

Abstract
Dolutegravir (DTG), an important active pharmaceutical ingredient (API) used in combination therapy for the treatment of HIV, has been synthesized in continuous flow. By adapting the reported GlaxoSmithKline process chemistry batch route for Cabotegravir, DTG was produced in 4.5 h in sequential flow operations from commercially available materials. Key features of the synthesis include rapid manufacturing time for pyridone formation, one‐step direct amidation of a functionalized pyridone, and telescoping of multiple steps to avoid isolation of intermediates and enable for greater throughput.
Keywords: amidation, continuous flow, HIV, multistep synthesis, pyridone
HIV is a disease that currently affects around 37 million people.1 A number of innovative medicines has made HIV a manageable disease; however, the cost of treatment is still prohibitive for many patients in lower income countries.2 As part of the “Medicines for All” initiative funded by the Bill and Melinda Gates Foundation,3 our research groups are focused on providing greater access to essential medicines for serious diseases such as HIV, tuberculosis, and malaria. The initiative has led to a number of reported studies.4 We became interested in Dolutegravir due to its importance in HIV combination therapy.
Dolutegravir (DTG) 1 (Figure 1) is an HIV integrase inhibitor co‐developed by GlaxoSmithKline (GSK) and Shinogi that was approved by the Food and Drug Administration (FDA) in 2013. Integrase inhibitors prevent the HIV virus from inserting into cellular DNA by blocking transesterification, a process that is vital for replication and spread of the disease.5 Raltegravir 2 and Elvitegravir 3 were the first integrase inhibitors to be approved and used in combination therapy; however, these two drugs require large doses and a pharmacokinetic booster, respectively, and have shown vulnerability to HIV virus mutations.6 DTG is an un‐boosted, once daily 50 mg tablet that is recommended as a universal first‐line treatment in combination therapy due to its low dosage and limited side effects.7 Only minimal resistance has been observed thus far, which has not led to significant spread of HIV virus after mutation.6 The high resistance and minimal side effects have led DTG to be placed on the World Health Organization's List of Essential Medicines.8 Recently, the first two drug combination therapy for HIV (Dolutegravir and Rilpivirine) was approved by the FDA.9 A number of DTG analogues are currently in clinical trials, including Cabotegravir 4 and Bictegravir 5; the latter was recently approved by the FDA in a single tablet, three drug regimen (Figure 1).10 These analogues differ from Dolutegravir 1 in the oxazine ring size; thus, a synthesis for DTG should also be amenable to 4 or 5 if they emerge as the integrase inhibitor of choice in the future.
Figure 1.
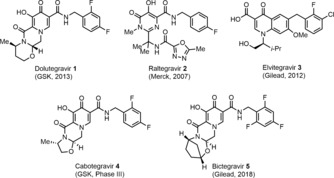
Integrase inhibitors for HIV treatment.
GSK and Shinogi disclosed a number of approaches to the synthesis of 1 and its analogues.11 Initial medicinal chemistry routes exploited commercially available heterocycles such as nicotinic acid and maltol, and subsequently installed the requisite functionalized N‐H pyridone in ten or more steps.11b–11d Wang and co‐workers from GSK later published a highly efficient, chromatography‐free approach to Cabotegravir 4 through rapid formation of the functionalized pyridone core 8 and subsequent cyclization with (S)‐alaninol 11 to synthesize the 5‐membered oxazine ring (Scheme 1).11e,11f We wished to optimize and adapt the synthesis to a continuous flow system in order to streamline manufacturing of the API.12 Continuous flow reactions benefit from increased mixing due to high surface area to volume ratio and the ability to heat solvents well past their boiling point.13 By developing an efficient flow synthesis and telescoping steps to avoid purifications, we felt we could achieve a short, scalable synthesis of DTG 1.
Scheme 1.
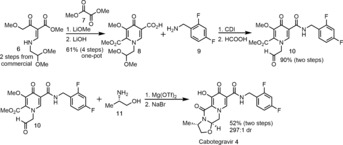
GSK process synthesis of Cabotegravir 4.
