

Abstract
Prostaglandin (PG) D2 is the ligand for the G‐protein coupled receptors DP1 (D‐type prostanoid receptor 1) and DP2 (also known as chemoattractant receptor homologous molecule, expressed on Th2 cells; CRTH2). Both, DP1 and DP2 are expressed on the cellular surface of eosinophils; although it has become quite clear that PGD2 induces eosinophil migration mainly via DP2 receptors, the role of DP1 in eosinophil responses has remained elusive. In this study, we addressed how DP1 receptor signaling complements the pro‐inflammatory effects of DP2. We found that PGD2 prolongs the survival of eosinophils via a DP1 receptor‐mediated mechanism that inhibits the onset of the intrinsic apoptotic cascade. The DP1 agonist BW245c prevented the activation of effector caspases in eosinophils and protected mitochondrial membranes from depolarization which—as a consequence—sustained viability of eosinophils. DP1 activation in eosinophils enhanced the expression of the anti‐apoptotic gene BCL‐XL, but also induced pro‐inflammatory genes, such as VLA‐4 and CCR3. In HEK293 cells that overexpress recombinant DP1 and/or DP2 receptors, activation of DP1, but not DP2, delayed cell death and stimulated proliferation, along with induction of serum response element (SRE), a regulator of anti‐apoptotic, early‐response genes. We conclude that DP1 receptors promote the survival via SRE induction and induction of pro‐inflammatory genes. Therefore, targeting DP1 receptors, along with DP2, may contribute to anti‐inflammatory therapy in eosinophilic diseases.
Keywords: allergy, apoptosis, inflammation, prostanoids
Short abstract
The PGD2‐DP1 axis impedes caspase activity, protects mitochondrial function and consequently sustains eosinophil survival by controlling SRE induction and upregulation of pro‐inflammatory and anti‐apoptotic genes.
Abbreviations
- Δ12‐PGJ2
Δ12‐prostaglandin J2
- 15‐deoxy‐PGJ2
15‐deoxy‐Δ12,14‐ prostaglandin J2
- Bcl‐XL
B‐cell lymphoma‐extra‐large
- BLT1/2
leukotriene B4 receptor 1/2
- C5A‐R1
complement component C5a receptor 1
- CCCP
carbonyl cyanide m‐chlorophenyl hydrazon
- CCR3
C‐C chemokine receptor type 3
- DKPGD2
13,14di‐hydro 15‐keto PGD2
- DP1
D‐type prostanoid receptor 1
- DP2
D‐type prostanoid receptor 2 [also chemoattractant receptor homologous molecule of Th2 cells (CRTH2)]
- ECIS
electric cell‐substrate impedance sensing
- HEK293
human embryonic kidney cells 293
- PG
prostaglandin
- PI
propidium iodide
- PPAR
peroxisome proliferator‐activated receptor
- SRE
serum response element
- SRF
serum response factor
- VLA‐4
very late antigen 4
1. INTRODUCTION
Numerous factors drive the progression of allergic conditions, affecting either the immediate, early, or the late phase of the allergic response. In the pivotal step of the inflammatory cascade mast cells release mediators such as histamine, prostaglandin (PG) D2, leukotriene C4, TNF‐α, and many others,1 which start off the allergic response in the first place, induce the recruitment of inflammatory cells into the tissue and finally stimulate the surrounding and infiltrating cells to drive the transition from early to late phase, resulting in tissue damage. Eosinophils are considered as crucial effector cells in chronic allergic inflammation since they are involved in increasing epithelial‐to‐mesenchymal transition,2, 3 thickening of airway walls, airway hyperresponsiveness, and angiogenesis.4, 5 Therefore, eosinophils are of major therapeutic interest in allergic diseases.
Concomitant with allergic inflammation, the life span of immune cells such as macrophages, Th2 cells, or eosinophils is known to be prolonged.6 Accordingly, blocking the prominent pro‐survival cytokine IL‐5 using the monoclonal antibodies mepolizumab or reslizumab profoundly reduced eosinophil numbers and effectively prevented exacerbations in cases of severe, highly eosinophilic asthma.7, 8, 9 This demonstrated that targeting mechanisms that regulate eosinophil survival is a clinically relevant approach in allergic diseases.
PGD2 is the major lipid mediator released by mast cells following crosslinking of allergen‐specific IgE molecules displayed on their surface in sensitized individuals as part of the early allergic reaction.10, 11 Furthermore, eosinophils, dendritic cells, macrophages, and endothelial cells12 produce PGD2 and substantial amounts have been detected in tissues affected by allergic reactions, such as lung,13 skin,14, 15 and esophagus. 16 PGD2 is the ligand for two 7‐transmembrane G protein‐coupled receptors, named D‐type prostanoid (DP) receptor 1 and chemoattractant receptor homologous molecule expressed on Th2 cells (CRTH2, also termed DP2),17 that are co‐expressed on the cell surface of eosinophils.18 Although DP1 and DP2 bind PGD2 with similar avidity,17 DP2 shares greater homology with classical chemoattractant receptors (e.g., BLT1/2, C5a‐R1, CCR3, FPR) than with other prostanoid receptors.19, 20
PGD2 and selective DP2 agonists cause chemotaxis and pro‐inflammatory activation of eosinophils including cellular responses, such as Ca2+ flux, CD11b upregulation, respiratory burst, and eosinophil cationic protein release,20, 21, 22, 23 via binding to DP2. In contrast, the functional responses and exact signaling pathways triggered by DP1 activation in eosinophils have remained unclear up to now. We have previously shown in eosinophils and heterologous expression systems that the DP1 receptor augments the signaling and functionality of the DP2 receptor, including intracellular Ca2+ flux, chemotaxis, and oxidative burst.18, 24, 25 Defining the role of DP1 on eosinophils might hence contribute to the full understanding of the role of PGD2 in allergic disease and asthma and explain the limited efficacy of DP2 antagonists in clinical studies.12 One single report described that activation of DP1 on eosinophils protected them from undergoing apoptosis21; whether this resulted in extended life‐span of eosinophils and was due to activation of pro‐survival signaling pathways has still remained unresolved. Another study, however, presented opposing data arguing for a pro‐apoptotic effect of higher concentrations (10 μM) of PGD2 that was attributed to inhibition of IkB degradation in eosinophils by the PGD2 metabolites 15‐deoxy‐PGJ2 and Δ12‐PGJ2.26
In order to unequivocally decipher the role of PGD2 and its receptors in the regulation of eosinophil survival, this study investigated how DP1 regulates eosinophil survival and operates as a transcriptional regulator in a pro‐inflammatory setting. Thus, we propose that DP1 antagonists could have beneficial effects by accelerating the resolution of allergic inflammation.
2. METHODS
2.1. Materials
Prostaglandin D2 receptor agonists PGD2, BW245c, 13,14‐di‐hydro 15‐keto PGD2 (DK‐PGD2), and antagonists MK0524, Cay10471, and BW A868c were purchased from Cayman, Ann Arbor, USA, and rosiglitazone, BH3I‐1, and HA14.1 were purchased from Sigma–Aldrich, St. Louis, USA.
2.2. Preparation of human peripheral blood eosinophils
Human eosinophils were isolated from blood samples of healthy volunteers according to a protocol approved by the Institutional Review Board of the Medical University of Graz or Cincinnati Children's Hospital Medical Center as previously described.27 In brief, erythrocytes were removed by dextran sedimentation and polymorphonuclear leukocytes were separated from mononuclear cells by density gradient centrifugation (Histopaque 1077, Sigma–Aldrich). Eosinophils were separated from neutrophils in the polymorphonuclear leukocyte fraction by negative magnetic selection using the MACS cell separation system (Eosinophil Isolation Kit, Milteny Biotech, Bergisch Gladbach, Germany) with a resulting purity of typically ≥98%.
2.3. Human embryonic kidney cells
Previously described Human embryonic kidney 293 (HEK293) cells lines stably expressing DP1, DP2, or both receptors18 are referred to as HEK‐DP1, HEK‐DP2+DP1, and HEK‐DP2. HEK293 cells were kept in serum containing selection medium (DMEM + 10% FBS) with either neomycin (0.2%) or zeocin (0.4%), or both. Cells were propagated in 75 cm2 cell culture flasks and medium was changed every second day. Cells were harvested after 3–4 days of culturing when they have grown to 90% confluence. Zeocin and Neomycin (Geneticin, G418), DMEM, and FBS were purchased from Thermo Fisher Scientific, Waltham, MA, USA.
2.4. Reporter gene assays
HEK‐DP1, HEK‐DP2, or HEK‐DP1 + DP2 cells were seeded in 96‐well plates at a density of 6 × 105 cells/well and grown to approximately 90% confluence in non‐selective medium (DMEM + 10% FCS). Using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) cells were transiently transfected with pSRE‐Luc reporter plasmid (100 ng/well) according to the manufacturer's protocol. Twenty‐four hours post transfection cells were pre‐treated with antagonists for 20 min (or the respective vehicle) followed by agonist stimulation for 3 h. Luciferase activity was determined using the Steadylite Plus Kit (PerkinElmer, Waltham, USA), as previously described.28, 29 Chemiluminescence was measured on a TopCount NXT device (Perkin Elmer/Packard Bioscience).
