

Abstract
C–N cross-coupling is an important class of reactions with far-reaching impacts across chemistry, materials science, biology, and medicine. Transition metal complexes can elegantly orchestrate diverse aminations but typically require demanding reaction conditions, precious metal catalysts, or oxygen-sensitive procedures. Here, we introduce a mild nickel-catalyzed C–N cross-coupling methodology that operates at room temperature using an inexpensive nickel source (NiBr2·3H2O), is oxygen tolerant, and proceeds through direct irradiation of the nickel–amine complex. This operationally robust process was employed for the synthesis of diverse C–N-coupled products (40 examples) by irradiating a solution containing an amine, an aryl halide, and a catalytic amount of NiBr2·3H2O with a commercially available 365 nm LED at room temperature without added photoredox catalyst and the amine substrate serving additional roles as the ligands and base. Density functional theory calculations and kinetic isotope effect experiments were performed to elucidate the observed C–N cross-coupling reactivity.
Graphical abstract
INTRODUCTION
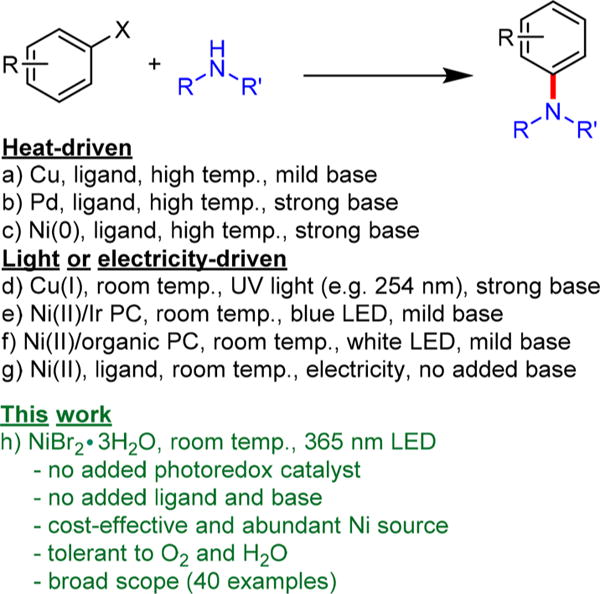
Aryl carbon–nitrogen (C–N) bonds are ubiquitous across a wide range of natural products and medicinally relevant compounds,1,2 making aminations one of the most important and frequently used reactions in medicinal chemistry.3 Copper-catalyzed Ullmann condensations constitute one of the oldest methods to construct an aryl C–N bond but commonly require elevated temperatures (e.g., 200 °C) that can limit reaction scope (Scheme 1).4,5 In the past two decades, however, there has been renewed interest in Ullmann-type cross-coupling reactions, largely due to the significant advances in new Cu/ligand systems that have drastically lowered reaction temperatures (e.g., 100 °C) and have employed catalytic, rather than stoichiometric, amounts of Cu species.6,7
Scheme 1.
Historical Development of the C–N Cross-Coupling Reaction
Since the development of Ullmann-type coupling, the field of transition-metal-catalyzed C–N bond formation has evolved to provide a plethora of approaches for efficient aminations. Notably, palladium-catalyzed C–N cross-coupling has become the predominant method for constructing aryl C–N bonds.2,8,9 As such, the potential to use abundant nickel catalysts10 has received significant interest.11–13 The widespread use of Ni is, however, hampered by the required use of high temperatures, strong alkoxide bases, and air-sensitive Ni(0) compounds.14 Although methods that implement air-stable Ni(II) complexes have been developed,15,16 excess reductants or strong bases are required to generate the Ni(0) in situ, which can severely limit functional group tolerance.
In recent years a new paradigm has arisen in aryl C–N bond formation as methods have emerged that are driven by light or electricity.17 For example, photoinduced Ullmann C–N cross-coupling proceeds through light irradiation of the Cu–amine complex and an aryl halide, which participate in a single electron transfer event to facilitate aryl C–N bond formation at room temperature.18,19 Although a significant advance, the reaction typically requires a strong alkoxide base and high-energy UV irradiation (e.g., 254 nm), and has a limited reaction scope.
More recently, dual photoredox systems driven by light through a union of photoredox catalysts (PCs) and Ni catalysis for C–N cross-couplings have been reported.20,21 The use of precious-metal-based PCs (e.g., Ir) in these systems, however, raises sustainability and cost concerns for the proliferation of such technology. As such, we have focused on developing strongly reducing organic PCs and have demonstrated dihydrophenazine22 and phenoxazine23 organic PCs as sustainable replacements for the precious metal PCs, achieving dual Ni/photoredox catalysis for aryl C–N bond formation under similar mild reaction conditions.24 These organic PCs were also shown to be effective in replacing Ir PCs in applications such as light-driven atom transfer radical polymerization, trifluoromethylation, and C–S cross-coupling.24–26
Herein, we describe a light-driven and Ni-catalyzed C–N cross-coupling methodology that does not use an added PC. The amine substrate further serves as the ligand in Ni complexes, significantly blue-shifting the UV–vis spectrum, and acts as the base to neutralize the acid byproduct. The exclusion of the added PC is enabled through direct photoexcitation of the nickel–amine complex (vide infra). This work establishes that the catalytically active Ni state for C–N cross-coupling can be efficiently accessed through electronic excitation of a nickel–amine complex without the aid of a supplementary PC to effect electron or energy transfer.27,28 By eliminating the need for a PC, this work is expected to further contribute to the mechanistic understanding of Ni-catalyzed cross-coupling chemistry29–33 in addition to improving the potential and sustainability of this technology. Comparable success was achieved with Ni-catalyzed C–C and C–O bond formation;34,35 however, the mechanistic steps involved in these coupling reactions are expected to be distinct from the aryl C–N bond formation described here.
