to IAPP‐induced β‐cell dysfunction and apoptosis are not yet fully defined.9, 116, 117, 118, 119, 120, 121 There are likely to be multiple mechanisms of toxicity and a variety have been reported; their relative contributions could depend upon cellular conditions. Defects in autophagy, increased production of pro‐inflammatory cytokines, endoplasmic reticulum (ER) stress, permeabilization of cell membranes, mitochondrial membrane damage, activation of Calpain‐2, receptor‐mediated mechanisms including Fas and RAGE activation linked to induction of cell stress and apoptotic signaling pathways, have all been proposed to contribute to IAPP‐induced β‐cell cytotoxicity.2, 7, 8, 9, 111, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132
Impairment of autophagy, and ER stress in hIAPP‐induced β‐cell proteotoxicity
Defects in autophagy play a role in the toxicity of a range of amyloidogenic proteins. In neurodegenerative diseases, for example, upregulation of autophagy acts as a protective adaptation to the accumulation of toxic amyloidogenic aggregates. However, lysosomal degradation of amyloidogenic polypeptides and autophagocytosis may not always be completely successful. The resulting accumulation of amyloidogenic aggregates can lead to autophagy‐mediated lysosomal cell death or dysfunction. Along these lines, the overexpression of hIAPP in β‐cells has been reported to lead to impairment in autophagy.125, 129, 130 Stimulation of autophagy has been shown to protect against IAPP toxicity, while inhibition of autophagy‐lysosomal degradation has been shown to enhance hIAPP‐induced β‐cell apoptosis.125, 130, 133, 134, 135 ER stress, defects in the unfolded protein response (UPR) and in ER‐associated protein degradation have all been proposed to contribute to hIAPP‐induced β‐cell death in T2D. ProIAPP and partially processed proIAPP could contribute to toxicity in cases where toxicity arises from intracellular aggregates since proIAPP mis‐processing occurs in T2D, and processing is completed in the Golgi and insulin secretory granules.77, 86 It is important to note, however, that the role of ER stress in hIAPP‐mediated toxicity is still controversial. Studies using exogenously added hIAPP and Tg mice that overexpress hIAPP have reported that ER stress is a mechanism of hIAPP‐induced β‐cell dysfunction,111, 124 while ER stress was not detected in studies employing cultured islets that produce more physiologically relevant levels of IAPP.127
Membrane disruption by hIAPP aggregates
Perturbation of membrane integrity by hIAPP has also been proposed to contribute to toxicity89, 128, 132 and there is a large literature on in vitro studies of the ability of hIAPP to disrupt model membranes.89, 136 Unfortunately, it is not clear if there is a direct one‐to‐one correlate between in vitro investigations and behavior in vivo.89, 137 The effect of hIAPP depends on the lipid to peptide ratio, the lipid composition, ionic strength and the pH. Many commonly used model systems that lack cholesterol contain a much higher percentage of anionic lipids than is found in the β‐cell membrane.138 These are important factors since cholesterol modulates hIAPP/membrane interactions and high percentages of anionic lipids significantly promote IAPP/membrane interactions.139
Toxic hIAPP aggregates cause pancreatic islet inflammation
hIAPP‐induced β‐cell proteotoxicity is linked to local islet inflammatory processes.7, 122, 140, 141 Activation of the inflammasome by hIAPP aggregates can contribute to local islet inflammation and β‐cell dysfunction.7, 131 Inflammasomes are multiprotein intracellular assemblies that sense a diverse range of pathogenic stimuli and regulate the production of active caspase‐1. In turn, caspase‐1 activates the pro‐inflammatory cytokines interleukin‐1b (IL‐1b) and IL‐18 by cleaving of their pro‐forms into mature sequences. The role of IL‐1b in hIAPP‐induced β‐cell death and dysfunction is currently a major focus in the field.7, 131 The depletion of islet‐resident macrophages, key modulators of inflammation, has been shown to improve glucose intolerance and increase islet amyloid accumulation, consistent with reports that amyloid fibrils are not toxic; suggesting that toxicity may be due to the activation of phagocytic cells that attempt to remove cytotoxic IAPP aggregates from the islet.141 Interdisciplinary approaches combining studies in human diabetic pancreas, mouse models, cell physiology and molecular biophysics show that toxic prefibrillar form(s) of hIAPP (and not nontoxic monomers and not amyloid fibrils) are ligands of RAGE, and that hIAPP‐induced upregulation and activation of RAGE leads to the induction of NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX), production of reactive oxygen species (ROS) and subsequent oxidative stress, a proximate mechanism preceding the development of β‐cell inflammation, dysfunction, and ultimately, cleavage of caspases, and β‐cell apoptosis.2, 110 A direct relationship between loss of β‐cell area and β‐cell stress, apoptosis and RAGE expression; and a lack of correlation between these parameters and islet amyloid area in a Tg hIAPP mouse model, support in vitro and in vivo findings that toxic pre‐amyloid hIAPP intermediates are more deleterious than amyloid fibrils.2 Macrophage chemo‐attractants (Ccl2, Cxcl1 and Cxcl2) and other pro‐inflammatory mRNA transcripts (Il1b and Il18) in the pancreas of Tg hIAPP mice with islet amyloidosis were significantly upregulated, while inhibition of hIAPP/RAGE interactions protected pancreatic islets, cells and Tg hIAPP mice from IAPP‐induced stress, inflammatory gene expression, metabolic dysfunction, islet amyloid deposition and loss of β‐cell area.2 Together, these findings establish RAGE as a mediator of pathological β‐cell signaling during islet amyloidosis in T2D, and highlight the interaction of RAGE and pre‐amyloid hIAPP intermediates as a primary target for β‐cell preservation in metabolic disease.
