


Abstract
Chemical communication in nematodes such as the model organism Caenorhabditis elegans is modulated by a variety of glycosides based on the dideoxysugar l-ascarylose. Comparative ascaroside profiling of nematode exometabolome extracts using a GC-EIMS screen reveals that several basic components including ascr#1 (asc-C7), ascr#2 (asc-C6-MK), ascr#3 (asc-ΔC9), ascr#5 (asc-ωC3), and ascr#10 (asc-C9) are highly conserved among the Caenorhabditis. Three novel side chain hydroxylated ascaroside derivatives were exclusively detected in the distantly related C. nigoni and C. afra. Molecular structures of these species-specific putative signaling molecules were elucidated by NMR spectroscopy and confirmed by total synthesis and chemical correlations. Biological activities were evaluated using attraction assays. The identification of (ω)- and (ω − 2)-hydroxyacyl ascarosides demonstrates how GC-EIMS-based ascaroside profiling facilitates the detection of novel ascaroside components and exemplifies how species-specific hydroxylation of ascaroside aglycones downstream of peroxisomal β-oxidation increases the structural diversity of this highly conserved class of nematode signaling molecules.
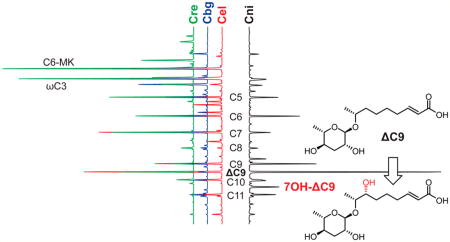
Graphical abstract
INTRODUCTION
Research with the model organism Caenorhabditis elegans and the development of novel analytical techniques promoted the characterization of ascarosides, a modular glycolipid library based on the 3,6-dideoxysugar l-ascarylose linked to fatty acid derived aglycones (Scheme 1).1 In C. elegans, ascarosides modulate a large diversity of biological responses, including dauer formation,2–6 reproduction,6,7 stress resistance,7,8 lifespan,8,9 and behavior.10–14 The diversity of biological responses modulated by ascarosides is paralleled by their large structural diversity. Even small changes in molecular structures,7,14,15 synergistic effects,10,11,16 and variations in ascaroside compositions7,17,18 can dramatically alter their biological activity. Homologous series originate from chain shortening of very long chain precursors upon peroxisomal β-oxidation (Scheme 1A) to furnish ascarosides carrying acyl (1, asc-C#), enoyl (2, asc-ΔC#), and (3R)-hydroxyacyl (3, asc-βOH-C#) aglycones.19–23 2-Ketoalkyl (6, asc-C#-MK) and 2-hydroxyalkyl (7, asc-C#-OH) aglycones are presumably produced via decarboxylation and subsequent reduction of labile 3-ketoacyl intermediates (4, X = OH). Furthermore, (ω)-ascarosides such as asc-ωC3 (8, ascr#5) demonstrate that (ω − 1) and (ω)-linked components enter the peroxisomal β-oxidation cycle. Downstream of the peroxisomal β-oxidation cycle, the resulting basic ascaroside skeletons serve as scaffolds for the attachment of additional structural units derived from primary metabolic pathways to furnish a modular library of species-specific signaling components such as the indole ascaroside IC-asc-C5 (9, icas#9) (Scheme 1B).5,12,16,22,24
Scheme 1. Ascaroside Diversity Originating from (A) Side Chain Modification and Shortening via Peroxisomal β-Oxidation, (B) Subsequent Attachment of Additional Metabolic Units from Primary Metabolism, or (C) Specific (ω), (ω − 2), or (ω − 3)-Hydroxylation Downstream of Peroxisomal β-Oxidation.

Ascaroside signaling is highly conserved in intraspecies nematode communication16,25–29 and also involved in intergenotypic competition30,31 and cross-kingdom interactions,32–34 demonstrating that ascarosides represent key regulators of nematode chemical ecology. Considering the small amounts of ascarosides present in nematode exometabolomes, the large diversity of homologous structures, and the complexity of the background matrix, mass spectrometric techniques are indispensable for comprehensive ascaroside analysis. Mass spectrometric screens that employ specific fragment ions as markers are capable to highlight putative nematode-derived ascarosides and their biosynthetic precursors and mask the background matrix. We previously developed an HPLC-ESI-(−)-MS/MS precursor ion screen that employs an ascarylose-derived fragment ion at m/z 73.1 [C3H5O2]− for the selective detection of known as well as yet unidentified components.22 ESI-(−)-MS/MS precursor ion screening has been employed in various studies22,25–27,29,33,34 but ultimately requires a triple quadrupole instrument. Considering the importance of ascaroside signaling in nematode chemical ecology, we recently developed a complementary GC-EIMS technique35 that employs an ascarylose-derived K1-fragment ion at m/z 130.1 [C6H14OSi]+• along with A1 and A2 fragment ions at m/z 275.1 [C12H27O3Si2]+ and m/z 185.1 [C9H17O2Si]+ as characteristic markers to facilitate selective ascaroside profiling in trimethylsilyl (TMS) derivatized crude nematode exometabolome extracts (Scheme 2). Furthermore, aglycone-specific fragment ions for a rearranged oxonium ion (J1) at [M − 173]+ and a carbocation (J2) at [M − 291]+ facilitate the identification of compound specific side chains. Here, we employ the GC-EIMS screen for comparative ascaroside profiling in a variety of Caenorhabditis species to demonstrate its potential for the discovery of novel ascaroside components in crude unfractionated nematode exometabolome extracts.
Scheme 2. EI-Induced Fragmentation of TMS-Derivatized Ascarosides.

RESULTS AND DISCUSSION
Following a phylometabolomic approach, we performed a GC-EIMS-based comparative analysis of exometabolome extracts from 13 Caenorhabditis species with a strong focus on the Elegans group that harbors the model organism C. elegans.36,37 Liquid cultures were established in S-medium, and propagating nematodes were fed with concentrated E. coli OP50 for 7 days, after which cultures were starved for another 7 days.38 The media supernatant representing the exometabolome was collected, lyophilized, and extracted with methanol. Crude exometabolome extracts were converted into their TMS derivatives using N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) and subsequently analyzed by GC-EIMS to show a large diversity of primary and secondary metabolites, some of which could be tentatively identified using the NIST 14 mass spectral library (Figure 1A). Putative ascarosides were detected by inspection of the extracted ion chromatograms for the highly characteristic ascarylose-derived K1 fragment ion at m/z 130.1 [C6H14OSi]+• (Figures 1B and S1). Individual ascaroside structures were identified based on their aglycone-specific J1 [M− 173]+ and J2 [M − 291]+ fragment ion signals and Kovats retention indices by comparison with a collection of more than 200 components that carry (ω − 1)- or (ω)-linked acyl (1), enoyl (2), (3R)-hydroxyacyl (3), 2-ketoalkyl (6), or 2-hydroxyalkyl (7) side chains previously characterized in C. elegans wild-type and peroxisomal β-oxidation mutants.35
Figure 1.

Ascaroside profiling of the C. nigoni exometabolome. (A) Total ion chromatogram (TIC) of the TMS-derivatized C. nigoni crude exometabolome; (B) extracted ion chromatogram (EIC) for the K1 fragment at m/z 130.1 [C6H14OSi]+•; and (C) HPLC-ESI-MS/MS precursor ion screen for m/z 73.1 [C3H5O2]−.
These analyses demonstrate that basic ascarosides with side chains ranging from 3 to 11 carbons are highly conserved in all Caenorhabditis wild-type isolates tested, although their relative compositions vary significantly between the different species (Figure 2). A set of five compounds, asc-C7 (1, n = 2, X = OH; ascr#1, daumone#1),2 asc-C9 (1, n = 4, X = OH; ascr#10),12 asc-ΔC9 (2, n = 4, X = OH; ascr#3, daumone#3),3 asc-C6-MK (6, n = 2; ascr#2, daumone#2),3 and asc-ωC3 (8, asc#5)4 consistently represent the dominating components and, taken together, account for more than 60% of the total ascarosides identified. An exception is the C. portoensis metabolome that predominantly contains asc-C11 (1, n = 6, X = OH; ascr#18). Furthermore, asc-C6-MK (6, n = 2; ascr#2) is absent in C. nigoni, C. japonica, C. afra, and C. portoensis, whereas asc-ωC3 (8, asc#5) is absent in C. nigoni, C. japonica, and C. portoensis. Asc-C6-OH (7, n = 2; ascr#6) was exclusively detected alongside asc-C6-MK (6, n = 2; ascr#2), supporting the assumption of a common biosynthetic origin (Scheme 1). While these basic ascarosides are common in most Caenorhabditis species tested, GC-EIMS-based ascaroside profiling also revealed some highly species-specific components, including a yet unidentified component with a J2 fragment at m/z 186.1 from C. elegans, asc-βOH-C13 (3, n = 8, X = OH; bhas#22) from C. japonica, and asc-C5-EA (1, n = 0, X = NHCH2CH2OH; easc#9) from C. portoensis.35
Figure 2.

Comparative GC-EIMS ascaroside profiling of 13 Caenorhabditis exometabolomes.
In addition, we observed 3 putative ascarosides that accounted for around 10% of the total ascarosides detected in the exometabolome of C. nigoni (Figures 2 and 1B) but did not match any of the more than 200 basic ascaroside structures previously identified in C. elegans wild-type and mutant metabolomes.35 Inspection of their 70 eV EIMS spectra (Figure 3) revealed dominating K1-fragment ion signals at m/z 130.1 [C6H14OSi]+•, along with A1-fragments at m/z 275.2 [C12H27O3Si2]+ and A2-fragments at m/z 185.1 [C9H17O2Si]+ that are characteristic for the ascarylose unit (Scheme 2). Several aglycone-specific signals for J1 fragment ions at m/z 433.2 [C19H41O5Si3]+, J2 fragment ions at m/z 315.2 [C15H31O3Si2]+, and (J2 − TMSOH) fragment ions at m/z 225.1 [C12H21O2Si]+ indicated nine carbon side chains with one additional unit of unsaturation and one trimethylsilyloxy moiety for compounds 10 and 12. The third compound 11 displayed diagnostic signals for a J1 fragment at m/z 435.3 [C19H43O5Si3]+, a J2 fragment at m/z 317.2 [C15H33O3Si2]+, and a (J2 − TMSOH) fragment at m/z 227.1 [C12H23O2Si]+ indicative for a trimethylsilyloxy-substituted nine carbon side chain. Comparison with the known (3R)-hydroxylated asc-βOH-C9 (3, n = 4, X = OH; bhas#10), previously characterized in the exometabolome of Panagrellus redivivus,26,35 demonstrated that both compounds are different (Figure S2) and excluded a β-oxidation-derived 3-hydroxyacyl aglycone due to the lack of the characteristic fragment ion at m/z 233.1 [C9H21O3Si2]+ derived from α-cleavage. However, the identification of a homologous fragment ion signal at m/z 289.2 [C13H29O3Si2]+ suggested a 7-hydroxyacyl structure for asc-7OH-C9 (11) from C. nigoni, demonstrating how EIMS fragmentation can aid in structure assignment.