RESULTS AND DISCUSSION
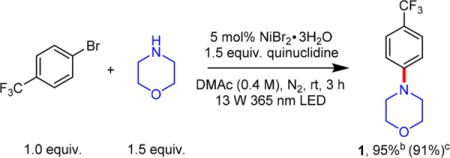
We hypothesized that such a C–N coupling reaction could utilize air-stable Ni(II) salts for a photoinduced electron transfer reaction between an electron-rich amine and the electron-poor Ni(II) to generate an amino radical36,37 and Ni(I),38 the proposed active species for C–N cross-coupling. During reaction development, we determined that C–N cross-coupling between 4-bromobenzotrifluoride and morpholine was efficient under irradiation with a 365 nm ultraviolet LED in a DMAc solution containing 5 mol% NiBr2·3H2O and 1.5 equiv of quinuclidine under a nitrogen atmosphere at room temperature. Active fan cooling of the LED maintained the reaction temperature <30 °C (see Supporting Information, Figure S11). After 3 h of irradiation, C–N-coupled product 1 was obtained in 95% yield (determined from 19F NMR, Figure S1) and was isolated at 91% yield (Table 1).
Table 1.
Reaction Development and Control Experimentsa
| ||
|---|---|---|
|
| ||
| entry | deviation from conditions above | yieldb (%) |
| 1 | no light (rt or 80 °C) | 0 |
| 2 | 13 W 405 nm LED | 93 |
| 3 | no nickel salt | 0 |
| 4 | 5 mol% NiBr2·glyme | 95 |
| 5 | 5 mol% NiCl2·6H2O | 95 |
| 6 | no quinuclidine base | 55 |
| 7 | no quinuclidine, 3.5 equiv morpholine | 94 (87)c |
| 8 | 1.5 equiv DBU base | 2 |
| 9 | same as entry 7, presence of oxygend | 91 |
| 10 | 1 h | 72 |
0.4 mmol scale. Abbreviations: DMAc, N,N-dimethylacetamide; rt, room temperature; LED, light-emitting diode; DBU, 1,8-diazabicyclo-[5.4.0]undec-7-ene.
Yield determined by 19F NMR.
Isolated yield.
Deoxygenated reaction mixture sparged with air for 2 min prior to light irradiation.
Control experiments revealed that no reaction occurred in the absence of light at either room temperature or 80 °C (Table 1, entry 1). Irradiation using a visible light 405 nm LED was similarly effective at promoting aryl C–N bond formation (93%, entry 2) but proceeded at a slower rate (Figure S2). The nickel salt is crucial for amination as no reaction was observed when it was omitted (entry 3). At 95% yield, both hydrated nickel salts NiBr2·3H2O and NiCl2·6H2O (entry 5) gave identical yield to NiBr2·glyme (entry 4), which was used in previous light-driven or electrochemically driven C–N cross-coupling reactions (Scheme 1e–g).17,20,21,24 Markedly, hydrated nickel salts are at least 2 orders of magnitude cheaper than NiBr2·glyme, thus rendering this aryl C–N cross-coupling methodology economically attractive.
The choice of base significantly impacts the success of the reaction. Quinuclidine outperformed other organic bases (Figure S5) such as triethylamine, N,N-diisopropylethylamine, and DABCO (1,4-diazabicyclo[2.2.2]octane), while the stronger base DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) almost completely halted the reaction (2% yield, entry 8). Unexpectedly, 55% yield was obtained in the absence of quinuclidine base (entry 6), where excess morpholine presumably acted to neutralize the HBr byproduct. As such, using a larger excess of morpholine (3.5 equiv, entry 7), the yield improved to 94% (87% isolated). In addition to water tolerance (through use of NiBr2·3H2O), the presence of oxygen also did not appreciably affect the yield (91%, entry 9). Notably, oxygen tolerance was similarly observed in previously reported light- or electricity-driven C–N cross-coupling systems catalyzed by Ni salts.17,20 Kinetically, this C–N cross-coupling reaction is reasonably fast, reaching 72% after 1 h of irradiation (entry 10).
