

Abstract
Rationale
The fundamental level of stable isotopic knowledge lies at specific atomic positions within molecules but existing methods of analysis require lengthy off‐line preparation to reveal this information. An automated position‐specific isotope analysis (PSIA) method is presented to determine the stable carbon isotopic compositions of the carboxyl groups of amino acids (δ 13CCARBOXYL values). This automation makes PSIA measurements easier and routine.
Methods
An existing high‐performance liquid chromatography (HPLC) gas handling interface/stable isotope ratio mass spectrometry system was modified by the addition of a post‐column derivatisation unit between the HPLC system and the interface. The post‐column reaction was optimised to yield CO2 from the carboxyl groups of amino acids by reaction with ninhydrin.
Results
The methodology described produced δ 13CCARBOXYL values with typical standard deviations below ±0.1 ‰ and consistent differences (Δ 13CCARBOXYL values) between amino acids over a 1‐year period. First estimates are presented for the δ 13CCARBOXYL values of a number of internationally available amino acid reference materials.
Conclusions
The PSIA methodology described provides a further dimension to the stable isotopic characterisation of amino acids at a more detailed level than the bulk or averaged whole‐molecule level. When combined with on‐line chromatographic separation or off‐line fraction collection of protein hydrolysates the technique will offer an automated and routine way to study position‐specific carboxyl carbon isotope information for amino acids, enabling more refined isotopic studies of carbon uptake and metabolism.
1. INTRODUCTION
There is a wide ranging interest in the isotopic analysis of amino acids, from medicine to meteorites.1 For example, there is a long‐term interest in the isotopic compositions of amino acids as monitors of metabolism, including indicators of serious medical conditions such as diabetes2 and disease such as breast cancer.3 New methods dealing with amino acids are thus timely and of widespread interest.
A number of previous studies have described the stable carbon isotopic analysis of individual amino acids, typically derived by hydrolysis of proteins, from various biological sources.4, 5, 6, 7, 8, 9 This compound‐specific isotope analysis (CSIA) is achieved either by gas chromatography of derivatised amino acids coupled with isotope ratio mass spectrometry (IRMS) or by high‐pressure liquid chromatography (HPLC) of underivatised amino acids coupled to IRMS using chemical oxidation to convert the carbon present in organic compounds into CO2 (LC/CO/IRMS).10, 11 The use of LC/CO/IRMS for CSIA is now a relatively mature technique, especially with regard to the separation of underivatised amino acids using mixed‐mode HPLC columns and aqueous mobile phases.12 Recent research has shown that the CSIA δ 13C values of underivatised amino acids, derived from hydrolysis of hair samples, can indicate metabolic trends associated with factors such as age and obesity in humans.13, 14
While the CSIA approach has yielded important information, it measures the average isotopic information available from amino acids which contain between two (glycine) and 11 (tryptophan) carbon atoms. These atoms are not all equal in 13C/12C ratios and it has long been recognised that the distribution of stable isotopes within amino acids is neither random nor homogeneous.15, 16 It is also generally recognised that the fundamental level of stable isotopic information lies at specific positions within molecules.17, 18 Therefore, it seems that the time is right to take the technology to ‘the‐next‐level’ and develop a more detailed position‐specific isotope analysis (PSIA) methodology for amino acids.
A number of IRMS techniques have previously been described to measure the stable isotopic composition at specific positions within small molecules. These methods typically require tedious chemical or enzymatic degradations that fragment the molecule and then separate and analyse the fragments for their δ 13C composition.17 Methods for PSIA of small molecules based on Stable Isotope Natural Fractionation–Nuclear Magnetic Resonance (SNIF‐NMR or 2H NMR) have also been developed although these techniques are typically applied to compounds that can be prepared in large amounts and high purity, for example ethanol distilled from European wines.19 These techniques now provide the basis for the authentication of a number of natural products, and reference materials (RMs) exist for the PSIA of ethanol with certified 2H/1H ratios of both methyl and methylene groups (e.g. European Commission, Joint Research Centre BCR‐656 and BCR‐660). More recently, 13C NMR has been developed for PSIA studies of (bio)chemical and physiochemical pathways but, unlike SNIF‐NMR, this has the disadvantage of not being linked to international RMs.20 A detailed discussion of these techniques is beyond the scope of this study but can be found in the citations above. The aims of this study were:
-
‐
to interface existing pieces of commercially available equipment to produce a practical flow‐injection analysis (FIA)–ninhydrin reaction–IRMS (FIA/NR/IRMS) instrument,
-
‐
to develop robust chemistry for the consistent yield of CO2 from amino acid carboxyl groups,
-
‐
to provide a first estimate of the carboxyl carbon isotopic compositions (δ 13CCARBOXYL values) of internationally available amino acid RMs.
The PSIA of the carboxyl group of amino acids was an easy choice because the ninhydrin (2,2‐dihydroxyindane‐1,3‐dione) reaction required to produce CO2 from the carboxyl groups of amino acids is already well established. The development of Ruhemann's purple (RP) (diketohyrindamine‐diketohydrindylidene enolate) from the reaction of ninhydrin with amino acids was first described over a century ago21 and the method has been widely used to visualise amino acids, peptides and proteins.22 Commercial systems are now available for the post‐column derivatisation (PCD) of amino acids separated by HPLC. These systems aim for consistent colour development although the reaction may only proceed to 30% completion in 30 s.23 A red‐coloured compound, hydrindantin (a dimer of ninhydrin), also makes a minor contribution to the colour formed during the ninhydrin reaction.
Although some controversy still exists as to the exact mechanism of the reaction of amino acids with ninhydrin it is generally accepted that CO2 is liberated at a very early stage of the reaction and before any colour development, as shown in Figure 1.22, 24, 25, 26 Although the formation of CO2 might be regarded as a side‐effect of the ninhydrin reaction, CO2 from this reaction has been used to quantify amino acids27, 28, 29 and for both radiocarbon dating and stable isotope analysis of peptide‐bonded carbon in protinaceous materials.30, 31, 32 Early studies showed that the reaction of amino acids with ninhydrin at boiling temperatures can liberate carboxyl CO2 at 100.0 ± 0.2% yields, with higher pH (4.7) conditions being optimum for most of the 20 common amino acids and lower pH (1) being optimum for cystine, glutamate, and lysine.27 This quantitative aspect has encouraged the use of the ninhydrin reaction for isotope studies, with no isotope fractionation expected when both 13C and 12C are completely converted into CO2.
Figure 1.