2.5. Annexin V/propidium iodide co‐staining
Isolated eosinophils (5 × 105/ml) were kept in RPMI (Thermo Fisher Scientific) supplemented with 1% FBS and PenStrep (Sigma–Aldrich) and stimulated with 1 μM PGD2, DK‐PGD2, or BW245c for 18 h at 37°C. After washing, cells were incubated with Annexin V and propidium iodide (PI) according to the manufacturer's protocol (Annexin V‐FITC Apoptosis Detection Kit I, BD Pharmingen) and subsequently analyzed on a FACSCalibur flow cytometer (BD, Franklin Lakes, NJ, USA).
2.6. Determination of mitochondrial integrity
Eosinophils were incubated with the potential‐dependent JC‐1 dye (5 μg/ml) (Sigma–Aldrich) for 20 min at 37°C. The percentage of cells with depolarized mitochondrial membrane potential was determined by the loss of red fluorescence. Carbonyl cyanide m‐chlorophenyl hydrazon (CCCP) destroys the mitochondrial membrane potential by uncoupling oxidative phosphorylation in mitochondria and hence increasing membrane permeability to ions.
MitoTracker Red CMXRos dye (75 nM) (Life Technologies, Carlsbad, CA, USA) was added to the cell suspension for 45 min and mitochondrial membrane integrity was detected by fluorescence microscopy (Zeiss Axiovert 40 CFL microscope, Olympus DP50‐CU digital camera and Olympus CellˆP software (Olympus, Lake Success, NY, USA).
2.7. Cell viability assay
HEK‐DP2+DP1, HEK‐DP1, and HEK‐DP2 cells were seeded in 96well plates at a density of 3 × 105 cells/ml and grown to confluence. Subsequently, cells were starved (OptiMEM, Thermo Fisher Scientific), treated with indicated concentrations of PGD2 and viability was determined as previously described using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega; Madison, WI, USA)30 at 24 and 48 h of PGD2 treatment.
2.8. Caspase 3/7 Glo assay
Eosinophils were seeded in 96‐well plates at a density of 0.5 × 105 cells/well in serum reduced medium (RPMI, 1% FBS, 1% PenStrep), and incubated for 18 h. Caspase‐Glo 3/7 assay was performed according to the manufacturer's protocol (Promega). Relative light units were detected with a chemiluminescence plate reader (TopCount NTX, Perkin Elmer/Packard Bioscience).
2.8.1. Electric cell‐substrate impedance sensing
Growth performance of HEK‐DP2+DP1, HEK‐DP1, and HEK‐DP2 was monitored using the electric cell‐substrate impedance sensing (ECIS) system (Applied Biophysics, Troy, NY, USA). Cells (3 × 104 cells/well) were seeded in gelatin coated 96W1E + polycarbonate arrays equipped with gold microelectrodes and allowed to settle overnight. Medium was changed to serum free (OptiMEM) medium 2 h prior to treatment with vehicle, PGD2 (0.1–10 μM) or FBS (10%). Impedance was measured at 4000 Hz and monitored continuously over ≥60 h.
2.8.2. Microscopy
HEK‐DP2+DP1, HEK‐DP1, and HEK‐DP2 cells were seeded in 96‐well plates at a density of 3 × 105 cells/ml and grown to confluency. Subsequently, cells were starved (OptiMEM) and treated with indicated concentrations of PGD2. Phase contrast images were taken after 48 h of culture in serum free media on a Zeiss Axiovert 40 CFL microscope and Zeiss LD A‐Plan 20×/0.30 Ph1 lens, using a Hamamatsu ORCA‐03G digital camera. Data show representative images of 4 independent experiments.
2.9. Gene expression analysis
Isolated peripheral blood eosinophils were lysed in TriPure and total RNA was extracted with RNeasy Mini Kit (Qiagen, Hilden, Germany) after incubation in RPMI (1% FBS, 1% PenStrep) for 3 h in the presence of 1 μM of PGD2, DK‐PGD2, or BW245c or 10 ng/ml of IL‐5. cDNA was synthesized from isolated RNA with iScript (BioRad, Hercules, CA, USA) according to manufacturer's instructions and used as the template for quantitative real time PCR (CFX, BioRad) and the use of SYBR Green Master Mix (BioRad). Relative expression of mRNA levels was calculated by normalizing to 18 s rRNA (2−Δct). Data are depicted as arbitrary units of vehicle control cells. The following primer pairs were used:
CCR3 (5′‐CATTGTCCATGCTGTGTTTGC‐3′; 5′‐AGGTGACGATGCTGGTGATGA‐3′), VLA‐4; (5′‐GTCCTTGTTTAATGCTGGAGATG AT‐3′; 5′‐GCTTCTCTTCCAGCTCTAAAATCTT‐3′), bcl‐xl (5′‐GCGTA GACAAGGAGATGCAGGT‐3′; 5′‐GGTCATTCAGGTAAGTGGCCAT‐3′), bax (5′‐AAAGATGGTCACGGTCTGCC‐3′; 5′‐TCCAAGACCAGGGT GGTTG‐3′),31 18S (5′‐GTGGAGCGATTTGTCTGGT‐3′; 5′‐GGACA TCTAAGGCATCACAG‐3′).
2.10. Flow cytometric analysis of CCR3 and VLA‐4 expression
Isolated eosinophils (5 × 105/ml) were kept in RPMI (Thermo Fisher Scientific) supplemented with 1% FBS and PenStrep (Sigma–Aldrich) and stimulated with 1 μM DK‐PGD2 or BW245c for 18 h at 37°C. Cells were stained with mouse anti‐human CD193 (CCR3)–BV421 (BioLegend, 5E8) and mouse anti‐human CD49d (VLA‐4)‐PE (BD Biosciences, 9F10), or the respective isotype controls. Human TruStain FcX (BioLegend) was used as Fc receptor blocking solution. Data were acquired in a BD FACS Canto II.
2.11. Statistical analysis
Data are shown as means ± sem or as individual data values. Statistical analysis was carried out with Graph Pad Prism® 7 (GraphPad Software, Inc. CA, USA). Differences between groups were tested by one‐way or two‐way ANOVA followed by Dunnett's, Tukey's, or Sidak's posttest. P values ≤ 0.05 were considered significant and are indicated as *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; and ****P ≤ 0.0001.
3. RESULTS
3.1. DP1 — but not DP2 activation promotes survival of eosinophils
We first tested the potential pro‐survival effect of the two PGD2 receptors DP1 and DP2 on eosinophils under ex vivo culture conditions. Isolated peripheral blood eosinophils were cultured in media containing 1% FBS for 18 h. Concurrently, eosinophils were treated with 1 μM of PGD2, the selective DP1 agonist BW245c, the selective DP2 agonist DK‐PGD2, or IL‐5 [100 pM]. IL‐5 is well known for its pro‐survival stimulus on eosinophils.32
The specific DP1 agonist BW245c significantly enhanced the percentage of viable cells (Annexin V−/PI−) from 25 to 50% of all eosinophils; by comparison, IL‐5 maintained 59% of the cells viable (Fig. 1A). BW245c concentration‐dependently inhibits apoptosis of eosinophils, with a half maximal efficacy (EC50) of 0.826 μM (Supplementary Fig. 1). PGD2 itself moderately increased the percentage of viable cells to 39%. In contrast, the DP2 agonist DK‐PGD2 at the same concentration as BW245c led only to a minor enhancement of the percentage of viable cells when compared to vehicle controls.
Figure 1.
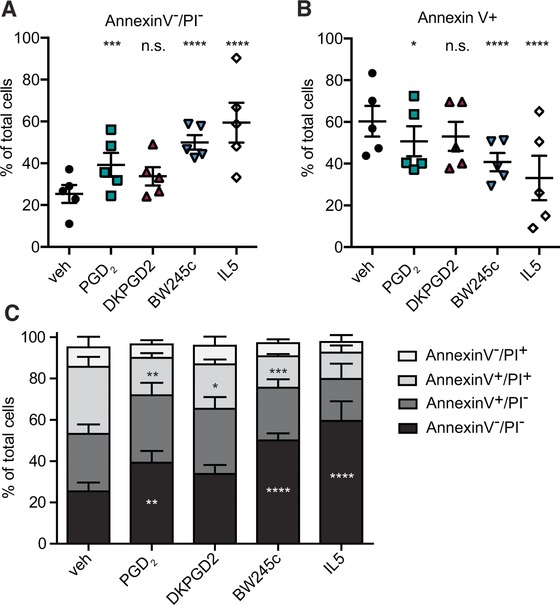
DP1 receptor activation promotes survival of eosinophils. Isolated eosinophils were cultured with or without 1 μM of PGD2, DK‐PGD2, BW245c, or IL‐5 [100 pM] for 18 h. BW245c, PGD2, and IL‐5 significantly enhanced the portion of annexin V−/PI− eosinophils (A) and PGD2, BW245c, DK‐PGD2, and IL‐5 decreased the annexin V+ population (B) as compared to vehicle‐treated cells. (C) Shows the percentage of annexin V−/PI−, annexin V+/PI−, annexin V+/PI+, annexin V−/PI+ populations of total eosinophils at 18 h. Data show mean ± sem of 5 individual experiments using eosinophils from different donors
Under the same experimental conditions, the apoptotic (Annexin V+) population of cultured eosinophils was reduced from 60% (vehicle control cells) to 41% (BW245c), to 51% (PGD2) and to 33% (IL‐5), respectively (Fig. 1B). Figure 1C discriminates between early and late apoptotic cells and depicts the distribution of single and double‐positive stained populations. Twenty‐five percent of vehicle‐treated control cells were identified as viable (Annexin V−/PI−), 28% as early apoptotic (Annexin V+/PI−), 33% were positive for both (Annexin V+/PI+), while the necrotic population (Annexin V−/PI+) represented 9% (Fig. 1C).