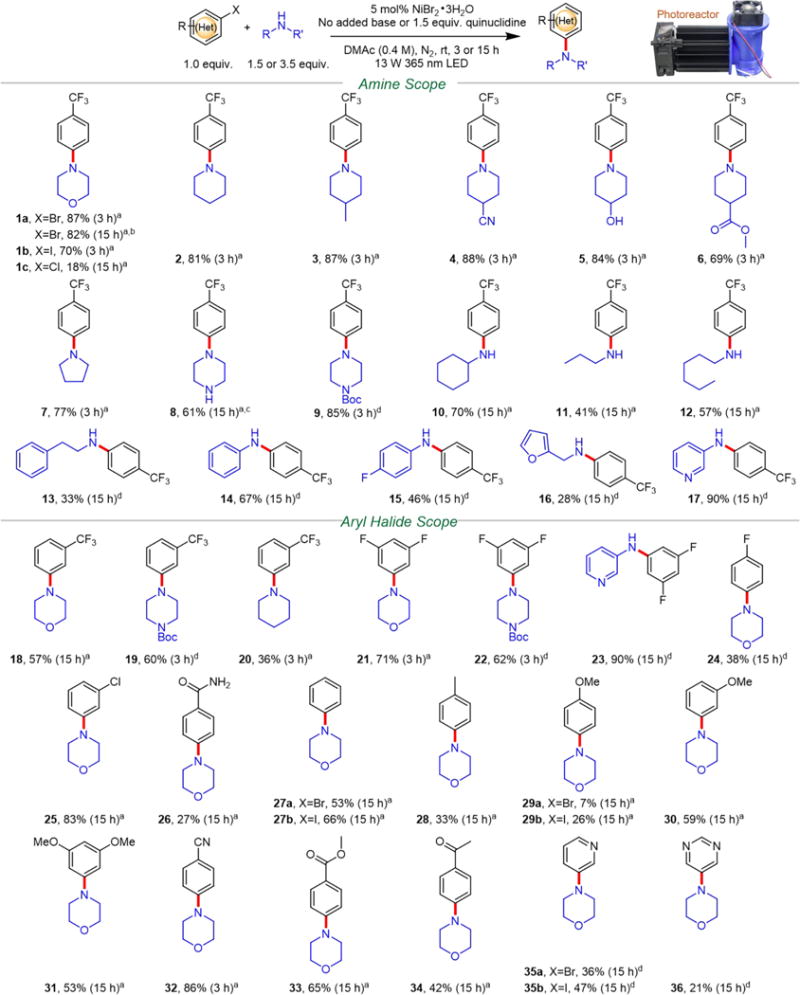
With the optimized conditions at hand, we sought to investigate the amine and aryl halide scope (Scheme 2). Secondary (Scheme 2, species 1–9), primary alkyl (10–13), and primary (hetero)aryl amines (14–17) were all successfully coupled with 4-bromobenzotrifluoride to yield the corresponding C–N products. For morpholine, in addition to aryl bromide (1a, 87%), 4-iodobenzotrifluoride (1b) was also effectively coupled in 70% isolated yield in 3 h. 4-Chlorobenzotrifluoride (1c), in contrast, was proven to be a challenging substrate and only gave 18% yield after 15 h of irradiation. C–N cross-coupling between morpholine and 4-bromobenzotrifluoride was further scaled to 6.4 mmol and isolated in 82% yield (1.21 g) after 15 h of irradiation using the same photoreactor setup.
Scheme 2.
C–N Cross-Coupling via Photoexcitation of Nickel–Amine Complexes: Amine and Aryl Halide Scope
Unless otherwise specified the reaction was conducted at 0.4 mmol scale and aryl bromide was used as the coupling partner. Percent isolated yield is reported next to the product’s boldface number. A photograph depicts the 3D-printed photoreactor equipped with the 365 nm LED used in this study (Figures S8 and S9). The LED consumes 13 W with 3.3 W radiant flux at 700 mA and 15.5 V (top right). Abbreviations: DMAc, N,N-dimethylacetamide; rt, room temperature; LED, light-emitting diode; Boc, tert-butyloxycarbonyl. a3.5 equiv amine used with no added base. b6.4 mmol scale reaction. cDimethyl sulfoxide (DMSO) used as solvent. d1.5 equiv amine used with 1.5 equiv quinuclidine base. See Supporting Information for further details.
Piperidine (2, 81%) and pyrrolidine (7, 77%) were both coupled in high yield without added base. A variety of functional groups were tolerated under these reaction conditions. For example, piperidine derivatives containing methyl (3, 87%), cyano (4, 88%), hydroxyl (5, 84%), and ester (6, 69%) functional groups were efficiently coupled. Highlighting the tolerance to oxygen, 3 was isolated in 86% yield when the solvent and reagents were used as received, without degassing. Significantly, hydroxyl groups are tolerated by this C–N coupling condition as a strong base (e.g., alkoxide) is not employed. The efficacious coupling of unprotected piperazine (8, 61%), not demonstrated in previous photochemical or electrochemical approaches,17,20,21 is particularly important39 as the aryl C–N-coupled piperazine moiety is prevalent among therapeutic compounds40 such as aripiprazole and flibanserin. The Boc-protected piperazine (9) was shown to be more reactive, yielding the C–N product in 85%.