Insulin Misfolding and Other Signals of Proteotoxicity in Diabetes
IAPP is co‐produced and co‐secreted with insulin by pancreatic β‐cells. The biosynthesis of insulin by β‐cells, like that of IAPP, is a multistep process, and mutations in the pre‐proinsulin gene, or disruptions in posttranslational modifications at any major stage, can result in the formation of proteotoxic aggregates and activation of cellular stress mechanisms leading to β‐cell toxicity and diabetes.142, 143, 144, 145, 146 Insulin is initially translated in the cytosol as a pre‐prohormone (pre‐proinsulin), which is translocated across the ER membrane where it is enzymatically cleaved to form proinsulin.147 Misprocessing during this early step results in the accumulation of pre‐proinsulin in the juxtanuclear compartment and induction of heat shock protein 70 (HSP70), which promotes human β‐cell apoptosis.147 Inside the oxidizing environment of the ER, generation of three evolutionarily conserved disulfide bonds facilitate the proper protein folding of proinsulin (PI). Disruption of this key process leads to PI misfolding and its aggregation into cytotoxic species. For example, in the male Akita murine model of diabetes, a PI‐C(A7)Y mutation in the A chain of PI prevents the formation of a critical disulfide bond, leading to proteotoxic aggregation, ER stress, increased XBP1 mRNA splicing, β‐cell dysfunction, and the development of postnatal diabetes within weeks.143, 148 Proper posttranslational modification of the A‐chain epitope of PI is also required for β‐cell recognition and induction of adaptive immune attack by human T‐cells.149, 150 Taken together, these data highlight the importance of posttranslational modification in insulin maturation and signaling, and indicate that defects in insulin processing can lead to PI proteotoxicity and/or autoimmunity, and subsequent β‐cell apoptosis.
Elevated levels of PI have been shown to be correlated to the degree of β‐cell secretory impairment in T2D patients.151 In the β‐cell, transport‐competent secretory peptide precursors, including PI, are regulated by autophagy, whereas efficient clearance of transport‐incompetent mutated forms of PI by alternative degradative pathways may be necessary to avoid β‐cell proteotoxicity.146, 152, 153 Insulin resistant Akita and db/db mice, as well as Zucker diabetic fatty rats display increased autophagosome flux, and disrupting autophagy in these models drives β‐cell UPR stress and the progression of diabetes.154, 155, 156, 157 Reduction of PI autophagic degradation increases its residency in the secretory pathway and enhances its secretion in response to stimuli.152, 154, 158 The production and overproduction of insulin in and of itself, drives cellular stress. Specifically, mitochondrial ROS have been implicated as an obligatory signal for GSIS,159, 160, 161, 162 and pharmacological inhibition of ROS diminishes insulin secretion. In the context of glucolipotoxicity, in which basal insulin hypersecretion and pronounced impairments in GSIS are known to occur, ROS stimulates insulin secretion from β‐cells in a concentration‐dependent manner.163 This phenomenon is bidirectional, as NOX, a major driver of ROS production, is upregulated prior to β‐cell dysfunction.163 These reactive molecules, which play an adaptive role in cellular oxidative signaling, must be rapidly removed or else they elicit β‐cell oxidative stress and metabolic defects.163 Insulin stimulated insulin secretion has also been demonstrated in pancreatic β‐cells,164, 165 providing another feed‐forward mechanism by which augmentation of insulin production by β‐cells may promote increased oxidative stress, further insulin secretion, more pronounced proteotoxicity and ROS production in a deleterious cycle, ultimately promoting β‐cell failure and diabetes.
Conclusion
Protein aggregation and amyloid formation have long been recognized as key events in a range of neurodegenerative diseases, including AD and Parkinson's disease, and in the systemic amyloidoises, but their role in T2D is less appreciated. The various investigations highlighted in this review develop a strong case for the role of aberrant protein misprocessing, modification and aggregation in the pathophysiology of T2D. Aggregation of IAPP to form islet amyloid contributes to β‐cell death and dysfunction in T2D, while aggregation of misprocessed insulin has been implicated to contribute to proteotoxicity in the disease. The co‐secretion of insulin and IAPP suggests the potential forging of shared molecular and biochemical mechanisms of toxicity in pathological microenvironments. AGE formation is likely to play a significant role in proteotoxicity in T2D, given that the diabetic milieu is favorable for the generation of AGEs. AGE formation is thought to play a role in other protein deposition diseases and engagement of RAGE with toxic aggregates derived from different amyloidogenic proteins may provide a common unifying theme. It is not known whether RAGE plays a role in β‐cell function; more work is needed to determine whether acute activation of RAGE in β‐cells may be beneficial. But chronic activation of RAGE by its ligands, including those linked to protein aggregation or AGEs, can be detrimental. Compounds that interfere with the interaction of RAGE with its ligands, or suppress RAGE‐mediated cellular signaling triggered by binding of RAGE ligands, especially in the setting of pathological ligand conformation and/or concentration, may thus have therapeutic potential for a range of inflammatory diseases, including diabetes.2, 166
Acknowledgments
We thank Rehana Akter for assistance with preparation of figures, and Latoya Woods for assistance with preparation of the manuscript.