Figure 3.

GC-EIMS spectra of TMS-derivatized (ω) and (ω − 2) hydroxyacyl ascarosides from C. nigoni.
In conclusion, comparative GC-EIMS-based ascaroside screening revealed three species-specific, side chain hydroxylated compounds in the C. nigoni exometabolome. The same components with molecular ion signals at m/z 317.2 [M − H]− for 10 and 12 and m/z 319.2 [M − H]− for 11 were also detected using the HPLC-ESI-(−)-MS/MS precursor ion screen for m/z 73.1 [C3H5O2]− (Figure 1C), demonstrating that the GC-EIMS and HPLC-MS/MS methods complement each other. However, in contrast to GC-EIMS, the MS/MS precursor ion screen also revealed additional derivatives, including large amounts of indole ascarosides such as IC-asc-C5 (9, icas#9), a male attractant in C. nigoni,16 demonstrating that GC-EIMS is restricted to the most basic ascaroside compounds.
To identify the molecular structures of the species-specific ascarosides (10–12), the exometabolome extract of 1.6 L C. nigoni liquid culture supernatant was fractionated by solid phase extraction (RP-C18-SPE) using a 10%-stepwise gradient of aqueous methanol as eluent. Fractions were screened for ascarosides by GC-EIMS (Figure S3) and 1H NMR spectroscopy (Figure S4) using the K1 fragment ion at m/z 130.1 [C6H14OSi]+• and the anomeric proton at approximately δH 4.65 ppm (s, 1H) as characteristic markers, respectively. These analyses confirmed the assignment of several known ascarosides and traced the target compounds to a fraction eluted with 40% methanol (Figure S3) that contained predominantly asc-C7 (1, n = 2, X = OH; ascr#1) along with a diversity of additional metabolites such as indole acetic acid (IAA, auxine) and anthranilic acid (Figure S5). Subsequent separation by semipreparative HPLC using a C18 column furnished fractions of sufficient purity to facilitate structure assignment.
The molecular formula of C15H26O7 for compound 10 (~275 µg) was established by HR-EIMS. Inspection of one-and two-dimensional NMR spectra (1H NMR, dqf-COSY, HSQC) confirmed an α-configured ascarylose moiety along with an (ω − 1)-linked α,β-unsaturated C9 side chain (Table 1). Furthermore, the (ω − 2)-position of the additional hydroxy group was deduced based on dqf-COSY correlations from the terminal (ω)-methyl group at δH 1.14 ppm (d, J = 6.3 Hz, 3H), δC 14.5 ppm to the (ω − 1)-oxymethine group at δH 3.74 ppm (dq, J = 3.9 Hz, J = 6.3 Hz, 1H), δC 75.5 ppm and further on to the (ω − 2)-position at δH 3.53 ppm (m, 1H), δC 74.8 ppm. While all (ω − 1)-linked homologous ascarosides that have been identified so far share the same (R)-configuration at the penultimate carbon,1 the stereochemistry of the (ω − 2)-hydroxymethine group of asc-7OH-ΔC9 (10) could not be unambiguously assigned based solely on the vicinal H,H-coupling constant of 3.9 Hz.39
Table 1.
NMR Data for (ω) and (ω − 2)-Hydroxyacyl Ascarosides (10–12) Isolated from C. nigoni (400 MHz, CD3OD)
| threo-asc-7OH-ΔC9 (10) | threo-asc-7OH-C9 (11) | asc-9OH-ΔC9 (12) | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| position | δCa | δHb, mult (J, Hz) | δCa | δHb, mult (J, Hz) | δCa | δHb, mult (J, Hz) | |||
| 1 | nd | C | – | nd | C | – | nd | C | – |
| 2 | 124.8 | CH | 5.83 d (15.5) | 37.5 | CH2 | 2.21 t (7.6) | 122.9 | CH | 5.81 d (15.6) |
| 3 | 148.1 | CH | 6.86 dt (15.5, 7.0) | 26.9 | CH2 | 1.63 m | 150.7 | CH | 6.95 dt (15.6, 6.9) |
| 4 | 32.7 | CH2 | 2.25 m | 30.4 | CH2 | 1.40 m | 33.1 | CH2 | 2.25 m |
| 5 | 25.6 | CH2 | 1.54–1.68 m | 27.0 | CH2 | 1.40 m | 29.3 | CH2 | 1.52 m |
| 6 | 32.8 | CH2 | 1.48–1.62 m | 32.9 | CH2 | 1.48–1.53 m | 26.3 | CH2 | 1.50 m |
| 7 | 74.8 | CH | 3.53 m | 75.2 | CH | 3.52 m | 32.8 | CH2 | 1.59 m |
| 8 | 75.5 | CH | 3.74 dq (3.9, 6.3) | 75.6 | CH | 3.73 dq (3.8, 6.2) | 78.6 | CH | 3.69 m |
| 9 | 14.5 | CH3 | 1.14 d (6.3) | 14.6 | CH3 | 1.14 d (6.2) | 64.6 | CH2 | 3.50 dd (11.7, 5.6) 3.60 dd (11.7, 4.2) |
| 1′ | 97.9 | CH | 4.65 s | 97.8 | CH | 4.65 s | 99.8 | CH | 4.75 s |
| 2′ | 69.7 | CH | 3.76 s.br | 69.8 | CH | 3.76 s.br | 69.6 | CH | 3.84 s.br |
| 3′ax | 35.8 | CH2 | 1.95 dt (13.0, 3.8) | 35.8 | CH2 | 1.95 dt (13.0, 3.8) | 35.9 | CH2 | 1.95 dt (13.0, 3.8) |
| 3′eq | 1.80 ddd (13.0, 11.4, 3.0) | 1.81 ddd (13.0, 11.4, 3.0) | 1.78 ddd (13.0, 11.2, 3.1) | ||||||
| 4′ | 68.2 | CH | 3.52 ddd (11.3, 9.3, 4.3) | 68.4 | CH | 3.51 ddd (11.4, 9.5, 4.4) | 68.4 | CH | 3.53 m |
| 5′ | 71.3 | CH | 3.64 dq (9.3, 6.3) | 71.4 | CH | 3.66 dq (9.5, 6.3) | 71.4 | CH | 3.67 m |
| 6′ | 17.8 | CH3 | 1.22 d (6.2) | 17.8 | CH3 | 1.22 d (6.2) | 18.1 | CH3 | 1.22 d (6.1) |
From HSQC spectrum.
From 1H NMR and dqf-COSY spectra.
Compound 11 (~110 µg) with a molecular formula of C15H28O7 according to ESI-HRMS exhibits almost identical NMR data for the ascarylose unit and the (ω)-part of the side chain but displays a triplet signal at 2.21 (t, J = 7.6 Hz, 2H) instead of the signals for an α,β-unsaturated enoyl moiety (Table 1), thus suggesting the corresponding dihydro-derivative structure asc-7OH-C9 (11).
Compound 12 (~130 µg) with a molecular formula of C15H26O7 according to ESI-HRMS was obtained as a 1:1 mixture with asc-7OH-C9 (11). Comparative analysis of their dqf-COSY spectra indicated an ascarylose unit with considerably different chemical shifts (Table 1). Furthermore, an α,β-unsaturated side chain was identified due to δH 5.81 (d, JE = 15.6 Hz, 1H) and 6.95 (dt, JE = 15.6 Hz, J = 6.9 Hz, 1H), along with a hydroxymethylene group at δH 3.50 (dd, 2J = 11.7 Hz, 3J = 5.6 Hz, 1H), 3.60 (dd, 2J = 11.7 Hz, 3J = 4.2 Hz, 1H), and δC 64.6 ppm that displayed vicinal H,H-coupling correlation to the (ω − 1)-oxymethine proton at δH 3.69 (m, 1H) and δC 78.6 ppm, thus indicating the terminal (ω)-position for the hydroxylation of the aglycone in asc-9OH-ΔC9 (12). In conclusion, analysis of one and two-dimensional NMR spectra revealed three novel side chain modified ascarosides that carry additional hydroxy functions at the 7-position (ω − 2) and the 9-position (ω), the structures of which were finally established by total synthesis and chemical correlations.
Both diastereomeric (ω − 2)-hydroxy ascarosides (7R,8R)-threo-10a and (7S,8R)-erythro-10b were synthesized as shown in Scheme 3. (R)-Methyl lactate (14) was converted to the para-methoxybenzyl (PMB) ether 15 and reduced to the aldehyde 16 using DIBAL-H. Addition of 4-pentenylmagnesium bromide afforded (6R,7R)-threo-6-hydroxy-7-PMB-O-1-octene (17) with a diastereomeric excess (de) of 92% due to asymmetric induction via a Cram chelate complex.40 Esterification of 17 with benzoyl chloride and pyridine or benzoic acid under Mitsunobu conditions (PPh3, DIAD) afforded the diastereomeric benzoates (6R,7R)-threo-18a or (6S,7R)-erythro-18b, respectively, in excellent diastereomeric purities of de > 99% after column chromatography (Figure S6). Cross metathesis with ethyl acrylate using Grubbs second generation catalyst41 furnished the corresponding ethyl 7-Bz-O-8-PMB-O-(2E)-nonenoates (7R,8R)-threo-19a or (7S,8R)-erythro-19b that were subsequently deprotected using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)42 to afford (7R,8R)-threo-20a or (7S,8R)-erythro-20b. Coupling with 2,4-di-O-benzoyl protected ascarylose via the trichloroacetimidate route43 furnished the diastereomeric ascarosides (22a or 22b) that were finally deprotected using alkaline hydrolysis to give the desired (ω − 2)-hydroxy ascarosides (7R,8R)-threo-10a or (7S,8R)-erythro-10b. In addition, an undesired intramolecular cyclization product (23a or 23b) was obtained. Comparison of the NMR (Figure S7) and GC-EIMS (Figure S8) data of diastereomeric (7R,8R)-threo-10a and (7S,8R)-erythro-10b with those of the natural product (10) isolated from the C. nigoni exometabolome confirmed its (7R,8R)-threo configuration. Furthermore, the (7R,8R)-threo-configuration of the dihydro-derivative asc-7OH-C9 (11) from C. nigoni was unambiguously established by comparison of the mass spectra and GC retention times with those of the hydrogenation products of (7R,8R)-threo-10a and (7S,8R)-erythro-10b (Figure S9).