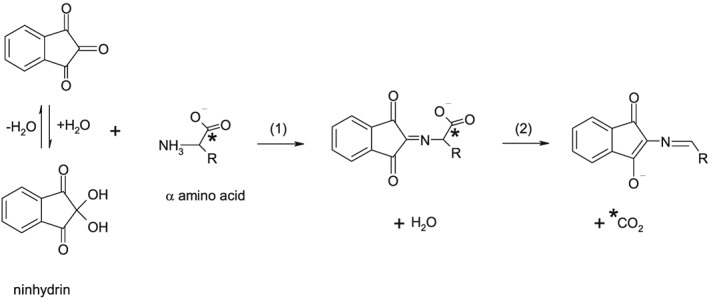
The initial reaction of ninhydrin with a generic α‐amino acid with a side group (R). The reaction proceeds by (1) nucleophillic attack and (2) elimination of the carboxyl group as carbon dioxide (*). Based on Joullié et al24
Generally, the carboxyl groups of amino acids show isotopic enrichment patterns specific to their metabolism and provide a means to test labelling expectations such as the metabolic uptake of bicarbonate. For example, carbon isotopic analysis of 15 samples of glutamic acid showed little variation in average composition whereas large and significant differences existed in the δ 13CCARBOXYL values.16 Other studies showed that δ 13CCARBOXYL patterns were characteristic of different growth conditions and enzyme pathways involved in carbon by autotrophs and heterotrophs.33, 34 More recently, a study of biopsy tissue concluded that bulk cellular 13C composition provided a marker for breast cancer, with a characteristic 13C enrichment linked to the urea cycle and arginine build‐up.3 PSIA of the arginine carboxyl group might well provide a more sensitive cancer indicator than isotope measurements of the whole tissue.
It also seems likely that individual carboxyl groups will be classified as those that do not change through the food web (carboxyl groups of ‘essential’ or ‘indispensable’ amino acids) while carboxyl groups of other amino acids (‘non‐essential’ or ‘dispensable’ amino acids) may change much more than average and be more dramatic metabolic indicators. Thus, amino acid PSIA might record the diet and the metabolic response to the diet in highly differentiated ways.
Although the LC/IRMS interface was intended to be used in conjunction with chromatographic separations it is frequently used for FIA, without HPLC separation, for the analysis of pre‐purified analytes such as amino acids and sugars.35 This manuscript describes an automated FIA method to determine the stable carbon isotopic composition of the carboxyl group of pre‐purified amino acids. The main automated work was undertaken in Brisbane, Australia, and additional off‐line calibration work with L‐alanine RMs was performed in Tokyo, Japan.
2. EXPERIMENTAL
2.1. Reference materials
Samples of L‐glutamic acid, glycine and L‐valine RMs were purchased from the US Geological Survey Stable Isotope Laboratory (Reston, VA, USA). These materials (Table 1) are internationally available and have well‐characterised CSIA δ 13CVPDB values.36, 37
Table 1.
Amino acid reference materials used in this study and their compound‐specific isotope analysis (CSIA) delta values
| Reference material | Nature | CSIA δ 13CVPDB (‰) | |
|---|---|---|---|
| mean | SD | ||
| USGS40 | L‐glutamic acid | −26.39 | ± 0.04 |
| USGS41a | L‐glutamic acid | +36.55 | ± 0.08 |
| USGS64 (G1) | glycine | −40.81 | ± 0.04 |
| USGS65 (G2) | glycine | −20.29 | ± 0.04 |
| USGS66 (G3) | glycine | −0.67 | ± 0.04 |
| D01 a | L‐valine | −10.97 | |
| USGS73 (V1) | L‐valine | −20.03 | ± 0.04 |
| USGS74 (V2) | L‐valine | −9.03 | ± 0.04 |
| USGS75 (V3) | L‐valine | +0.49 | ± 0.07 |
| TTAa | L‐alanine | −23.30 | ± 0.12 |
| TTSa | L‐alanine | −18.65 | ± 0.11 |
| TTWa | L‐alanine | −19.64 | ± 0.18 |
see section 2.1.
A sample of natural abundance L‐valine (D01) that was used to prepare L‐valine RMs USGS74 and 75 was kindly supplied by colleagues from Indiana University (Bloomington, IN, USA).37
Three samples of L‐alanine (TTA, TTS, TTW) were, respectively, purchased from Aldrich Chemical Co., Inc. (St Louis, MO, USA; Lot No. CA03519DO, certified purity >99%), from Sigma Chemical Co. (St Louis, MO, USA; Lot No. 078 K1099, certified purity >98.5%) and from Wako Pure Chemical Industries (Osaka, Japan; Lot No. EWL2621, certified purity >99%). In Tokyo, CSIA δ 13CVPDB values were characterised using an off‐line method. The method involved sealed tube combustion followed by cryogenic separation of the resulting CO2 using a high vacuum line system, then measurement of the purified CO2 by dua‐inlet IRMS. The δ 13CCARBOXYL values were characterised using an off‐line method consisting of a 1‐h boiling reaction with 3.5% ninhydrin in phosphoric acid (pH 2) followed by cryogenic separation of the resulting CO2 and measurement of the purified CO2 by dual‐inlet‐IRMS. Both CSIA and PSIA dual‐inlet measurements were made using a Delta XL isotope ratio mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Data were normalised to the VPDB‐LSVEC scale using CO2 liberated from NBS 19 (δ 13C = +1.95 ‰) and LSVEC (δ 13C = −46.6 ‰) (International Atomic Agency, Vienna, Austria) by reaction with phosphoric acid.
Although the four amino acids used in this study (Table 1) are a small subset of the 20 common amino acids, they represented a cross‐section of ninhydrin reaction chemistry. For example, glutamic acid is reported to readily over‐yield CO2 at higher pH (4.7) whereas alanine and glycine under‐yield at low pH (1) and L‐valine gives consistent yields across a range of reaction chemistries.27, 28
For the automated work in Brisbane, 5 mM solutions of individual amino acids were prepared by dissolving weighed aliquots in 1 mM sulphuric acid (approximately pH 3). Solutions were then degassed by ultrasonication under vacuum to remove any dissolved atmospheric CO2. This ‘sonivac’ (sonication under vacuum) process proved especially important for isotopically labelled materials for which the dissolved CO2 was very different in carbon isotope composition from the 13C‐labelled carboxyl group.
A number of in‐house quality control amino acids (glycine, phenylalanine and aspartic acid) were analysed together with each batch of samples. These amino acids were purchased from Sigma (Castle Hill, NSW, Australia) with certified purities >98%.
The stretch factor for the Brisbane IRMS instrument was assessed using inorganic carbon standards; LSVEC lithium carbonate (−46.6 ‰) and a sodium bicarbonate (LSUB, −3.28 ± 0.06 ‰) which was calibrated against NBS 19 at the Louisiana State University (Baton Rouge, LA, USA) by the first author. Both LSVEC and LSUB were prepared as 5 mM solutions by dissolving the dry salts in high‐purity water.