References
- 1. Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511. [DOI] [PubMed] [Google Scholar]
- 2. Abedini A, Cao P, Plesner A, Zhang J, He M, Derk J, Patil SA, Rosario R, Lonier J, Song F, Koh H, Li H, Raleigh DP, Schmidt AM (2018) RAGE binds preamyloid IAPP intermediates and mediates pancreatic beta cell proteotoxicity. J Clin Invest 128:682–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sturchler E, Galichet A, Weibel M, Leclerc E, Heizmann CW (2008) Site‐specific blockade of RAGE‐Vd prevents amyloid‐beta oligomer neurotoxicity. J Neurosci 28:5149–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt AM, Sahagan B, Nelson RB, Selmer J, Rothlein R, Bell JM (2009) The role of RAGE in amyloid‐beta peptide‐mediated pathology in Alzheimer's disease. Curr Opin Investig Drugs 10:672–680. [PubMed] [Google Scholar]
- 5. Law E, Lu S, Kieffer TJ, Warnock GL, Ao Z, Woo M, Marzban L (2010) Differences between amyloid toxicity in alpha and beta cells in human and mouse islets and the role of caspase‐3. Diabetologia 53:1415–1427. [DOI] [PubMed] [Google Scholar]
- 6. Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, Kindy M (2000) Receptor‐dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 6:643–651. [DOI] [PubMed] [Google Scholar]
- 7. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O'Neill LA (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL‐1beta in type 2 diabetes. Nat Immunol 11:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zraika S, Hull RL, Udayasankar J, Aston‐Mourney K, Subramanian SL, Kisilevsky R, Szarek WA, Kahn SE (2009) Oxidative stress is induced by islet amyloid formation and time‐dependently mediates amyloid‐induced beta cell apoptosis. Diabetologia 52:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subramanian SL, Hull RL, Zraika S, Aston‐Mourney K, Udayasankar J, Kahn SE (2012) cJUN N‐terminal kinase (JNK) activation mediates islet amyloid‐induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 55:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Despa S, Sharma S, Harris TR, Dong H, Li N, Chiamvimonvat N, Taegtmeyer H, Margulies KB, Hammock BD, Despa F (2014) Cardioprotection by controlling hyperamylinemia in a “humanized” diabetic rat model. J Am Heart Assoc 3:e001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denroche HC, Verchere CB (2018) IAPP and type 1 diabetes: implications for immunity, metabolism and islet transplants. J Mol Endocrinol 60:R57–R75. [DOI] [PubMed] [Google Scholar]
- 12. International Diabetes Federation . (2017). IDF Diabetes Atlas, 8th ed., Brussels, Belgium: http://www.diabetesatlas.org [Google Scholar]
- 13. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, Cavan D, Shaw JE, Makaroff LE (2017) IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 128:40–50. [DOI] [PubMed] [Google Scholar]
- 14. The Center for Disease Control . National Diabetes Statistics Report, 2014. https://www.cdc.gov/diabetes/pdfs/data/2014-report-estimates-of-diabetes-and-its-burden-in-the-united-states.pdf
- 15. American Diabetes Association (2018) Classification and diagnosis of diabetes: standards of medical care in diabetes‐2018. Diabetes Care 41:S13–S27. [DOI] [PubMed] [Google Scholar]
- 16. Goh S‐Y, Cooper ME (2008) The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrin Metabol 93:1143–1152. [DOI] [PubMed] [Google Scholar]
- 17. Singh R, Barden A, Mori T, Beilin L (2001) Advanced glycation end‐products: a review. Diabetologia 44:129–146. [DOI] [PubMed] [Google Scholar]
- 18. Nishikawa T, Edelstein D, Du XL, Yamagishi S‐i, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes H‐P, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787. [DOI] [PubMed] [Google Scholar]
- 19. Ramasamy R, Yan SF, Schmidt AM (2011) Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann NY Acad Sci 1243:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chibber R, Molinatti PA, Rosatto N, Lambourne B, Kohner EM (1997) Toxic action of advanced glycation end products on cultured retinal capillary pericytes and endothelial cells: relevance to diabetic retinopathy. Diabetologia 40:156–164. [DOI] [PubMed] [Google Scholar]
- 21. Cohen G, Rudnicki M, Walter F, Niwa T, Horl WH (2001) Glucose‐modified proteins modulate essential functions and apoptosis of polymorphonuclear leukocytes. J Am Soc Nephrol 12:1264–1271. [DOI] [PubMed] [Google Scholar]
- 22. Gasic‐Milenkovic J, Loske C, Deuther‐Conrad W, Münch G (2001) Protein “AGEing"–cytotoxicity of a glycated protein increases with its degree of AGE‐modification. Z Gerontol Geriatr 34:457–460. [DOI] [PubMed] [Google Scholar]
- 23. Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K (1999) Neurotoxicity of methylglyoxal and 3‐deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J Neurosci Res 57:280–289. [DOI] [PubMed] [Google Scholar]
- 24. Loske C, Neumann A, Cunningham AM, Nichol K, Schinzel R, Riederer P, Munch G (1998) Cytotoxicity of advanced glycation endproducts is mediated by oxidative stress. J Neural Transm (Vienna) 105:1005–1015. [DOI] [PubMed] [Google Scholar]
- 25. Lyons TJ, Li W, Wojciechowski B, Wells‐Knecht MC, Wells‐Knecht KJ, Jenkins AJ (2000) Aminoguanidine and the effects of modified LDL on cultured retinal capillary cells. Invest Ophthalmol Vis Sci 41:1176–1180. [PubMed] [Google Scholar]
- 26. Min C, Kang E, Yu SH, Shinn SH, Kim YS (1999) Advanced glycation end products induce apoptosis and procoagulant activity in cultured human umbilical vein endothelial cells. Diabetes Res Clin Pract 46:197–202. [DOI] [PubMed] [Google Scholar]
- 27. Takeuchi M, Bucala R, Suzuki T, Ohkubo T, Yamazaki M, Koike T, Kameda Y, Makita Z (2000) Neurotoxicity of advanced glycation end‐products for cultured cortical neurons. J Neuropathol Exp Neurol 59:1094–1105. [DOI] [PubMed] [Google Scholar]
- 28. Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M, Makita Z (2002) Advanced glycation end product‐induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein‐1 in human‐cultured mesangial cells. J Biol Chem 277:20309–20315. [DOI] [PubMed] [Google Scholar]