We began our investigation with the condensation reaction of methyl 4‐methoxyacetoacetate 12 and dimethylformamide dimethylacetal (DMF‐DMA) 13. Our first attempt at the analogous flow reaction proceeded with 60 % conversion to 14 when an equimolar ratio of the neat reactants 12 and 13 were streamed through a T‐mixer at 30 °C with a residence time (t R) of 10 min. Further optimization led to the discovery that an elevated reaction temperature of 85 °C and 1.6 equiv of DMF‐DMA 13 resulted in full conversion of 12 to 14 via HPLC with the same 10 min residence time.14 With an efficient approach to the dimethyl vinylogous amide 14, we next sought to telescope Steps 1 and 2 in continuous flow (Scheme 2). This was achieved by connecting Reactor I with a T‐mixer and adding neat aminoacetaldehyde dimethylacetal 15 directly to the output of Reactor I. Due to the high crystallinity of the product 6, we conducted Step 2 at 85 °C to increase solubility and avoid clogging. The optimized telescoped flow process produced 6 in an isolated yield of 95 % and a throughput of 43 g h−1.
Scheme 2.
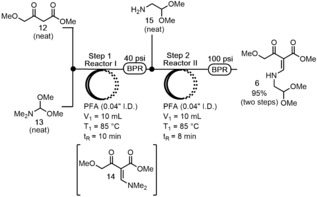
Telescoped flow synthesis of vinylogous amide 6. PFA=perfluoroalkoxy, I.D.=inside diameter.
The next step in the reaction sequence was the pyridone 16 formation (Scheme 3). Given the complexity of adapting to continuous flow, we first examined the independent flow process by starting with purified vinylogous amide 6. The choice of solvent for the reaction proved to be crucial given both 6 and dimethyl oxalate 7 are solids. A solvent screen in batch showed that CH3CN afforded the highest conversion (93 %) to product 16 compared to N‐methylpyrrolidinone and MeOH (both 85 %); however the CH3CN condition suffered from poor solubility that led to clogging in flow.14 In addition, when examining different bases for the deprotonation/cyclization sequence, NaOMe in MeOH had much better solubility and conversion to 16 than LiOMe, which was utilized in the GSK synthesis.11e,11f These screening results in batch led us to investigate Step 3 in flow using NaOMe as base and MeOH as solvent in order to simplify the system through the use of a single solvent. Following optimization of residence time and temperature,14 flow conditions of 85 °C and 30 min t R led to a 91 % isolated yield of 16 (Scheme 3).
Scheme 3.
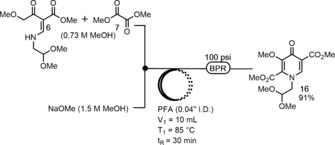
Pyridone 16 formation in flow.
Next, we examined the synthesis of pyridone 16 from 12 in a three‐step telescope process to obviate time consuming purification operations and work towards our goal of a fully continuous synthesis (Scheme 4). After minor modifications to the previously optimized flow conditions from Scheme 2 and Scheme 3, the telescoped synthesis of 16 was achieved. In this setup, several 40 psi back pressure regulators (BPR) were utilized as check valves and Reactor III was used to facilitate premixing of solution 7 with the output from Reactor II prior to the addition of NaOMe. A 55 min residence time in Reactor IV was required for the reaction to reach completion. The three‐step telescoped synthesis of pyridone 16 led to a 56 % isolated yield in a total residence time of 74 min with a throughput of 3.4 g h−1.
Scheme 4.
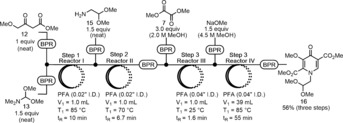
Three‐step telescoped synthesis of pyridone 16.
When considering ways to shorten the reported GSK synthesis, the ester saponification to form 8 and subsequent amide coupling stood out as an opportunity. Specifically, in GSK's synthesis of Cabotegravir 4 (Scheme 1),11e,11f ester 16 was saponified to the 5‐carboxylic acid 8 and then treated with carbonyldiimidazole (CDI) and difluorobenzylamine 9 to form amide product 17. The process required a filtration and extraction, and took 17 h in total. We were encouraged by a similar direct amidation that was reported in the midst of our own studies.15 Application of Kumar and co‐workers’ AcOH‐catalyzed conditions led to high yield and chemoselectivity for amidation at the 5‐position, producing amide 17 after 10 h (Scheme 5 a). We reason that there is both an electronic and steric effect which led to the selectivity. When the corresponding N‐H pyridone was reacted under the same conditions, a 2:1 selectivity for 5‐amidation over 2‐amidation was observed. The only byproducts observed on a gram scale batch reaction of 16 were ring opened starting material 6 and the analogous difluorobenzyl vinylogous amide (<5 % each). An ethyl ester substrate gave comparable yield and AcOH was found to be necessary for practical reaction time.16 A brief screen of Brønsted and Lewis acids revealed that the batch reaction could be accelerated compared to the AcOH condition, but with a decrease in isolated yield due to other byproducts and generally poorer solubility.