Scheme 3. Synthesis of (ω − 2)-Hydroxyacyl Ascarosides threo-asc-7OH-ΔC9 (10a) and erythro-asc-7OH-ΔC9 (10b).

Reagents and conditions: (i) 4-methoxybenzyl trichloroacetimidate, trimethylsilyl triflate, CH2Cl2, 0 °C, 3 h, 62%; (ii) DIBAL-H, CH2Cl2, −78 °C, 0.5 h, 100%; (iii): 4-pentenylmagnesium bromide, Et2O, 0 °C, 1 h, 58%; (iv) benzoyl chloride, pyridine, CH2Cl2, 0 °C, 12 h, 87% (18a); (v) benzoic acid, PPh3, DIAD, THF, 4 h, 55% (18b); (vi): ethyl acrylate, Grubbs second generation catalyst, CH2Cl2, 40 °C, 9 h, 76% (19a) and 81% (19b); (vii): DDQ, CH2Cl2/H2O, 1 h, 70% (20a) and 72% (20b); (viii): 2,4-di-O-benzoyl-ascarosyl-1-(2,2,2-trichloroacetimidate) (21), trimethylsilyl triflate, CH2Cl2, 0 °C, 3 h, 68% (22a) and 64% (22b); (ix): lithium hydroxide, water, MeOH, 12 h, 36% (10a), 46% (10b), 39% (23a), and 42% (23b)
The (ω)-hydroxylated ascaroside asc-9OH-ΔC9 (12) was synthesized as shown in Scheme 4. Copper(I)-catalyzed addition of pentenyl magnesium bromide to tert-butyldimethylsilyl (S)-glycidyl ether (24) afforded (S)-8-tert-butyldimethylsilyloxy-7-hydroxy-1-octene (25). Cross metathesis with ethyl acrylate using Grubbs second generation catalyst41 gave the corresponding (8S,2E)-ethyl 8-hydroxy-9-tert-butyldimethylsilyloxy-2-nonenoate (26). Coupling of 26 to 2,4-di-O-benzoyl-ascarosyl trichloroacetimidate (21) furnished 27, which was subsequently deprotected using tetrabutylammonium fluoride (TBAF) to give the alcohol 28, followed by alkaline hydrolysis to give asc-9OH-ΔC9 (12) identical to the natural product from C. nigoni as shown by comparison of the GC-EIMS and NMR data (Figures S8 and S10). In addition, an undesired intramolecular cyclization product 29 with an oxacyclooctan ring was obtained.
Scheme 4. Synthesis of the (ω)-Hydroxyacyl Ascaroside asc-9OH-ΔC9 (12).

Reagents and conditions: (i) 4-pentenylmagnesium bromide, copper(I)iodide, THF, 0 °C, 3 h, 94%; (ii) ethyl acrylate, Grubb’s second generation catalyst, CH2Cl2, 40 °C, 9 h, 81%; (iii) 2,4-di-O-benzoylascarosyl-1-(2,2,2-trichloroacetimidate) (21), trimethylsilyl triflate, CH2Cl2, 0 °C, 3 h, 53%; (iv) tetrabutylammonium fluoride, THF, 3 h, 78%; (v) lithium hydroxide, water, MeOH, 12 h, 25% (12) and 69% (29).
Using the synthetic compounds as reference standards, threo-asc-7OH-ΔC9 (10a), threo-asc-7OH-C9 (11a), and asc-9OH-ΔC9 (12) could also be identified in the exometabolome extract of Caenorhabditis afra (sp. 7) strain JU1286 from Ghana (Figures S11 and S12), a member of the distantly related Japonica group (Figure 2),36,37 suggesting that (ω) and (ω − 2)-hydroxylation in C. nigoni and C. afra has a polyphyletic origin. Targeted screening for homologous compounds using GC-EIMS and HPLC-MS demonstrated that (ω) and (ω − 2)-hydroxylation in C. nigoni and C. afra is tightly controlled and exclusively affects C9 and ΔC9 aglycones, indicating that this species-specific modification of ascaroside aglycones occurs downstream of peroxisomal β-oxidation.
Aiming to characterize the biological functions of the novel ascarosides, behavioral response of C. nigoni males or females to 1 µM asc-(7R)−OH-ΔC9 (10a), asc-(7S)−OH-ΔC9 (10b), asc-9OH-ΔC9 (12), as well as asc-ΔC9 (2, n = 4, X = OH; ascr#3) was evaluated using a spot attraction assay. While these analyses demonstrate that the common asc-ΔC9 (2, n = 4, X = OH; ascr#3) acts as a potent male attractant in C. nigoni, reminiscent of the attraction of C. elegans males,10 the species-specific (ω − 2) and (ω)-hydroxylated derivatives (10a, 10b, and 12) are not active (Figure S13). Additional experiments are required to clarify the biological functions of these compounds and unravel the ecological significance of species-specific (ω) and (ω − 2)-hydroxylation in C. nigoni and C. afra.
CONCLUSION
Our results demonstrate that comparative GC-EIMS-based ascaroside profiling represents a powerful technique to characterize ascaroside diversity and detect novel species-specific components in TMS-derivatized crude nematode exometabolomes. We isolated three new (ω) and (ω − 2)-hydroxylated ascarosides from C. nigoni and determined their structures using NMR spectroscopy. Structure assignments were unambiguously established by total synthesis and chemical correlations. Comparative analysis of 13 Caenorhabditis species demonstrates that within the Elegans group, both (ω)- and (ω − 2)-hydroxylated ascarosides (10–12) are highly specific for C. nigoni (sp. 9), but the same compounds were also detected in the rather distantly related C. afra (sp. 7), a member of the Japonica group, thus suggesting a polyphyletic origin for the hydroxylation steps. In addition, we found that (ω)- and (ω − 2)-hydroxylation in C. nigoni and C. afra is highly specific for ascarosides carrying C9 and ΔC9 side chains, strongly suggesting that the hydroxylation step occurs downstream of peroxisomal β-oxidation. While traces of (3R)-hydroxyacyl ascarosides, intermediates of the peroxisomal β-oxidation cycle, are widespread in nematode exometabolomes, side chain hydroxylation downstream of β-oxidation such as (ω)- and (ω − 2)-hydroxylation in C. nigoni and C. afra has so far been described only as male-specific (ω − 3)-hydroxylation in Panagrellus redivivus (Scheme 1C).26 However, while the dihydroxylated dhas#18 (13) represents a male-produced female-attractant in Panagrellus redivivus,26 the hydroxy ascarosides (10–12) did not attract C. nigoni males or females. Additional research is required to elucidate the biological functions of these species-specific components and decipher the ecological significance of (ω)- and (ω − 2)-hydroxylation of ascaroside aglycones as a means to further increase structural diversity of this highly conserved class of nematode signaling molecules.
EXPERIMENTAL SECTION
Preparation of Exometabolome Extracts
Wild-type isolates of 13 Caenorhabditis species were cultivated at 23 °C on NGM agar seeded with E. coli OP50: C. elegans N2 (Bristol), C. nigoni (sp. 9) JU1422, C. briggsae AF16, C. sinica (sp. 5) JU727, C. remanei PB4641, C. tropicalis (sp. 11) JU1373, C. wallacei (sp. 16) JU1904, C. doughertyi (sp. 10) JU1771, C. brenneri (sp. 4) PB2801, C. japonica DF5081, C. afra (sp. 7) JU1286, C. portoensis (sp. 6) EG4788, and C. n. sp. 8 (sp. 8) QX1182. Mixed stage nematodes from five 10 cm plates collected in M9 buffer served as inoculums for liquid cultures grown in 100 mL S-medium at 23 °C and 150 rpm. Concentrated E. coli OP50 bacteria pellet from an overnight culture in LB medium at 37 °C and 170 rpm was provided as food from days 1–7, after which the cultures were starved for 7 days. After 14 days, nematodes were separated by centrifugation (5 min at 5000g). The filtered supernatant representing the exometabolome was frozen at −80 °C, lyophilized, and extracted with 3 × 100 mL methanol for 12 h each. The combined extract was filtered, concentrated to dryness at 40 °C under reduced pressure, and reconstituted in 1 mL methanol, and aliquots were analyzed by HPLC-HRMS, HPLC-MS/MS precursor ion screening for m/z 73.1, and GC-EIMS. All experiments were performed in triplicate.
Preparation of Trimethylsilyl (TMS) Derivatives for GC-EIMS Analysis
Aliquots of crude nematode exometabolome extracts, C. nigoni exometabolome fractions, and synthetic ascaroside standards were concentrated to dryness. The residues were treated with 10 µL N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) at 60 °C for 40 min and diluted with 10 µL DCM, and 1 µL of the solution was analyzed by GC-EIMS.
Gas Chromatography-Electron Impact-Mass Spectrometry (GC-EIMS)
Separation of volatile TMS derivatives and acquisition of their 70 eV electron impact mass spectra was performed using a Trace GC 2000 series (Thermo Scientific) equipped with a Zebron ZB-5 Guardian column (15 m, 0.25 mm ID, 0.25 µm film thickness; with 10 m guardian end) coupled to a single quadrupole ThermoQuest Trace MS (Finnigan). Helium was used as the carrier gas at a flow rate of 1 mL/min. A temperature program starting at 130 °C for 5 min, followed by a linear gradient of +10 °C/min to 350 °C was applied. A total volume of 1 µL was injected using a 10:1 split ratio and an injector temperature of 250 °C. Electron ionization (EI, 70 eV) mass spectra were acquired from m/z 35–650 amu. Data were analyzed with the Xcalibur 3.1 software (Thermo Fisher Scientific).
Liquid Chromatography-Electrospray Ionization-High Resolution-Mass Spectrometry (HPLC-ESI-HRMS)
HPLC-ESI-HRMS analysis of crude nematode exometabolome extracts and C. nigoni exometabolome fractions was performed using a Dionex UltiMate 3000 HPLC instrument coupled to a Bruker Maxis ultrahigh resolution (UHR) qTOF mass spectrometer equipped with an electrospray ionization (ESI) unit operated in positive or negative mode. Chromatographic separations were achieved using an Agilent ZORBAX Eclipse XDB-C18 column (250 × 3 mm, 5 µm particle diameter) with a flow rate of 400 µL/min and gradient elution starting at 3% acetonitrile in 0.5% aqueous acetic acid for 5 min followed by a linear increase to 100% acetonitrile with 0.5% acetic acid within 35 min. Data were analyzed with the Compass DataAnalysis 4.3 software (Bruker).