- 29. Iannuzzi C, Maritato R, Irace G, Sirangelo I (2013) Glycation accelerates fibrillization of the amyloidogenic W7FW14F apomyoglobin. PLos One 8:e80768. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Bertoletti L, Regazzoni L, Altomare A, Colombo R, Colzani M, Vistoli G, Marchese L, Carini M, De Lorenzi E, Aldini G (2014) Advanced glycation end products of beta2‐microglobulin in uremic patients as determined by high resolution mass spectrometry. J Pharm Biomed Anal 91:193–201. [DOI] [PubMed] [Google Scholar]
- 31. Ernest A, Tibor D, Abdu A (2013) Alzheimer disease and diabetes mellitus: do they have anything in common?. Curr Alzheimer Res 10:609–617. [DOI] [PubMed] [Google Scholar]
- 32. Bouma B, Kroon‐Batenburg LM, Wu YP, Brunjes B, Posthuma G, Kranenburg O, de Groot PG, Voest EE, Gebbink MF (2003) Glycation induces formation of amyloid cross‐beta structure in albumin. J Biol Chem 278:41810–41819. [DOI] [PubMed] [Google Scholar]
- 33. Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 91:4766–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sasaki N, Takeuchi M, Chowei H, Kikuchi S, Hayashi Y, Nakano N, Ikeda H, Yamagishi S, Kitamoto T, Saito T, Makita Z (2002) Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt‐Jakob disease with prion plaques. Neurosci Lett 326:117–120. [DOI] [PubMed] [Google Scholar]
- 35. Kapurniotu A, Bernhagen J, Greenfield N, Al‐Abed Y, Teichberg S, Frank RW, Voelter W, Bucala R (1998) Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur J Biochem 251:208–216. [DOI] [PubMed] [Google Scholar]
- 36. Gomes R, Sousa Silva M, Quintas A, Cordeiro C, Freire A, Pereira P, Martins A, Monteiro E, Barroso E, Ponces Freire A (2005) Argpyrimidine, a methylglyoxal‐derived advanced glycation end‐product in familial amyloidotic polyneuropathy. Biochem J 385:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li J, Liu D, Sun L, Lu Y, Zhang Z (2012) Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci 317:1–5. [DOI] [PubMed] [Google Scholar]
- 38. Miranda HV, Outeiro TF (2010) The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol 221:13–25. [DOI] [PubMed] [Google Scholar]
- 39. Moro ML, Phillips AS, Gaimster K, Paul C, Mudher A, Nicoll JAR, Boche D (2018) Pyroglutamate and isoaspartate modified Amyloid‐Beta in ageing and Alzheimer's disease. Acta Neuropathol Commun 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takeuchi M, Yamagishi S‐i (2008) Possible involvement of advanced glycation end‐products (AGEs) in the pathogenesis of Alzheimer's disease. Curr Pharm Des 14:973–978. [DOI] [PubMed] [Google Scholar]
- 41. Ko S‐Y, Ko H‐A, Chu K‐H, Shieh T‐M, Chi T‐C, Chen H‐I, Chang W‐C, Chang S‐S (2015) The possible mechanism of advanced glycation end products (AGEs) for Alzheimer's disease. Plos One 10:e0143345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kummer MP, Heneka MT (2014) Truncated and modified amyloid‐beta species. Alzheimers Res Ther 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, Ervin FR, Snigdha S, Cotman CW, Saido TC, Vassar RJ, George‐Hyslop PS, Ikezu T, Schilling S, Demuth H‐U, Lemere CA (2013) Pyroglutamate‐3 amyloid‐β deposition in the brains of humans, non‐human primates, canines, and Alzheimer disease–like transgenic mouse models. Am J Pathol 183:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Atwood CS, Martins RN, Smith MA, Perry G (2002) Senile plaque composition and posttranslational modification of amyloid‐β peptide and associated proteins. Peptides 23:1343–1350. [DOI] [PubMed] [Google Scholar]
- 45. Thornalley P (1998) Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol 44:1013–1023. [PubMed] [Google Scholar]
- 46. Schmidt AM, Yan SD, Yan SF, Stern DM (2000) The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta Mol Cell Res 1498:99–111. [DOI] [PubMed] [Google Scholar]
- 47. Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M (2001) Abeta‐induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 158:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El‐Khoury JB (2002) CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta‐amyloid fibrils. Am J Pathol 160:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Husemann J, Loike JD, Kodama T, Silverstein SC (2001) Scavenger receptor class B type I (SR‐BI) mediates adhesion of neonatal murine microglia to fibrillar beta‐amyloid. J Neuroimmunol 114:142–150. [DOI] [PubMed] [Google Scholar]
- 50. Krieger M, Stern DM (2001) Series introduction: multiligand receptors and human disease. J Clin Invest 108:645–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ (2001) Familial amyloid polyneuropathy: receptor for advanced glycation end products‐dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci 21:7576–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kranenburg O, Bouma B, Kroon‐Batenburg LM, Reijerkerk A, Wu YP, Voest EE, Gebbink MF (2002) Tissue‐type plasminogen activator is a multiligand cross‐beta structure receptor. Curr Biol 12:1833–1839. [DOI] [PubMed] [Google Scholar]
- 53. Sun‐Ho H, Yoon Hee K, Inhee M‐J (2011) RAGE: the beneficial and deleterious effects by diverse mechanisms of actions. Mol Cells 31:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park H, Adsit FG, Boyington JC (2010) The 1.5 A crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J Biol Chem 285:40762–40770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G (2010) Structural basis for ligand recognition and activation of RAGE. Structure 18:1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rai V, Maldonado AY, Burz DS, Reverdatto S, Yan SF, Schmidt AM, Shekhtman A (2012) Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C‐terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem 287:5133–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M (1992) Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem 267:14987–14997. [PubMed] [Google Scholar]