Scheme 5.
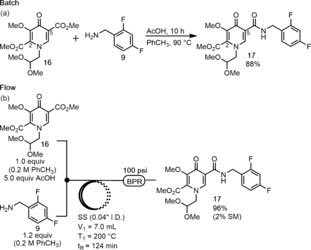
Direct amidation comparison. SS=stainless steel.
The next step was to adapt the process to a flow setup. Initially, we examined the reaction at 120 °C with a residence time of 1 h. Gratifyingly, product 17 was obtained with the use of a 100 psi BPR albeit in low conversion of starting material 16. A systematic evaluation of temperature and residence time led to good conversion at 150 °C with a 1 h residence time and at 180 °C with a 30 min residence time.14 We screened other solvents and found that dioxane and CH3CN were viable, but gave lower conversion than PhCH3 in the flow setup. DCE led to clogging in the system, and DMF and MeOH failed to produce any desired amide 17 after 1 h. Under the optimal conditions of 200 °C with a residence time of 124 min, we obtained 96 % isolated yield of amide 17 (2 % recovered 16) on a 3 mmol scale, which amounted to 3.5 g h−1 (Scheme 5 b).
The analogous base‐promoted amidation17 was also feasible using either LiOMe or NaOMe as base in a MeOH/PhCH3 mixed solvent system. The optimized flow process led to both shorter residence time and milder temperature compared to the acid‐mediated method (Scheme 6). However, attempts to telescope the basic amidation into the subsequent downstream process led to extensive clogging issues.
Scheme 6.
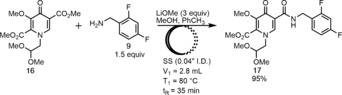
Base‐promoted direct amidation in flow.
Next, we examined the acetal deprotection of 17 and cyclization with (R)‐3‐aminobutan‐1‐ol 18 in flow. Initial attempts were conducted on purified pyridone amide 17 with an acid additive and amino alcohol 18. Ultimately, we found that formic acid was required as a co‐solvent to observe any conversion of 17. Our best result afforded an 8 % yield of DTG‐OMe 19 as a 5:1 mix of diastereomers favoring the desired product (Scheme 7).18 The major byproducts were ring‐opened 20, produced through elimination of the hemiaminal ether functionality, and the deprotected pyridone aldehyde 10. We reasoned that a two‐step flow procedure in which an acid would deprotect acetal 17 in one reactor and then meet amino alcohol 18 in a subsequent reactor may be more fruitful for conversion and milder conditions. Gratifyingly, separating the steps gave full conversion and allowed for stoichiometric amounts of p‐TsOH⋅H2O instead of neat formic acid with no observation of elimination byproduct 20.
Scheme 7.
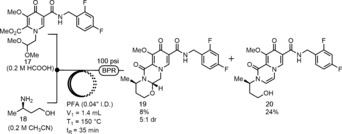
Initial cyclization attempts.
We next sought to telescope the acid‐mediated direct amidation into the deprotection/cyclization steps to form a three‐step telescoped sequence. Initially, incomplete conversion was observed due to issues with PhCH3 from Step 4 inhibiting the subsequent steps; however, it was eventually found that more concentrated reaction solutions and longer residence times in Steps 5 and 6 led to full conversion of intermediate amide 17 and aldehyde 10. The optimized setup proceeded in a total residence time of just over 3 h and a 48 % isolated yield of DTG‐OMe 19 in 7:1 dr (Scheme 8). The major diastereomer was separated by silica gel chromatography to give analytically pure material.
Scheme 8.
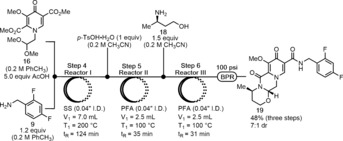
Telescoped synthesis of DTG‐OMe 19.