Primary alkyl amines were typically less reactive than secondary amines, generally resulting in lower yield while requiring longer irradiation times (e.g., 15 h). Nonetheless, cyclohexylamine (10, 70%), propylamine (11, 41%), hexylamine (12, 57%), and phenethylamine (13, 33%) were successfully coupled to 4-bromobenzotrifluoride in moderate to good yield. It is noteworthy that phenethylamine and its analogues are naturally occurring alkaloids that are commonly found in psychoactive drugs.41 Furfuryl amine (16, 28%) and aromatic amines such as aniline (14, 67%), 4-fluoroaniline (15, 46%) and 3-aminopyridine (17, 90%) were also effectively coupled. Since aromatic amines are less basic than primary or secondary alkyl amines, 1.5 equiv of quinuclidine as the base was required to obtain appreciable yields.
In a previous photochemical system employing Ni and Ir PC, the addition of 10 mol% of pyrrolidine was required to effect the coupling of aniline and therefore resulted in a mixture of two C–N-coupled products derived from both pyrrolidine and aniline.21 On the contrary, our approach does not require the addition of a secondary amine or elevated temperature (e.g., 55 °C) to promote coupling of aniline, thus avoiding undesirable product mixtures. For example, coupling under our approach proceeds effectively at room temperature for 9, 10, and 14 where 55 °C was required previously.21 For 3-aminopyridine, few examples have been reported using Ni to catalyze C–N cross-coupling. A light-driven Ni/Ir PC system was employed for the cross-coupling of 3-aminopyridine and 4-iodotoluene with 33% yield,20 which is increased to 90% yield in our approach using 4-bromobenzotrifluoride (17) as the coupling partner. Furthermore, Pd catalysis has not been implemented for the synthesis of compound 17 using 3-aminopyridine. Cu was previously used to catalyze formation of 17;42 however, an aryl boronic acid was used as the coupling partner instead of an aryl bromide as demonstrated here.
Regarding the scope of aryl halides, this C–N cross-coupling method is compatible with aryl halides containing trifluoromethyl (Scheme 2, species 1–20), fluoro (21–24), chloro (25), amide (26), methyl (28), methoxy (29–31), cyano (32), ester (33), and carbonyl (34) functional groups. In addition, heteroaryl halides containing pyridine (35) and pyrimidine (36) were also successfully coupled. Generally, aryl halides containing electron-withdrawing groups are more reactive than their electron-neutral or electron-donating counterparts. For example, when comparing substituents in the para position of an aryl bromide under similar reaction conditions, the yield of cyano (32, 86%) > hydrogen (27a, 53%) > methoxy (29a, 7%). The use of aryl iodides such as iodobenzene (27b, 66%), 4-iodoanisole (29b, 26%), and 3-iodopyridine (35b, 47%) resulted in increased yields relative to using aryl bromides.
To further establish the utility of this C–N cross-coupling method, it was employed in multistep syntheses (Figure 1A,B). Recently, we reported a visible light-driven aryl C–S cross-coupling methodology that proceeds under mild conditions to synthesize a wide range of diaryl thioether products through white LED irradiation of a solution containing (hetero)aryl thiol, (hetero)aryl halide and Cs2CO3 in DMSO at room temperature in the absence of catalysts.43 Using this method, we synthesized aryl thioether 37 at 56% yield and subsequently subjected it to the C–N cross-coupling conditions developed here, coupling it with 3-aminopyridine to yield novel compound 38 in 88% yield (Figure 1A). This example highlights two industrially important processes, namely aryl C–S and C–N cross-couplings, that can be driven by light irradiation under mild conditions to achieve molecular complexity.
Figure 1.