Of the BW245c treated eosinophils 50% were viable, 25% early‐ and 15% late‐apoptotic, and 6% were necrotic after 18 h. IL‐5 treatment led proportionally to a similar result with a slightly more pronounced effect on the increase of live cells (59% viable, 20% early‐, 13% late‐apoptotic, 8% necrotic eosinophils). Annexin V/PI staining at 0 and 3 h revealed that at these early time points there were no significant differences between these treatment groups (data not shown).
We conclude that activation of the DP1 receptor functions as a pro‐survival stimulus for eosinophils and reduces the portion of Annexin V+ and PI+ cell populations.
3.2. DP1 signaling enhances eosinophil survival by inhibiting the intrinsic apoptosis pathway
Since the DP1 agonist BW245c prolonged the survival of eosinophils, we aimed to identify the pro‐survival signals that were induced by DP1 receptor activation and to assess whether this function of PGD2 included the activation of programmed cell death pathways. Therefore, we investigated the potential involvement of DP1‐mediated signaling on the onset of the apoptotic cascade in terms of effector caspase 3/7 activation, mitochondrial membrane potential, and involvement of the anti‐apoptotic protein Bcl‐XL. Pore formation in the mitochondrial membrane leads to a loss of the mitochondrial membrane potential ( Δψm) and subsequently to the release of cytochrome C; inhibition of this pathway was shown to be directly linked to the recruitment and/or stabilization of anti‐apoptotic proteins of the Bcl‐2 family.33
BW245c significantly decreased the activity of effector caspases 3 and 7 by 27% in eosinophils aged in serum‐reduced media for 18 h, as compared to vehicle‐treated control cells (Fig. 2A). The DP1 receptor antagonist MK0524 prevented BW245c‐induced protection from caspase 3/7 activation in a concentration‐dependent manner (Fig. 2B). PGD2 failed to significantly decrease effector caspase activity by itself but, of note, after pharmacological blocking of DP2 receptors by the antagonist Cay10471, PGD2 gained the capacity to decrease caspase 3/7 activity (Fig. 2C).
Figure 2.
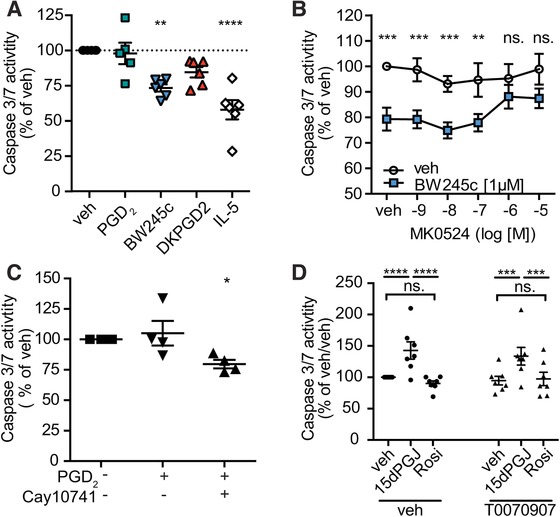
Activation of the DP1 receptor inhibits the intrinsic apoptotic pathway in eosinophils. Caspase 3/7 activity in eosinophils was assessed in a luminescent assay after 18 h incubation with vehicle, 1 μM of PGD2, BW245c, DK‐PGD2, or 15d‐PGJ2, Rosiglitazone (Rosi), or 100 pM of IL‐5. Cells were pretreated with 1 μM of vehicle, Cay10471 or T0070907 or increasing concentrations of MK0524 prior to agonist treatment. (A) BW245c protected from effector caspase 3/7 activity in eosinophils cultured for 18 h in serum‐reduced media (RPMI 1% FBS, 1% PenStrep). Data show means ± SEM of 5–6 individual experiments. (B) DP1 receptor antagonist MK0524 reversed the BW245c‐induced protection from caspase 3/7 activation. Data show mean ± sem of 9 individual experiments. (C) Blockade of DP2 (Cay10471) unmasks PGD2 as a potent inhibitor of caspase 3/7 activation. Data show mean ± sem of 4 individual experiments. (D) 15dPGJ2, but not rosiglitazone (Rosi), increased caspase 3/7 activity, which was not altered by the PPAR‐α antagonist T0070907 (n = 7)
PGD2 is a short‐lived molecule that can rapidly be metabolized to J‐metabolites such as 15‐deoxy‐Δ12 , Δ14‐PGJ2 (15dPGJ2), which has been reported to have a higher affinity for DP2 than DP1.17 15dPGJ2 also activates the intracellular peroxisome proliferator‐activated receptor (PPAR)‐γ at micromolar concentrations, thereby inducing apoptosis of eosinophils.34 In our hands, 1 μM of 15dPGJ2 significantly increased caspase 3/7 activity in a PPAR‐γ‐independent manner, since 1 μM of the PPARγ T0070907 antagonist did not reverse the 15dPGJ2‐induced caspase 3/7 activation. Moreover, the PPAR‐γ agonist rosiglitazone (1 μM) did not mimic the 15dPGJ2‐induced caspase activation (Fig. 2D). T0070907 had no effect on basal caspase 3/7 activity.
3.3. BW245c, PGD2 and IL‐5 protect eosinophils from mitochondrial membrane depolarization
Mitochondrial membrane depolarization is an early step of the intrinsic apoptotic cascade and can be determined by staining cells with ΔΨm‐dependent fluorescent dye JC‐1, whose fluorescent characteristics depend on the integrity of the mitochondrial membrane potential (ΔΨm). In cells with intact ΔΨm the dye binds and forms J‐aggregates at the mitochondrial membrane, and emits light at 590 nm after excitation at 527 nm. Due to the low ΔΨm in early apoptotic cells J‐aggregate formation is prevented and allows JC‐1 to be present in its monomeric form which emits light at 527 nm.35 Therefore, this initiating step in the onset of the intrinsic apoptotic cascade can be detected by the loss of fluorescence intensity at 590 nm. Indeed, a smaller proportion of eosinophils showed a depolarized mitochondrial membrane when cultured for 18 h in presence of PGD2 (22%), BW245c (18%), or IL‐5 (20%) compared to vehicle‐treated eosinophils (37%). The mitochondrial uncoupler CCCP was added to the cells for the last 30 min of culture and was used as a control for collapsed ΔΨm (Fig. 3A and B). DK‐PGD2 did not have a significant impact on mitochondrial membrane depolarization of eosinophils.
Figure 3.
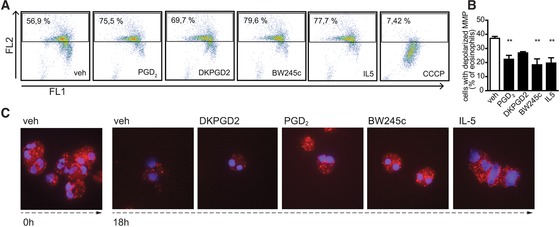
Activation of the DP1 receptor maintains mitochondrial function in aging eosinophils. (A, B) Flow cytometric dot plot analysis of eosinophils stained with JC1 after 18 h incubation with 1 μM of vehicle, PGD2, BW245c, DK‐PGD2, or 50 pM of IL‐5. Depolarized mitochondrial membrane potential (MMP) is shown by the loss in yellow fluorescence (FL2). (A) Shows representative dot plots and (B) means of n = 3–4 individual experiments ± sem. **P ≤ 0.01. (C) MitoTracker Red CMXRos staining of eosinophils incubated with 1 μM of vehicle, PGD2, BW245c, and DK‐PGD2, and 100 pM of IL‐5 up to 18 h. Representative images of 3 independent experiments are shown
Additionally, eosinophils incubated with PGD2, BW245c, or IL‐5 showed more intense staining of intact mitochondria than vehicle‐treated cells when identified by mitochondria‐selective probes (MitoTracker Red CMXRos), which selectively accumulate and are oxidized at the mitochondrial membranes with intact membrane potential (Fig. 3C).
3.4. BCL‐XL protein inhibitors abrogate DP1‐mediated anti‐apoptotic signaling
To investigate whether the pro‐survival effect of DP1 includes the anti‐apoptotic proteins of the Bcl‐2 family we tested the Bcl‐2 family protein inhibitors BH3I‐1 and HA14.1 with regard to their impact on apoptosis and effector caspase activation in eosinophils. Bcl‐XL belongs to the family of Bcl‐2 proteins. The protein is expressed in human eosinophils and is involved in the pro‐survival signaling initiated by IL‐5 and GM‐CSF.36
The Bcl‐2 family inhibitor HA‐14.1 has been characterized as a ligand of a Bcl‐2 surface pocket and inhibits the binding of anti‐apoptotic Bcl‐2 proteins on the mitochondrial membrane. The small molecule compound was shown to induce apoptosis associated with mitochondrial membrane depolarization and caspase activation.37 Hence, we used HA14.1 to test whether Bcl‐2/Bcl‐XL pathways are activated in the setting of BW245c‐mediated survival of eosinophils. A second small molecule inhibitor, BH3I‐1, acts by preventing the interaction between pro‐ and anti‐apoptotic members of the Bcl‐2 family via the BH3 domain of Bcl‐XL.38
In eosinophil preparations cultured for 5 h, BH3I‐1 increased the percentage of apoptotic cells and abrogated the viability‐enhancing effects of BW245c and IL‐5 (Fig. 4A). At this early time point BW245c and IL‐5 increased the portion of annexin V‐/PI‐cells while BW245c but not IL‐5 significantly decreased the potion of annexin V+ or annexin V+/PI‐ (Supplementary Fig. 2). Similarly, HA14.1 enhanced the number of apoptotic cells in BW245c‐supplemented cultures, but it also induced apoptosis by itself already after 3 h (Fig. 4B). Moreover, 10 μM HA14.1 prevented the inhibitory effect of caspase 3/7 activity in BW235c‐treated cells (Fig. 4C) while BH3I‐1 did not interfere with BW245c‐induced caspase 3/7 inhibition at 18 h.