Finally, we examined the demethylation step in continuous flow as a discrete operation using purified 19. Following a batch screen of different demethylating reagents, it was found that GSK's published reaction using LiBr gave the best conversion and lowest amount of byproducts.11e,11f Reaction temperatures higher than 120 °C led to formation of a similar elimination byproduct to 20. The batch conditions translated well to flow, with 89 % yield of DTG observed at 100 °C with a residence time of 31 min (Scheme 9). The reaction concentration proved crucial to reproducible, extended running of the continuous flow reactor. DTG 1 was insoluble upon cooling to room temperature, which led to clogging at concentrations higher than 0.5 m THF. Attempts to telescope this final demethylation with the previous three‐step sequence in Scheme 8 led to an 8 % isolated yield over four steps; however, the same clogging issue meant the system could not be run for more than 10 h at a time.
Scheme 9.
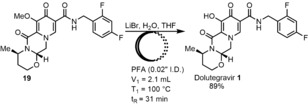
Demethylation to form DTG 1.
In conclusion, we have developed a continuous flow synthesis of the HIV integrase inhibitor Dolutegravir 1. The optimized process described involved seven total steps in three separate flow operations in 24 % overall yield (37 % overall when Step 3 was run as a separate flow operation).19 The key features of the flow route are rapid manufacturing time, direct amidation of ester 16 to reduce the step count, and separation of the acetal deprotection/oxazine formation flow reactors to attain high reactivity and selectivity for tricyclic product DTG‐OMe 19. Importantly, our synthesis should be adaptable to both Cabotegravir 4 and Bictegravir 5 by switching the benzylamine and amino alcohol used in the synthesis. Further studies will focus on the telescoping of all steps to achieve an end‐to‐end continuous flow synthesis as well as formulation of the final API as its sodium salt and to produce cGMP formulations in an engineered system without the use of silica gel chromatography.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Bill & Melinda Gates Foundation (“Medicines for All” intitiative) for funding. We thank Dr. Rachel L. Beingessner and Dr. Justin Lummiss (MIT) as well as Matt Graham, Pooja Panchal, Megan Freeman and Dr. Daniel Cook (VCU) for helpful discussions.
R. E. Ziegler, B. K. Desai, J.-A. Jee, B. F. Gupton, T. D. Roper, T. F. Jamison, Angew. Chem. Int. Ed. 2018, 57, 7181.
Contributor Information
Prof. Dr. Thomas D. Roper, Email: tdroper@vcu.edu
Prof. Dr. Timothy F. Jamison, Email: tfj@mit.edu.
References
- 1. Vitoria M., Hill A. M., Ford N. P., Doherty M., Khoo S. H., Pozniak A. L., J. Int. AIDS Soc. 2016, 19, 20504–20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gupta A., Juneja S., Vitoria M., Habiyambere V., Nguimfack B. D., Doherty M., Low-Beer D., PLoS One 2016, 11, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medicines For All website: https://medicines4all.vcu.edu.
- 4.For recent publications, see:
- 4a. Ocampo C. E., Lee D., Jamison T. F., Org. Lett. 2015, 17, 820–823; [DOI] [PubMed] [Google Scholar]
- 4b.A. Sevenich, G.-Q. Liu, A. J. Arduengo 3rd, B. F. Gupton, T. Opatz, J. Org. Chem 2017, 82, 1218–1223; [DOI] [PubMed]
- 4c. Verghese J., Kong C. J., Rivalti D., Yu E. C., Krack R., Alcazar J., Manley J. B., McQuade D. T., Ahmad S., Belecki K., Gupton B. F., Green Chem. 2017, 19, 2986–2991; [Google Scholar]
- 4d. Britton J., Jamison T. F., Angew. Chem. Int. Ed. 2017, 56, 8823–8827; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8949–8953; [Google Scholar]
- 4e. Britton J., Jamison T. F., Eur. J. Org. Chem. 2017, 6566–6574. [Google Scholar]
- 5.For recent reviews, see:
- 5a. Su M., Tan J., Lin C.-Y., Drug Discovery Today 2015, 20, 1337–1348; [DOI] [PubMed] [Google Scholar]
- 5b. Gu S.-X., Xue P., Ju X.-L., Zhu Y.-Y., Bioorg. Med. Chem. 2016, 24, 5007–5016; [DOI] [PubMed] [Google Scholar]
- 5c. Meanwell N. A., Bioorg. Med. Chem. Lett. 2017, 27, 5355–5372; [DOI] [PubMed] [Google Scholar]
- 5d. Psichogiou M., Poulakou G., Basoulis D., Paraskevis D., Markogiannakis A., Daikos G. L., Curr. Pharm. Des. 2017, 23, 2552–2567; [DOI] [PubMed] [Google Scholar]
- 5e. Han Y., Mesplede T., Wainberg M. A., Expert Opin. Invest. Drugs 2017, 26, 1207–1213. [DOI] [PubMed] [Google Scholar]
- 6. Wainberg M. A., Mesplede T., J. Int. AIDS Soc. 2015, 18, 20824–20826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cahn P., Curr. Opin. HIV AIDS 2017, 12, 318–323. [DOI] [PubMed] [Google Scholar]
- 8.WHO Model List of Essential Medicines. 20th list, March 2017.