2.2. Instrumentation
The FIA/NR/IRMS instrument is shown schematically in Figure 2. Stable carbon isotope ratio measurements were made using a Delta V Advantage isotope ratio mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
Figure 2.
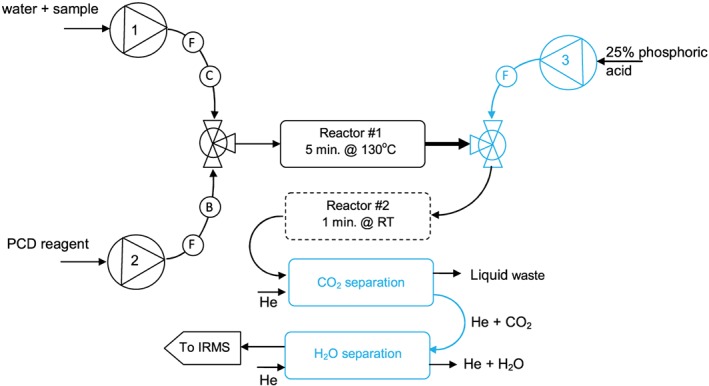
Schematic representation of the FIA/NR/IRMS instrumentation; components in blue show original elements of the LC IsoLink. (B) = back‐pressure regulator, (C) = non‐metallic check‐value, (F) = frit filter. Reactor #2 is advisable to prevent blockages but can be removed to improve peak width. The small‐volume reaction coil of the LC IsoLink was not used in this application. RT = room temperature [Color figure can be viewed at http://wileyonlinelibrary.com]
Aliquots of the amino acid solutions were injected into a flow of high‐purity water (18.2 MΩ, 0.15 mL min−1) using an Accela 600 high‐pressure liquid chromatography pump (Figure 2, pump #1) and autosampler (Thermo Fisher Scientific, Waltham, MA, USA). The minimum sample sizes were equivalent to approximately 180 ng of carboxyl carbon per injection.
The HPLC eluent passed through a small‐volume (37 μL) non‐metallic check valve (Upchurch Scientific/Idex, Rohnert Park, CA, USA) and was combined with a 0.15 mL min−1 flow from a modular PCD system (Rigas Labs, Sindos, Greece) (Figure 2, pump #2). The detailed preparation of the PCD reagent is described below. The PCD unit was modified by replacing the 0.5‐mL PEEK reaction coil with a stainless steel coil with a nominal internal volume of 1.5 mL (Figure 2, reactor #1), to allow longer reaction times at elevated temperatures. The total residence time in the PCD coil was approximately 5 min. The mixing tee in the PCD unit was replaced with a smaller mixing tee with 2‐μL internal volume (VICI AG International, Schenkon, Switzerland).
The outlet from the PCD unit was connected to an LC IsoLink interface (Thermo Fisher Scientific, Bremen, Germany) which added 25 μL min−1 of 25% phosphoric acid (Figure 2. pump #3) via another low‐volume mixing tee. The overall flow through the CO2 separation membrane in the LC IsoLink was maintained below the 0.4 mL min−1 upper limit considered optimum for quantitative transfer of CO2.38 Acid was added primarily to decrease the solubility of CO2 but had the added effect of solubilising the RP that formed along with CO2; RP tended to block and sometimes break the CO2 separation membrane. Frit filters (5 μm pore size, VICI #1110‐5P‐5) were used on all liquid input lines (Figure 2); these filter required only infrequent replacement, every 3–6 months. The small‐volume reaction coil (0.2‐mL oxidation reactor) of the LC IsoLink did not provide sufficient residence time for the ninhydrin reaction and was not used in this application. Gas from the LC IsoLink was transferred to the mass spectrometer via two fused‐silica capillaries for sample (1.5 m × 100 μm id) and working gas (1.5 m × 50 μm i.d.).
3. RESULTS AND DISCUSSION
3.1. Notes on method development
The first problem to be addressed by this research was how to link the four analytical instruments; HPLC system, PCD unit, LC IsoLink and mass spectrometer. The three liquid‐handling units were connected with 0.005″ i.d. PEEK tubing, with the exception of a 30 mm length of larger‐bore 0.03″ i.d. PEEK tubing used to connect the PCD reaction coil outlet to the mixing tee where acid was added (shown in bold in Figure 2). This larger‐bore tubing was used to prevent clogging from the RP that was most likely to form at this point, as the reaction solution cooled and before downstream acid addition that began to dissolve the RP. Initial experiments resulted in many clogging problems due to RP and a length of tubing was added downstream of the acid addition to allow 1–2 min for RP to dissolve (Figure 2, reactor #2). Later experiments dispensed with this extra reactor when it was found that clogging was minimised if relatively small samples (<8 V peak heights) were analysed, a practice that became routine and also resulted in narrower peaks. Using relatively small samples and adding 25% phosphoric acid immediately following the PCD reaction coil allowed routine analysis over several days, with cleaning typically once a week. Cleaning mostly involved removing hydrindantin that precipitated as a side‐product of the RP reaction. The cleaning regime involved cooling the PCD reaction coil to room temperature then sequential flushing with 1% potassium hydroxide, 100% water, 100% acetone, and 10% nitric acid. Finally, three separate flushes of high‐purity water were used to remove the cleaning agents (especially acetone) that could enter the mass spectrometer via the LC IsoLink CO2 separation membrane and disturb the background signals of m/z 44, 45 and 46 for many hours. The system required some maintenance each day and regular cleaning at least once a week. Having a spare CO2 separation membrane and PCD reaction coil proved useful.
The second challenge was to optimise the PCD reaction chemistry to produce consistent yields of CO2 and reproducible δ 13CCARBOXYL values. The PCD reaction presented a large number of parameters to be optimised including the concentrations of ninhydrin, organic solvent and acid in the PCD solution, the overall pH and buffer strength of the reagent plus the reaction temperature and time in the coil. Faster (more complete) reactions have been reported at higher pH, higher temperature, longer reaction times and greater concentrations of ninhydrin.27, 28
Although commercial reagents are formulated for consistent colour development and fast analysis, not necessarily complete reaction, we began with conditions common to many commercial systems: pH 5.8 and 130°C reaction. An increased concentration of organic solvent (sulfolane) facilitated higher concentrations of ninhydrin in the reagent that had a concomitant effect of faster reaction times. The use of organic solvents such as sulfolane is avoided in LC/CO/IRMS in which any organic carbon present is oxidised to CO2, but organic solvents such as sulfolane could be tolerated in the PSIA method that specifically targeted carboxyl groups without oxidation. Some organic solvents were found to decompose at high temperatures and sulfolane was chosen for its high boiling point and good thermal stability, i.e. low CO2 background in the mass spectrometer. The reagent was buffered using phosphate to stabilise the chemistry, with the amounts of sulfolane and phosphate adjusted such that all components would dissolve overnight to a clear, yellow solution. The final PCD solution was prepared as follows (per litre): sulfolane (200 mL), deionised water (800 mL), ninhydrin (14 g), sodium dihydrogen phosphate dihydrate (28.905 g) and disodium phosphate anhydrous (2.659 g). The solution was sparged overnight with helium to remove residual CO2 from this slightly acidic (pH 5.8) phosphate‐buffered solution.