- 58. Hudson BI, Kalea AZ, del Mar Arriero M, Harja E, Boulanger E, D'Agati V, Schmidt AM (2008) Interaction of the RAGE cytoplasmic domain with diaphanous‐1 is required for ligand‐stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 283:34457–34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Natale G, Ferrucci M, Lazzeri G, Paparelli A, Fornai F (2011) Transmission of prions within the gut and towards the central nervous system. Prion 5:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Monteiro FA, Cardoso I, Sousa MM, Saraiva MJ (2006) In vitro inhibition of transthyretin aggregate‐induced cytotoxicity by full and peptide derived forms of the soluble receptor for advanced glycation end products (RAGE). FEBS Lett 580:3451–3456. [DOI] [PubMed] [Google Scholar]
- 62. Arumugam T, Simeone DM, Schmidt AM, Logsdon CD (2004) S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J Biol Chem 279:5059–5065. [DOI] [PubMed] [Google Scholar]
- 63. Sparvero LJ, Asafu‐Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT (2009) RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Geroldi D, Falcone C, Minoretti P, Emanuele E, Arra M, D'Angelo A (2006) High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J Am Geriatric Soc 54:1149–1150. [DOI] [PubMed] [Google Scholar]
- 65. Forbes JM, Söderlund J, Yap FYT, Knip M, Andrikopoulos S, Ilonen J, Simell O, Veijola R, Sourris KC, Coughlan MT, Forsblom C, Slattery R, Grey ST, Wessman M, Yamamoto H, Bierhaus A, Cooper ME, Groop PH (2011) Receptor for advanced glycation end‐products (RAGE) provides a link between genetic susceptibility and environmental factors in type 1 diabetes. Diabetologia 54:1032–1042. [DOI] [PubMed] [Google Scholar]
- 66. Hudson BI, Stickland MH, Grant PJ (1998) Identification of polymorphisms in the receptor for advanced glycation end products (RAGE) gene: prevalence in type 2 diabetes and ethnic groups. Diabetes 47:1155. [DOI] [PubMed] [Google Scholar]
- 67. Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, Bucciarelli LG, Moser B, Moxley G, Itescu S, Grant PJ, Gregersen PK, Stern DM, Schmidt AM (2002) RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun 3:123. [DOI] [PubMed] [Google Scholar]
- 68. Osawa M, Yamamoto Y, Munesue S, Murakami N, Sakurai S, Watanabe T, Yonekura H, Uchigata Y, Iwamoto Y, Yamamoto H (2007) De‐N‐glycosylation or G82S mutation of RAGE sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta Gen Subj 1770:1468–1474. [DOI] [PubMed] [Google Scholar]
- 69. Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, Ma ZA (2009) Advanced glycation end products inhibit glucose‐stimulated insulin secretion through nitric oxide‐dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 150:2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee B, Chae HY, Kwon SJ, Park SY, Ihm J, Ihm S (2010) RAGE ligands induce apoptotic cell death of pancreatic β‐cells via oxidative stress. Int J Mol Med 26:813–818. [PubMed] [Google Scholar]
- 71. Han D, Yamamoto Y, Munesue S, Motoyoshi S, Saito H, Win MTT, Watanabe T, Tsuneyama K, Yamamoto H (2013) Induction of receptor for advanced glycation end products by insufficient leptin action triggers pancreatic β‐cell failure in type 2 diabetes. Genes Cells 18:302–314. [DOI] [PubMed] [Google Scholar]
- 72. Zhu Y, Shu T, Lin Y, Wang H, Yang J, Shi Y, Han X (2011) Inhibition of the receptor for advanced glycation endproducts (RAGE) protects pancreatic β‐cells. Biochem Biophys Res Commun 404:159–165. [DOI] [PubMed] [Google Scholar]
- 73. Tajiri Y, Möller C, Grill V (1997) Long term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function1. Endocrinology 138:273–280. [DOI] [PubMed] [Google Scholar]
- 74. Costal F, Oliveira E, Raposo A, Machado‐Lima A, Peixoto E, Roma L, Santos L, Lopes Faria JB, Carpinelli AR, Giannella‐Neto D, Passarelli M, Correa‐Giannella ML (2013) Dual effect of advanced glycation end products in pancreatic islet apoptosis. Diabetes/Metab Res Rev 29:296–307. [DOI] [PubMed] [Google Scholar]
- 75. Shu T, Zhu Y, Wang H, Lin Y, Ma Z, Han X (2011) AGEs decrease insulin synthesis in pancreatic beta‐cell by repressing Pdx‐1 protein expression at the post‐translational level. PLoS One 6:e18782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jiang Y, Zhang W, Xu S, Lin H, Sui W, Liu H, Peng L, Fang Q, Chen L, Lou J (2017) Transplantation of human fetal pancreatic progenitor cells ameliorates renal injury in streptozotocin‐induced diabetic nephropathy. J Transl Med 15:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Westermark P, Andersson A, Westermark GT (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91:795–826. [DOI] [PubMed] [Google Scholar]
- 78. Lukinius A, Wilander E, Westermark GT, Engstrom U, Westermark P (1989) Co‐localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia 32:240–244. [DOI] [PubMed] [Google Scholar]
- 79. Marzban L, Trigo‐Gonzalez G, Verchere CB (2005) Processing of pro‐islet amyloid polypeptide in the constitutive and regulated secretory pathways of beta cells. Mol Endocrinol 19:2154–2163. [DOI] [PubMed] [Google Scholar]
- 80. Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF (1988) An islet amyloid peptide is derived from an 89‐amino acid precursor by proteolytic processing. J Biol Chem 263:17243–17246. [PubMed] [Google Scholar]
- 81. Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA, Verchere CB (2006) Impaired NH2‐terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 55:2192–2201. [DOI] [PubMed] [Google Scholar]
- 82. Park K, Verchere CB (2001) Identification of a heparin binding domain in the N‐terminal cleavage site of pro‐islet amyloid polypeptide. Implications for islet amyloid formation. J Biol Chem 276:16611–16616. [DOI] [PubMed] [Google Scholar]
- 83. Paulsson JF, Westermark GT (2005) Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes 54:2117–2125. [DOI] [PubMed] [Google Scholar]