Figure 4.
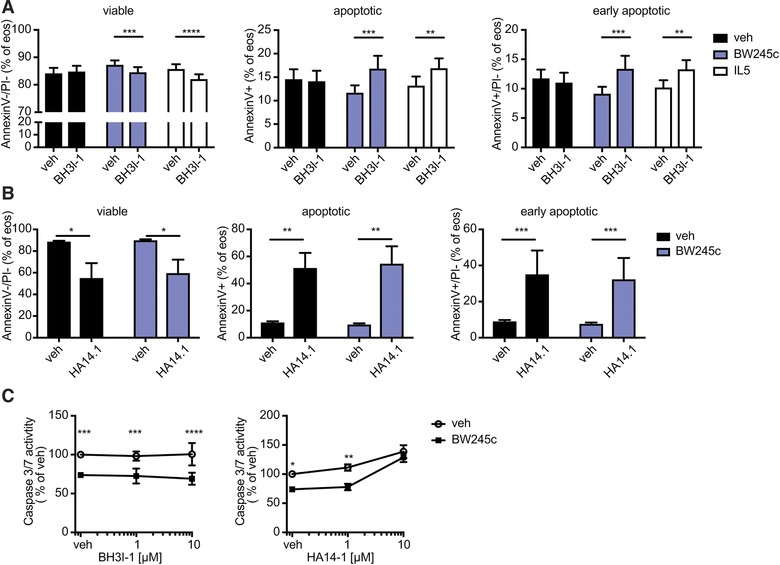
Bcl‐2 family protein inhibitor BH3I‐1 reverses the anti‐apoptotic effect of DP1 receptor activation. Eosinophils isolated from peripheral blood were incubated with BW245c [1 μM] in combination with or without Bcl‐XL inhibitors (A and C) BH3l‐1 [50 μM] for 5h or (B) HA14.1 [10 μM] for 3 h; (C) 18 h. (A) BH3l‐1 prevented the reduction of annexin V+ apoptotic cells induced by BW245c or IL‐5. (B) HA14.1 induced apoptosis of veh‐ and BW245c‐treated eosinophils. (C) In contrast to BH3I‐1, HA14.1 reversed the BW245c‐induced inhibition of caspase 3/7 activity. Data show mean ± sem of 5 individual experiments
3.5. DP1 receptor expression facilitates increased viability of PGD2‐treated HEK293 cells
Because eosinophils naturally expressed both, DP1 and DP2 receptors we used a HEK293 cell system, with stably overexpressed DP1, DP2, or both receptors in combination (HEK‐DP1, HEK‐DP2, HEK‐DP1 + DP2) and performed reporter gene assays. First, we tested whether the pro‐survival signaling of DP1 is conserved also in the HEK293 cell lines by formazan‐based viability tests (MTS). When treated with PGD2, cells expressing DP1 (HEK‐DP1 and HEK‐DP1+DP2) had an advantage in maintaining viability under starving conditions (Opti‐MEM) as compared to cells lacking DP1 (HEK‐DP2) (Fig. 5A). DP1 antagonist MK0524, but not DP2 antagonist Cay10471, reversed the observed pro‐survival stimulus provided by 100 nM PGD2 (Fig. 5B). Further, DP1 expressing cells showed a morphologically visible advantage in growth and formation of monolayers when cultured with PGD2 in starving conditions as compared to HEK‐DP2 cells (Fig. 6A).
Figure 5.
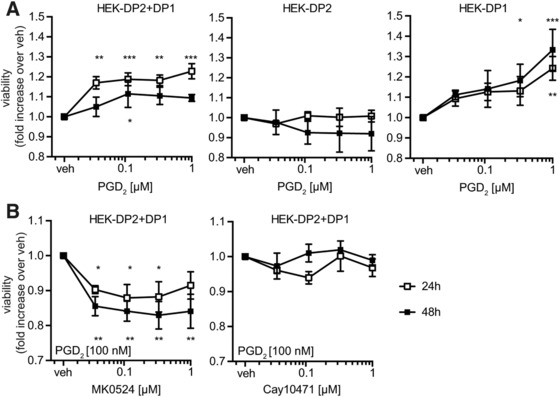
PGD2 enhances the viability of HEK‐DP2+DP1 and HEK‐DP1 but not of HEK‐DP2 cells. (A) HEK293 cell lines were starved in OptiMEM for 4 h and incubated with increasing concentrations of PGD2 for 24 of 48 h. Viability was detected by MTS assay (n = 5). (B) MK0524 but not Cay10471 antagonized the viability‐enhancing action of PGD2 on HEK‐DP2+DP1. (n = 3). Data show means of 3–5 independent experiments ± sem
Figure 6.
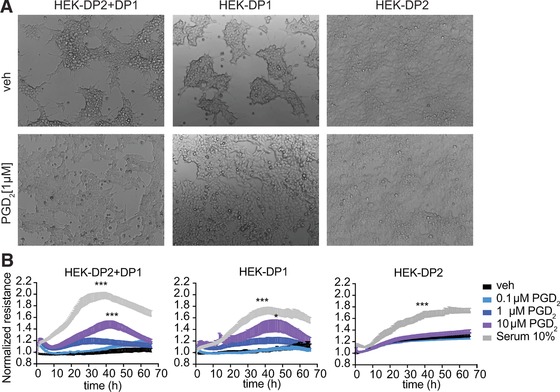
PGD2 treatment causes a growth advantage of HEK‐DP2+DP1 and HEK‐DP1 but not of HEK‐DP2 cells. (A) PGD2 treatment of DP1‐expressing cells led to morphologically visible growth advantage compared to HEK‐DP2 cells. Data show representative images of 4 independent experiments. (B) Cultures of serum‐starved HEK‐DP2+DP1, HEK‐DP1, or HEK‐DP2 cells were treated with increasing concentrations of PGD2 or 10% FBS and their electrical resistance was monitored for more than 60 h on an ECIS device. Data show means of 5 independent experiments + sem
Next, we assessed the growth of HEK293 monolayers expressing DP1 and/or DP2 that were treated with PGD2 (0.1–10 μM), or 10% FBS as positive control stimulus. PGD2 increased the growth rate of HEK‐DP1 and HEK‐DP2+DP1 but not HEK‐DP2 at day 2 (Fig. 6B) as assessed by ECIS. The increase in resistance has been shown to directly correlate with the growth rate of adherent cells.39 Thus, besides promoting viability, in cells capable of proliferating DP1 receptor expression leads to increased growth in response to PGD2. Although PGD2 did not change the resistance of HEK‐DP2, we observed an increased basal resistance of HEK‐DP2 when compared to HEK‐DP1 or HEK‐DP2+DP1. This was probably consistent with faster proliferation of this cell line which we also observed during cell culture handling (Supplementary Fig. 4).
3.6. DP1 but not DP2 is a transcriptional regulator and induces serum response element
Programmed cell death can be prevented by the induction of anti‐apoptotic genes, which counteract the initiation of the apoptotic cascade. To further elucidate the pro‐survival signaling of the DP1 receptor and test the hypothesis that DP1 signaling modulates eosinophil function on a transcriptional level, we studied the potential of DP1 and DP2 to induce activation of serum response element (SRE). SRE induces the expression of anti‐apoptotic genes in the Bcl‐2 family40 which could further promote allergic inflammation due to prolonged survival of eosinophils. Moreover, SRE and its activator, serum response factor (SRF) might be particularly important in eosinophil migration and adhesion as SRE is a transcriptional regulator in the promotor region of genes encoding for proteins regulating the cytoskeleton such as c‐Fos or profillin.41 SRE was shown to regulate migration of other innate immune cells such as neutrophils42 and macrophages43 but its function in eosinophils has not been investigated yet.
In a reporter gene assay, DP1 but not DP2 was capable of inducing SRE activation in HEK‐DP1 and HEK‐DP2+DP1 cells (Fig. 7A). In contrast, no SRE activation was observed in HEK‐DP2 cells. Remarkably, however, the potency of BW245c to activate SRE was reduced when DP2 was co‐expressed (Fig. 7B). Although the DP2 agonist DK‐PGD2 was unable to induce SRE in HEK‐DP1+DP2 cells (Fig. 7C), blocking DP2 with the specific antagonist Cay10471 led to reduced potency of PGD2 and BW245c to induce SRE (Fig. 7D–E). The respective EC50 and IC50 values are shown in Supplementary Table 1. This finding highlights the crosstalk of DP1 and DP2 in a jointly regulated receptor signaling unit and suggests a modulator role of DP2 on DP1 signaling.