Liquid Chromatography Electrospray Ionization Precursor Ion Screening
HPLC-MS/MS precursor ion screening for m/z 73.1 was performed using an Agilent 1260 HPLC instrument (Agilent Technologies) coupled to an API5000 Triple Quadrupole LC/MS/MS mass spectrometer (AB Sciex, Darmstadt) equipped with an electrospray ionization (ESI) unit operated in negative mode. A CID energy of −34 was applied. Chromatographic separations were achieved using an Agilent ZORBAX Eclipse XDB-C18 column (50 × 4.6 mm, 1.8 µm particle diameter) (Agilent Technologies) with a flow rate of 1.1 mL/min and gradient elution starting at 5% acetonitrile in 0.05% aqueous formic acid followed by a linear increase to 95% acetonitrile with 0.05% formic acid within 10 min. Data were analyzed with the Analyst 1.6 software (AB Sciex).
NMR Spectroscopy
NMR spectra were recorded in CD3OD or CDCl3 at 400 MHz for 1H and 100 MHz for 13C using a Bruker AMX400 instrument. Residual solvent signals were used as internal standard with 1H at 3.31 ppm and 13C at 49.05 ppm for CD3OD or 1H at 7.26 ppm and 13C at 77.16 ppm for CDCl3. Two-dimensional homonuclear double quantum filtered (dqf)-COSY spectra were recorded using phase cycling for coherence selection. For the isolated compounds a total of 32 scans were acquired using a time domain of 8k in F2 (acquisition time of 1.2 s) and 512 increments in F1. For two-dimensional heteronuclear HSQC spectra 96 scans were acquired using a time domain of 1k in F2 and 256 increments in F1. Spectra were zero-filled to 8k × 4k (COSY) or 4k × 2k (HSQC) prior to Fourier transformation, phased manually, and baseline corrected using the Topspin 3.2 (Bruker) and MNova 9.0 (Mestrelab Research) software.
Spot Retention Assay
Assays were performed as described previously.10,44 50–60 larval-stage 4 (L4) worms were segregated by sex and stored at 20 °C for 5 h to overnight to be assayed as young adults. 0.6 µL of vehicle control or ascaroside solution was placed in each scoring region. As the working stock of ascaroside was made in Milli-Q-purified ultrapure H2O, this was used as the vehicle control. Five animals were placed on each “X” of the assay plate, which was then transferred to a microscope containing a camera and recorded for 20 min. Each sex and compound was assayed over three plates per day on at least three different days.
Isolation of Hydroxy Ascarosides from the C. nigoni Exometabolome
Hydroxyacyl ascarosides of C. nigoni were isolated from 1.6 L of the liquid culture supernatant. The filtered supernatant was frozen at −80 °C, lyophilized, and the residue extracted with 3 × 100 mL methanol for 12 h each. The filtered extract was concentrated to dryness under reduced pressure and the resulting C. nigoni exometabolome extract was adsorbed onto 2 g of Celite and fractionated by reverse phase chromatography on 5 g RP-C18–SPE cartridges (Chromabond, Macherey-Nagel) using increasing concentrations of methanol in water as eluent to afford 10 fractions (20 mL each). Aliquots of 10 µL were concentrated to dryness under reduced pressure, treated with 10 µL MSTFA at 60 °C for 30 min, diluted with 10 µL DCM, and analyzed by GC-EIMS (Figure S3). Fractions were concentrated to dryness under reduced pressure and analyzed by 1H NMR spectroscopy (Figure S4). The 40% methanol fraction containing the target components according to GC-EIMS was subsequently submitted to semipreparative HPLC using an Agilent HP-1100 HPLC instrument equipped with a Grom-Sil 120 ODS-4 HE column (250 × 8 mm, 5 µm) coupled to a Gilson 206 Abimed fraction collector. A flow rate of 2 mL/min with gradient elution was used starting at 3% acetonitrile in 0.5% aqueous acetic acid for 3 min, followed by a linear increase to 100% acetonitrile with 0.5% acetic acid within 30 min. Aliquots of 10 µL were analyzed by GC-EIMS and HPLC-ESI-(−)-HR-MS as described before. Fractions containing the target compounds were concentrated to dryness, dissolved in 650 µL CD3OD, and analyzed by one- and two-dimensional NMR spectroscopy.
(7R,8R,2E)-threo-8-[(3′,6′-Dideoxy-α-l-arabino-hexopyranosyl)-oxy]-7-hydroxy-2-nonenoic Acid (threo-Asc-7OH-ΔC9, 10)
Isolated from the C. nigoni exometabolome (275 µg, c = ~540 nmol/L), for 1H and 13C NMR data see Table 1; HRMS (ESI-TOF) m/z (M − H)− calcd for C15H25O7 317.1606, found 317.1619.
(7R,8R)-threo-8-[(3′,6′-Dideoxy-α-l-arabino-hexopyranosyl)oxy]-7-hydroxynonanoic Acid (threo-Asc-7OH-C9, 11)
Isolated from the C. nigoni exometabolome (110 µg, ~215 nmol/L), for 1H and 13C NMR data see Table 1; HRMS (ESI-TOF) m/z (M − H)− calcd for C15H27O7 319.1762, found 319.1771.
(2E,8S)-8-[(3,6-Dideoxy-α-l-arabino-hexopyranosyl)oxy]-9-hydroxy-2-nonenoic Acid (Asc-9OH-ΔC9, 12)
Isolated from the C. nigoni exometabolome (130 µg, ~255 nmol/L), for 1H and 13C NMR data see Table 1; HRMS (ESI-TOF) m/z (M − H)− calcd for C15H25O7 317.1606, found 317.1617.
(R)-Methyl 2-(4-methoxybenzyloxy)propanoate (15).45
Under argon atmosphere a solution of (R)-(+)-methyl 2-hydroxypropanoate (14) (1.04 g, 10 mmol) and 4-methoxybenzyl 2,2,2-trichloroacetimidate (2.8 g, 10 mmol) in dry DCM (15 mL) at 0 °C was treated with trimethylsilyl triflate (10 µL). After being stirred at 0 °C for 3 h, the reaction was quenched by addition of saturated NaHCO3 solution (1 mL), and the mixture was diluted with DCM (15 mL), washed with saturated NaHCO3 solution (2 × 10 mL), dried over Na2SO4, and concentrated under reduced pressure. The product was isolated by column chromatography (silica gel, 9/1 v/v hexane/ethyl acetate elution, Rf = 0.22) to afford 15 (1.38 g, 6.2 mmol, 62%) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 4.61 (d, J = 11.3 Hz, 1H), 4.38 (d, J = 11.3 Hz, 1H), 4.05 (q, J = 6.8 Hz, 1H), 3.79 (s, 3H), 3.75 (s, 3H), δ 1.41 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.9, 159.5, 129.72, 129.70, 113.9, 73.7, 71.8, 55.4, 52.0, 18.8.45
(R)-2-(4-Methoxybenzyloxy)propanal (16).45
Under argon atmosphere a solution of 15 (672 mg, 3 mmol) in dry DCM (10 mL) at −78 °C was treated dropwise with a 1 M DIBAL-H solution (3.3 mL, 3.3 mmol) in toluene. After stirring at −78 °C for 30 min the reaction was quenched with methanol (0.5 mL) and saturated sodium potassium tartrate solution (10 mL) and stirred for 1 h and extracted with DCM (2 × 20 mL). The combined organic phase was dried over Na2SO4 and concentrated under reduced pressure. Column chromatography (silica gel, DCM elution) afforded 16 (580 mg, 3 mmol, 100% yield) as a colorless oil that was directly used for the next step. 1H NMR (400 MHz, CDCl3) δ 9.63 (d, J = 1.8 Hz, 1H), 7.29 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 4.57 (d, J = 11.4 Hz, 1H), 4.54 (d, J = 11.4 Hz, 1H), 3.87 (dq, J = 1.8 Hz, J = 7.0 Hz, 1H), 3.80 (s, 3H), 1.31 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 203.7, 159.7, 129.8, 129.5, 114.1, 79.3, 71.9, 55.4, 15.4.45
(2R,3R)-threo-3-Hydroxy-2-(4-methoxybenzyloxy)-7-octene (17)
Under argon atmosphere, a solution of 4-pentenylmagnesium bromide (6 mmol) in diethyl ether (10 mL) prepared from 5-bromo-1-pentene (900 mg, 6 mmol) and magnesium (150 mg, 6.2 mmol) was cooled to 0 °C and treated dropwise with 16 (580 mg, 3 mmol) in Et2O (2 mL) over the course of 5 min. The resulting mixture was stirred at 0 °C for 1 h and quenched with saturated aqueous NH4Cl solution (10 mL), and the aqueous layer extracted with ethyl acetate (2 × 10 mL). The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The product was isolated by column chromatography (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.42) to afford 17 (460 mg, 1.74 mmol, 58% yield) with a diastereoisomeric excess of de = 92% as determined by 1H NMR spectroscopy. 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.80 (ddt, J = 17.2 Hz, J = 10.2 Hz, J = 6.7 Hz, 1H), 5.00 (dbr, J = 17.2 Hz, 1H), 4.94 (dbr, J = 10.3 Hz, 1H), 4.60 (d, 2J = 11.1 Hz, 1H), 4.36 (d, 2J = 11.1 Hz, 1H), 3.80 (s, 3H), 3.40 (m, 1H), 3.34 (dq, J = 6.0, J = 6.3 Hz, 1H), 2.09 (m, 2H), 1.62 (m, 1H), 1.47 (m, 1H), 1.44 (m, 2H), 1.17 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.4, 138.9, 130.6, 129.5, 114.6, 114.0, 78.2, 74.9, 70.8, 55.3, 33.8, 32.4, 24.9, 15.7.
(2R,3R)-threo-3-Benzoyloxy-2-(4-methoxybenzyloxy)-7-octene (18a)
A solution of 17 (316.5 mg, 1.2 mmol) and dry pyridine (290 µL, 3.6 mmol) in dry DCM (2 mL) at 0 °C was treated with benzoyl chloride (280 µL, 2.4 mmol) in dry DCM (1 mL). After stirring at RT for 12 h the mixture was diluted with DCM (10 mL), washed with 1 M HCl (10 mL), saturated aqueous NaHCO3 solution (10 mL), dried over Na2SO4, and concentrated under reduced pressure. Column chromatography of the residue (silica gel, 9/1 v/v hexane/ethyl acetate elution, Rf = 0.37) afforded (2R,3R)-threo-18a (386 mg, 1.05 mmol, 87% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.6 Hz, 2H), 7.59 (m, 1H), 7.47 (m, 2H), 7.26 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 5.79 (ddt, J = 17.0 Hz, J = 10.3 Hz, J = 6.7 Hz, 1H), 5.25 (m, 1H), 5.01 (d, J = 17.1 Hz, 1H), 4.96 (d, J = 10.3 Hz, 1H), 4.62 (d, J = 11.5 Hz, 1H), 4.49 (d, J = 11.5 Hz, 1H), 3.81 (s, 3H), 3.74 (dq, J = 4.9 Hz, J = 6.3 Hz, 1H), 2.10 (m, 2H), 1.77 (m, 2H), 1.46 (m, 2H), 1.23 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.4, 159.3, 138.5, 133.0, 130.7, 129.8, 129.4, 129.0, 128.5, 114.9, 113.9, 76.1, 74.6, 70.9, 55.4, 33.7, 29.0, 25.0, 15.5.