- 84. Meng F, Abedini A, Song B, Raleigh DP (2007) Amyloid formation by pro‐islet amyloid polypeptide processing intermediates: examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry 46:12091–12099. [DOI] [PubMed] [Google Scholar]
- 85. Wang H, Raleigh DP (2014) The ability of insulin to inhibit the formation of amyloid by pro‐islet amyloid polypeptide processing intermediates is significantly reduced in the presence of sulfated glycosaminoglycans. Biochemistry 53:2605–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Akter R, Cao P, Noor H, Ridgway Z, Tu LH, Wang H, Wong AG, Zhang X, Abedini A, Schmidt AM, Raleigh DP (2016) Islet amyloid polypeptide: structure, function, and pathophysiology. J Diabetes Res 2016:2798269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hull RL, Westermark GT, Westermark P, Kahn SE (2004) Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab 89:3629–3643. [DOI] [PubMed] [Google Scholar]
- 88. Jaikaran ET, Clark A (2001) Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta 1537:179–203. [DOI] [PubMed] [Google Scholar]
- 89. Raleigh D, Zhang X, Hastoy B, Clark A (2017) The beta‐cell assassin: IAPP cytotoxicity. J Mol Endocrinol 59:R121–R140. [DOI] [PubMed] [Google Scholar]
- 90. Abedini A, Raleigh DP (2005) The role of His‐18 in amyloid formation by human islet amyloid polypeptide. Biochemistry 44:16284–16291. [DOI] [PubMed] [Google Scholar]
- 91. Jha S, Snell JM, Sheftic SR, Patil SM, Daniels SB, Kolling FW, Alexandrescu AT (2014) pH dependence of amylin fibrillization. Biochemistry 53:300–310. [DOI] [PubMed] [Google Scholar]
- 92. Abedini A, Raleigh DP (2006) Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: is there a critical amyloidogenic domain in human IAPP?. J Mol Biol 355:274–281. [DOI] [PubMed] [Google Scholar]
- 93. Koo BW, Hebda JA, Miranker AD (2008) Amide inequivalence in the fibrillar assembly of islet amyloid polypeptide. Protein Eng Des Sel 21:147–154. [DOI] [PubMed] [Google Scholar]
- 94. Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kistler J, Cooper G, Aebi U (2003) Full‐length rat amylin forms fibrils following substitution of single residues from human amylin. J Mol Biol 326:1147–1156. [DOI] [PubMed] [Google Scholar]
- 95. Wang H, Abedini A, Ruzsicska B, Raleigh DP (2014) Rationally designed, nontoxic, nonamyloidogenic analogues of human islet amyloid polypeptide with improved solubility. Biochemistry 53:5876–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Williamson JA, Miranker AD (2007) Direct detection of transient alpha‐helical states in islet amyloid polypeptide. Protein Sci 16:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jayasinghe SA, Langen R (2005) Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 44:12113–12119. [DOI] [PubMed] [Google Scholar]
- 98. Nanga RP, Brender JR, Vivekanandan S, Ramamoorthy A (2011) Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys Acta 1808:2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Abedini A, Raleigh DP (2009) A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng Des Sel 22:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Williamson JA, Loria JP, Miranker AD (2009) Helix stabilization precedes aqueous and bilayer‐catalyzed fiber formation in islet amyloid polypeptide. J Mol Biol 393:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wiltzius JJ, Sievers SA, Sawaya MR, Eisenberg D (2009) Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci 18:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Abedini A, Raleigh DP (2009) A role for helical intermediates in amyloid formation by natively unfolded polypeptides?. Phys Biol 6:015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Luca S, Yau WM, Leapman R, Tycko R (2007) Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid‐state NMR. Biochemistry 46:13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wiltzius JJ, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, Eisenberg D (2008) Atomic structure of the cross‐beta spine of islet amyloid polypeptide (amylin). Protein Sci 17:1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lutz TA (2010) The role of amylin in the control of energy homeostasis. Am J Physiol Regul Integr Comp Physiol 298:R1475–R1484. [DOI] [PubMed] [Google Scholar]
- 106. Wielinga PY, Lowenstein C, Muff S, Munz M, Woods SC, Lutz TA (2010) Central amylin acts as an adiposity signal to control body weight and energy expenditure. Physiol Behav 101:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Morris DL, Rui L (2009) Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab 297:E1247–E1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Trevaskis JL, Parkes DG, Roth JD (2010) Insights into amylin‐leptin synergy. Trends Endocrinol Metab 21:473–479. [DOI] [PubMed] [Google Scholar]
- 109. Abedini A, Schmidt AM (2013) Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett 587:1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Abedini A, Plesner A, Cao P, Ridgway Z, Zhang J, Tu LH, Middleton CT, Chao B, Sartori DJ, Meng F, Wang H, Wong AG, Zanni MT, Verchere CB, Raleigh DP, Schmidt AM (2016) Time‐resolved studies define the nature of toxic IAPP intermediates, providing insight for anti‐amyloidosis therapeutics. Elife 5:e12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O'Brien TD, Glabe CG, Butler PC (2010) Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol 176:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhao HL, Sui Y, Guan J, He L, Gu XM, Wong HK, Baum L, Lai FM, Tong PC, Chan JC (2009) Amyloid oligomers in diabetic and nondiabetic human pancreas. Transl Res 153:24–32. [DOI] [PubMed] [Google Scholar]