Figure 7.
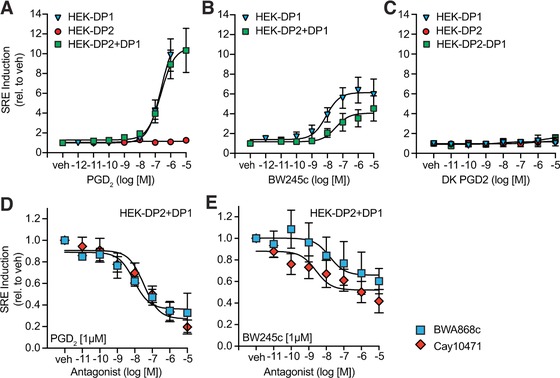
DP1 induces SRE activation. (A) PGD2 activates SRE in HEK‐DP2 + DP1 and HEK‐DP1 but not in HEK‐DP2. (B) DP1 agonist BW245c induces SRE activation in HEK‐DP2 + DP1 and HEK‐DP1. (C) DP2 agonist DK‐PGD2 does not induce SRE activation. (D) Antagonists of DP1 (BWA868c) and DP2 (Cay10471) block PGD2‐ and (E) BW245c induced SRE activation in HEK‐DP2 + DP1. Data are shown as mean ± sem of 3–6 independent experiments
3.7. Gene expression in eosinophils is regulated by DP1
The transcription factor SRF regulates actin cytoskeleton remodeling, cell contact maintenance and adhesion44, 45, 46 and was shown to promote the expression of anti‐apoptotic Bcl‐XL (B‐cell lymphoma‐extra‐large) and Bcl‐2 (B‐cell lymphoma 2).46 Thus, we tested whether DP1‐mediated signaling can affect eosinophil expression of Bcl‐XL and the major chemokine receptor, CCR3, and adhesion molecule, VLA‐4. Eosinophils were treated with PGD2 receptor agonists or IL‐5 10 ng/ml for 3 h (RPMI 1% FBS, 37°C, 5% CO2), total RNA was extracted from cell lysates and mRNA expression was analyzed by qRT‐PCR. Indeed, BW245c significantly increased both, mRNA levels and protein surface expression of VLA‐4 and CCR3 in human eosinophils, while DK‐PGD2 was ineffective in altering the levels of expression (Fig. 8A and B). Similar, BW245c increased mRNA levels of Bcl‐XL but did not enhance the level of protein expression significantly as shown by intracellular flow cytometry staining (Supplementary Fig. 5A and B).
Figure 8.
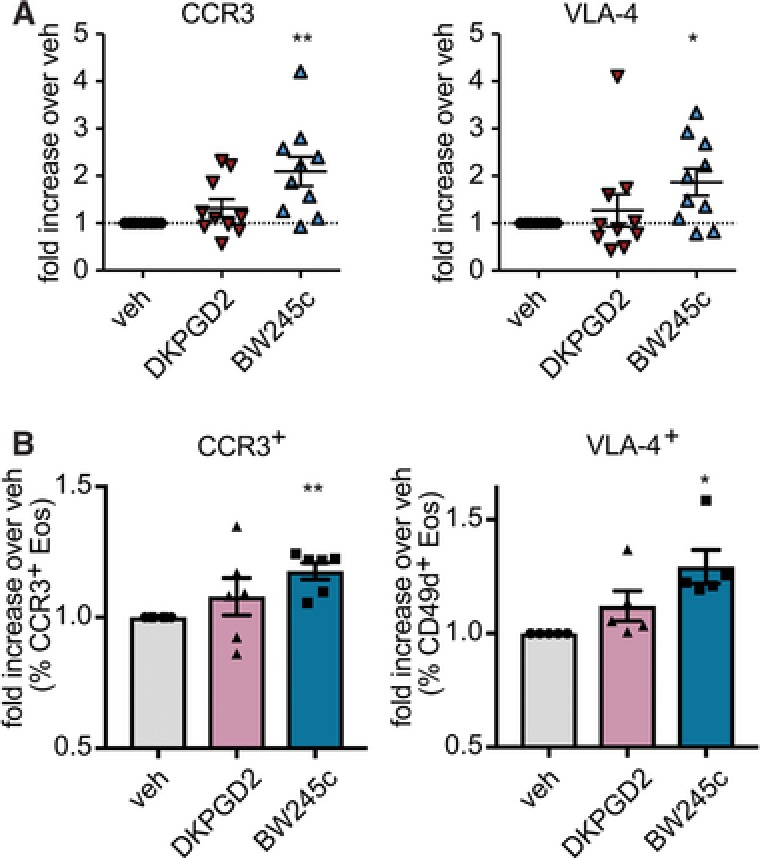
DP1 receptor agonist BW245c enhances (A) mRNA expression of CCR3 and VLA‐4 and (B) upregulates surface expression of CCR3 and VLA‐4. Isolated eosinophils (5 × 106/ml) were incubated with vehicle (EtOH), DK‐PGD2 [1 μM], or BW245c [1 μM] for 3 h (A) or 18h (B) (RPMI, 1% FBS, 1%, PenStrep, 37°C). mRNA expression was measures by qRT PCR (A). Surface expression was determined by flow cytometric analysis (B). Data show mean ± sem (n = 5–10)
The ratio of pro‐apoptotic BAX to anti‐apoptotic Bcl‐2 or Bcl‐XL reflects the pro‐survival or pro‐apoptotic status of a cell and can predict whether programmed cell death has been initiated or not. A ratio of BCL‐2 to BAX above 1 was shown to correlate with caspase 3 activation whereas a low value (<1) has been described for cells where the onset of apoptosis is suppressed, e.g., in cancer cells.47 Here, IL‐5 significantly decreased the BAX/Bcl‐XL ratio in eosinophils whereas BW245c moderately lowered the mean ratio to a value below 1 (Supplementary Fig. 6).
Thus, we conclude that activation of DP1 contributes to a pro‐inflammatory status by enhancing VLA‐4 and CCR3 expression and delays the onset of apoptosis in eosinophils by enhancing Bcl‐XL expression.
4. DISCUSSION
In the present study, we report a mechanism through which the PGD2 receptor DP1 leads to a prolonged survival of human eosinophils. In detail, we show that the DP1 agonist BW245c suppresses the activation of effector caspases in eosinophils, and protects mitochondrial membranes from depolarization which consequently sustains viability of eosinophils under minimal culture conditions. In a recombinant cell line, DP1 induces the activation of SRE. In human eosinophils, mRNA level of Bcl‐XL, and the expression of CCR3 and VLA‐4 is induced by DP1 activation. Thus, the DP1 receptor might not only promote the survival and, hence, persistence of eosinophils at sites of inflammation, but also modulate the course of allergic reactions by upregulating pro‐inflammatory genes, such as VLA‐4 and CCR3 or the anti‐apoptotic gene Bcl‐XL. Both, VLA‐438 and CCR3 are crucial in adhesion and migration, and CCR3 being the major receptor chemoattractant receptor (binding to the ligands CCL11, CCL24, CCL26, CCL7, CCL13, CCLl5, and CCL5) on eosinophils also positively regulates their survival.49
PGD2 has been shown to regulate apoptosis in a cell type specific manner. It protects Th2 cells from cytokine deprivation‐induced apoptosis via DP2 signaling,50 and antagonizes PGE2‐mediated Bax activation and hence inhibits the induction of apoptosis in glioma cells.51, 52 In contrast, PGD2 induces apoptosis in human osteoclasts,53 leukemia cells54 and non‐small cell lung carcinoma cell lines.55, 56 In eosinophils, PGD2 has been suggested to decrease the portion of late apoptotic eosinophils at nanomolar concentrations,21 but to induce apoptosis in micromolar concentrations (≥10 μM).26
In the current study, we show that DP1 prolongs the survival of eosinophils by protecting the mitochondria from the onset of intrinsic apoptosis. The DP1 agonist BW245c reduced mitochondrial membrane depolarization and decreased caspase 3/7 activation while pharmacological blockade with the DP1 antagonist reversed the inhibitory action on caspase 3/7. In general, apoptosis of eosinophils can be induced intrinsically, or extrinsically mediated via Fas receptor.57, 58 The mechanism of DP1 induced survival is consistent with the mechanism of eosinophil‐specific survival stimuli, such as IL‐5,32 that has been shown to inhibit the intrinsic apoptosis pathway by activating anti‐apoptotic proteins of the Bcl2 family.59 Additionally, DP1 receptor expression promoted viability and increased the growth of PGD2‐treated HEK293 monolayers. Because this finding shows that DP1 has a growth stimulating role on proliferating cells, DP1‐signaling might contribute to the differentiation and growth of eosinophil progenitor cells in addition to IL‐5.60 Future experiments might therefore address the role of PGD2 in the differentiation and proliferation of eosinophil progenitors.