(2R,3S)-erythro-3-Benzoyloxy-2-(4-methoxybenzyloxy)-7-octene (18b)
Under argon atmosphere a solution of 17 (294.5 mg, 1.11 mmol), triphenylphosphine (668.8 mg, 2.55 mmol), and benzoic acid (300.4 mg, 2.46 µmol) in dry THF (6 mL) at 0 °C was treated with 440 µL diisopropyl azodicarboxylate (DIAD, 440 µL, 2.23 mmol). After stirring at RT for 4 h the solvent was removed under reduced pressure. Column chromatography of the residue (silica gel, 9/1 v/v hexane/ethyl acetate elution, Rf = 0.37) afforded (2R,3S)-erythro-18b (223.5 mg, 606.6 µmol, 55% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 7.6 Hz, 2H), 7.59 (m, 1H), 7.47 (m, 2H), 7.27 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.80 (ddt, J = 17.0 Hz, J = 10.2 Hz, J = 6.7 Hz, 1H), 5.28 (m, 1H), 5.03 (d, J = 17.1 Hz, 1H), 4.98 (d, J = 10.3 Hz, 1H), 4.59 (d, J = 11.5 Hz, 1H), 4.54 (d, J = 11.5 Hz, 1H), 3.79 (s, 3H), 3.74 (dq, J = 3.8 Hz, J = 6.3 Hz, 1H,), 2.11 (m, 2H), 1.80 (m, 2H), 1.52 (m, 2H), 1.28 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.3, 159.2, 138.5, 132.9, 130.68, 130.74, 129.8, 129.4, 128.5, 114.9, 113.9, 76.2, 75.4, 70.8, 55.3, 33.7, 29.3, 25.0, 16.0.
(7R,8R,2E)-threo-Ethyl 7-Benzoyloxy-8-(4-methoxybenzyloxy)-2-nonenoate (19a)
Under argon atmosphere a solution of 18a (368.5 mg, 1 mmol) and ethyl acrylate (545 µL, 5 mmol) in DCM (30 mL) was treated with Grubbs-II catalyst (50 mg, 59 µmol) and stirred at 40 °C for 9 h. The solution was concentrated under reduced pressure and the residue purified by chromatography (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.40) to afford 19a (335.7 mg, 762 µmol, 76% yield) as a green oil. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.2 Hz, 2H), 7.57 (m, 1H), 7.47 (m, 2H), 7.25 (d, J = 8.6 Hz, 2H), 6.93 (dt, J = 15.6, J = 7.0 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 5.82 (d, J = 15.6 Hz, 1H), 5.24 (m, 1H), 4.62 (d, J = 11.5 Hz, 1H), 4.48 (d, J = 11.5 Hz, 1H), 4.19 (q, J = 7.0 Hz, 2H), 3.81 (s, 3H), 3.73 (dq, J = 4.6, J = 6.4 Hz, 1H), 2.23 (m, 2H), 1.78 (m, 2H), 1.52 (m, 2H), 1.30 (t, J = 6.9 Hz, 3H), 1.23 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 166.4, 159.3, 148.6, 133.1, 130.7, 130.4, 129.8, 129.5, 128.5, 121.8, 113.9, 75.8, 74.4, 70.9, 60.3, 55.4, 32.1, 29.1, 24.2, 15.4, 14.4.
(7S,8R,2E)-erythro-Ethyl 7-Benzoyloxy-8-(4-methoxybenzyloxy)-2-nonenoate (19b)
Under argon atmosphere a solution of 18b (223.5 mg, 607 µmol) and ethyl acrylate (330 µL, 3 mmol) in DCM (20 mL) was treated with Grubbs-II catalyst (30 mg, 35 µmol) and stirred at 40 °C for 9 h. The solution was concentrated under reduced pressure and the residue purified by chromatography (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.40) to afford 19b (218.2 mg, 495 µmol, 81% yield) as a brownish oil. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.2 Hz, 2H), 7.59 (m, 1H), 7.47 (m, 2H), 7.26 (d, J = 8.5 Hz, 2H), 6.94 (dt, J = 15.6, J = 6.9 Hz, 1H), 6.84 (d, J = 8.5 Hz, 2H), 5.82 (d, J = 15.6 Hz, 1H), 5.24 (dt, J = 9.1 Hz, J = 3.9 Hz, 1H), 4.55 (s, 2H), 4.19 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.72 (dq, J = 3.9 Hz, J = 6.4 Hz, 1H), 2.25 (m, 2H), 1.79 (m, 2H), 1.56 (m, 2H), 1.29 (t, J = 7.0 Hz, 3H), 1.26 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 166.2, 159.2, 148.5, 133.0, 130.6, 130.5, 129.7, 129.4, 128.5, 121.8, 113.8, 76.0, 75.3, 70.8, 60.2, 55.3, 32.0, 29.3, 24.2, 16.0, 14.4.
(7R,8R,2E)-threo-Ethyl 7-Benzoyloxy-8-hydroxy-2-nonenoate (20a)
A solution of 19a (330 mg, 750 µmol) in DCM (5 mL) was treated with water (260 µL) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (187 mg, 825 µmol). After stirring at RT for 1 h the mixture was quenched with water (1 mL) and the aqueous phase extracted with DCM (5 × 1 mL). The combined organic phase was dried over Na2SO4 and concentrated under reduced pressure. Column chromatography (silica gel, 2/1 v/v hexane/ethyl acetate elution, Rf = 0.42) afforded 20a (168.7 mg, 527 µmol, 70% yield) as a yellowish oil.
1H NMR (400 MHz, CDCl3) δ 8.05 (m, 2H); 7.57 (m, 1H), 7.45 (m, 2H), 6.91 (dt, J = 15.7, J = 7.0 Hz, 1H), 5.80 (d, J = 15.7 Hz, 1H), 5.05 (dt, J = 7.8 Hz, J = 5.0 Hz, 1H), 4.16 (q, J = 7.2 Hz, 2H), 3.94 (dq, J = 4.8, J = 6.4 Hz, 1H), 2.23 (m, 2H), 1.77 (m, 2H), 1.55 (m, 2H), 1.26 (t, J = 7.0 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 166.6, 148.4, 133.3, 130.1, 129.8, 128.6, 121.9, 77.9, 69.0, 60.3, 32.0, 30.2, 24.1, 19.6, 14.4; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C18H28NO5 338.1962, found 338.1979.
(7S,8R,2E)-erythro-Ethyl 7-Benzoyloxy-8-hydroxy-2-nonenoate (20b)
A solution of 19b (218.2 mg, 495 µmol) in DCM (5 mL) was treated with water (260 µL) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (124 mg, 545 µmol). After being stirred at RT for 1 h, the mixture was quenched with 1 mL water, and the aqueous phase was extracted with DCM (5 × 1 mL). The combined organic phase was dried over Na2SO4 and concentrated under reduced pressure. Column chromatography (silica gel, 2/1 v/v hexane/ethyl acetate elution, Rf = 0.42) afforded 20b (113.5 mg, 354 µmol, 72% yield) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 8.05 (m, 2H), 7.57 (m, 1H), 7.45 (m, 2H), 6.91 (dt, J = 15.7, J = 6.9 Hz, 1H), 5.80 (d, J = 15.7 Hz, 1H), 5.11 (dt, J = 9.3 Hz, J = 3.7 Hz, 1H), 4.15 (q, J = 7.2 Hz, 2H), 4.00 (dq, J = 3.8, J = 6.4 Hz, 1H), 2.24 (m, 2H), 1.76 (m, 2H), 1.57 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H), 1.23 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 166.7, 148.4, 133.2, 130.1, 129.7, 128.5, 121.9, 78.2, 69.3, 60.2, 31.9, 29.2, 24.2, 18.3, 14.3; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C18H28NO5 338.1962, found 338.1977.
O-(2,4-Di-O-benzoyl-3,6-dideoxy-α-l-arabino-hexopyranosyl) Trichloroacetimidate (21)
A solution of 2,4-di-O-benzoyl-ascarylose (53.5 mg, 150 µmol) in DCM (1 mL) was treated with trichloroacetonitrile (32 µL, 320 µmol) and 1,8-diazabicyclo[5.4.0]-undec-7-ene (5 µL, 33.4 µmol). After being stirred for 30 min, the yellowish solution was concentrated under reduced pressure. Column chromatography of the residue (silica gel, 4/1 v/v hexane/ethyl acetate elution) afforded 20 (61.2 mg, 122.4 µmol, 81% yield) as a colorless oil that was directly used for the next steps.
(7R,8R,2E)-threo-Ethyl 7-Benzoyloxy-8-[(2,4-di-O-benzoyl-3,6-dideoxy-α-l-arabino-hexopyranosyl)oxy]-2-nonenoate (22a)
A solution of 20a (14.7 mg, 45.9 µmol) and 21 (15.3 mg, 30.6 µmol) in dry DCM (1 mL) at 0 °C was treated with trimethylsilyl triflate (5 µL) and stirred for 3 h. The reaction was quenched with sat. NaHCO3 solution (100 µL), dried over Na2SO4, and concentrated under reduced pressure. Column chromatography (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.24) afforded 22a (13.8 mg, 20.9 µmol, 68% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.12 (m, 2H), 8.10 (m, 2H), 7.80 (d, J = 7.6 Hz, 2H), 7.58 (m, 3H), 7.47 (m, 2H), 7.46 (m, 2H), 7.42 (m, 2H), 6.94 (dt, J = 15.7, J = 7.0 Hz, 1H), 5.83 (d, J = 15.6 Hz, 1H), 5.31 (m, 1H), 5.15 (s.br, 1H), 5.10 (ddd, J = 11.0, J = 9.8 Hz, J = 4.7 Hz, 1H), 4.98 (s, 1H), 4.16 (q, J = 7.1 Hz, 2H), 4.08 (m, 1H), 4.01 (dq, J = 9.7 Hz, J = 6.3 Hz, 1H), 2.41 (dt, J = 13.7 Hz, J = 3.8 Hz, 1H), 2.28 (m, 2H), 2.19 (m, 1H), 1.80 (m, 2H), 1.61 (m, 2H), 1.29 (d, J = 6.5 Hz, 3H), 1.26 (t, J = 7.0 Hz, 3H), 1.03 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.6, 166.1, 165.9, 165.7, 148.3, 133.4, 133.2, 133.2, 130.4, 130.3, 130.0, 129.91, 129.88, 129.7, 128.6, 128.5, 122.0, 93.3, 75.9, 72.5, 71.1, 70.4, 67.3, 60.3, 32.0, 29.9, 29.8, 24.0, 17.8, 15.0, 14.4; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C38H46NO10 676.3116, found 676.3133.