- 113. Glabe CG (2008) Structural classification of toxic amyloid oligomers. J Biol Chem 283:29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, Fraser PE, Raleigh DP, Kahn SE (2010) Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence?. Diabetologia 53:1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Aston‐Mourney K, Hull RL, Zraika S, Udayasankar J, Subramanian SL, Kahn SE (2011) Exendin‐4 increases islet amyloid deposition but offsets the resultant beta cell toxicity in human islet amyloid polypeptide transgenic mouse islets. Diabetologia 54:1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta‐cell apoptosis prevents adaptive increase in beta‐cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52:2304–2314. [DOI] [PubMed] [Google Scholar]
- 117. Cooper GJ, Aitken JF, Zhang S (2010) Is type 2 diabetes an amyloidosis and does it really matter (to patients)?. Diabetologia 53:1011–1016. [DOI] [PubMed] [Google Scholar]
- 118. Matveyenko AV, Butler PC (2006) Beta‐cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 55:2106–2114. [DOI] [PubMed] [Google Scholar]
- 119. Park YJ, Lee S, Kieffer TJ, Warnock GL, Safikhan N, Speck M, Hao Z, Woo M, Marzban L (2012) Deletion of Fas protects islet beta cells from cytotoxic effects of human islet amyloid polypeptide. Diabetologia 55:1035–1047. [DOI] [PubMed] [Google Scholar]
- 120. Saafi EL, Konarkowska B, Zhang S, Kistler J, Cooper GJ (2001) Ultrastructural evidence that apoptosis is the mechanism by which human amylin evokes death in RINm5F pancreatic islet beta‐cells. Cell Biol Int 25:339–350. [DOI] [PubMed] [Google Scholar]
- 121. Zhang S, Liu J, Dragunow M, Cooper GJ (2003) Fibrillogenic amylin evokes islet beta‐cell apoptosis through linked activation of a caspase cascade and JNK1. J Biol Chem 278:52810–52819. [DOI] [PubMed] [Google Scholar]
- 122. Westwell‐Roper C, Dai DL, Soukhatcheva G, Potter KJ, van Rooijen N, Ehses JA, Verchere CB (2011) IL‐1 blockade attenuates islet amyloid polypeptide‐induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol 187:2755–2765. [DOI] [PubMed] [Google Scholar]
- 123. Bram Y, Frydman‐Marom A, Yanai I, Gilead S, Shaltiel‐Karyo R, Amdursky N, Gazit E (2014) Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes‐associated specific antibodies. Sci Rep 4:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, Novials A (2007) Impairment of the ubiquitin‐proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta‐cell apoptosis. Diabetes 56:2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gupta D, Leahy JL (2014) Islet amyloid and type 2 diabetes: overproduction or inadequate clearance and detoxification?. J Clin Invest 124:3292–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Huang CJ, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S, Wu X, Butler AE, Butler PC (2010) Calcium‐activated calpain‐2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem 285:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hull RL, Zraika S, Udayasankar J, Aston‐Mourney K, Subramanian SL, Kahn SE (2009) Amyloid formation in human IAPP transgenic mouse islets and pancreas, and human pancreas, is not associated with endoplasmic reticulum stress. Diabetologia 52:1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mirzabekov TA, Lin MC, Kagan BL (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem 271:1988–1992. [DOI] [PubMed] [Google Scholar]
- 129. Morita S, Sakagashira S, Shimajiri Y, Eberhardt NL, Kondo T, Kondo T, Sanke T (2011) Autophagy protects against human islet amyloid polypeptide‐associated apoptosis. J Diabetes Investig 2:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rivera JF, Gurlo T, Daval M, Huang CJ, Matveyenko AV, Butler PC, Costes S (2011) Human‐IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta‐cells: protective role of p62‐positive cytoplasmic inclusions. Cell Death Differ 18:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ (2013) CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 14:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC (1999) The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate‐sized toxic amyloid particles. Diabetes 48:491–498. [DOI] [PubMed] [Google Scholar]
- 133. Kim J, Cheon H, Jeong YT, Quan W, Kim KH, Cho JM, Lim YM, Oh SH, Jin SM, Kim JH, Lee MK, Kim S, Komatsu M, Kang SW, Lee MS (2014) Amyloidogenic peptide oligomer accumulation in autophagy‐deficient beta cells induces diabetes. J Clin Invest 124:3311–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC (2014) Autophagy defends pancreatic beta cells from human islet amyloid polypeptide‐induced toxicity. J Clin Invest 124:3489–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Shigihara N, Fukunaka A, Hara A, Komiya K, Honda A, Uchida T, Abe H, Toyofuku Y, Tamaki M, Ogihara T, Miyatsuka T, Hiddinga HJ, Sakagashira S, Koike M, Uchiyama Y, Yoshimori T, Eberhardt NL, Fujitani Y, Watada H (2014) Human IAPP‐induced pancreatic beta cell toxicity and its regulation by autophagy. J Clin Invest 124:3634–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hebda JA, Miranker AD (2009) The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu Rev Biophys 38:125–152. [DOI] [PubMed] [Google Scholar]
- 137. Cao P, Abedini A, Wang H, Tu LH, Zhang X, Schmidt AM, Raleigh DP (2013) Islet amyloid polypeptide toxicity and membrane interactions. Proc Natl Acad Sci USA 110:19279–19284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Seeliger J, Weise K, Opitz N, Winter R (2012) The effect of Abeta on IAPP aggregation in the presence of an isolated beta‐cell membrane. J Mol Biol 421:348–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhang X, St Clair JR, London E, Raleigh DP (2017) Islet amyloid polypeptide membrane interactions: effects of membrane composition. Biochemistry 56:376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Meier DT, Morcos M, Samarasekera T, Zraika S, Hull RL, Kahn SE (2014) Islet amyloid formation is an important determinant for inducing islet inflammation in high‐fat‐fed human IAPP transgenic mice. Diabetologia 57:1884–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Westwell‐Roper CY, Ehses JA, Verchere CB (2014) Resident macrophages mediate islet amyloid polypeptide‐induced islet IL‐1beta production and beta‐cell dysfunction. Diabetes 63:1698–1711. [DOI] [PubMed] [Google Scholar]