Furthermore, we observed that DP1 signaling induces SRE, which in turn regulates the expression of genes that are crucial for the formation of the cytoskeleton and survival. Together with ternary complex factors, serum response factors (SRF) bind and activate SRE in the promotor of immediate early genes. Besides being crucial for the cytoskeleton integrity by regulating profilin expression, SRE activation drives anti‐apoptotic proteins, such as MCL1 or Bcl‐2.40, 41, 46 In B‐cells SRF is inactivated by cleavage of effector caspases in order to facilitate apoptosis pathways.61 Interestingly, the DP2 antagonist could block DP1‐mediated gene transcription but a DP2 agonist did not activate it by itself. This is consistent with our previous finding that the signaling of DP1 and DP2 is interlinked and can be different when DP1 and DP2 are expressed alone or in combination.18
The mechanism behind PGD2‐regulated gene transcription has remained largely unknown and it is not clear which particular DP1‐ or DP2‐dependent pathway leads to the observed SRE induction and at which level of the cascade the interaction between DP1 and DP2 may happen. DP1 has been shown to signal via activation of Gαs thereby activating adenylate cyclase and raise cAMP levels. However, we previously demonstrated that DP1 cannot only couple to Gαs‐,62, 63 but also to the Gαq18 subunit of heteromeric G‐proteins. Moreover, the Ca2+ signaling of DP2 is mediated by Gαi coupling, which significantly loses potency when Gαq is inhibited and DP1 co‐expressed.18 DP2 signaling can also be regulated by β arrestins64 but the signaling cascades that are directly activated by the Gβγ subunit linked to DP1 or DP2 have been barely investigated yet.
Moreover, we report that the DP1 agonist induced Bcl‐XL mRNA upregulation and pharmacological inhibition of Bcl‐XL reversed its pro‐survival effect. It is known that Bcl‐XL is expressed in eosinophils and contributes to the pro‐survival function of IL‐5 and GM‐CSF,36, 59 and is transcriptionally regulated by SRF.46 Bcl‐XL mRNA expression is upregulated in eosinophils from atopic dermatitis patients in comparison to eosinophils from healthy controls.31 Therefore, our finding supports the significance of Bcl‐XL in enhanced eosinophils survival.
The BcL‐XL inhibitor HA14.1 is a strong inducer of apoptosis, and although HA14.1 completely reversed the pro‐survival effect of BW245c, we cannot rule out an unspecific effect of HA14.1 since the inhibitor induced apoptosis in all samples regardless of treatment. Hence, it is difficult to decide whether HA14.1 inhibits the signaling cascades activated by BW245c. On the other hand, BH3I‐1 specifically reversed the increase of viable cells by BW245c treatment to levels of vehicle‐treated cells. Moreover, both BH3I‐1 and HA14.1 abrogated DP1‐mediated caspase 3/7 inhibition. A total of 10 μM HA14.1 induced effector caspase activation by itself which, at this concentration, could have potentially masked BW245c signaling at this concentration.
Polymorphisms, methylation, and expression patterns of PTGDR, the gene encoding the DP1 receptor, have been associated with asthma.65, 66 Therefore, altered DP1 receptor signaling might be crucial in allergic inflammation. Indeed, we could show that the DP1 receptor agonist upregulated CCR3 and VLA‐4 mRNA expression. Besides inducing chemotaxis toward eotaxin, CCR3 was shown to enhance eosinophil viability49 and to be transcriptionally upregulated in allergic asthma.67 Eosinophils bind to endothelial cells mainly via VLA‐4–VCAM interaction68 which—along with CCR3 activation—might be a critical regulator of eosinophil extravasation.
Enhanced survival of immune cells at inflammatory sites has been observed in the following allergic conditions6: (i) peripheral blood eosinophils from asthmatic patients survive longer compared to eosinophils from healthy control subjects,69 (ii) apoptosis rate of sputum eosinophils correlates with exhaled nitric oxide in asthmatic children,70 and (iii) delayed apoptosis contributes to tissue eosinophilia in nasal polyps.71 Moreover, there is substantial literature on the beneficial effects of the reduction of circulating eosinophils in eosinophilic asthma.7 In fact, depletion of eosinophils by antibody‐dependent cell‐mediated cytotoxicity (ADCC) has been recently shown to effectively improve airway resistance and disease scores in asthma patients with ≥300 eosinophils/μl blood.72, 73 The anti‐IL‐5Rα antibody benralizumab (MEDI‐563) binds to circulating eosinophils and induces apoptosis and ADCC of eosinophils and basophils in vitro. The compound depletes human bone marrow derived mononuclear cells of eosinophil precursors and eosinophils of peripheral blood of nonhuman primates and binds to eosinophils in nonhuman primate lung tissue (bronchi, small airways, bronchial parenchyma).74
In the present study, we observed that PGD2 did not decrease effector caspase activity and only moderately enhanced eosinophil viability compared to the DP1 agonist, but gained in anti‐apoptotic capacity when DP2 signaling was blocked by the respective antagonist. Furthermore, we confirm that the DP2 selective PGD2 metabolite 15dPGJ2 induces caspase activity in eosinophils in a PPAR‐ɣ independent manner.26 15dPGJ2 was shown to induce apoptosis in cardiomyocytes via activation of DP2/MAPK/TNF‐α75 and via ROS formation in non‐small lung cancer cells.76
Our data supports previous literature demonstrating the cooperative pro‐inflammatory signaling of DP1 and DP2. PGD2 induces leukotriene C4 production in eosinophils only sufficiently by simultaneous activation of DP1 and DP277. In guinea pigs, the mobilization of eosinophils from the bone marrow can be stimulated by both, DP1 and DP225. We have shown previously that DP1 receptors are profoundly involved in the DP2‐triggered Ca2+ signaling and that DP1 and DP2 receptors are co‐localized and form heteromeric units in HEK‐DP2 + DP1 cells.18
In contrast to cooperative pro‐inflammatory functions of DP1/DP2, there is evidence that DP1 receptor counteracts CD11b upregulation in human eosinophils,22 and, in mice, DP1 diminishes allergic inflammation in OVA models of experimental asthma as shown by decreased lung‐eosinophilia and airway hyperresponsiveness after intratracheal administration of BW245c.78, 79 However, in mice, DP1 activation was also shown to promote allergic inflammation since DP1‐deficient mice have reduced number of eosinophils and cytokine levels (IL‐4, IL‐5, IL‐13) in the BAL fluid in OVA models of experimental asthma.13 Hence, we tested if PGD2 receptors directly regulate survival of ex vivo differentiated murine bone marrow‐derived eosinophils (bmEos). We found that neither PGD2 nor BW245c or DK‐PGD2 significantly changed the in vitro survival of bmEos significantly (Supplementary Fig. 7). Therefore, the reported roles of DP1 in vivo could result from indirect effects mediated through other cell types such as lung dendritic cells and regulatory T‐cells71 and it is likely that the functions of DP1 differs between human and murine eosinophils.
For the development of future therapies based on DP2 antagonists, it is hence important to consider that such compounds could potentially enhance the survival of eosinophils by shunting the signaling cascade towards anti‐apoptotic DP1‐dependent pathways. Since targeting the trafficking and, hence, the infiltration of eosinophils to the sites of inflammation seems insufficient, additionally targeting the survival of circulating and tissue‐resident eosinophils could be accomplished with dual DP1/DP2 antagonists which should be addressed in future studies.
Targeting eosinophil survival by DP1 receptor antagonists may interfere with recently discovered homeostatic functions of eosinophils. Eosinophils modulate the functions of several other immune cell types, such as T cells, B cells, mast cells, macrophages, and neutrophils, to promote a Th2‐type of inflammation80 but they have also been shown to directly exert antiviral and antimicrobial host defense mechanisms and modulate the functions of lymphocytes. Thereby eosinophils support the resolution of inflammatory conditions.81 In addition, a population of homeostatic eosinophils (hEos) that differs phenotypically and functionally from inflammatory eosinophils (iEos) has been discovered recently. hEos in the lung express genes that negatively regulate Th2 cell functions to maintain lung immune homeostasis.82 These findings might be important to consider in a potential therapy where DP1 receptors are targeted to prevent eosinophil survival in tissues.
On the other hand, no negative side effects of eosinophil depletion by benralizumab74 have been reported so far. Benralizumab is generally well tolerated and effective in patients with severe eosinophilic asthma.83
Interestingly, in a recent proteome analysis DP2, cyclooxygenase‐1, and prostaglandin E2 synthase, but neither DP1 nor EP2, EP4, or IP receptors were detected in peripheral blood eosinophils of allergic donors.84 This finding confirms the low expression level of these GPCRs which we found in eosinophils from healthy donors by Western blot and flow cytometry.85, 86, 87
In conclusion, our data demonstrate that the PGD2 receptor DP1 has a critical role in regulating the survival of eosinophils by inhibiting the onset of the intrinsic apoptotic cascade.
Hence, PGD2 has a dual role in allergic inflammation, that is, besides directly recruiting eosinophils to afflicted tissues, this lipid mediator might also counteract the resolution of the allergic response by prolonging the survival of eosinophils. Consequently, DP1 receptor antagonists—in addition to DP2 antagonists—or dual DP1/DP2 antagonists might be useful therapeutic tools to reduce eosinophils infiltration and activation at sites of allergic inflammation.
Supporting information
supplementary information
ACKNOWLEDGMENTS
We thank Kathrin Rohrer for the excellent technical assistance. M.P., D.R., and M.S. were funded by the Austrian Science Fund (FWF W1241, DK‐MOLIN). A.T. is a recipient of a DOC Fellowship of the Austrian Academy of Science at the Otto Loewi Research Center for Vascular Biology, Immunology and Inflammation, Division of Pharmacology.