The normal working temperature for the PCD reactor (130°C) was found to be adequate for routine work with the FIA/NR/IRMS system. To illustrate, Figure 3 shows the relative CO2 yields and Δ 13CCARBOXYL (Δ = δ 1 – δ 2) values at reaction temperatures of 90, 100 and 130°C, with Δ 13CCARBOXYL representing the difference between the measured δ 13CCARBOXYL values and values obtained at 130°C. At lower temperatures the CO2 yields dropped dramatically while the Δ 13CCARBOXYL values changed only slightly in the −3 to +1 ‰ range compared with results at 130°C. As the reaction temperature approached 130°C both yields and Δ 13CCARBOXYL values reached asymptotic values that were consistent above this temperature (e.g. Table 2). These patterns were also typical of other amino acids. These relatively minor deviations in the Δ 13CCARBOXYL values (Figure 3) indicate that yields were not too critical in the PSIA reactions, and were consistent with reaction 1 (Figure 1) mostly limiting reaction kinetics, in agreement with a previous study.22 Larger isotope effects and larger Δ 13CCARBOXYL value offsets than those documented for low yields in Figure 3 would be expected if decarboxylation (Figure 1, reaction 2) was rate‐limiting.
Figure 3.
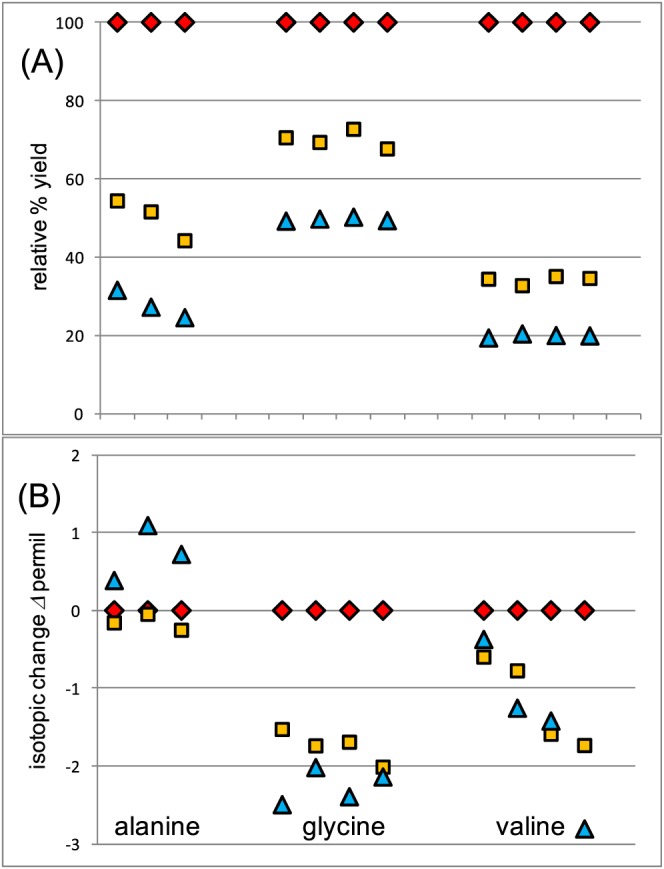
The effect of increasing reaction temperature on (A) yield of CO2 and (B) CO2 carbon isotopic composition for a number of samples of alanine [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 2.
– Carboxyl carbon isotopic compositions (δ 13CCARBOXYL values (‰)) versus VPDB for four glycine samples measured over a 1‐year period and at different reaction temperatures
| Date | Mar. 2016 | Feb. 2017 | Mar. 2017 | Mar. 2017 | ||
|---|---|---|---|---|---|---|
| Temperature (°C) | 150 | 130 | 130 | 170 | Mean | Predictedb |
| WHG | −27.5 ± 0.16 | −28.1 ± 0.2 | −27.8 ± 0.2 | −26.1 ± −0.2 | ||
| USGS64 (G1) | −38.0 ± 0.2 | −38.8 ± 0.2 | −38.3 ± 0.3 | −36.3 ± 0.2 | ||
| USGS65 (G2) | −24.9 ± 0.1 | −25.3 ± 0.1 | −24.9 ± 0.1 | −23.0 ± 0.2 | ||
| USGS66 (G3) | +14.3 ± 0.2 | +13.6 ± 0.2 | +13.5 ± 0.1 | +16.0 ± 0.4 | ||
| G1a | −37.5 | −37.7 | −37.6 | −37.3 | −37.5 ± 0.2 | |
| G2a | −24.2 | −24.3 | −24.5 | −24.0 | −24.2 ± 0.2 | |
| G3a | +14.2 | +14.7 | +14.7 | +15.1 | +14.6 ± 0.4 | |
| Δ G2‐G1 | 13.4 | 13.5 | 13.0 | 13.3 | 13.3 ± 0.2 | 13.4 |
| Δ G3‐G2 | 38.4 | 39.0 | 39.3 | 39.1 | 38.9 ± 0.4 | 39.2 |
| Δ G3‐G1 | 51.7 | 52.4 | 52.3 | 52.4 | 52.2 ± 0.3 | 52.6 |
δ scale has been shifted based on measurements of in‐house QC glycine WHG.
From calculations based on the reported formulations of these materials presented in the supporting information, assuming 96% purity of enriched materials used in preparations.
The PCD reaction coil typically required 2 h to stabilise after which the δ 13CCARBOXYL values became very precise and repeatable; the average standard deviation (SD) for multiple isotope measurements was typically <0.1 ‰ (n = 3) so the system was precise. There were, however, day‐to‐day offsets or variations in accuracy, for reasons that were usually unclear but might include incomplete degassing of CO2 from reaction solutions or incomplete transfer of CO2 across a contaminated diffusion membrane to the mass spectrometer. Regardless of the detailed explanation, it became apparent that measuring isotopic differences between samples proved to be very reproducible across reaction conditions, even when the absolute daily values varied somewhat. Table 2 illustrates some of the reaction conditions explored over a 1‐year period for three glycine standards (USGS 64–66 also known as G1‐G3). To correct for varying reaction conditions, a number of in‐house quality control materials including ‘Woods Hole glycine’ (WHG) were analysed together with each batch of samples. Using this in‐house standard to normalise data, results were very comparable across more than 1 year of work and under several time and temperature variations in reaction conditions.