- 142. Nishi M, Nanjo K (2011) Insulin gene mutations and diabetes. J Diabetes Invest 2:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, Beccaria L, Monciotti C, Toni S, Pedersen O, Hansen T, Federici L, Pesavento R, Cadario F, Federici G, Ghirri P, Arvan P, Iafusco D, Barbetti F The Early Onset Diabetes Study Group of the Italian Society of Pediatric E, Diabetes (2008) Seven mutations in the human insulin gene linked to permanent neonatal/infancy‐onset diabetes mellitus. J Clin Invest 118:2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, Damm P, Bergholdt R, Pociot F, Pisinger C, Barbetti F, Lebl J, Pedersen O, Hansen T (2010) Further evidence that mutations in INS can be a rare cause of Maturity‐Onset Diabetes of the Young (MODY). BMC Med Genet 11:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, Fetita S, Tarasov AI, Guillausseau P‐J, Boesgaard TW, Pedersen O, Hansen T, Polak M, Gautier J‐F, Froguel P, Rutter GA, Vaxillaire M (2010) Insulin gene mutations resulting in early‐onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 59:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sheng Q, Xiao X, Prasadan K, Chen C, Ming Y, Fusco J, Gangopadhyay NN, Ricks D, Gittes GK (2017) Autophagy protects pancreatic beta cell mass and function in the setting of a high‐fat and high‐glucose diet. Sci Rep 7:16348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Guo H, Xiong Y, Witkowski P, Cui J, Wang L‐j, Sun J, Lara‐Lemus R, Haataja L, Hutchison K, Shan S‐o, Arvan P, Liu M (2014) Inefficient translocation of preproinsulin contributes to pancreatic β cell failure and late‐onset diabetes. J Biol Chem 289:16290–16302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu M, Hodish I, Rhodes CJ, Arvan P (2007) Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA 104:15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, Kay TWH, Rossjohn J, Falk BA, Nepom GT, Purcell AW (2005) The insulin A‐chain epitope recognized by human T cells is posttranslationally modified. J Exper Med 202:1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, Gomez‐Touriño I, Arif S, Peakman M, Drijfhout JW, Roep BO (2014) Posttranslational modification of HLA‐DQ binding islet autoantigens in Type 1 diabetes. Diabetes 63:237. [DOI] [PubMed] [Google Scholar]
- 151. Røder ME, Porte JD, Schwartz RS, Kahn SE (1998) Disproportionately elevated proinsulin levels reflect the degree of impaired B cell secretory capacity in patients with noninsulin‐dependent diabetes mellitus1. J Clin Endocrinol Metab 83:604–608. [DOI] [PubMed] [Google Scholar]
- 152. Riahi Y, Wikstrom J, Bachar‐Wikstrom E, Polin N, Zucker H, Lee M‐S, Quan W, Haataja L, Liu M, Arvan P, Cerasi E, Leibowitz G (2016) Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 59:1480–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, Marchetti P (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52:1083–1086. [DOI] [PubMed] [Google Scholar]
- 154. Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, Kawamori R, Fujitani Y, Watada H (2008) Autophagy is important in islet homeostasis and compensatory increase of Beta cell mass in response to high‐fat diet. Cell Metab 8:325–332. [DOI] [PubMed] [Google Scholar]
- 155. Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon K‐H, Kim J‐W, Jeong YT, Han MS, Lee M‐K, Kim K‐W, Shin J, Lee M‐S (2008) Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab 8:318–324. [DOI] [PubMed] [Google Scholar]
- 156. Quan W, Hur KY, Lim Y, H, Oh S, Lee JC, Kim KH, H Kim G, Kim S‐W, L, Kim H, Lee MK, Kim K, Kim J, Komatsu M, Lee MS (2012) Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 55:392–403. [DOI] [PubMed] [Google Scholar]
- 157. Bachar‐Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, Leibowitz G (2013) Stimulation of autophagy improves endoplasmic reticulum stress–induced diabetes. Diabetes 62:1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Goginashvili A, Zhang Z, Erbs E, Spiegelhalter C, Kessler P, Mihlan M, Pasquier A, Krupina K, Schieber N, Cinque L, Morvan J, Sumara I, Schwab Y, Settembre C, Ricci R (2015) Insulin secretory granules control autophagy in pancreatic β cells. Science 347:878. [DOI] [PubMed] [Google Scholar]
- 159. Leloup C, Tourrel‐Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani A‐L, Ktorza A, Casteilla L, Pénicaud L (2009) Mitochondrial reactive oxygen species are obligatory signals for glucose‐induced insulin secretion. Diabetes 58:673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Affourtit C, Jastroch M, Brand MD (2011) Uncoupling protein‐2 attenuates glucose‐stimulated insulin secretion in INS‐1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Rad Biol Med 50:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Robson‐Doucette CA, Sultan S, Allister EM, Wikstrom JD, Koshkin V, Bhattacharjee A, Prentice KJ, Sereda SB, Shirihai OS, Wheeler MB (2011) β‐Cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 60:2710–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Munhoz AC, Riva P, Simões D, Curi R, Carpinelli AR (2016) Control of insulin secretion by production of reactive oxygen species: study performed in pancreatic islets from fed and 48‐hour fasted Wistar rats. PLoS One 11:e0158166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PIH (2016) Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J 473:4527. [DOI] [PubMed] [Google Scholar]
- 164. Aspinwall CA, Lakey JRT, Kennedy RT (1999) Insulin‐stimulated insulin secretion in single pancreatic Beta cells. J Biol Chem 274:6360–6365. [DOI] [PubMed] [Google Scholar]
- 165. Halperin F, Lopez X, Manning R, Kahn CR, Kulkarni RN, Goldfine AB (2012) Insulin augmentation of glucose‐stimulated insulin secretion is impaired in insulin‐resistant humans. Diabetes 61:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Borg DJ, Forbes JM (2016) Targeting advanced glycation with pharmaceutical agents: where are we now?. Glycoconj J 33:653–670. [DOI] [PubMed] [Google Scholar]