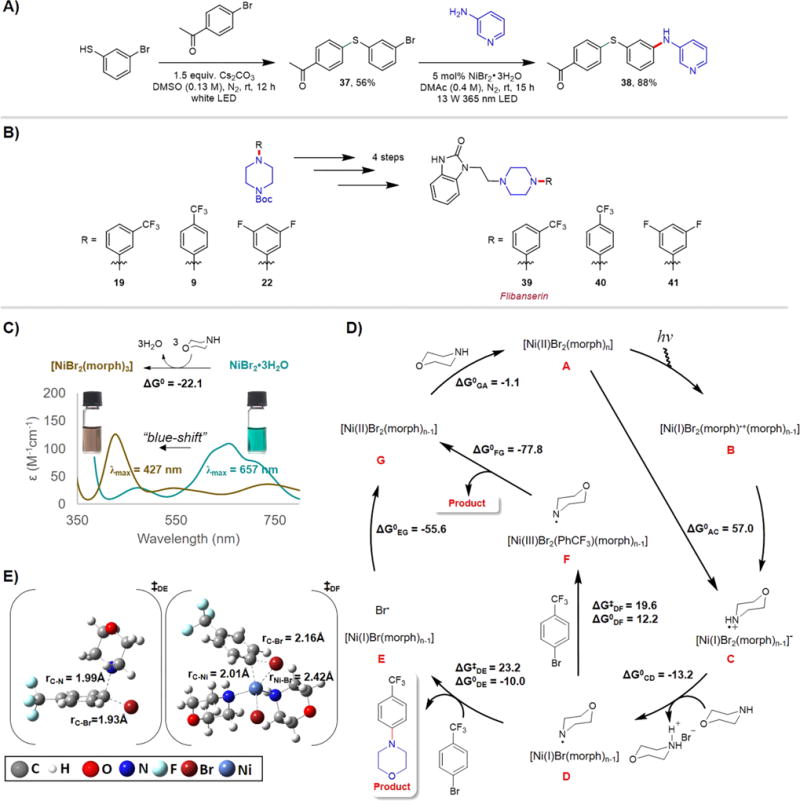
Synthetic applications and mechanistic studies. (A) Light-driven sequential C–S and C–N cross-couplings to construct molecular complexity. For the synthesis of 37, 1.0 equiv of 4′-bromoacetophenone and 1.5 equiv of 3-bromothiophenol were used; for the synthesis of 38, 1.5 equiv of 3-aminopyridine and 1.5 equiv of quinuclidine base were used. DMSO = dimethyl sulfoxide. (B) Synthesis of flibanserin and two structurally related derivatives; the yields of 19, 9, and 22 are shown in Scheme 2. Using 19, 9, and 22 as the reagents, 39, 40, and 41 were obtained in the yields of 50%, 40%, and 70%, respectively. (C) UV–vis spectra of NiBr2·3H2O and NiBr2·3H2O + morpholine in DMAc; 70 equiv of morpholine was added with respect to NiBr2·3H2O in accordance to our standard reaction conditions. Photographs showing the teal color of NiBr2·3H2O solution in DMAc transformed to brownish yellow upon morpholine addition. (D) Proposed C–N cross-coupling mechanism derived from density functional theory (DFT) calculations for n = 3. Reported free energies (in kcal/mol at 298 K and 1 M in solution) were computed at uM06/6-311+G(d,p)//uM06/6-31+G(d,p) level of theory with CPCM-described solvation in DMAc solvent. (E) Computed transition state structures for steps DE and DF (for n = 3). morph = morpholine, PhCF3Br = 4-bromobenzotrifluoride, CPCM = conductor-like polarizable continuum model; λmax is the maximum absorption wavelength and ε the molar absorptivity.
The piperazine functionality is abundant across pharmaceutical products (vide supra).40 Using established methods,44 in four synthetic steps we converted aryl-coupled piperazine derivatives 19, 9, and 22 to flibanserin (39) and two flibanserin derivatives (40 and 41, Figure 1B). We note that 40 can also be accessed from 8, therefore eliminating both Boc protection and deprotection steps. These examples illuminate the prospect of efficient and sustainable access to medicinally relevant precursors using this C–N coupling procedure for the development and manufacturing of pharmaceutical products.
To gain insight into this mechanism, density functional theory (DFT) calculations were performed to compute the energetics of intermediates involved in the proposed lowest energy potential energy surface (Figure 1C–E).26,45 Specifically, to construct possible mechanistic pathways the mechanism to produce 1a was investigated.
A proposed mechanism commences with formation of nickel–amine complexes [NiBr2(morph)n], where n = 2 or 3 and morph = morpholine (Figure 1C,D). The λmax of a NiBr2·3H2O solution in DMAc blue-shifted from 657 nm to ~550 nm as 1–4 equiv of morpholine (relative to Ni) was sequentially added (Figure S18). We assign the species absorbing at ~550 nm as NiBr2(morph)2. Further, as 8–70 equiv of morpholine was successively added to the Ni solution, the ~550 nm peak gradually decreased while the peak at ~430 nm increased. We assign the absorption peak at ~430 nm to [NiBr2(morph)3]. These assignments are based on previous reported [NiBr2(morph)2] and [NiBr2(morph)3] complexes, which displayed λmax = 580 and 454 nm, respectively.46 Notably, these λmax values, which were recorded as Nujol mulls, are systematically offset from our values in a DMAc solution by ~30 nm.
Under these reaction conditions, containing 70 equiv of morpholine relative to Ni, we suggest that [NiBr2(morph)3] exists as the predominant species in equilibrium with [NiBr2-(morph)2]; this is supported by the relatively strong peak absorbance at 427 nm and weak absorbance at 543 nm (Figure 1C). In addition, [NiBr2(morph)3] is a brownish yellow solid that is very distinct from the deep blue color of [NiBr2-(morph)2].46 Thus, the observed color change from the teal NiBr2·3H2O solution in DMAc (λmax = 657 nm) to brownish yellow (λmax = 427 nm) upon addition of 70 equiv of morpholine further supports the presence of [NiBr2(morph)3] as the predominant nickel–amine complex (Figure 1C). Importantly, the blue-shifting of the UV–vis spectrum upon amine addition was similarly observed for basic alkyl amines such as pyrrolidine, piperidine, and cyclohexyl amine (Figure S19). Here, we further note that the UV–vis spectrum in the region from 350–800 nm remained identical upon addition of 4-bromobenzotrifluoride to a solution containing NiBr2·3H2O and morpholine, demonstrating that only the nickel–amine complexes are responsible for photon absorption in the region of 350–800 nm to effect C–N cross-coupling (Figures S12–S17).