Previous studies have shown that ninhydrin reactions at higher pH (4.7) can result in low yields of CO2 for the amino acid cystine and 5 to 50% over‐yields for glutamic acid and lysine.27, 28 This study confirmed these results and showed that USGS41a glutamic acid was especially sensitive to over‐yield because the α carboxyl group was highly 13C‐enriched (approximately +280 ‰) versus natural carbon in the reaction mix. For this amino acid, over‐yield of CO2 resulted in δ 13CCARBOXYL values of approximately +150 ‰ compared with an expected value of approximately +280 ‰. Earlier studies27, 28 showed that cystine, glutamic acid and lysine give quantitative yields under controlled low pH conditions, and we found that adding 50 mM sulphuric acid to the sample and to the HPLC solvent gave 100% yields, the expected δ 13CCARBOXYL values for USGS41a, and consistent δ 13CCARBOXYL values for lysine and cystine.
3.2. Notes on system performance
The water background (m/z 18) of the FIA/NR/IRMS system was typically 30 to 45 V in the middle Faraday collector. Although greater than the 12 V specified in the LC IsoLink operating manual this background appeared to be comparable with the water background reported by other LC/IRMS users. The background of CO2 (m/z 44) was between 0.4 and 2.2 V and was typically less than the 2.0 V background reported for LC/CSIA applications. The reduced m/z 44 background allowed observations of 13C/12C isotope ratio swings that were typically 3 to 6 ‰ across peaks.
Injections of 3 to 24 μL of 5 mM amino acid solutions yielded CO2 with typical peak heights of 3 to 6 V with amplitude/width ratios between 75 and 150. Blank injections of high‐purity water produced peaks with <1% of the height of sample injections.
The stretch factor of the mass spectrometer was assessed by injecting solutions of inorganic carbon standards that had known δ 13C values; LSVEC lithium carbonate (−46.6 ‰) and LSUB sodium bicarbonate (−3.28 ± 0.06 ‰). Typically, the measured δ 13C values were in close agreement with the expected values and no stretch correction was applied.
Peak size correction (linearity) was assessed by injecting varying amounts of amino acid solutions and was found to be approximately 0.5 ‰ across the range of peak sizes and for a number of amino acids. A linearity correction factor was applied as previously described.39
The average yields for the RMs were somewhat variable and ranged from 96 to 108% (Table 3) although the range always included 100%. Common amino acids, other than those listed in Table 3, had similar, near‐100% yields – aspartic acid, proline, threonine, phenylalanine, leucine, etc. – such that the ninhydrin chemistry appeared generally robust for the quantitative recovery of carboxyl CO2, as previously observed.27, 28
Table 3.
– Yields of CO2 derived from on‐line, automated reaction of amino acids with ninhydrin reagent
| Amino acid | Yield (%) relative to LSVEC carbonate solution (± 1 sd) | |
|---|---|---|
| L‐alanine | 102 ± 9 | n = 12 |
| L‐glutamic acid | 108 ± 20 | n = 6 |
| glycine | 103 ± 3 | n = 27 |
| L‐valine | 96 ± 6 | n = 15 |
Because many metabolic studies involving amino acids are conducted using labelled materials the authors considered the possibility of carry‐over from the isotopic composition of previous, potentially labelled, compounds. Alternating injections of labelled (USGS41a) and natural abundance (USGS40) glutamic acid, with a difference in their δ 13CCARBOXYL values of approximately 310 ‰, showed that carry‐over was not readily detectable and was no more than 0.2 ‰. This test indicated a very small amount of carry‐over, <0.1% of the true isotope difference between subsequent samples.
3.3. δ 13CCARBOXYL measurements of international RMs
Repeated measurements of glycine samples made over a 1‐year period at reaction temperatures of 130 to 170°C showed that the between‐sample Δ 13CCARBOXYL values were consistent across time (Table 2), and that these differences were in reasonable agreement with the theoretical values of 13.4, 39.2 and 52.6 ‰ (see supporting information). The consistency of the results over a long period (Table 2) and the low sensitivity to yield (Figure 3) indicated that the automated system was robust for PSIA determinations. A final step was to perform an inter‐laboratory calibration to test whether similar results were obtained by independent laboratories and to establish a common anchor or reference point for PSIA δ 13CCARBOXYL measurements.
The inter‐laboratory calibration involved three L‐alanine samples that were measured in both Tokyo and Brisbane. The two IRMS systems were independently calibrated with inorganic carbon standards, and the ninhydrin reaction conditions were optimised for high yield. The measurements in Tokyo involved 1‐h off‐line ninhydrin incubations that were much longer than the 3–7‐min heating times used in the FIA/NR/IRMS procedure in Brisbane. The Tokyo measurements occurred with >95% yields and consistent δ 13CCARBOXYL values were obtained, with replicates (n = 5) for each L‐alanine sample. The difference between laboratories for the average of the three L‐alanines was small, <0.3 ‰, so that there was overall good agreement between independent laboratories regarding the L‐alanine PSIA values. The average of these L‐alanine values was used as the best available anchor for the δ 13CCARBOXYL data at this time, with Brisbane data adjusted to match the average measured in Tokyo.
Using this common anchor approach, the values shown in bold in Table 4 from the automated system are a first estimate of the δ 13CCARBOXYL values for internationally available RMs. The uncertainties associated with these values were calculated as a combination of:
-
‐
the uncertainty in the certified CSIA values of the materials,
-
‐
the standard deviation derived from replicate off‐line PSIA measurements of L‐alanine,
-
‐
the standard deviation derived from replicate FIA PSIA measurements.
Table 4.