- 9. Capetti A. F., Cossu M. V., Paladini L., Rizzardini G., Expert Opin. Pharmacother. 2018, 19, 65–77. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gallant J., Lazzarin A., Mills A., Orkin C., Podzamczer D., Tebas P., Girard P.-M., Brar I., Daar E. S., Wohl D., Rockstroh J., Wei X., Custodio J., White K., Martin H., Cheng A., Quirk E., Lancet 2017, 390, 2063–2072; [DOI] [PubMed] [Google Scholar]
- 10b. Sax P. E., Pozniak A., Montes M. L., Koenig E., DeJesus E., Stellbrink H.-J., Antinori A., Workowski K., Slim J., Reynes J., Garner W., Custodio J., White K., SenGupta D., Cheng A., Quirk E., Lancet 2017, 390, 2073–2082; [DOI] [PubMed] [Google Scholar]
- 10c. Markham A., Drugs 2018, 78, 601–606. [DOI] [PubMed] [Google Scholar]
- 11.For a review, see:
- 11a. Schreiner E., Richter F., Nerdinger S., Top. Heterocycl. Chem. 2016, 44, 187–208; for selected articles, see: [Google Scholar]
- 11b. Kawasuji T., Johns B. A., Yoshida H., Taishi T., Taoda Y., Murai H., Kiyama R., Fuji M., Yoshinaga T., Seki T., Kobayashi M., Sato A., Fujiwara T., J. Med. Chem. 2012, 55, 8735–8744; [DOI] [PubMed] [Google Scholar]
- 11c. Kawasuji T., Johns B. A., Yoshida H., Weatherhead J. G., Akiyama T., Taishi T., Taoda Y., Mikamiyama-Iwata M., Murai H., Kiyama R., Fuji M., Tanimoto N., Yoshinaga T., Seki T., Kobayashi M., Sato A., Garvey E. P., Fujiwara T., J. Med. Chem. 2013, 56, 1124–1135; [DOI] [PubMed] [Google Scholar]
- 11d. Johns B. A., Kawasuji T., Weatherhead J. G., Taishi T., Temelkoff D. P., Yoshida H., Akiyama T., Taoda Y., Murai H., Kiyama R., Fuji M., Tanimoto N., Jeffrey J., Foster S. A., Yoshinaga T., Seki T., Kobayashi M., Sato A., Johnson M. N., Garvey E. P., Fujiwara T., J. Med. Chem. 2013, 56, 5901–5916; [DOI] [PubMed] [Google Scholar]
- 11e. Goodman S. N., Kowalski M. D., Mans D. M., Wang H., US 8889877B220141118, 2014;
- 11f. Wang H., Kowalski M. D., Lakdawala A. S., Vogt F. G., Wu L., Org. Lett. 2015, 17, 564–567. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews, see:
- 12a. Malet-Sanz L., Susanne F., J. Med. Chem. 2012, 55, 4062–4098; [DOI] [PubMed] [Google Scholar]
- 12b. Baumann M., Baxendale I. R., Beilstein J. Org. Chem. 2015, 11, 1194–1219; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Porta R., Benaglia M., Puglisi A., Org. Process Res. Dev. 2016, 20, 2–25; [Google Scholar]
- 12d. Hughes D. L., Org. Process Res. Dev. 2018, 22, 13–20; for recent examples, see: [Google Scholar]
- 12e. Hopkin M. D., Baxendale I. R., Ley S. V., Org. Biomol. Chem. 2013, 11, 1822–1839; [DOI] [PubMed] [Google Scholar]
- 12f. Hartwig J., Ceylan S., Kupracz L., Coutable L., Kirschning A., Angew. Chem. Int. Ed. 2013, 52, 9813–9817; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9995–9999; [Google Scholar]
- 12g. Mascia S., Heider P. L., Zhang H., Lakerveld R., Benyahia B., Barton P. I., Braatz R. D., Cooney C. L., Evans J. M. B., Jamison T. F., Jensen K. F., Myerson A. S., Trout B. L., Angew. Chem. Int. Ed. 2013, 52, 12359–12363; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12585–12589; [Google Scholar]
- 12h. Tsubogo T., Oyamada H., Kobayashi S., Nature 2015, 520, 329–332; [DOI] [PubMed] [Google Scholar]