Computationally, the displacement of three water molecules by three morpholine molecules to generate [NiBr2(morph)3] was determined to be exergonic by 22.1 kcal/mol (Figure 1C). In addition, the ground state of [NiBr2(morph)3] was computationally determined to be a triplet that is 14.0 kcal/mol more stable than the corresponding singlet (Figure S25). Corroborating DFT predictions, [NiBr2(morph)3] was reported to exhibit a magnetic moment of 2.95 μB, reaffirming the triplet ground state.46
We propose that catalytic activity for aryl C–N bond formation begins with photon absorption by [NiBr2(morph)n] (A), where n = 2 or 3 (Figure 1D); for n = 3, λmax = 427 nm and εmax = 126 M−1 cm−1. Using 365 nm as the excitation wavelength, we were able to measure the emission spectrum of a DMAc solution containing predominantly the proposed [NiBr2(morph)3] species (Figure S20), which features sharp and broad emission peaks at 386 and 484 nm, respectively. Interestingly, measuring the emission intensity (at λ = 484 nm) as a function of excitation wavelength revealed that the maximum emission intensity at this wavelength occurs with excitation at 359 nm (Figure S21). Thus, despite the low molar absorptivity at 365 nm (Figure 1C), excitation is efficient using this irradiation wavelength.
Under 365 nm LED irradiation, we propose that photo-induced electron transfer from electron-rich morpholine to the electron-poor Ni(II) metal center results in the reduced Ni(I) and oxidized morpholine radical cation (B), which can subsequently dissociate into the corresponding ion pairs (C). Thermodynamically, the free energy cost to produce C from A (ΔG°AC) is endergonic by 57.0 kcal/mol (for n = 3), which is energetically supplied by photon absorption (365 nm or 78.3 kcal/mol). The proton of the morpholine radical cation is relatively acidic and the bromide anion of Ni(I) complex C is also comparatively labile such that excess morpholine in solution can act as a base to neutralize the HBr to form D ( kcal/mol, n = 3).
The Ni(I) species and morpholine radical in D are both reactive intermediates that can react with 4-bromobenzotrifluoride through either step DE or DF. In step DE, the morpholine radical adds to 4-bromobenzotrifluoride to form the desired product 1a through bromine atom displacement (E). The DFT-predicted free energy of activation for this step is 23.3 kcal/mol, while the free energy of reaction is thermodynamically favored by 10.0 kcal/mol. The Ni(I) species and the bromine atom in E can then quench ( kcal/mol, n = 3) to form the closed-shell [NiBr2(morph)n‑1] complex (G). Subsequently, G can associate with morpholine in solution ( kcal/mol, n = 3) to re-enter the catalytic cycle as A.
Alternatively, 4-bromobenzotrifluoride can oxidatively add to Ni(I) species in D to form a Ni(III) intermediate (F) ( kcal/mol and kcal/mol, n = 3). This Ni(III) and the morpholine radical can then react energetically ( kcal/mol, n = 3) to eliminate the C–N product 1a while forming the aforementioned G. The DFT-predicted transition-state structures for steps DE and DF are shown in Figure 1E. We note that the commonly employed entropy evaluations within the rigid rotor, harmonic oscillator, and ideal gas approximations normally overestimate the entropic cost (>5 kcal/mol) for reactions occurring in solution phase45,47,48 such that the predicted and values are likely overestimated.
Investigating the reaction in the presence of 0.2 equiv of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), the production of 1a was significantly decreased such that only 59% conversion was observed after 15 h of irradiation. Addition of 0.5 equiv of TEMPO or more completely halted the C–N coupling reaction. These observations suggest that radical intermediates are involved in the mechanism of this transformation.
Next, we compared the rate of C–N product formation between piperidine and piperidine-d11 using 4-bromobenzotrifluoride as the coupling partner (Figure 2) in order to determine the kH/kD ratio to investigate the presence of a kinetic isotope effect (KIE).49–51 In monitoring the reactions, we observed an inhibition period of 6 min for piperidine and much longer 52 min for piperidine-d11. The involvement of O2 in this inhibition was ruled out because the solutions were carefully deoxygenated, and the cause of this inhibition period is currently unknown. With incorporation of the inhibition period values, we numerically solved the second-order rate equation to obtain kobs(H) = 0.054 M−1 min−1 and kobs(D) = 0.011 M−1 min−1 for piperidine and piperidine-d11, respectively. As a result, the KIE value was determined to be 4.9. The large KIE = 4.9 value is consistent with a primary KIE for dissociating a N–H(D) bond in the rate-determining step prior to forming a C–N bond.
Figure 2.