– The measured and calculated carbon isotopic composition of amino acid α‐carboxyl groups. Values in bold are for internationally available RMs developed for inter‐laboratory calibration R
| Reference material | Nature | δ 13CCARBOXYL ×1000, VPDB | |||
|---|---|---|---|---|---|
| expecteda | measured | MU (k = 2)b | |||
| 99% | 96% | ||||
| USGS40 | L‐glutamic acid | −30.0 | ± 0.9 | ||
| USGS41a | L‐glutamic acid | +284.9 | +275.2 | +276.8 | ± 0.9 |
| USGS64 | glycine G1 | −37.7 | ± 0.3 | ||
| USGS65 | glycine G2 | −23.9 | −24.3 | −24.3 | ± 0.3 |
| USGS66 | glycine G3 | +16.6 | +14.9 | +14.7 | ± 0.3 |
| D01 | L‐valine | −5.6 | ± 0.6 | ||
| USGS73 | L‐valine V1 | −18.5 | ± 0.5 | ||
| USGS74 | L‐valine V2 | −2.8 | −2.9 | −2.7 | ± 0.6 |
| USGS75 | L‐valine V3 | +33.3 | +32.1 | +31.6 | ± 0.3 |
| TTA | L‐alanine | −31.60 ± 0.28 c | −31.58 ± 0.3 d | ||
| TTS | L‐alanine | −27.98 ± 0.08 c | −28.06 ± 0.1 d | ||
| TTW | L‐alanine | −28.85 ± 0.03 c | −28.80 ± 0.2 d | ||
Calculated from information supplied by Qi36 or Schimmelmann37 assuming either 99 or 96% purity of the α‐carboxyl labelled materials used.
Measurement uncertainty (MU) was determined from three sources, as explained in the text, using a coverage factor (k) of two. The reported MU is an error‐propagated SD, and multiplied by 2 to approximate a 95% confidence range for the reported mean value.
Values determined by off‐line reaction with ninhydrin.
Uncertainty for these materials is reported as ± one standard deviation for FIA/NR/IRMS anslysis.
The CSIA uncertainty was included to represent possible physical inhomogeneity in the RMs and resulting variability in isotope ratios.37 The δ 13CCARBOXYL value assigned to USGS41a will probably be subject to larger uncertainty than the 0.9 ‰ given in Table 4 because the value has an associated extrapolation uncertainty as it fell far outside the calibration range of the underlying LSVEC and LSUB RMs.
Figure 4 shows the relationship between the calculated and measured δ 13CCARBOXYL values for the internationally available RMs. The measured values were corrected for linearity and anchored to the Tokyo (L‐alanine) scale, as explained above. Several of these RMs were prepared by mixing 13C‐enriched amino acids with natural abundance amino acids. Using the production notes involved in these preparations, and the measured values of the starting natural abundance RMs (USGS40, G1, V1 and D01), it was possible to estimate δ 13CCARBOXYL values for the 13C‐enriched RMs (see supporting information) These estimates allowed the comparison shown in Figure 4; these results show an overall close agreement between the calculated and measured δ 13CCARBOXYL values for a number of chemically different compounds (the solid line shows a 1:1 relationship).
Figure 4.
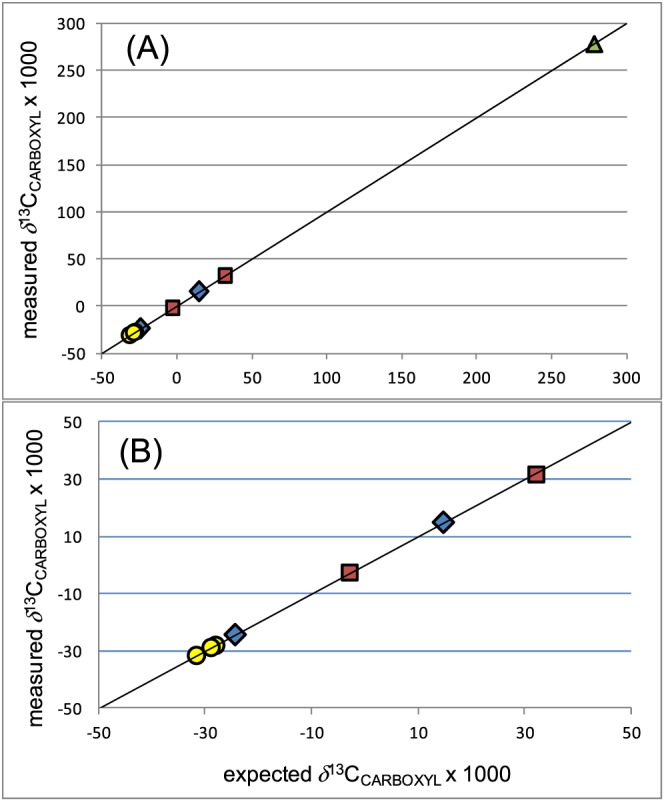
Measured versus expected δ 13CCARBOXYL values for amino acids (from Table 4) showing (A) full data range and (B) excluding USGS41a. Triangle (green) = glutamic acid, circles (yellow) = alanine, diamonds (blue) = glycine, squares (red) = valine. Solid line shows a 1:1 relationship. Expected values were either measured off‐line or calculated based on production notes for mixtures of unlabelled and enriched compounds assuming 96% label at the α‐carboxyl position, as detailed in the supporting information. Alternative calculations assuming 99% labels are also presented in Table 4 and shown in the supporting information [Color figure can be viewed at http://wileyonlinelibrary.com]
Although it is widely recommended that stable isotope measurements are traceable to international reporting scales via two‐point calibration, PSIA is an emerging technique and the initial values reported here are based on a single‐point calibration, the average of the L‐alanine samples in Table 4. It may well become good practice for two‐point calibrations to use the approximately 50 ‰ differences that exist between RMs such as the two glycine samples (USGS64 and 66) or the two L‐valine sample (USGS73 and 75). We have analysed all the RMs successfully, only varying the reaction conditions somewhat, in particular using a more acidic (50 mM sulphuric acid) carrier and sample diluent for the glutamic acid RMs and avoiding similar acid conditions for the alanine and glycine RMs in Table 4.
4. CONCLUSIONS
The methodology described provided a means to measure the carbon isotopic composition of the carboxyl group (δ 13CCARBOXYL values) of amino acids and has proved to be reproducible over many months of operation. The methodology was found to be very forgiving of most changes in reaction temperature, chemistry and CO2 yield. Exceptions were found for glutamic acid and lysine that over‐yielded CO2 with biased and low δ 13CCARBOXYL values. More acidic conditions in the sample and carrier solvents were needed for good isotope results for glutamic acid and lysine. Cystine is also better analysed under acid conditions.
This study presents the first estimates for the δ 13CCARBOXYL values for the carboxyl groups of a number of internationally available amino acid RMs. The results from the automated method were found to be in close agreement with values determined both by off‐line measurements and by calculations based on descriptions of how enriched and natural abundance compounds were mixed to produce these RMs. Readers are invited to review and improve on the values presented in Table 4.
Using the method described, the analysis of an individual amino acid can be performed in approximately 7.5 min, such that the isotopic profile of the 17 amino acids, typically present in hair, can be obtained in a little over 2 h, provided that these amino acids have been separated and purified prior to PSIA.