AUTHORSHIP
M.P., D.R., and A.T. performed experiments and analyzed data. M.P., M.S., R.S., G.M., E.M.S., I.S., M.E.R., and A.H. interpreted the results; A.H. and M.E.R. supervised the study.
DISCLOSURE
M.E.R. is a consultant for Pulm One, Spoon Guru, Celgene, Shire, Astra Zeneca, GlaxoSmithKline, Allakos, and Novartis and has an equity interest in the first two listed and Immune Pharmaceuticals, and royalties from reslizumab (Teva Pharmaceuticals). M.E.R. is an inventor of patents, owned by Cincinnati Children's. A.H. received consultancy fees from AstraZeneca. The other authors declare no conflicts of interest.
Peinhaupt M, Roula D, Theiler A, et al. DP1 receptor signaling prevents the onset of intrinsic apoptosis in eosinophils and functions as a transcriptional modulator. J Leukoc Biol. 2018;104:159–171. https://doi.org/10.1002/JLB.3MA1017-404R
REFERENCES
- 1. Modena BD, Dazy K, White AA. Emerging concepts: mast cell involvement in allergic diseases. Transl Res. 2016;174:98–121. [DOI] [PubMed] [Google Scholar]
- 2. Kagalwalla AF, Akhtar N, Woodruff SA, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129:1387–1396. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yasukawa A, Hosoki K, Toda M, et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One. 2013;8:e64281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aceves SS, Broide DH. Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Curr Mol Med. 2008;8:350–358. [DOI] [PubMed] [Google Scholar]
- 5. Wilson SJ, Rigden HM, A WardJ, et al. The relationship between eosinophilia and airway remodelling in mild asthma. Cea. 2013;43:1342–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohta K, Yamashita N. Apoptosis of eosinophils and lymphocytes in allergic inflammation. J Allergy Clin Immunol. 1999;104:14–21. [DOI] [PubMed] [Google Scholar]
- 7. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198–1207. [DOI] [PubMed] [Google Scholar]
- 8. Flood‐Page PT, Menzies‐Gow AN, Kay AB, Robinson DS. Eosinophil's role remains uncertain as anti–interleukin‐5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. [DOI] [PubMed] [Google Scholar]
- 9. Castro M, Mathur S, Hargreave F, et al. Reslizumab for poorly controlled, eosinophilic asthma. Am J Respir Crit Care Med. 2011;184:1125–1132. [DOI] [PubMed] [Google Scholar]
- 10. Naclerio RM, Meier HL, Kagey‐Sobotka A, et al. Mediator release after nasal airway challenge with allergen. Am Rev Respir Dis. 1983;128:597–602. [DOI] [PubMed] [Google Scholar]
- 11. Lewis RA, Soter NA, Diamond PT, et al. Prostaglandin D2 generation after activation of rat and human mast cells with anti‐IgE. J Immunol. 1982;129:1627–1631. [PubMed] [Google Scholar]
- 12. Peinhaupt M, Sturm EM, Heinemann A. Prostaglandins and their receptors in eosinophil function and as therapeutic targets. Front Med. 2017;4:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsuoka T, Hirata M, Tanaka H, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. [DOI] [PubMed] [Google Scholar]
- 14. Shen Z‐J, Esnault S, Schinzel A, Borner C, Malter JS. The peptidyl‐prolyl isomerase Pin1 facilitates cytokine‐induced survival of eosinophils by suppressing Bax activation. Nat Immunol. 2009;10:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pienkowski MM, Adkinson NF, Plaut M, Norman PS, Lichtenstein LM. Prostaglandin D2 and histamine during the immediate and the late‐phase components of allergic cutaneous responses. J Allergy Clin Immunol. 1988;82:95–100. [DOI] [PubMed] [Google Scholar]
- 16. Zhang S, Wu X, Yu S. Prostaglandin D2 receptor d‐type prostanoid receptor 2 mediates eosinophil trafficking into the esophagus. Dis Esophagus. 2014;27:601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sawyer N, Cauchon E, Chateauneuf A, et al. Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br J Pharmacol. 2002;137:1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sedej M, Schröder R, Bell K, et al. D‐type prostanoid receptor enhances the signaling of chemoattractant receptor‐homologous molecule expressed on T H2 cells. J Allergy Clin Immunol. 2012;129:492–500. e9. [DOI] [PubMed] [Google Scholar]
- 19. Nagata K, Hirai H. The second PGD2 receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins, Leukot Essent Fatty Acids. 2003;69:169–177. [DOI] [PubMed] [Google Scholar]
- 20. Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven‐transmembrane receptor CRTH2. J Exp Med. 2001;193:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gervais FG, Cruz RP, Chateauneuf A, et al. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol. 2001;108:982–988. [DOI] [PubMed] [Google Scholar]
- 22. Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–1948. [DOI] [PubMed] [Google Scholar]
- 23. Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Delta 12‐prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J Immunol. 2003;170:4752–4758. [DOI] [PubMed] [Google Scholar]
- 24. Royer JF, Schratl P, Lorenz S, et al. A novel antagonist of CRTH2 blocks eosinophil release from bone marrow, chemotaxis and respiratory burst. Allergy Eur J Allergy Clin Immunol. 2007;62:1401–1409. [DOI] [PubMed] [Google Scholar]
- 25. Schratl P, Royer JF, Kostenis E, et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J Immunol. 2007;179:4792–4799. [DOI] [PubMed] [Google Scholar]
- 26. Ward C, Dransfield I, Murray J, et al. Prostaglandin D2 and its metabolites induce caspase‐dependent granulocyte apoptosis that is mediated via inhibition of IκBα degradation using a peroxisome proliferator‐activated receptor‐γ‐independent mechanism. J Immunol. 2002;168:6232–6243. [DOI] [PubMed] [Google Scholar]
- 27. Hartnell A, Heinemann A, Conroy DM, et al. Identification of selective basophil chemoattractants in human nasal polyps as insulin‐like growth factor‐1 and insulin‐like growth factor‐2. J Immunol. 2004;173:6448–6457. [DOI] [PubMed] [Google Scholar]
- 28. Henstridge CM, Balenga NAB, Ford LA, et al. The GPR55 ligand L‐alpha‐lysophosphatidylinositol promotes RhoA‐dependent Ca2+ signaling and NFAT activation. FASEB J. 2009;23:183–193. [DOI] [PubMed] [Google Scholar]
- 29. Balenga NA, Martínez‐Pinilla E, Kargl J, et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br J Pharmacol. 2014;171:5387–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kargl J, Haybaeck J, Stančić A, et al. O‐1602, an atypical cannabinoid, inhibits tumor growth in colitis‐associated colon cancer through multiple mechanisms. J Mol Med. 2013;91:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogawa K, Hashida R, Miyagawa M, et al. Analysis of gene expression in peripheral blood eosinophils from patients with atopic dermatitis and in vitro cytokine‐stimulated blood eosinophils. Clin Exp Immunol. 2003;131:436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamaguchi Y, Hayashi Y, Sugama Y, et al. Highly purified murine interleukin 5 (IL‐5) stimulates eosinophil function and prolongs in vitro survival. J Exp Med. 1988;167:1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kluck RM, Bossy‐wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria : A primary site for bcl‐2 regulation of apoptosis. Science. 1997;1275:1132–1136. [DOI] [PubMed] [Google Scholar]
- 34. Ueki S, Kato H, Kobayashi Y, et al. Anti‐ and Proinflammatory effects of 15‐Deoxy‐Δ12,14‐Prostaglandin J2(15d‐PGJ2) on human eosinophil functions. Int Arch Allergy Immunol. 2007;143:15–22. [DOI] [PubMed] [Google Scholar]
- 35. Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC‐1, but not DiOC 6 (3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77–82. [DOI] [PubMed] [Google Scholar]
- 36. Dibbert B, Daigle I, Braun D, et al. Role for Bcl‐xL in delayed eosinophil apoptosis mediated by granulocyte‐macrophage colony‐stimulating factor and interleukin‐5. Blood. 1998;92:778–783. [PubMed] [Google Scholar]
- 37. Wang J‐LL, Liu D, Zhang Z‐JJ, et al. Structure‐based discovery of an organic compound that binds Bcl‐2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA. 2000;97:7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Degterev A, Lugovskoy A, Cardone M, et al. Identification of small‐molecule inhibitors of interaction between the BH3 domain and Bcl‐x L. Nat Cell Biol. 2001;3:173–182. [DOI] [PubMed] [Google Scholar]
- 39. Zudaire E, Cuesta N, Murty V, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vickers ER, Kasza A, Kurnaz IA, et al. Ternary complex factor‐serum response factor complex‐regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol. 2004;24:10340–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70–81. [DOI] [PubMed] [Google Scholar]
- 42. Taylor A, Tang W, Bruscia EM, et al. SRF is required for neutrophil migration in response to inflammation. Blood. 2014;123:3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sullivan AL, Benner C, Heinz S, et al. Serum response factor utilizes distinct promoter‐ and enhancer‐based mechanisms to regulate cytoskeletal gene expression in macrophages. Mol Cell Biol. 2011;31:861–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ragu C, Elain G, Mylonas E, et al. The transcription factor Srf regulates hematopoietic stem cell adhesion. Blood. 2010;116:4464–4473. [DOI] [PubMed] [Google Scholar]