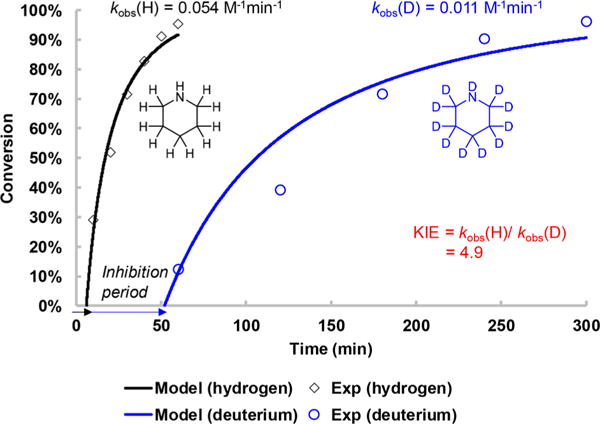
Kinetic Isotopic Effect (KIE) Experiment. The rate of C–N cross-coupling was compared between piperidine and piperidine-d11 using 4-bromobenzotrifluoride as the coupling partner. Reactions conditions: 1.0 equiv of 4-bromobenzotrifluoride (0.4 mmol), 3.5 equiv of piperidine (H or D), 5 mol% NiBr2·3H2O, and irradiation at 365 nm. The observed rate constant (kobs) was determined by solving a second-order rate equation model via numerical integration (RK4 method, Figures S23 and S24); inhibition periods of 6 and 52 min were included in the model fitting of piperidine and piperidine-d11, respectively.
CONCLUSIONS
We have developed a light-driven and nickel-catalyzed C–N cross-coupling methodology that proceeds via direct photoexcitation of nickel–amine complexes. This work reveals that catalytically active nickel states can be efficiently accessed without requiring energy or electron transfer mechanisms from a supplemental photoredox catalyst. By irradiating a solution containing an amine, an aryl halide, and a catalytic amount of NiBr2·3H2O with a commercially available 365 nm LED at room temperature, this operationally simple process was applied for the coupling of secondary, primary alkyl, or primary (hetero)aryl amines and aryl halides with diverse electronics (40 examples) without added photoredox catalyst or ligand and, in most cases, without added base. The effectiveness of this method was highlighted by the successive use of light-driven C–S/C–N cross-couplings to synthesize complex structures as well as en route to the synthesis of flibanserin and structurally related derivatives. DFT calculations suggest the production of an amino radical through irradiation of the nickel–amine complex. Concomitantly, the large H/D kinetic isotope effect value of 4.9 (for piperidine) suggests the dissociation of a N–H(D) bond as the rate-determining step to generate the amino radical for the observed C–N cross-coupling reactivity.
Supplementary Material
Acknowledgments
This work was supported by Colorado State University. Research reported in this publication was supported by the National Institute of General Medical Sciences (Award Number R35GM119702) of the National Institutes of Health. C.-H.L. is grateful for an NIH F32 Postdoctoral Fellowship (F32GM122392). We acknowledge the use of computational resources provided by the XSEDE - Comet supercomputer (NSF ACI-1053575).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03744.
General information; Figures S1–S25 and Tables S1 and S2, detailing reaction development and optimization, photoreactor development, UV–visible spectroscopy, fluorimetry, kinetic isotope effect, computational details, and characterizations; NMR spectra; and coordinates of molecular structures (PDF)
ORCID
Chern-Hooi Lim: 0000-0003-1823-6305
Garret M. Miyake: 0000-0003-2451-7090
Notes
The authors declare no competing financial interest.
References
- 1.Bariwal J, Van der Eycken E. Chem Soc Rev. 2013;42:9283. doi: 10.1039/c3cs60228a. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz-Castillo P, Buchwald SL. Chem Rev. 2016;116:12564. doi: 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown DG, Boström J. J Med Chem. 2016;59:4443. doi: 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- 4.Ullmann F. Ber Dtsch Chem Ges. 1903;36:2382. [Google Scholar]
- 5.Monnier F, Taillefer M. Angew Chem, Int Ed. 2009;48:6954. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 6.Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337. [Google Scholar]
- 7.Ley SV, Thomas AW. Angew Chem, Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 8.Hartwig JF. Angew Chem, Int Ed. 1998;37:2046. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe JP, Buchwald SL. J Org Chem. 1996;61:1133. [Google Scholar]
- 10.Wolfe JP, Buchwald SL. J Am Chem Soc. 1997;119:6054. [Google Scholar]
- 11.Ramgren SD, Silberstein AL, Yang Y, Garg NK. Angew Chem, Int Ed. 2011;50:2171. doi: 10.1002/anie.201007325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavoie CM, MacQueen PM, Rotta-Loria NL, Sawatzky RS, Borzenko A, Chisholm AJ, Hargreaves BKV, McDonald R, Ferguson MJ, Stradiotto M. Nat Commun. 2016;7:11073. doi: 10.1038/ncomms11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green RA, Hartwig JF. Angew Chem, Int Ed. 2015;54:3768. doi: 10.1002/anie.201500404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marín M, Rama RJ, Nicasio MC. Chem Rec. 2016;16:1819. doi: 10.1002/tcr.201500305. [DOI] [PubMed] [Google Scholar]