Typical metabolic studies of amino acids analyse mixtures of compounds liberated from biological tissues by hydrolysis with hydrochloric acid whereas the methodology presented here is applicable only to individual amino acids. A next step will be to modify the instrument design to a system compatible with on‐line HPLC separation. Using this approach it may be possible to combine carbon and nitrogen CSIA (amino acid) and PSIA (carboxyl group) data to enhance the specificity and power of stable isotopes to elucidate sources and metabolic cycling of amino acids.
Supporting information
Data S1 Supporting information item
ACKNOWLEDGEMENTS
This work was partly funded by ARC Discovery Project DP150104006, QHFSS research grant RSS15‐ 005 and Kiban‐S grant‐in‐aid 17H06105 by the Japan Society for the Promotion of Science. Thermo Fisher Scientific provided replacement parts for the LC IsoLink. Arndt Schimmelmann and Jon Fong of Indiana University discussed the preparation of reference amino acids and supplied the starting material (D01 valine) used to prepare V2 and V3. Pantelis Rigas of Rigas Labs and Maria Orfitserova of Pickering Laboratories provided helpful advice about post‐column derivatisation. Urs Wermuth of QHFSS deepened our understanding of the ninhydrin reaction and Carol Church of QHFSS helped with literature review and citations. The authors thank the reviewers of this work for their careful reading and insightful comments that have greatly improved this manuscript. The authors also thank John Hayes for insightful discussions about the isotopic compositions of carboxyl carbon atoms.
Fry B, Carter JF, Yamada K, Yoshida N, Juchelka D. Position‐specific 13C/12C analysis of amino acid carboxyl groups – automated flow‐injection analysis based on reaction with ninhydrin. Rapid Commun Mass Spectrom. 2018;32:992‐1000. https://doi.org/10.1002/rcm.8126
REFERENCES
- 1. Scott JH, O'Brien D, Emerson D. An examination of the carbon isotope effects associated with amino acid biosynthesis. Astrobiology. 2006;6(6):867‐880. https://doi.org/10.1089/ast.2006.6.867 [DOI] [PubMed] [Google Scholar]
- 2. Rashaid AH, Harrington PB, Jackson GP. Profiling amino acids of Jordanian scalp hair as a tool for diabetes mellitus diagnosis: a pilot study. Anal Chem. 2015;87(14):7078‐7084. https://doi.org/10.1021/acs.analchem.5b00460 [DOI] [PubMed] [Google Scholar]
- 3. Tea I, Martineau E, Antheaume I, et al. 13C and 15N natural isotope abundance reflects breast cancer cell metabolism. Sci Rep. 2016;6:34251 https://doi.org/10.1038/srep34251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCullagh JS, Juchelka D, Hedges RE. Analysis of amino acid 13C abundance from human and faunal bone collagen using liquid chromatography/isotope ratio mass spectrometry. Rapid Commun Mass Spectrom. 2006;20(18):2761‐2768. https://doi.org/10.1002/rcm.2651 [DOI] [PubMed] [Google Scholar]
- 5. McCullagh J, Gaye‐Siessegger J, Focken U. Determination of underivatized amino acid δ 13C by liquid chromatography/isotope ratio mass spectrometry for nutritional studies: the effect of dietary non‐essential amino acid profile on the isotopic signature of individual amino acids in fish. Rapid Commun Mass Spectrom. 2008;22(12):1817‐1822. https://doi.org/10.1002/rcm.3554 [DOI] [PubMed] [Google Scholar]
- 6. Smith CI, Fuller BT, Choy K, Richards MP. A three‐phase liquid chromatographic method for δ 13C analysis of amino acids from biological protein hydrolysates using liquid chromatography–isotope ratio mass spectrometry. Anal Biochem. 2009;390(2):165‐172. https://doi.org/10.1016/j.ab.2009.04.014 [DOI] [PubMed] [Google Scholar]
- 7. Raghavan M, McCullagh JS, Lynnerup N, Hedges RE. Amino acid δ 13C analysis of hair proteins and bone collagen using liquid chromatography/isotope ratio mass spectrometry: Paleodietary implications from intra‐individual comparisons. Rapid Commun Mass Spectrom. 2010;24(5):541‐548. https://doi.org/10.1002/rcm.4398 [DOI] [PubMed] [Google Scholar]
- 8. Abaye DA, Morrison DJ, Preston T. Strong anion exchange liquid chromatographic separation of protein amino acids for natural 13C‐abundance determination by isotope ratio mass spectrometry. Rapid Commun Mass Spectrom. 2011;25(3):429‐435. https://doi.org/10.1002/rcm.4844 [DOI] [PubMed] [Google Scholar]
- 9. Lynch AH, McCullagh JSO, Hedges REM. Liquid chromatography/isotope ratio mass spectrometry measurement of δ 13C of amino acids in plant proteins. Rapid Commun Mass Spectrom. 2011;25(20):2981‐2988. https://dx.doi.org/10.1002/rcm.5142 [DOI] [PubMed] [Google Scholar]
- 10. Dunn PJH, Honch NV, Evershed RP. Comparison of liquid chromatography–isotope ratio mass spectrometry (LC/IRMS) and gas chromatography–combustion–isotope ratio mass spectrometry (GC/C/IRMS) for the determination of collagen amino acid δ 13C values for palaeodietary and palaeoecological reconstruction. Rapid Commun Mass Spectrom. 2011;25(20):2995‐3011. https://doi.org/10.1002/rcm.5174 [DOI] [PubMed] [Google Scholar]
- 11. Tea I, Tcherkez G. Natural isotope abundance in metabolites: Techniques and kinetic isotope effect measurement in plant, animal, and human tissues. Methods Enzymol. 2017;596:113‐147. https://doi.org/10.1016/bs.mie.2017.07.020 [DOI] [PubMed] [Google Scholar]
- 12. McCullagh JS. Mixed‐mode chromatography/isotope ratio mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(5):483‐494. https://doi.org/10.1002/rcm.4322 [DOI] [PubMed] [Google Scholar]
- 13. Jackson GP, An Y, Konstantynova KI, Rashaid AH. Biometrics from the carbon isotope ratio analysis of amino acids in human hair. Sci Justice. 2015;55(1):43‐50. https://doi.org/10.1016/j.scijus.2014.07.002 [DOI] [PubMed] [Google Scholar]