(7S,8R,2E)-erythro-Ethyl 7-Benzoyloxy-8-[(2,4-di-O-benzoyl-3,6-dideoxy-α-l-arabino-hexopyranosyl)oxy]-2-nonenoate (22b)
A solution of 20b (14.7 mg, 45.9 µmol) and 21 (15.3 mg, 30.6 µmol) in dry DCM (1 mL) at 0 °C was treated with trimethylsilyl triflate (5 µL) and stirred for 3 h. The reaction was quenched with sat. NaHCO3 solution (100 µL), dried over Na2SO4 and concentrated under reduced pressure. The residue was chromatographed (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.26) to afford 22b (12.9 mg, 19.5 µmol, 64% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.12 (m, 2H), 8.09 (m, 2H), 7.99 (d, J = 7.8 Hz, 2H), 7.58 (m, 3H), 7.46 (m, 6H), 6.96 (dt, J = 15.8, J = 7.2 Hz, 1H), 5.85 (d, J = 15.5 Hz, 1H), 5.22 (dt, J = 15.2 Hz, J = 4.0 Hz, 1H), 5.15 (s.br, 1H), 5.12 (m, 1H), 4.94 (s, 1H), 4.16 (q, J = 7.1 Hz, 2H), 4.16 (dq, J = 3.8 Hz, J = 6.4 Hz, 1H), 4.13 (m, 1H), 2.41 (dt, J = 13.2 Hz, J = 3.8 Hz, 1H), 2.30 (m, 2H), 2.19 (ddd, J = 13.0 Hz, J = 11.7 Hz, J = 2.8 Hz, 1H), 1.93 (m, 1H), 1.82 (m, 1H), 1.64 (m, 2H), 1.29 (d, J = 6.5 Hz, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.03 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 166.2, 165.9, 165.7, 148.4, 133.4, 133.3, 133.2, 130.4, 130.1, 130.0, 129.8, 128.6, 128.5, 122.0, 93.6, 76.6, 72.5, 71.0, 70.6, 67.2, 60.3, 32.1, 29.8, 28.6, 24.4, 17.7, 14.8, 14.4; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C38H46NO10 676.3116, found 676.3129.
(7R,8R,2E)-threo-8-[(3′,6′-Dideoxy-α-l-arabino-hexopyranosyl)-oxy]-7-hydroxy-2-nonenoic Acid ((7R,8R)-threo-10a)
A solution of 22a (13.8 mg, 20.9 µmol) in methanol (2 mL) was treated with LiOH monohydrate (7.0 mg, 167 µmol) in H2O (100 µL). After stirring for 12 h the reaction mixture was acidified with acetic acid and concentrated under reduced pressure. The product was isolated by a combination of column chromatography on silica gel using a mixture of 15% methanol in dichloromethane with 0.1% acetic acid and solid phase extraction on reverse phase C18 using increasing concentrations of methanol in water as eluent to afford threo-10a (2.4 mg, 7.5 µmol, 36% yield) along with the intramolecular cyclization product 23a (2.5 mg, 7.9 µmol, 39% yield).
(7R,8R)-threo-10a
1H NMR (400 MHz, CD3OD) δ 6.95 (dt, J = 15.6 Hz, J = 7.0 Hz, 1H), 5.82 (d, J = 15.6 Hz, 1H), 4.65 (s, 1H), 3.75 (s.br, 1H), 3.74 (dq, J = 3.9 Hz, J = 6.1 Hz, 1H), 3.64 (dq, J = 9.3 Hz, J = 6.2 Hz, 1H), 3.53 (m, 1H), 3.52 (m, 1H), 2.27 (m, 2H), 1.95 (dt, J = 13.1 Hz, J = 3.8 Hz, 1H), 1.80 (ddd, J = 13.1 Hz, J = 11.0 Hz, J = 3.0 Hz, 1H), 1.70 (m, 1H), 1.54 (m, 3H), 1.22 (d, J = 6.2 Hz, 3H), 1.14 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 170.6, 150.5, 123.2, 97.8, 75.3, 74.9, 71.4, 69.9, 68.3, 35.9, 33.1, 33.0, 25.9, 18.1, 14.7; HRMS (ESI-TOF) m/z (M - H)− calcd for C15H25O7 317.1606, found 317.1618.
2-((6R)-6-((R)-1-[(3,6-Dideoxy-α-l-arabino-hexopyranosyl)oxy]-ethyl)tetrahydro-2H-pyran-2-yl)acetic Acid (23a)
1H NMR (400 MHz, CD3OD) δ 4.66 (s, 1H), 3.76 (s.br, 1H), 3.65(m, 1H), 3.74 (m, 1H), 3.65 (m, 1H), 3.52 (m, 2H), 2.50 (ddd, J = 15.0 Hz, J = 7.2 Hz, J = 2.6 Hz, 1H), 2.41 (ddd, J = 15.0 Hz, J = 5.6 Hz, J = 2.7 Hz, 1H), 1.95 (dt, J = 13.1 Hz, J = 3.8 Hz, 1H), 1.81 (ddd, J = 13.1 Hz, J = 11.3 Hz, J = 3.0 Hz, 1H), 1.58 (m, 3H), 1.51 (m, 2H), 1.46 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H), 1.14 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 175.6, 97.8, 79.32/79.37, 75.33/75.38, 75.1, 71.4, 69.9, 68.4, 40.4, 35.9, 35.0, 33.4, 22.83/22.87, 18.1, 14.7; HRMS (ESI-TOF) m/z (M - H)− calcd for C15H25O7 317.1606, found 317.1614.
(7S,8R,2E)-erythro-8-[(3′,6′-Dideoxy-α-l-arabino-hexopyranosyl)-oxy]-7-hydroxy-2-nonenoic Acid ((7S,8R)-erythro-10b)
A solution of 22b (12.9 mg, 19.5 µmol) in methanol (2 mL) was treated with LiOH monohydrate (6.6 mg, 156 µmol) in H2O (100 µL). After being stirred for 12 h, the reaction mixture was acidified with acetic acid and concentrated under reduced pressure. The product was isolated by a combination of column chromatography on silica gel using a mixture of 15% methanol in dichloromethane with 0.1% acetic acid and solid phase extraction on reverse phase C18 using increasing concentrations of methanol in water as eluent to afforded erythro-10b (2.9 mg, 9.1 µmol, 46% yield) along with the intramolecular cyclization product 23b (2.6 mg, 8.2 µmol, 42% yield).
(7S,8R)-erythro-10b
1H NMR (400 MHz, CD3OD) δ 6.96 (dt, J = 15.5 Hz, J = 7.1 Hz, 1H), 5.82 (d, J = 15.5 Hz, 1H), 4.66 (s, 1H), 3.75 (s.br, 1H), 3.66 (m, 1H), 3.63 (dq, J = 9.5 Hz, J = 6.2 Hz, 1H), 3.53 (m, 1H), 3.52 (m, 1H), 2.27 (m, 2H), 1.95 (dt, J = 13.3 Hz, J = 3.7 Hz, 1H), 1.80 (ddd, J = 13.1 Hz, J = 11.1 Hz, J = 3.0 Hz, 1H), 1.65 (m, 2H), 1.54 (m, 1H), 1.43 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H), 1.15 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 170.4, 150.6, 123.0, 97.6, 75.6, 75.2, 71.3, 69.8, 68.4, 35.9, 33.6, 33.0, 25.6, 18.1, 14.2; HRMS (ESI-TOF) m/z (M - H)− calcd for C15H25O7 317.1606, found 317.1615.
2-((6S)-6-((R)-1-[(3,6-Dideoxy-α-l-arabino-hexopyranosyl)oxy]-ethyl)tetrahydro-2H-pyran-2-yl)acetic Acid (23b)
1H NMR (400 MHz, CD3OD) δ 4.66 (s, 1H), 3.75 (s.br, 1H), 3.66 (m, 2H), 3.64 (m, 1H), 3.52 (m, 2H), 2.50 (ddd, J = 15.0 Hz, J = 7.1 Hz, J = 2.1 Hz, 1H), 2.41 (ddd, J = 15.1 Hz, J = 5.4 Hz, J = 2.2 Hz, 1H), 1.95 (1H, dt, J = 13.1 Hz, J = 3.9 Hz, 1H), 1.80 (ddd, J = 13.1 Hz, J = 11.3 Hz, J = 3.1 Hz, 1H), 1.59 (m, 1H), 1.57 (m, 2H), 1.44 (m, 1H), 1.41 (m, 2H), 1.22 (d, J = 6.2 Hz, 3H), 1.15 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 175.7, 97.6, 79.3, 75.57/75.61, 75.4, 71.3, 69.8, 68.4, 40.4, 35.9, 34.9, 34.1, 22.6, 18.1, 14.1; HRMS (ESI-TOF) m/z (M - H)− calcd for C15H25O7 317.1606, found 317.1622.
(7R,8R)-threo- or (7S,8R)-erythro-8-[(3′,6′-Dideoxy-α-l-arabino-hexopyranosyl) oxy]-7-hydroxynonanoic Acid (threo-11a or erythro-11b)
Aliquots (10 µg) of synthetic (7R,8R)-threo-asc-7OH-ΔC9 (10a) or (7S,8R)-erythro- asc-7OH-ΔC9 (10b) in methanol (500 µL) were treated with 10% palladium on carbon (10 mg) and hydrogenated under atmospheric pressure for 1 h. The mixture was filtered over a small patch of silica, concentrated to dryness, and the resulting (7R,8R)-threo-asc-7OH-C9 (11a) or (7S,8R)-erythro- asc-7OH-C9 (11b) submitted to TMS derivatization for chemical correlation with the natural product isolated from C. nigoni.