- 45. Medjkane S, Perez‐Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin‐related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schratt G, Philippar U, Hockemeyer D, et al. SRF regulates Bcl‐2 expression and promotes cell survival during murine embryonic development. EMBO J. 2004;23:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu F‐T, Goff LK, Hao J‐H, Newland AC, Jia L. Increase in the ratio of mitochondrial Bax/Bcl‐XL induces Bax activation in human leukemic K562 cell line. Apoptosis. 2004;9:377–384. [DOI] [PubMed] [Google Scholar]
- 48. Das AM, Williams TJ. Lobb R, Nourshargh S. Lung eosinophilia is dependent on IL‐5 and the adhesion molecules CD18 and VLA‐4, in a guinea‐pig model. Immunology. 1995;84:41–46. [PMC free article] [PubMed] [Google Scholar]
- 49. Shinagawa K, Trifilieff A, Anderson GP. Involvement of CCR3‐reactive chemokines in eosinophil survival. Int Arch Allergy Immunol. 2003;130:150–157. [DOI] [PubMed] [Google Scholar]
- 50. Xue L, Fergusson J, Salimi M, et al. Prostaglandin D2 and leukotriene E4 synergize to stimulate diverse TH2 functions and TH2 cell/neutrophil crosstalk. J Allergy Clin Immunol. 2015;135:1358–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lalier L, Cartron P‐F, Pedelaborde F, et al. Increase in PGE2 biosynthesis induces a Bax dependent apoptosis correlated to patients’ survival in glioblastoma multiforme. Oncogene. 2007;26:4999–5009. [DOI] [PubMed] [Google Scholar]
- 52. Lalier L, Cartron P‐F, Olivier C, et al. Prostaglandins antagonistically control Bax activation during apoptosis. Cell Death Differ. 2011;18:528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yue L, Haroun S, Parent J‐L, de Brum‐Fernandes AJ. Prostaglandin D2 induces apoptosis of human osteoclasts through ERK1/2 and Akt signaling pathways. Bone. 2014;60:112–121. [DOI] [PubMed] [Google Scholar]
- 54. Chen Y‐C, Shen S‐C, Tsai S‐H. Prostaglandin D2 and J2 induce apoptosis in human leukemia cells via activation of the caspase 3 cascade and production of reactive oxygen species. Biochim Biophys Acta. 2005;1743:291–304. [DOI] [PubMed] [Google Scholar]
- 55. Wang J, Mak O. Induction of apoptosis in non‐small cell lung carcinoma A549 cells by PGD2 metabolite, 15d‐PGJ 2. Cell Biol Int. 2011;35:1089–1096. [DOI] [PubMed] [Google Scholar]
- 56. Ramer R, Heinemann K, Merkord J, et al. COX‐2 and PPAR‐g confer cannabidiol‐induced apoptosis of human lung cancer cells. Mol Cancer Ther. 2013;12:69–82. [DOI] [PubMed] [Google Scholar]
- 57. Segal M, Niazi S, Simons MP, Galati SA, Zangrilli JG. Bid activation during induction of extrinsic and intrinsic apoptosis in eosinophils. Immunol Cell Biol. 2007;85:518–524. [DOI] [PubMed] [Google Scholar]
- 58. Tsuyuki S, Bertrand C, Erard F, et al. Activation of the Fas receptor on lung eosinophils leads to apoptosis and the resolution of eosinophilic inflammation of the airways. J Clin Invest. 1995;96:2924–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schwartz C, Willebrand R, Huber S, et al. Eosinophil‐specific deletion of IkBa in mice reveals a critical role of NF‐kB–induced Bcl‐xL for inhibition of apoptosis. Blood. 2015;125:3896–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clutterbuck EJ, Sanderson CJ. Human Eosinophil Hematopoiesis studied in vitro by means of murine eosinophil differentiation factor (1l5): production of functionally active eosinophils from normal human bone marrow. Blood. 1988;71:646–651. [PubMed] [Google Scholar]
- 61. Drewett V, Devitt A, Saxton J, et al. Serum response factor cleavage by caspases 3 and 7 linked to apoptosis in human bjab cells. J Biol Chem. 2001;276:33444–33451. [DOI] [PubMed] [Google Scholar]
- 62. Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004;103:147–166. [DOI] [PubMed] [Google Scholar]
- 63. Hirata M, A Kakizuk, Aizawa M, Ushikubi F, Narumiya S. Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc Natl Acad Sci USA. 1994;91:11192–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schröder R, Merten N, Mathiesen JM, et al. The C‐terminal Tail of CRTH2 is a key molecular determinant that constrains gα i and downstream signaling cascade activation. J Biol Chem. 2009;284:1324–1336. [DOI] [PubMed] [Google Scholar]
- 65. Isidoro‐García M, Sanz C, García‐Solaesa V, et al. PTGDR gene in asthma: a functional, genetic, and epigenetic study. Allergy. 2011;66:1553–1562. [DOI] [PubMed] [Google Scholar]
- 66. Oguma T, Palmer LJ, Birben E, et al. Role of prostanoid DP receptor variants in susceptibility to asthma. N Engl J Med. 2004;351:1752–1763. [DOI] [PubMed] [Google Scholar]
- 67. Ying S, Robinson DS, Meng Q, et al. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. association with airway hyperresponsiveness and predominant co‐localization of eotaxin mRNA to bronchial epithelial and endothelial cells. Eur J Immunol. 1997;27:3507–3516. [DOI] [PubMed] [Google Scholar]
- 68. Dobrina A, Menegazzi R, Carlos TM, et al. Mechanisms of eosinophil adherence to cultured vascular endothelial cells. Eosinophils bind to the cytokine‐induced ligand vascular cell adhesion molecule‐1 via the very late activation antigen‐4 integrin receptor. J Clin Invest. 1991;88:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kankaanranta H, Lindsay MA, Giembycz MA, et al. Delayed eosinophil apoptosis in asthma. J Allergy Clin Immunol. 2000;106:77–83. [DOI] [PubMed] [Google Scholar]
- 70. Pontin J, Blaylock MG, Walsh GM, Turner SW. Sputum eosinophil apoptotic rate is positively correlated to exhaled nitric oxide in children. Pediatr Pulmonol. 2008;43:1130–1134. [DOI] [PubMed] [Google Scholar]
- 71. Simon HU, Yousefi S, Schranz C, et al. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol. 1997;158:3902–3908. [PubMed] [Google Scholar]
- 72. FitzGerald JM, Bleecker ER, Nair P, et al. Benralizumab, an anti‐interleukin‐5 receptor α monoclonal antibody, as add‐on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2016;388:2128–2141. [DOI] [PubMed] [Google Scholar]
- 73. Ferguson GT, FitzGerald JM, Bleecker ER, et al. Benralizumab for patients with mild to moderate, persistent asthma (BISE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Respir Med. 2017;2600:1–9. [DOI] [PubMed] [Google Scholar]
- 74. Kolbeck R, Kozhich A, Koike M, et al. MEDI‐563, a humanized anti‐IL‐5 receptor α mAb with enhanced antibody‐dependent cell‐mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344–1353. e2. [DOI] [PubMed] [Google Scholar]
- 75. Koyani CN, Windischhofer W, Rossmann C, et al. 15‐deoxy‐Δ12,14‐PGJ₂ promotes inflammation and apoptosis in cardiomyocytes via the DP2/MAPK/TNFα axis. Int J Cardiol. 2014;173:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J‐J, Mak O‐T. Induction of apoptosis by 15d‐PGJ2 via ROS formation: An alternative pathway without PPARγ activation in non‐small cell lung carcinoma A549 cells. Prostaglandins Other Lipid Mediat. 2011;94:104–111. [DOI] [PubMed] [Google Scholar]
- 77. Mesquita‐Santos FP, Bakker‐Abreu I, Luna‐Gomes, et al. Co‐operative signalling through DP(1) and DP(2) prostanoid receptors is required to enhance leukotriene C(4) synthesis induced by prostaglandin D(2) in eosinophils. Br J Pharmacol. 2011;162:1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Spik I, Brénuchon C, Angéli V, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–3708. [DOI] [PubMed] [Google Scholar]
- 79. Hammad H, Kool M, Soullié T, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med. 2007;204:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Travers J, Rothenberg ME. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015;8:464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mesnil C, Raulier S, Paulissen G, et al. Lung‐resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest. 2016;126:3279–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kupczyk M, Kuna P. Benralizumab: an anti‐IL‐5 receptor α monoclonal antibody in the treatment of asthma. Immunotherapy. 2018:2017–0161. [DOI] [PubMed] [Google Scholar]
- 84. Wilkerson EM, Johansson MW, Hebert AS, et al. The peripheral blood eosinophil proteome. J Proteome Res. 2016;15:1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sturm EM, Schratl P, Schuligoi R, et al. Prostaglandin E2 inhibits eosinophil trafficking through E‐prostanoid 2 receptors. J Immunol. 2008;181:7273–7283. [DOI] [PubMed] [Google Scholar]
- 86. Luschnig‐Schratl P, Sturm EM, Konya V, et al. EP4 receptor stimulation down‐regulates human eosinophil function. Cell Mol Life Sci. 2011;68:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Konya V, Sturm EM, Schratl P, et al. Endothelium‐derived prostaglandin I(2) controls the migration of eosinophils. J Allergy Clin Immunol. 2010;125:1105–1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information