- 14. Rashaid Rashaid AH, Harrington PB, Jackson GP. Amino acid composition of human scalp hair as a biometric classifier and investigative lead. Anal Methods. 2015;7(5):1707‐1718. https://doi.org/10.1039/C4AY02588A [Google Scholar]
- 15. Abelson PH, Hoering T. Carbon isotope fractionation in formation of amino acids by photosynthetic organisms. Proc Natl Acad Sci U S A. 1961;47(5):623‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Savidge WB, Blair NB. Intramolecular carbon isotopic composition of monosodium glutamate: Biochemical pathways and product source identification. J Agric Food Chem. 2005;53:197‐201. https://doi.org/10.1021/jf040200k [DOI] [PubMed] [Google Scholar]
- 17. Corso TN, Brenna JT. High‐precision position‐specific isotope analysis. Proc Natl Acad Sci U S A. 1997;94(4):1049‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meier‐Augenstein W. Stable Isotope Forensics – Methods and Forensic Applications of Stable Isotope Analysis. 2nd ed. Oxford: Wiley‐Blackwell; 2018. ISBN 978‐11‐1‐908020‐6. [Google Scholar]
- 19. Jézéquel T, Joubert V, Giraudeau P, Gérald S, Remauda GS, Akokaa S. The new face of isotopic NMR at natural abundance. Magn Reson Chem. 2017;55:77‐90. https://doi.org/10.1002/mrc.4548 [DOI] [PubMed] [Google Scholar]
- 20. Bayle K, Grand M, Chaintreau A, et al. Internal referencing for 13C position‐specific isotope analysis measured by NMR spectrometry. Anal Chem. 2015;87:7550‐7554. https://doi.org/10.1021/acs.analchem.5b02094 [DOI] [PubMed] [Google Scholar]
- 21. Ruhemann S. CCXII. – Triketohydrindene hydrate. J Chem Soc Trans. 1910;97:2025‐2031. [Google Scholar]
- 22. Friedman M. Applications of the ninhydrin reaction for analysis of amino acids, peptides, and proteins to agricultural and biomedical sciences. J Agric Food Chem. 2004;52(3):385‐406. https://doi.org/10.1021/jf030490p [DOI] [PubMed] [Google Scholar]
- 23. Rigas PG. Liquid chromatography–post‐column derivatization for amino acid analysis: Strategies, instrumentation, and applications. Instrum Sci Technol. 2012;40(2–3):161‐193. https://doi.org/10.1080/10739149.2011.651669 [Google Scholar]
- 24. Joullié MM, Thompson TR, Nemeroff NH. Ninhydrin and ninhydrin analogs. Syntheses and applications. Tetrahedron. 1991;47(42):8791‐8830. https://doi.org/10.1016/S0040-4020(01)80997-2 [Google Scholar]
- 25. MacFadyen DA, Fowler N. On the mechanism of the reaction of ninhydrin with a‐amino acids. J Biol Chem. 1950;186:13‐22. [PubMed] [Google Scholar]
- 26. McCaldin DJ. The chemistry of ninhydrin. Chem Rev. 1960;60(1):39‐51. https://doi.org/10.1021/cr60203a004 [Google Scholar]
- 27. Van Slyke DD, Dillon RT. Gasometric determination of carboxyl groups in amino acids. Exp Biol Med. 1936;34(3):362‐363. https://doi.org/10.3181/00379727-34-8621P [Google Scholar]
- 28. Van Slyke DD, Dillon RT, MacFadyen DA, Hamilton P. Gasometric determination of carboxyl groups in free amino acids. J Biol Chem. 1941;141(2):627‐669. [Google Scholar]
- 29. Hamilton PB, Van Slyke DD, Lemish S. The gasometric determination of free amino acids in blood filtrates by the ninhydrin‐carbon dioxide method. J Biol Chem. 1943;150:231‐250. [Google Scholar]
- 30. Nelson D. A new method for carbon isotopic analysis of protein. Science. 1991;251(4993):552‐554. https://doi.org/10.1126/science.1990430 [DOI] [PubMed] [Google Scholar]
- 31. Keeling C, Nelson E, Slessor K. Stable carbon isotope measurements of the carboxyl carbons in bone collagen. Archaeometry. 1999;41(1):151‐164. https://doi.org/10.1111/j.1475-4754.1999.tb00857.x [Google Scholar]
- 32. Keeling C, Nelson D. Changes in the intramolecular stable carbon isotope ratios with age of the European cave bear (Ursus spelaeus). Oecologia. 2001;127(4):495‐500. https://doi.org/10.1007/s004420000611 [DOI] [PubMed] [Google Scholar]
- 33. Savidge WB, Blair NE. Patterns of intermolecular carbon isotopic heterogeneity within amino acids of autotrophs and heterotrophs. Oecologia. 2004;139:178‐189. https://doi.org/10.1007/s00442-004-1500-z [DOI] [PubMed] [Google Scholar]
- 34. Savidge WB, Blair NE. Seasonal and within‐plant gradients in the intramolecular carbon isotopic composition of amino acids of Spartina alterniflora . J Exp Mar Biol Ecol. 2004;308:151‐167. https://doi.org/10.1016/j.jembe.2004.02.010 [Google Scholar]
- 35. Godin JP, Stellingwerff T, Actis‐Goretta L, Mermoud AF, Kochhar S, Rezzi S. The role of liquid chromatography and flow injection analyses coupled to isotope ratio mass spectrometry for studying human in vivo glucose metabolism. Rapid Commun Mass Spectrom. 2011;25(20):2989‐2994. https://doi.org/10.1002/rcm.5179 [DOI] [PubMed] [Google Scholar]
- 36. Qi H, Coplen TB, Mroczkowski SJ, et al. A new organic reference material, L‐glutamic acid, USGS41a, for δ 13C and δ 15N measurements – a replacement for USGS41. Rapid Commun Mass Spectrom. 2016;30(7):859‐866. https://doi.org/10.1002/rcm.7510 [DOI] [PubMed] [Google Scholar]
- 37. Schimmelmann A, Qi H, Coplen TB, et al. Organic reference materials for hydrogen, carbon, and nitrogen stable isotope‐ratio measurements: caffeines, n‐alkanes, fatty acid methyl esters, glycines, L‐valines, polyethylenes, and oils. Anal Chem. 2016;88(8):4294‐4302. https://doi.org/10.1021/acs.analchem.5b04392 [DOI] [PubMed] [Google Scholar]
- 38. Krummen M, Hilkert AW, Juchelka D, Pesch R. A new concept for isotope ratio monitoring liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2004;18(19):2260‐2266. https://doi.org/10.1002/rcm.1620 [DOI] [PubMed] [Google Scholar]
- 39. Carter JF, Fry B. Ensuring the reliability of stable isotope ratio data – beyond the principle of identical treatment. Anal Bioanal Chem. 2013;405(9):2799‐2814. https://doi.org/10.1007/s00216-012-6551-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting information item