(7S)-7-tert-Butyldimethylsilyloxy-6-hydroxy-1-octene (25)
Under argon atmosphere, a solution of 4-pentenylmagnesium bromide (1.68 mmol) in THF (2 mL) prepared from 5-bromo-1-pentene (250 mg, 1.68 mmol) and magnesium (45 mg, 1.88 mmol) was added slowly to a mixture of copper(I)iodide (32 mg, 168 µmol) and (S)-3-tert-butyldimethylsilyloxy-1,2-epoxypropane (24) (210 mg, 1.12 mmol) in THF (2 mL) at 0 °C. After being stirred at 0 °C for 3 h, the solution was quenched with saturated ammonium chloride solution and stirred for 1 h, and the aqueous phase extracted with diethyl ether. The organic phase was washed with brine and concentrated under reduced pressure, and the residue was chromatographed (silica gel, DCM elution, Rf = 0.43) to afford 25 (273 mg, 1.05 mmol, 94% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.76 (ddt, J = 17.0 Hz, J = 10.3 Hz, J = 6.7 Hz, 1H), 4.95 (ddt, J = 17.1 Hz, J = 2.0 Hz, J = 1.5 Hz, 1H), 4.89 (ddt, J = 10.2 Hz, J = 2.0 Hz, J = 1.5 Hz, 1H), 3.58 (m, 2H), 3.36 (m, 1H), 2.02 (dt, J = 6.7 Hz, J = 7.1 Hz, 2H), 1.26–1.48 (m, 6H), 0.87 (s, 9H), 0.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.9, 114.4, 71.8, 67.4, 33.8, 32.7, 29.1, 26.0, 25.1, 18.3, −5.3, −5.4.
(2E,8S)-Ethyl 9-tert-Butyldimethylsilyloxy-8-hydroxy-2-nonenoate (26)
Under argon atmosphere, a solution of 25 (258.5 mg, 1 mmol) and ethyl acrylate (545 µL, 5 mmol) in DCM (30 mL) was treated with Grubbs second generation catalyst (50 mg, 58.9 µmol) and stirred at 40 °C for 9 h. The solution was concentrated under reduced pressure, and the residue was chromatographed (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.51) to afford 26 (268 mg, 810 mmol, 81% yield) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ 6.95 (dt, J = 15.7 Hz, J = 6.9 Hz, 1H), 5.80 (d, J = 15.7 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.62 (m, 1H), 3.60 (dd, J = 10.6 Hz, J = 3.3 Hz, 1H), 3.37 (dd, J = 10.6 Hz, J = 8.3 Hz, 1H), 2.20 (m, 2H), 1.33–1.54 (m, 6H), 1.27 (t, J = 7.1 Hz, 3H), 0.89 (s, 9H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 166.9, 149.2, 121.6, 71.8, 67.4, 60.3, 32.7, 32.2, 28.3, 26.0, 25.3, 18.4, 14.4, −5.2, −5.3; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C17H38NO4Si 348.2565, found 348.2572.
(2E,8S)-Ethyl 8-[(2,4-Di-O-benzoyl-3,6-dideoxy-α-l-arabinohexopyranosyl) oxy]-9-tert-butyldimethylsilyloxy-2-nonenoate (27)
Under argon atmosphere, a solution of 26 (30.3 mg, 91.8 µmol) and 21 (30.6 mg, 61.2 µmol) in dry DCM (1 mL) at 0 °C was treated with trimethylsilyl triflate (5 µL) and stirred for 3 h. The reaction was quenched by addition of saturated aqueous NaHCO3 solution (100 µL), dried over Na2SO4, and concentrated under reduced pressure. The product was isolated by column chromatography (silica gel, 4/1 v/v hexane/ethyl acetate elution, Rf = 0.50) to afford 27 (21.7 mg, 32.4 µmol, 53% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.0 Hz, 2H), 8.03 (d, J = 7.9 Hz, 2H), 7.58 (m, 2H), 7.46 (m, 4H), 6.99 (dt, J = 15.6 Hz, J = 7.0 Hz, 1H), 5.85 (d, J = 15.6 Hz, 1H), 5.22 (s.br, 1H), 5.15 (ddd, J = 11.4 Hz, J = 10.3 Hz, J = 4.4 Hz, 1H), 5.10 (s, 1H), 4.16 (q, J = 7.1 Hz, 2H), 4.15 (m, 1H) 3.78 (m, 1H), 3.65 (dd, J = 10.5 Hz, J = 5.9 Hz, 1H), 3.61 (dd, J = 10.6 Hz, J = 5.0 Hz, 1H), 2.43 (dt, J = 13.3 Hz, J = 3.9 Hz, 1H), 2.26 (m, 2H), 2.19 (ddd, J = 13.5 Hz, J = 11.6 Hz, J = 3.0 Hz, 1H), 1.40–1.70 (m, 6H), 1.28 (d, J = 6.9 Hz, 3H), 1.26 (t, J = 7.1 Hz, 3H), 0.85 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.8, 165.8, 165.7, 149.1, 133.3, 130.2, 130.1, 130.0, 129.8, 128.6, 128.5, 121.7, 96.4, 78.4, 71.0, 70.8, 67.3, 65.5, 60.3, 32.3, 31.8, 29.8, 28.2, 25.9, 25.8, 25.3, 18.3, 18.0, 14.4, −5.3, −5.4; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C37H56NO9Si 686.3719, found 686.3735.
(2E,8S)-Ethyl 8-[(2,4-Di-O-benzoyl-3,6-dideoxy-α-l-arabinohexopyranosyl) oxy]-9-hydroxy-2-nonenoate (28)
Under argon atmosphere a solution of 27 (21.7 mg, 32.4 µmol) in dry THF (1 mL) was treated with 1 M tetrabutylammonium fluoride (50 µL, 50 µmol) in dry THF (1 mL) and stirred at RT for 3 h. The solution was concentrated under reduced pressure, and the residue was separated by column chromatography (silica gel, 2/1 v/v hexane/ethyl acetate elution) to afford 28 (14.0 mg 25.2 µmol, 78% yield). 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 7.8 Hz, 2H), 8.05 (d, J = 7.7 Hz, 2H), 7.59 (m, 2H), 7.46 (m, 4H), 6.97 (dt, J = 15.6 Hz, J = 6.9 Hz, 1H), 5.83 (d, J = 15.7 Hz, 1H), 5.20 (s.br, 1H), 5.19 (ddd, J = 11.3 Hz, J = 10.5 Hz, J = 4.5 Hz, 1H), 5.08 (s, 1H), 4.16 (q, J = 7.1 Hz, 2H), 4.13 (m, 1H), 3.82 (m, 1H), 3.76 (dd, J = 12.0 Hz, J = 2.9 Hz, 1H), 3.61 (dd, J = 12.0 Hz, J = 5.6 Hz, 1H), 2.44 (dt, J = 13.5 Hz, J = 3.9 Hz, 1H), 2.25 (m, 2H), 2.21 (ddd, J = 13.5 Hz, J = 11.6 Hz, J = 3.0 Hz, 1H), 1.40–1.75 (m, 6H), 1.29 (d, J = 6.8 Hz, 3H), 1.26 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.8, 166.1, 165.8, 148.9, 133.5, 133.4, 130.1, 130.0, 129.82, 129.79, 128.6, 121.7, 95.9, 79.3, 71.2, 70.6, 67.5, 64.5, 60.3, 32.2, 31.4, 29.8 28.2, 25.3, 18.0, 14.4; HRMS (ESI-TOF) m/z (M + NH4)+ calcd for C31H42NO9 572.2854, found 572.2871.
(2E,8S)-8-[(3,6-Dideoxy-α-l-arabino-hexopyranosyl)oxy]-9-hydroxy-2-nonenoic Acid (Asc-9OH-ΔC9) (12)
A solution of 28 (14.0 mg, 25.3 µmol) in methanol (2 mL) was treated with LiOH monohydrate (8.5 mg, 202.4 µmol) in water (100 µL) and stirred for 12 h. The mixture was acidified with acetic acid and concentrated to dryness under reduced pressure. The residue was separated by column chromatography (silica gel, 15% v/v methanol in dichloromethane with 0.1% acetic acid as eluent) and solid phase extraction (reverse phase C18, 10% stepwise increase of methanol in water as eluent) to afford 12 (2.0 mg, 6.3 µmol, 25% yield), identical to the natural product from C. nigoni, along with its intramolecular cyclization product 28 (5.6 mg, 17.6 µmol, 69% yield).
Asc-9OH-ΔC9 (12)
Colorless oil, 1H NMR (400 MHz, CD3OD) δ 6.95 (dt, J = 15.6 Hz, J = 6.9 Hz, 1H), 5.81 (d, J = 15.6 Hz, 1H), 4.75 (s, 1H), 3.84 (s.br, 1H), 3.69 (m, 1H), 3.67 (m, 1H), 3.60 (dd, J = 11.7 Hz, J = 4.2 Hz, 1H), 3.53 (m, 1H), 3.50 (dd, J = 11.7 Hz, J = 5.6 Hz, 1H), 2.25 (m, 2H), 1.95 (dt, J = 13.0 Hz, J = 3.8 Hz, 1H), 1.78 (ddd, J = 13.0 Hz, J = 11.2 Hz, J = 3.1 Hz, 1H), 1.59 (m, 2H), 1.52 (m, 2H), 1.50 (m, 2H), 1.22 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 170.3, 150.7, 122.9, 99.8, 78.6, 71.4, 69.6, 68.4, 64.6, 35.9, 33.1, 32.8, 29.3, 26.3 18.1; HRMS (ESI-TOF) m/z (M − H)− calcd for C15H25O7 317.1606, found 317.1614.
(7S)-7-[(3,6-Dideoxy-α-l-arabino-hexopyranosyl)oxy]oxocan-2-yl)-acetic Acid (29)
Colorless oil. 1H NMR (400 MHz, CD3OD) δ 4.75 (s, 1H), 3.84 (s.br, 1H), 3.69 (m, 1H), 3.68 (m, 1H), 3.65 (m, 1H), 3.53 (m, 1H), 3.52 (m, 1H), 2.49 (dd, J = 15.2 Hz, J = 7.2 Hz, 1H), 2.40 (dd, J = 15.2 Hz, 5.6 Hz, 1H), 1.95 (dt, J = 13.0 Hz, J = 4.0 Hz, 1H), 1.78 (m, 1H), 1.59 (m, 1H), 1.57 (m, 2H), 1.40–1.60 (m, 4H), 1.22 (d, J = 6.2 Hz, 3H), 13C NMR (100 MHz, CD3OD) δ 175.6, 99.8, 79.29/79.32, 78.73/78.77, 71.37/71.40, 69.6, 68.4, 64.6, 40.4, 36.0, 34.9, 32.8, 26.8, 26.3, 18.2; HRMS (ESI-TOF) m/z (M − H)− calcd for C15H25O7 317.1606, found 317.1619.
Supplementary Material
Acknowledgments
Financial support by the Max Planck Society (MPG), the Jena School of Microbial Communication (JSMC), and the Swiss National Science Foundation (SNSF) is gratefully acknowledged. This work was supported by grants from the NIH (R01DC016058) and startup funds from WPI to JS. We thank Dr. Michael Reichelt (Department for Biochemistry, MPICE, Jena) for acquisition of the MS/MS precursor ion screen and Kerstin Ploss (Department of Bioorganic Chemistry, MPICE, Jena) for technical assistance with the GC-EIMS measurements. All nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (Grant P40 OD010440).
Footnotes
ASSOCIATED CONTENT
- Supporting figures as indicted in the text; NMR spectra of isolated ascarosides (10–12) and synthetic compounds (10a, 10b, 12, and 15–29) (PDF)
The authors declare no competing financial interest.
References
- 1.For recent reviews see: Butcher RA. Nat. Prod. Rep. 2017;34:472. doi: 10.1039/c7np00007c.Butcher RA. Nat. Chem. Biol. 2017;13:577. doi: 10.1038/nchembio.2356.von Reuss SH, Schroeder FC. Nat. Prod. Rep. 2015;32:994. doi: 10.1039/c5np00042d.Schroeder FC. Chem. Biol. 2015;22:7. doi: 10.1016/j.chembiol.2014.10.012.Chute CD, Srinivasan J. Semin. Cell Dev. Biol. 2014;33:18. doi: 10.1016/j.semcdb.2014.06.002.Ludewig AH, Schroeder FC. WormBook. 2013;18:1. doi: 10.1895/wormbook.1.155.1.
- 2.Jeong PY, Jung M, Yim YH, Kim H, Park M, Hong E, Lee W, Kim YH, Kim K, Paik YK. Nature. 2005;433:541. doi: 10.1038/nature03201. [DOI] [PubMed] [Google Scholar]
- 3.Butcher RA, Fujita M, Schroeder FC, Clardy J. Nat. Chem. Biol. 2007;3:420. doi: 10.1038/nchembio.2007.3. [DOI] [PubMed] [Google Scholar]
- 4.Butcher RA, Ragains JR, Kim E, Clardy J. Proc. Natl. Acad. Sci. U. S. A. 2008;105:14288. doi: 10.1073/pnas.0806676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butcher RA, Ragains JR, Clardy J. Org. Lett. 2009;11:3100. doi: 10.1021/ol901011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wharam B, Weldon L, Viney M. BMC Evol. Biol. 2017;17:197. doi: 10.1186/s12862-017-1033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aprison EZ, Ruvinsky I. PLoS Genet. 2015;11:e1005729. doi: 10.1371/journal.pgen.1005729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ludewig AH, Izrayelit Y, Park D, Malik RU, Zimmermann A, Mahanti P, Fox BW, Bethke A, Doering F, Riddle DL, Schroeder FC. Proc. Natl. Acad. Sci. U. S. A. 2013;110:5522. doi: 10.1073/pnas.1214467110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maures TJ, Booth LN, Benayoun BA, Izrayelit Y, Schroeder FC, Brunet A. Science. 2014;343:541. doi: 10.1126/science.1244160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan J, Kaplan F, Ajredini R, Zachariah C, Alborn HT, Teal PE, Malik RU, Edison AS, Sternberg PW, Schroeder FC. Nature. 2008;454:1115. doi: 10.1038/nature07168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pungaliya C, Srinivasan J, Fox BW, Malik RU, Ludewig AH, Sternberg PW, Schroeder FC. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7708. doi: 10.1073/pnas.0811918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan J, von Reuss SH, Bose N, Zaslaver A, Mahanti P, Ho MC, O’Doherty OG, Edison AS, Sternberg PW, Schroeder FC. PLoS Biol. 2012;10:e1001237. doi: 10.1371/journal.pbio.1001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YK, Sanchez-Ayala MA, Sternberg PW, Srinivasan J, Schroeder FC. Org. Lett. 2017;19:2837. doi: 10.1021/acs.orglett.7b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izrayelit Y, Srinivasan J, Campbell SL, Jo Y, von Reuss SH, Genoff MC, Sternberg PW, Schroeder FC. ACS Chem. Biol. 2012;7:1321. doi: 10.1021/cb300169c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollister KA, Conner ES, Zhang XX, Spell M, Bernard GM, Patel P, de Carvalho ACGV, Butcher RA, Ragains JR. Bioorg. Med. Chem. 2013;21:5754. doi: 10.1016/j.bmc.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong CF, Dolke F, von Reuss SH. Org. Biomol. Chem. 2016;14:7217. doi: 10.1039/c6ob01230b. [DOI] [PubMed] [Google Scholar]
- 17.Aprison EZ, Ruvinsky I. Curr. Biol. 2017;27:2589. doi: 10.1016/j.cub.2017.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Aprison EZ, Ruvinsky I. Curr. Biol. 2016;26:2827. doi: 10.1016/j.cub.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Butcher RA, Ragains JR, Li W, Ruvkun G, Clardy J, Mak HY. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1875. doi: 10.1073/pnas.0810338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joo HJ, Yim YH, Jeong PY, Jin YX, Lee JE, Kim H, Jeong SK, Chitwood DJ, Paik YK. Biochem. J. 2009;422:61. doi: 10.1042/BJ20090513. [DOI] [PubMed] [Google Scholar]
- 21.Joo HJ, Kim KY, Yim YH, Jin YX, Kim H, Kim MY, Paik YK. J. Biol. Chem. 2010;285:29319. doi: 10.1074/jbc.M110.122663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Reuss SH, Bose N, Srinivasan J, Yim JJ, Judkins JC, Sternberg PW, Schroeder FC. J. Am. Chem. Soc. 2012;134:1817. doi: 10.1021/ja210202y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Feng L, Chinta S, Singh P, Wang Y, Nunnery JK, Butcher RA. Proc. Natl. Acad. Sci. U. S. A. 2015;112:3955. doi: 10.1073/pnas.1423951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panda O, Akagi AE, Artyukhin AB, Judkins JC, Le HH, Mahanti P, Cohen SM, Sternberg PW, Schroeder FC. Angew. Chem., Int. Ed. 2017;56:4729. doi: 10.1002/anie.201700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choe A, von Reuss SH, Kogan D, Gasser RB, Platzer EG, Schroeder FC, Sternberg PW. Curr. Biol. 2012;22:772. doi: 10.1016/j.cub.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choe A, Chuman T, von Reuss SH, Dossey AT, Yim J, Ajredini R, Kolawa AA, Kaplan F, Alborn HT, Teal PE, Schroeder FC, Sternberg PW, Ediso AS. Proc. Natl. Acad. Sci. U. S. A. 2012;109:20949. doi: 10.1073/pnas.1218302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bose N, Ogawa A, von Reuss SH, Yim JJ, Sommer RJ, Schroeder FC. Angew. Chem., Int. Ed. 2012;51:12438. doi: 10.1002/anie.201206797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noguez JH, Conner ES, Zhou Y, Ciche TA, Ragains JR, Butcher RA. ACS Chem. Biol. 2012;7:961. doi: 10.1021/cb300056q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhuri J, Bose N, Tandonnet S, Adams S, Zuco G, Kache V, Parihar M, von Reuss SH, Schroeder FC, PiresdaSilva A. Sci. Rep. 2016;5:17676. doi: 10.1038/srep17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bose N, Meyer JM, Yim JJ, Mayer MG, Markov GV, Ogawa A, Schroeder FC, Sommer RJ. Curr. Biol. 2014;24:1536. doi: 10.1016/j.cub.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz SA, Brunet V, Lloyd-Jones GC, Spinner W, Wharam B, Viney M. BMC Evol. Biol. 2014;14:46. doi: 10.1186/1471-2148-14-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsueh YP, Mahanti P, Schroeder FC, Sternberg PW. Curr. Biol. 2013;23:83–86. doi: 10.1016/j.cub.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manosalva P, Manohar M, von Reuss SH, Chen S, Koch A, Kaplan F, Choe A, Micikas RJ, Wang X, Kogel KH, Sternberg PW, Williamson VM, Schroeder FC, Klessig DF. Nat. Commun. 2015;6:7795. doi: 10.1038/ncomms8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao L, Zhang X, Wei Y, Zhou J, Zhang W, Qin P, Chinta S, Kong X, Liu Y, Yu H, Hu S, Zou Z, Butcher RA, Sun J. Nat. Commun. 2016;7:12341. doi: 10.1038/ncomms12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Reuss SH, Dolke F, Dong CF. Anal. Chem. 2017;89:10570. doi: 10.1021/acs.analchem.7b02803. [DOI] [PubMed] [Google Scholar]
- 36.Kiontke KC, Félix MA, Ailion M, Rockman MV, Braendle C, Pénigault JB, Fitch DHA. BMC Evol. Biol. 2011;11:339. doi: 10.1186/1471-2148-11-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Félix MA, Braendle C, Cutter AD. PLoS One. 2014;9:e94723. doi: 10.1371/journal.pone.0094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stiernagle T. WormBook. The C. elegans Research Community; 2006. Maintenance of C. elegans. http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J. Org. Chem. 1999;64:866. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 40.(a) Roush WR, Bennett CE, Roberts SE. J. Org. Chem. 2001;66:6389. doi: 10.1021/jo015756a. [DOI] [PubMed] [Google Scholar]; (b) Zhao Z, Hong L, Liu H. J. Am. Chem. Soc. 2005;127:7692. doi: 10.1021/ja042702k. [DOI] [PubMed] [Google Scholar]; (c) Hong L, Zhao Z, Melançon CE, Zhang H, Liu H. J. Am. Chem. Soc. 2008;130:4954. doi: 10.1021/ja0771383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yadav JS, Swamy T, Reddy BVS, Ravinder V. Tetrahedron Lett. 2014;55:4054. [Google Scholar]
- 41.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 42.(a) Oikawa Y, Yoshioka T, Yonemitsu O. Tetrahedron Lett. 1982;23:885. [Google Scholar]; (b) Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021. [Google Scholar]; (c) Chandrasekhar S, Sumithra G, Yadav JS. Tetrahedron Lett. 1996;37:1645. [Google Scholar]
- 43.(a) Schmidt RR, Michel J. Angew. Chem. 1980;92:763. [Google Scholar]; (b) Schmidt RR, Michel J. Angew. Chem., Int. Ed. Engl. 1980;19:731. [Google Scholar]
- 44.Narayan A, Venkatachalam V, Durak O, Reilly DK, Bose N, Schroeder FC, Samuel AD, Srinivasan J, Sternberg PW. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E1392–401. doi: 10.1073/pnas.1600786113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curti C, Zanardi F, Battistini L, Sartori A, Rassu G, Pinna L, Casiraghi G. J. Org. Chem. 2006;71:8552. doi: 10.1021/jo061521t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.