- 15.Brenner E, Fort Y. Tetrahedron Lett. 1998;39:5359. [Google Scholar]
- 16.Chen, Yang L-M. Org Lett. 2005;7:2209. doi: 10.1021/ol050608i. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Kawamata Y, Nakamura H, Vantourout JC, Liu Z, Hou Q, Bao D, Starr JT, Chen J, Yan M, Baran PS. Angew Chem, Int Ed. 2017;56:13088. doi: 10.1002/anie.201707906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- 19.Ziegler DT, Choi J, Muñoz-Molina JM, Bissember AC, Peters JC, Fu GC. J Am Chem Soc. 2013;135:13107. doi: 10.1021/ja4060806. [DOI] [PubMed] [Google Scholar]
- 20.Oderinde MS, Jones NH, Juneau A, Frenette M, Aquila B, Tentarelli S, Robbins DW, Johannes JW. Angew Chem, Int Ed. 2016;55:13219. doi: 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]
- 21.Corcoran EB, Pirnot MT, Lin S, Dreher SD, DiRocco DA, Davies IW, Buchwald SL, MacMillan DWC. Science. 2016;353:279. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theriot JC, Lim C-H, Yang H, Ryan MD, Musgrave CB, Miyake GM. Science. 2016;352:1082. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- 23.Pearson RM, Lim C-H, McCarthy BG, Musgrave CB, Miyake GM. J Am Chem Soc. 2016;138:11399. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y, Pearson RM, Lim C-H, Sartor SM, Ryan MD, Yang H, Damrauer NH, Miyake GM. Chem - Eur J. 2017;23:10962. doi: 10.1002/chem.201702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theriot JC, McCarthy BG, Lim C-H, Miyake GM. Macromol Rapid Commun. 2017;38:1700040. doi: 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim C-H, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM. J Am Chem Soc. 2017;139:348. doi: 10.1021/jacs.6b11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arias-Rotondo DM, McCusker JK. Chem Soc Rev. 2016;45:5803. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
- 28.Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 29.Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DW. Nat Rev Chem. 2017;1:0052. [Google Scholar]
- 30.Oderinde MS, Frenette M, Robbins DW, Aquila B, Johannes JW. J Am Chem Soc. 2016;138:1760. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]
- 31.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi Z-H, Ma J. ACS Catal. 2018;8:1456. [Google Scholar]
- 34.Ishida N, Masuda Y, Ishikawa N, Murakami M. Asian J Org Chem. 2017;6:669. [Google Scholar]
- 35.Shields BJ, Kudisch B, Scholes GD, Doyle AG. J Am Chem Soc. 2018;140:3035. doi: 10.1021/jacs.7b13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris SA, Wang J, Zheng N. Acc Chem Res. 2016;49:1957. doi: 10.1021/acs.accounts.6b00263. [DOI] [PubMed] [Google Scholar]
- 37.Xiong T, Zhang Q. Chem Soc Rev. 2016;45:3069. doi: 10.1039/c5cs00852b. [DOI] [PubMed] [Google Scholar]
- 38.Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reilly SW, Mach RH. Org Lett. 2016;18:5272. doi: 10.1021/acs.orglett.6b02591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 41.Dean BV, Stellpflug SJ, Burnett AM, Engebretsen KM. J Med Toxicol. 2013;9:172. doi: 10.1007/s13181-013-0295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazu TK, Etukala JR, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. Eur J Med Chem. 2011;46:2378. doi: 10.1016/j.ejmech.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Lim C-H, Miyake GM. J Am Chem Soc. 2017;139:13616. doi: 10.1021/jacs.7b07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F, Wu C, Li Z, Tian G, Wu J, Zhu F, Zhang J, He Y, Shen J. Org Process Res Dev. 2016;20:1576. [Google Scholar]
- 45.Lim C-H, Holder AM, Hynes JT, Musgrave CB. J Am Chem Soc. 2014;136:16081. doi: 10.1021/ja510131a. [DOI] [PubMed] [Google Scholar]
- 46.Palazón J, Gálvez J, García G, Lopez G. Polyhedron. 1983;2:1353. [Google Scholar]
- 47.Huang D, Makhlynets OV, Tan LL, Lee SC, Rybak-Akimova EV, Holm RH. Proc Natl Acad Sci U S A. 2011;108:1222. doi: 10.1073/pnas.1017430108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Y, Liu S, Xia Y, Li Y, Yu Z-X. Chem - Eur J. 2008;14:4361. doi: 10.1002/chem.200701725. [DOI] [PubMed] [Google Scholar]
- 49.Wiberg KB. Chem Rev. 1955;55:713. [Google Scholar]
- 50.Westheimer FH. Chem Rev. 1961;61:265. [Google Scholar]
- 51.Gómez-Gallego M, Sierra MA. Chem Rev. 2011;111:4857. doi: 10.1021/cr100436k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.