- 12i. Martin A. D., Siamaki A. R., Belecki K., Gupton B. F., J. Org. Chem. 2015, 80, 1915–1919; [DOI] [PubMed] [Google Scholar]
- 12j. Correia C. A., Gilmore K., McQuade D. T., Seeberger P. H., Angew. Chem. Int. Ed. 2015, 54, 4945–4948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5028–5032; [Google Scholar]
- 12k. Adamo A., Beingessner R. L., Benham M., Chen J., Jamison T. F., Jensen K. F., Monbaliu J.-C. M., Myerson A. S., Revalor E. M., Snead D. R., Stelzer T., Weeranoppanant N., Wong S. Y., Zhang P., Science 2016, 352, 61–67; [DOI] [PubMed] [Google Scholar]
- 12l. Yang J. C., Niu D., Karsten B. P., Lima F., Buchwald S. L., Angew. Chem. Int. Ed. 2016, 55, 2531–2535; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2577–2581; [Google Scholar]
- 12m. Suveges N. S., de Souza R. O. M. A., Gutmann B., Kappe C. O., Eur. J. Org. Chem. 2017, 6511–6517; [Google Scholar]
- 12n. Zhang P., Weeranoppanant N., Thomas D. A., Tahara K., Stelzer T., Russell M. G., O'Mahony M., Myerson A. S., Lin H., Kelly L. P., Jensen K. F., Jamison T. F., Dai C., Cui Y., Briggs N., Beingessner R. L., Adamo A., Chem. Eur. J. 2018, 24, 2776–2784. [DOI] [PubMed] [Google Scholar]
- 13.For selected recent reviews on the benefits of continuous flow, see:
- 13a. McQuade D. T., Seeberger P. H., J. Org. Chem. 2013, 78, 6384–6389; [DOI] [PubMed] [Google Scholar]
- 13b. Ley S. V., Fitzpatrick D. E., Ingham R. J., Myers R. M., Angew. Chem. Int. Ed. 2015, 54, 3449–3464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3514–3530; [Google Scholar]
- 13c. Gutmann B., Cantillo D., Kappe C. O., Angew. Chem. Int. Ed. 2015, 54, 6688–6728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6788–6832; [Google Scholar]
- 13d. Ley S. V., Fitzpatrick D. E., Myers R. M., Battilocchio C., Ingham R. J., Angew. Chem. Int. Ed. 2015, 54, 10122–10136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10260–10275; [Google Scholar]
- 13e. Morse P. D., Beingessner R. L., Jamison T. F., Isr. J. Chem. 2017, 57, 218–227; [Google Scholar]
- 13f. Britton J., Raston C. L., Chem. Soc. Rev. 2017, 46, 1250–1271; [DOI] [PubMed] [Google Scholar]
- 13g. Plutschack M. B., Pieber B., Gilmore K., Seeberger P. H., Chem. Rev. 2017, 117, 11796–11893. [DOI] [PubMed] [Google Scholar]
- 14.See the Supporting Information for complete experimental details.
- 15. Sankareswaran S., Mannam M., Chakka V., Mandapati S. R., Kumar P., Org. Process Res. Dev. 2016, 20, 1461–1468. [Google Scholar]
- 16.Less than 10 % conversion was observed when 9 and 16 were refluxed in PhCH3 overnight.
- 17.For a continuous flow direct amidation mediated by base, see: Vrijdag J. L., Delgado F., Alonso N., De Borggraeve W. M., Perez-Macias N., Alcazar J., Chem. Commun. 2014, 50, 15094–15097. [DOI] [PubMed] [Google Scholar]
- 18.5:1 is the thermodynamic ratio of diastereomers, see ref. [11f] for calculations and experimental results.
- 19.Note that reference 11f is for the synthesis of Cabotegravir. GSK's Dolutegravir synthesis has been noted in a patent (reference [11e]), but not described in full detail.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary