

Abstract
Chimeric antigen receptor (CAR)-expressing T cells targeting surface-bound tumor antigens have yielded promising clinical outcomes, with two CD19 CAR-T cell therapies recently receiving FDA approval for the treatment of B-cell malignancies. The adoption of CARs for the recognition of soluble ligands, a distinct class of biomarkers in physiology and disease, could significantly broaden the utility of CARs in disease treatment. In this study, we demonstrate that CAR-T cells can be engineered to respond robustly to diverse soluble ligands, including CD19 ectodomain, GFP variants, and transforming growth factor beta (TGF-β). We additionally show that CAR signaling in response to soluble ligands relies on ligand-mediated CAR dimerization, and that CAR responsiveness to soluble ligands can be fine-tuned by adjusting the mechanical coupling between the CAR’s ligand-binding and signaling domains. Our results support a role for mechanotransduction in CAR signaling and demonstrate an approach to systematically engineer immune-cell responses to soluble, extracellular ligands.
Introduction
Chimeric antigen receptors (CARs) are synthetic fusion proteins consisting of an extracellular ligand-binding domain linked via a spacer and transmembrane segment to intracellular signaling domains, which can include the CD3ζ T-cell–activation domain and co-stimulatory domains such as CD28 or 4–1BB1,2. This incorporation of native signaling domains enables CARs to interface with endogenous signaling pathways that lead to multifunctional T-cell effector outputs, including cytokine production, T-cell proliferation, and tumor-cell clearance. Consequently, T cells engineered with CD19-binding CARs have shown remarkable clinical efficacy against B-cell malignancies1,2, and CAR-T cells targeting other surface-bound antigens associated with cancer, viral infection, and autoimmunity are under active evaluation3–5.
As a T-cell engineering platform, CARs are highly versatile. CAR molecules are modular, which allows alternative parts to be used for each functional and structural domain in the fusion protein. For example, a variety of target-binding moieties, including antibody-derived single-chain variable fragments (scFvs) and nanobodies, can serve as the ligand-binding domain of CAR molecules. Furthermore, unlike the native T-cell receptor (TCR) complex, CARs can recognize antigens without the requirement of peptide presentation by major histocompatibility complex (MHC) molecules, enabling CARs to bind to a wider array of antigens—including soluble ligands. However, CAR engineering efforts thus far have focused on directing T-cell responses to surface-bound antigens, with no published examples of CARs designed specifically for soluble ligands. Although several CARs have been designed to target surface-bound antigens that also exist in shed, soluble forms, characterization efforts have focused on verifying that shed antigens do not inhibit CAR activation in response to surface-bound ligands6–13. In fact, studies on CARs targeting CD30, mesothelin, carcinoembryonic antigen (CEA), and Lewis Y antigen reported that the soluble form of each antigen does not have the ability to trigger CAR signaling6–10. As such, tumor-secreted cytokines, shed tumor antigens, and other soluble factors associated with pathologic microenvironments remain an untapped repertoire of potentially valuable therapeutic targets. The ability to engineer CAR-T cells to respond to these soluble antigens could create new opportunities in cell-based immunotherapy for numerous diseases.
Although CARs are typically designed to target surface-bound ligands, evidence supporting the possibility of engineering soluble-antigen–responsive CARs can be found in several early studies of CAR constructs that used soluble, crosslinking antibodies to initiate events that resembled proximal TCR signaling in CAR-T cells14–16. These studies confirmed that CARs can be triggered by soluble ligands, but it remains unclear whether such behavior is restricted to crosslinking antibodies or can be extended to other soluble antigens. To date, the design principles that govern the ability of a CAR to respond to soluble antigens remain undefined.
Here, we establish that CAR-T cells can be engineered to respond robustly to soluble ligands, provided that the ligands are capable of mediating CAR dimerization. We describe the construction of CARs that respond to a variety of soluble ligands, including transforming growth factor beta (TGF-β), and demonstrate the ability to effectively convert TGF-β from a potent immunosuppressive cytokine to a strong stimulant for primary human T cells. We further demonstrate that CAR responses to soluble ligands can be tuned by adjusting the mechanical coupling of the CAR’s extracellular ligand-binding domain and its intracellular signaling domains. Our results are consistent with a mechanotransduction model of CAR signaling and may serve as a guide for future efforts to engineer synthetic immunoreceptors to redirect immune-cell responses to soluble cues.
Results
Soluble CD19 ligand activates CD19-binding CARs
To verify whether soluble ligands could indeed activate CARs, we generated soluble CD19 ligands by secreting the CD19 ectodomain (CD19ecto) from transfected HEK293T cells. Application of concentrated supernatant containing soluble CD19ecto triggered CD69 upregulation in Jurkat T cells expressing a CD19 CAR (Supplementary Fig. 1a). Furthermore, the soluble ligand stimulated CD19 CAR-expressing primary human CD4+ T cells to produce immunostimulatory cytokines (Supplementary Fig. 1b). Interestingly, non-reducing western blot of CD19ecto showed that the soluble ligand existed in both monomeric and oligomeric forms (Supplementary Fig. 1c). This observation, combined with the previously noted knowledge that crosslinking antibodies (which are bivalent in nature) can stimulate CAR-T–cell activation, points to the possibility that a dimeric form of soluble ligand may be necessary for triggering CAR signaling.
Soluble-ligand–mediated dimerization activates GFP CARs
To determine the role of the multimericity of a soluble ligand in triggering CAR activation, we generated a green fluorescent protein (GFP)-binding CAR model system and compared two soluble ligands: monomeric superfolder GFP (sfGFP) versus homodimeric sfGFP linked by a disulfide bond introduced by a D117C substitution (Fig. 1a)17. The monomeric and dimeric states of the sfGFP ligands were verified by size-exclusion chromatography (Supplementary Fig. 2a).
Figure 1:
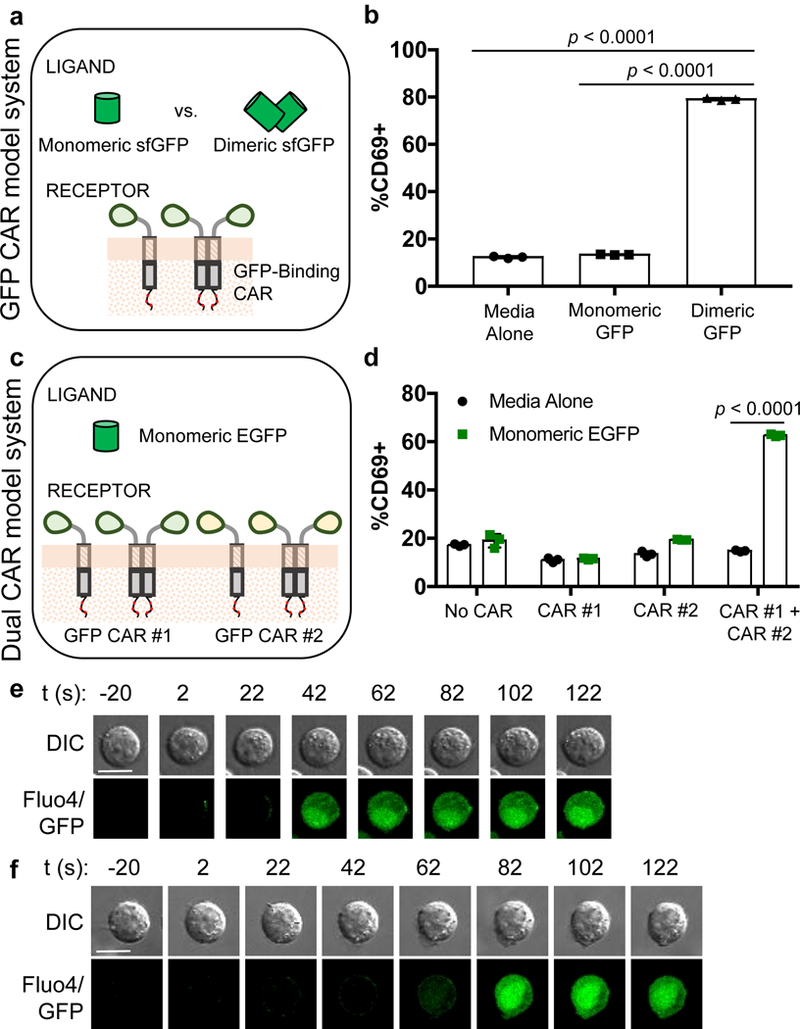
GFP-binding CARs can respond to soluble GFP ligands that are capable of dimerizing CARs. (a) The GFP CAR model system consists of a GFP-binding CAR and two ligands—a monomeric superfolder GFP (sfGFP) and a homodimeric sfGFP that is covalently dimerized by a cysteine introduced at position 117. (b) GFP CAR-expressing Jurkat cells upregulate CD69 in response to dimeric sfGFP. (c) Two GFP CARs have distinct GFP-binding domains that can simultaneously bind a monomeric EGFP molecule at two different epitopes. (d) Only Jurkat cells that co-express both GFP CARs upregulate CD69 in response to EGFP. (e) Dimeric sfGFP triggers Ca2+ flux in GFP CAR-Jurkat cells in a cell-contact–independent manner. (f) Monomeric EGFP triggers Ca2+ flux in dual-CAR Jurkats in a cell-contact–independent manner. Scale bars denote 10 μm in (e) and (f). Images representative of ≥5 cells across (e) 4 and (f) 2 independent cell cultures are shown. In all graphs, data points from n = 3 biologically independent cell cultures are plotted with means ± 1 standard deviation (SD). Statistical comparisons performed with the two-tailed Student’s t test and the Sidak correction for multiple comparisons.
We found that GFP-CAR–expressing Jurkat cells were activated by dimeric sfGFP, but not by monomeric sfGFP, suggesting ligand-mediated receptor dimerization as a mechanism of CAR triggering (Fig. 1b). Notably, the GFP CAR itself, which utilizes the “enhancer” GFP-binding nanobody18 as its ligand-binding domain and the hinge domain of human IgG4 as its extracellular spacer, was observed to exist as a mix of monomers and dimers in the absence of ligand (Supplementary Fig. 2b,c). The fact that the dimeric GFP CARs do not signal in the absence of dimeric GFP ligand indicates that CAR pre-dimerization in the absence of ligand binding does not trigger signaling. Taken together, these results suggest that the act of bringing together receptors at the cell plasma membrane to form a dimer can cause signaling, even though the formation of receptor dimers intracellularly (such as in the endoplasmic reticulum) prior to trafficking to the cell surface does not induce receptor activation.
To confirm the hypothesis that ligand-mediated receptor dimerization (as opposed to the intrinsic dimerism of the ligand itself) is critical to CAR triggering, we tested whether monomeric GFP could activate Jurkat cells that co-express two different GFP-binding CARs, which could simultaneously bind a single GFP monomer at separate epitopes. In this context, monomeric GFP would serve as a dimerizing ligand to bring together GFP CAR #1 (with the “enhancer” nanobody) and GFP CAR #2 (with the “GBP6” GFP-binding nanobody19; Fig. 1c and Supplementary Figs 2c-e). To accommodate the GBP6 nanobody, enhanced GFP (EGFP) was used as the soluble ligand. We found that monomeric EGFP activated dual-CAR Jurkat cells but not Jurkat cells expressing either of the GFP-binding CARs individually (Fig. 1d). These results support the conclusion that ligand-induced dimerization can trigger CAR signaling, and that CAR-T cells can respond to monomeric, soluble ligands as long as two appropriate ligand-binding domains can be identified to simultaneously bind different epitopes on the ligand.
Soluble ligands such as bispecific T-cell engagers (BiTEs) and fusion proteins that trigger T-cell/target-cell contact can induce T-cell activation20–22. This cell-cell contact mechanism could also explain the ability to trigger CAR-T cells with CAR-binding antibodies, which are bivalent in nature. Similarly, the GFP molecules used in the experiments above could conceivably bridge GFP CARs on separate cells in trans, thereby forming an immune synapse-like interface and activating T cells in a cell-contact–dependent manner. It was therefore unclear whether soluble ligands can trigger CAR signaling in a truly soluble manner, without forming cell-cell interfaces. To investigate this, we observed GFP-CAR and dual-CAR Jurkat cells under the microscope and found that, upon addition of dimerizing ligand, Ca2+ influx could occur in cells that never came into contact with another cell (Fig. 1e,f). These results confirm that cell-contact–independent activation does occur and that a dimerizing ligand can activate CARs by binding them in cis.
A TGF-β–binding CAR rewires the T-cell response to TGF-β
Next, we tested whether CARs can be engineered to respond to TGF-β, a soluble, homodimeric cytokine known to be a potent, overproduced immunosuppressive factor in a variety of solid tumors23,24. We generated three scFvs based on the sequences of three TGF-β–neutralizing antibodies that bind the active form of human TGF-β (US7151169B2, US008012482B2, US20140127230A1). Each scFv was confirmed to block TGF-β–induced SMAD2 phosphorylation in HepG2 cells (Supplementary Fig. 3a), and the most potent scFv was incorporated into a second-generation CAR containing a CD28 costimulatory domain (Fig. 2a).
Figure 2:
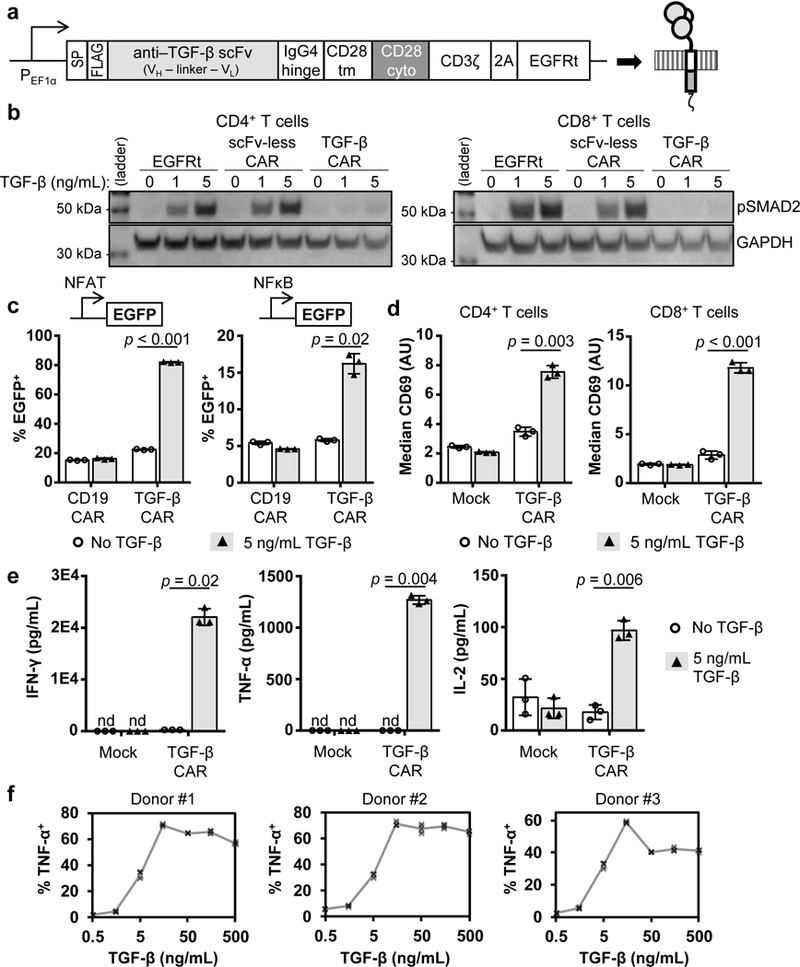
A TGF-β–binding CAR converts soluble TGF-β into a T-cell stimulatory molecule. (a) The TGF-β CAR contains an extracellular TGF-β–binding scFv linked to the CD28 and CD3ζ endodomains. SP, signal peptide; FLAG, DYKDDDDK epitope; VH, heavy-chain variable domain; VL, light-chain variable domain; tm, transmembrane; cyto, cytosolic; 2A, 2A “self-cleaving” peptide; EGFRt, truncated epidermal growth factor receptor. (b) TGF-β was added to CD4+ and CD8+ T cells expressing no CAR (EGFRt only), an scFv-less CAR, or the TGF-β CAR. A western blot for phosphorylated SMAD2 (pSMAD2) shows that the TGF-β CAR inhibits native TGF-β signaling (full blot in Supplementary Fig. 3d). (c) TGF-β was added to parental or CAR-engineered Jurkat cells carrying an EGFP reporter driven by an NFAT- or NFκB-responsive promoter. (d) CD69 surface staining of primary human CD4+ and CD8+ T cells expressing the TGF-β CAR, incubated with or without 5 ng/mL TGF-β. (e) A cytometric bead-array assay quantifies TGF-β CAR-T cell secretion of interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and interleukin (IL)-2 in response to TGF-β. (f) TGF-β dose-dependent production of TNF-α by TGF-β CAR-T cells, as quantified by intracellular staining of cell lines from three different healthy blood donors. In all graphs, data points from n = 3 biologically independent cell cultures are shown with means ± 1 SD. Statistical comparisons performed with the two-tailed Student’s t test and Sidak correction. n.d., not detected.
In primary human T cells, the TGF-β CAR was found to exist as a mix of monomers and dimers (Supplementary Fig. 3b), present to the cell surface (Supplementary Fig. 3c), and strongly inhibit endogenous TGF-β signaling (Fig. 2b). Utilizing fluorescent genetic reporters, we observed that TGF-β CAR Jurkat cells upregulated both NFAT and NFκB signaling pathways in the presence of soluble TGF-β (Fig. 2c), demonstrating that the TGF-β CAR can redirect the T-cell response away from endogenous TGF-β signaling and toward immunostimulatory pathways.
In both CD4+ and CD8+ primary human T cells, the TGF-β CAR triggered upregulation of CD69 in response to TGF-β (Fig. 2d). Furthermore, TGF-β CAR-T cells produced Th1 cytokines in response to TGF-β (Fig. 2e and Supplementary Fig. 4a), exhibiting a monotonic increase in cytokine production in response to 1.5 – 15 ng/mL TGF-β before reaching maximal cytokine output (Fig. 2f). Consistent dose-response curves were observed in cells obtained from multiple healthy donors (Fig. 2f), demonstrating that the TGF-β CAR behaves in a predictable manner across different genetic backgrounds and logarithmically distinguishes among TGF-β concentrations in a physiological range.
TGF-β CAR converts TGF-β into a growth-promoting cytokine
TGF-β has been reported to aid tumor immune escape by limiting T-cell proliferation25, and untransduced CD4+ and CD8+ primary T cells indeed showed a marked decrease in ex vivo expansion in the presence of soluble TGF-β, despite the support of irradiated feeder cells, interleukin (IL)-2, and IL-15 (Fig. 3a and Supplementary Fig. 4b). Similarly, T cells expressing an scFv-less CAR (which contains the same transmembrane and cytoplasmic components as the TGF-β CAR but lacks the TGF-β–binding scFv domain) exhibited a significant growth defect in response to TGF-β, indicating that simply overexpressing CD28 and CD3ζ domains could not confer resistance to TGF-β–mediated growth inhibition (Fig. 3b). In contrast, expression of the TGF-β CAR increased fold-expansion by an order of magnitude compared to the no-TGF-β control across 3 weeks of monitoring (Fig. 3b). Consistent proliferation results were observed in CD4+ T cells from multiple donors (Fig. 3c) as well as CD8+ primary human T cells (Supplementary Fig. 4c). Additionally, the growth response of TGF-β CAR-T cells increased monotonically with increasing TGF-β concentrations (Fig. 3d). Taken together, these results demonstrate that the TGF-β CAR can effectively convert TGF-β from an inhibitor to a stimulant of T-cell growth.
Figure 3:
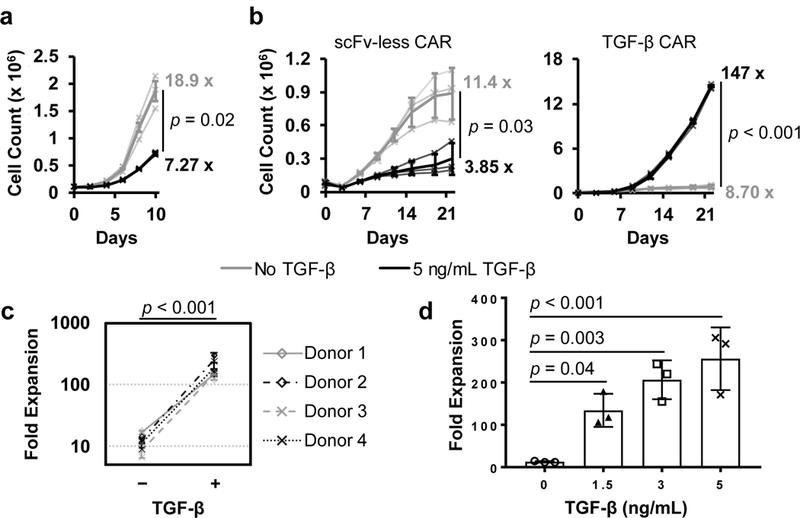
The TGF-β CAR converts TGF-β from a T-cell growth suppressant to a T-cell growth stimulant. Primary human CD4+ T cells were cultured in the presence of irradiated TM-LCL feeder cells plus IL-2, IL-15, and either no TGF-β or 5 ng/mL TGF-β supplemented every two days. (a) Expansion of unmodified CD4+ T cells. (b) Expansion of CD4+ T cells expressing an scFv-less CAR or TGF-β CAR. Numbers at the end of each trace denote fold-expansion from the initial to the final time-points, and the final time-point values are compared by the two-tailed Student’s t test. (c) Fold expansion of TGF-β CAR-T cells after 22 days. The addition of TGF-β resulted in a significant increase in expansion as determined by one-way, repeated-measure ANOVA (F = 173.2, df = 1). (d) Fold expansion of TGF-β CAR-T cells supplemented with varying levels of TGF-β across 22 days, with statistical comparison using Dunnett’s test for multiple comparisons. In all graphs, data points from n = 3 biologically independent cell cultures are shown with means ± 1 SD.
TGF-β CAR signaling does not cause bystander-cell lysis
In addition to cytokine production and proliferation, target-cell lysis is a major effector function associated with productive T-cell activation. However, TGF-β CAR-T cells do not form synapses when stimulated by soluble TGF-β, so it is unclear whether TGF-β–triggered TGF-β CAR-T cells may be activated to non-specifically attack off-target bystander cells. To examine this possibility, we co-incubated CD8+ TGF-β CAR-T cells with TM-LCL cells that do not express any cognate antigen and observed minimal lysis regardless of the presence of TGF-β (Supplementary Fig. 5a and Supplementary Data Set). In contrast, TGF-β CAR-T cells effectively lysed OKT3+ TM-LCL cells, which express a surface-bound version of the anti-CD3 antibody OKT3 that serves to stimulate the endogenous TCR on all T cells (Supplementary Fig. 5a and Supplementary Data Set). This cytotoxicity profile is indistinguishable from that of T cells bearing an scFv-less CAR (Supplementary Fig. 5b and Supplementary Data Set). Taken together, these results indicate that TGF-β CAR-T cells do not kill antigen-negative bystander cells even in TGF-β–rich environments, but they remain capable of forming proper immunological synapses with and killing target cells that present cognate membrane-bound antigens.
TGF-β can activate TGF-β CARs via dimerization in cis
Given the potential utility of CARs that respond to tumor-associated cytokines such as TGF-β, we next sought to establish mechanistic details underlying TGF-β CAR activation in order to understand its potential generalizability to other soluble antigens. First, we evaluated whether the TGF-β CAR supports the design principle that ligand-mediated receptor dimerization triggers CAR activation, as suggested by the aforementioned GFP CAR results.
The TGF-β CAR can exist in three distinct states in the presence of TGF-β: unbound, singly bound to TGF-β, or dimerized by TGF-β. Using biochemical modeling, we predicted the dose-response activation curves when either or both of the TGF-β–bound states were considered to be active (Fig. 4a, see Online Methods for modeling details). When either all ligand-bound receptors or only singly bound receptors were designated signaling-competent, the activation curve shape was monotonic and saturating (Fig. 4ai,ii). In contrast, if only ligand-dimerized receptors were designated signaling-competent, the activation curve reached a peak before dropping at high TGF-β levels (Fig. 4aiii). Using TGF-β CAR Jurkat cells, we empirically evaluated the dose-response behavior of NFAT activation (a signaling event just downstream of CAR signaling), and observed a dose-response curve consistent with the model where only ligand-dimerized receptors are in the active state (Fig. 4b). Notably, the dose-response behavior of a further downstream event, TNF-α production, also demonstrated a similarly shaped curve (Fig. 2f), supporting that the TGF-β–mediated receptor dimerization is critical for CAR activation.
Figure 4:
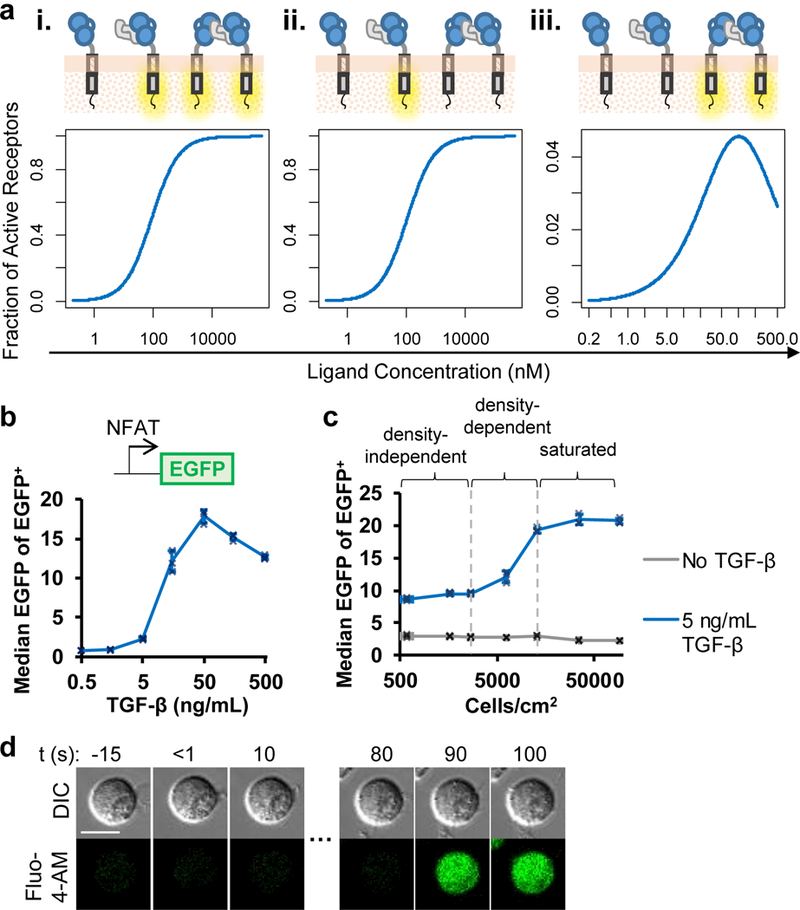
TGF-β–mediated CAR dimerization in cis can activate the TGF-β CAR. (a) Biochemical modeling predicts different dose-response behaviors for receptor activation depending on which receptor states are considered active: (i) all ligand-bound receptors, (ii) only single-receptor/ligand complexes, (iii) only ligand-dimerized receptors. See Online Methods for model parameters. (b) TGF-β dose response of an EGFP NFAT signaling reporter in a TGF-β CAR Jurkat cell line. (c) TGF-β CAR Jurkat cells expressing the NFAT-responsive EGFP reporter were seeded at varying cell densities (Supplementary Fig. 6c) in the presence or absence of soluble TGF-β. (d) TGF-β CAR Jurkat cells loaded with the Fluo-4-AM Ca2+ indicator were imaged every 10 s. After TGF-β addition at t = 0 s to the edge of the cell culture well, individual cells demonstrated Ca2+ influx in the absence of any cell-to-cell contact. All images are representative of ≥5 cells across 3 independent cell cultures. Scale bar denotes 10 μm. In all graphs, data points from n = 3 biologically independent cell cultures are shown with means ± 1 SD.
In principle, each TGF-β dimer can bind two TGF-β CARs on adjacent cells (in trans) (Supplementary Fig. 6a, top) or the same cell (in cis) (Supplementary Fig. 6a, middle) to trigger T-cell activation. Distinguishing between these specific signaling mechanisms could significantly impact the behavior of TGF-β CAR-T cells, since trans interactions would render CAR signaling strongly dependent on cell density—i.e., the probability of cell-cell contact and receptor signaling would increase with increasing cell concentration (Supplementary Fig. 6b, top). In contrast, receptor signaling should be largely independent of cell density if cis interactions were the dominant mechanism, provided that the soluble ligand concentration is not limiting relative to cell consumption rates (Supplementary Fig. 6b, middle).
Quantification of TGF-β–mediated NFAT reporter output in TGF-β CAR Jurkat cells seeded at varying cell densities revealed that both cell-density–dependent and cell-density–independent mechanisms are involved in triggering cell activation, demonstrating that contact-independent signaling can occur and that TGF-β–induced CAR signaling occurs even at low cell densities (Fig. 4c and Supplementary Fig. 6c). Fluorescence microscopy of Ca2+ flux in TGF-β CAR cells further confirmed that soluble TGF-β could indeed trigger T-cell activation in the absence of cell-cell contact (Fig. 4d). These results indicate that the TGF-β CAR-T cells can both respond to freely soluble TGF-β and be activated at a wide range of cell densities, with high local cell concentration serving as a means to boost CAR-signaling intensity. Multiple potential mechanisms may contribute to increased signaling at high cell densities, including cell-cell contacts mediated by CARs dimerizing in trans, as well as cell-density–dependent changes in the levels of extracellular proteins or small molecules that impact NFAT signaling.
TGF-β triggers actin-dependent TGF-β CAR clustering
To further understand how soluble TGF-β can trigger TGF-β CAR activation, we visualized the spatial distribution of TGF-β CARs in Jurkat cells and observed the formation of receptor clusters in response to TGF-β (Fig. 5a). These clusters, ~0.5–1 μm in diameter, were distributed throughout the cell periphery, reflecting a lack of polarity in the soluble TGF-β stimulus. Ca2+ influx was observable prior to complete CAR redistribution into clusters (Fig. 5a), indicating that widespread receptor clustering is not necessary for initial signal transduction. Furthermore, we observed that ZAP70 aggregated at TGF-β CAR clusters (Fig. 5b), suggesting that TGF-β CAR clusters serve as sites of signal transduction and may support sustained and productive signaling.
Figure 5:
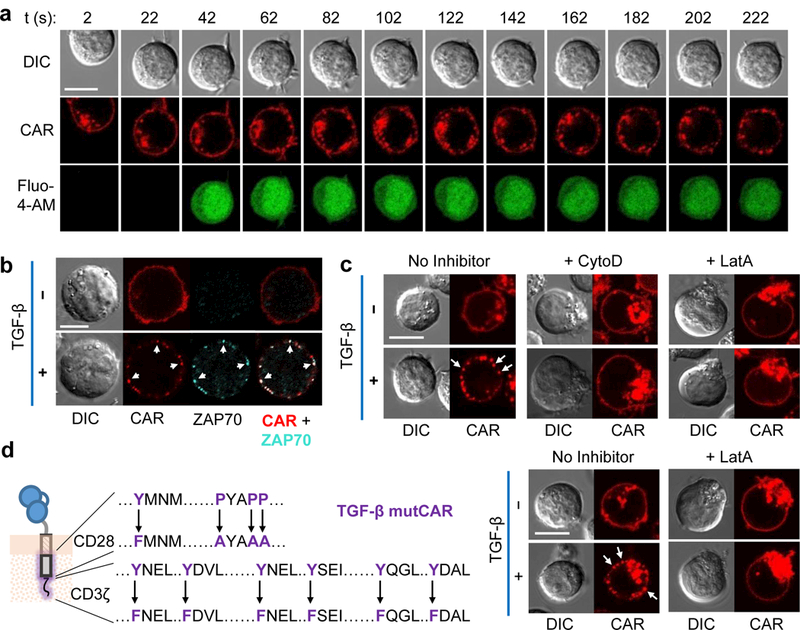
TGF-β CARs form actin-dependent clusters. (a) Time-lapse confocal microscopy of Jurkats loaded with the Fluo-4-AM Ca2+ indicator and expressing a TGF-β CAR-mCherry fusion. TGF-β was added at t = 0 s to the corner of the well and cells were followed at 20 s intervals. (b) TGF-β CAR-mCherry Jurkat cells were incubated with or without TGF-β for 15 min, fixed, and intracellularly stained for ZAP70. White arrows indicate sites where CAR and ZAP70 clusters spatially overlap. (c) TGF-β CAR-mCherry Jurkat cells were imaged 15 min after the addition of TGF-β and/or the actin-polymerization inhibitors cytochalasin D (CytoD) or latrunculin A (LatA). (d) Microscopy as in (c), but with Jurkat cells expressing the non-signaling TGF-β mutant CAR (mutCAR) with the indicated mutations. Images shown are representative of ≥5 cells across 2 independent cell cultures for the CytoD case in (c) and across 3 independent cell cultures for all other images. All scale bars denote 10 μm.
We next examined whether ligand-induced cluster formation was due to the multimerization of extracellular receptor domains. Western-blot analysis indicated TGF-β CARs exist as a mix of monomers and dimers (Supplementary Fig. 3b). The presence of two inter-chain disulfide bonds in the IgG4 hinge domain that serves as an extracellular spacer in the receptor is a main contributor of receptor dimerization, and the interaction between dimeric CARs and homodimeric TGF-β could conceivably result in a polymerization effect that allows a soluble homodimer such as TGF-β to cause receptor clustering (Supplementary Fig. 7a). C→A mutations that prevented covalent dimerization of the TGF-β CAR did not impact TGF-β–mediated Ca2+ influx and CAR clustering (Supplementary Fig. 7b-d), indicating that the disulfide-mediated pre-dimerization of CAR molecules is not essential to cluster formation or receptor signaling.
We next explored whether intracellular events may mediate TGF-β CAR clustering, with a focus on actin filaments given their role in TCR microcluster formation26,27. The actin-polymerization inhibitors cytochalasin D and latrunculin A both prevented TGF-β CAR clustering, suggesting that a functional actin cytoskeletal network helps orchestrate CAR clustering (Fig. 5c). Notably, mutated signaling-deficient TGF-β CARs, with inactivated CD28 and CD3ζ domains, still formed clusters in an actin-dependent manner (Fig. 5d). Taken together, these results suggest that TGF-β binding induces TGF-β CAR conformational changes that either directly or indirectly alter the receptor’s interaction with actin filaments, leading to receptor clustering that is independent of canonical CD28 and CD3ζ signaling.
Receptor structure tunes CAR response to soluble antigens
The observations that (1) ligand-mediated receptor dimerization triggers CAR signaling, (2) receptor pre-dimerization in the absence of ligand is not sufficient for CAR signaling, (3) ligand binding results in active, actin-dependent changes in CAR distribution, and (4) the resulting CAR clusters strongly co-localize with ZAP70 suggest that the dynamic process of receptor dimerization may be important to both initial and sustained CAR signaling. As TGF-β–mediated receptor dimerization necessarily exerts tensile force on the TGF-β CARs that are brought together, we hypothesized that this mechanical force may play a role in CAR activation, reminiscent of studies demonstrating force-actuated TCR signaling2,28–30 (Fig. 6a). Such a force-transmission model suggests that CAR signaling behavior could potentially be calibrated by modifying the mechanical coupling between the ligand-binding and signaling domains.
Figure 6:
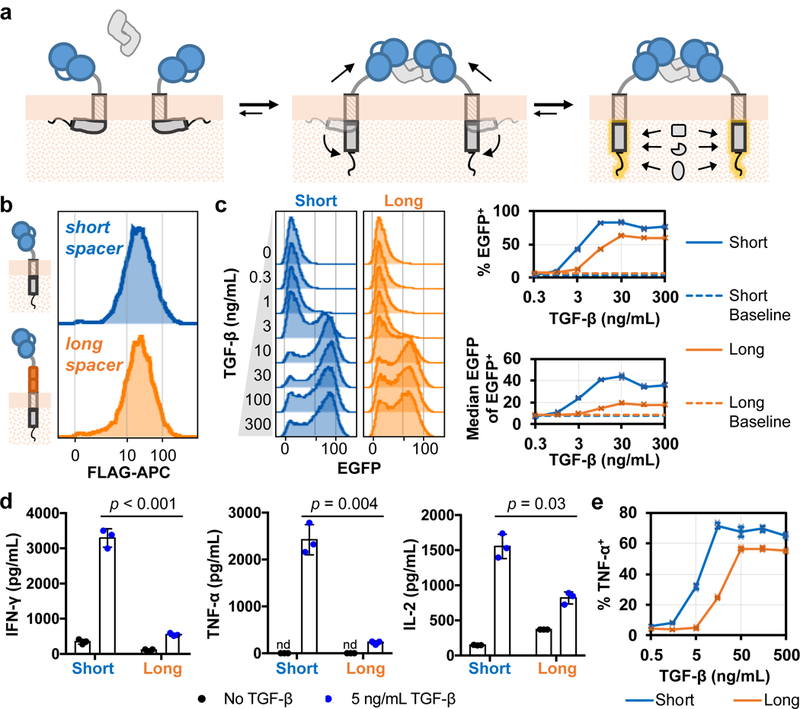
A mechanotransduction model for TGF-β CAR function and functional tuning. (a) The dynamic process of TGF-β–mediated TGF-β CAR dimerization can exert mechanical forces onto the intracellular signaling domains to elicit a shift towards the signaling-competent conformation. (b) The short and long TGF-β CAR extracellular spacers are the 12-aa IgG4 hinge and the 229-aa IgG4 hinge-CH2-CH3, respectively. T cells expressing the indicated receptor were surface-stained for their N-terminal FLAG tag and assessed for surface-expression by flow cytometry. Data are representative of biological triplicates. (c) Jurkat cells expressing the NFAT-responsive EGFP reporter were transduced with either short or long TGF-β CARs and exposed to varying TGF-β concentrations. Plots summarizing the frequency of NFAT triggering (% EGFP+) and extent of NFAT activation among triggered cells (median EGFP intensity of EGFP+ cells) in n = 3 biologically independent cell cultures are shown. (d-e) Short and long TGF-β CARs were expressed in primary CD4+ T cells and characterized in the presence or absence of TGF-β. (d) A cytometric bead-based assay was used to quantified Th1 cytokine production by short and long TGF-β CAR-T cells. Statistical comparisons performed with the two-tailed Student’s t test and the Sidak correction. (e) Intracellular staining for TNF-α production in response to varying TGF-β doses in short and long TGF-β CAR-T cells. In all graphs, data points from n = 3 biologically independent cell cultures are shown with means ± 1 SD.
Indeed, we found that lengthening the TGF-β CAR’s extracellular spacer affected signaling threshold and intensity (Fig. 6b-f). We replaced the 12-aa (IgG4 hinge) extracellular spacer in the original TGF-β CAR with a 229-aa (IgG4 hinge-CH2-CH3) spacer to loosen the coupling between the ligand-binding domain and the intracellular signaling domains. The increased spacer length did not significantly impact receptor surface-expression levels (Fig. 6b). However, compared to the original (short) TGF-β CAR, the long CAR required a substantially higher TGF-β input concentration before triggering NFAT signaling, and showed a lower maximal signaling intensity (Fig. 6c). Furthermore, primary T cells expressing the long TGF-β CAR produced significantly less Th1 cytokines and showed an upward shift in the TGF-β dose-response threshold that mirrored the NFAT response in Jurkat cells (Fig. 6d,e). These results highlight a means to tune receptor signaling intensity by rationally adjusting CAR structural properties.
Discussion
To our knowledge, this work provides the first demonstration of CARs that are specifically engineered to respond to freely soluble ligands. In this study, we showed that CARs can be employed to stimulate T-cell effector functions in response to a variety of soluble ligands, including CD19ecto, GFP, and TGF-β, an otherwise potent immunosuppressive molecule. Our results demonstrated that the dynamic process of receptor dimerization and the mechanical coupling between the ligand-binding and signaling domains of the CAR are both important for signal transduction, supporting a role for mechanical forces in actuating CAR activation. This view suggests a few features that may be important for CARs to be triggered by a soluble ligand. First, the soluble ligand must be able to bring together two or more CARs. Second, the CAR-ligand binding affinity must be strong enough to withstand the tensile force necessary for CAR activation. Third, both the soluble ligand and the CARs involved must be mechanically rigid enough to transmit tensile force to the CARs’ intracellular signaling domains. The first feature is consistent with past literature reporting that shed CD30 and mesothelin ligands, both thought to be monomeric31,32, cannot trigger CAR activation6,7; the latter two requirements may explain why shed, multimeric CEA, Lewis Y antigen, and Fc-fused CEA and CD30 were not observed to activate their cognate CARs6,8–10. Atomic force microscopy (AFM) has been used fruitfully to study the relationship between ligand-binding affinity and unbinding force33, as well as forces involved in TCR signaling and T-cell activation34. Thus, AFM may be useful in future quantitative studies elucidating the force required for CAR activation by soluble ligands.
Viewing the CAR as a mechanotransducing molecule is consistent with the known biology of the CAR’s individual signaling domains. In their native contexts, the intracellular signaling domains of CD28 and CD3ζ receptor both associate with the inner leaflet of the plasma membrane, and receptor ligation and activation (by immobilized ligand) coincide with the signaling domains’ dissociation from the membrane35,36. Although the detailed biophysical mechanisms underlying CD28 and CD3ζ conformational changes are unclear, mechanical tugging on the CD28 and CD3ζ receptors by immobilized ligand has been suggested to initiate receptor-tail conformational changes2,37. Our results suggest that the mechanical forces exerted by soluble-ligand–mediated CAR dimerization are sufficient to yield any CAR conformational changes required for CAR activation.
CD3ζ, CD28, and CARs themselves have been observed to form microclusters in T cells that are stimulated by immobilized ligands5,26,38. Our results demonstrate that a soluble ligand, with the help of actin polymerization, can also orchestrate CAR cluster formation and that CAR clusters strongly co-localize with ZAP70, suggesting a role in sustained CAR signaling. Furthermore, CAR clustering can occur independently of canonical CD28 and CD3ζ signal-transducing residues, indicating that cluster formation facilitates but does not depend on canonical CD28 and CD3ζ signaling. Further work is needed to elucidate additional molecular details that occur with CAR-T cell activation. Nevertheless, the observations that ligand-mediated receptor dimerization is required for CAR triggering, and that structural changes designed to loosen the physical connection between extracellular and intracellular receptor domains reduced signaling intensity, strongly support mechanotransduction as a model to guide CAR design.
Our data do not exclude the possibility that conformational changes via allosteric effects are important in CAR activation. Nevertheless, the mechanotransduction model of CAR activation is appealing in its consistency with the adaptability of the CAR structure to diverse ligand-binding domains: as long as the synthetic receptor maintains a reasonable mechanical stiffness, it can function despite wide variations in the extracellular domain’s specific affinity or shape. Indeed, a mechanotransductive mode of signaling appears to be particularly amenable to the modular modifications desired in synthetic biology, and may explain the CAR molecule’s successful track record as a synthetic biology tool39,40. Given the additional success of designing synthetic functions by engineering Notch receptors41–43, which also rely on mechanical forces for signal transduction, identifying and co-opting mechanotransductive protein architectures may prove to be a viable strategy to further expand the synthetic biology toolbox.
Our finding that synthetic receptors can be engineered to respond to soluble molecules via a dimerization-induced mechanotransduction mechanism complements current efforts to construct synthetic responses to soluble ligands using dimerization-induced proteolysis44. Together, our work highlights the opportunity in exploiting soluble factors, such as cytokines and chemokines, that play chief roles in cell-cell communication and serve as a rich source of local and systemic disease biomarkers45,46. For example, the TGF-β CAR described here effectively converts a potent immunosuppressive cytokine associated with solid-tumor microenvironments into a robust T-cell stimulant. Future work is needed to explore the therapeutic use of synthetic immunoreceptors in rewiring immune cell responses to this rich source of disease-microenvironment descriptors. Additional incorporation of logic-gate synthetic immunoreceptor architectures47–50 in targeting soluble factors could further increase the specificity and efficacy of engineered immune cell therapies for a broad range of diseases.
Online Methods
DNA constructs.
DNA encoding the GFP-binding nanobody in GFP CAR #1 was synthesized (Integrated DNA Technologies) based on a previously reported amino acid sequence18, and a second GFP-binding nanobody used in GFP CAR #2 was taken from pCAG-p65AD-GBP6, a gift from Dr. Connie Cepko (Addgene plasmid #49440). Anti-TGF-β scFvs were constructed by identifying the variable regions in the heavy and light chains (VH and VL, respectively) of TGF-β–binding antibodies and synthesizing gene fragments with the VH-(G4S)3-VL architecture (Integrated DNA Technologies). The CD19-binding scFv was constructed from the FMC63 antibody. GFP CAR, TGF-β CAR, and CD19 CAR plasmids were constructed by isothermal assembly using gene fragments encoding GFP-binding nanobodies, TGF-β–binding scFvs, or the CD19-binding scFv; IgG4 hinge, IgG4 hinge with C→A substitutions, IgG4 hinge-CH3, or IgG4 hinge-CH2-CH3 with L235E and N297Q substitutions to prevent binding to Fc receptors encountered in vivo51; the CD28 transmembrane domain; the CD28 cytosolic tail with GG mutations that enhance CAR surface expression52, with or without additional P208/211/212A and Y191F substitutions that prevent CD28 signal transduction53; and the CD3ζ cytosolic domain or corresponding version with Y→F substitutions in all three ITAM motifs to prevent CD3ζ signaling via ZAP70 binding54. The scFv-less CAR was similarly constructed, except with no N-terminal scFv domain. CAR constructs are directly fused to mCherry or linked by the T2A peptide to a truncated epidermal growth factor receptor (EGFRt), a transduction marker that also facilitates sorting of CAR-expressing cells. The CD19 ectodomain was amplified from a plasmid encoding full-length CD19, a gift from Dr. Donald B. Kohn (University of California, Los Angeles).
Cell lines.
Jurkat Clone E6–1 cells were obtained from ATCC (TIB-152). The EGFP NFAT reporter Jurkat cell line was a gift from Dr. Arthur Weiss (University of California, San Francisco). The EGFP NFκB reporter Jurkat cell line was a gift from Dr. Xin Lin (MD Anderson). The TM-LCL cell line, an Epstein-Barr virus (EBV)-transformed lymphoblastoid cell line, was a gift from Dr. Michael Jensen (Seattle Children’s Research Institute). The OKT3+ TM-LCL line was a gift from Dr. Stephen Forman (City of Hope National Medical Center). The above cell lines were maintained in complete RPMI (RPMI1640 (Lonza) + 10% heat-inactivated FBS (HI-FBS, Gibco)). HEK293T cells (ATCC; CRL-11268) were cultured in DMEM (HyClone) + 10% HI-FBS. Hep G2 cells (ATCC; HB-8065) were cultured in EMEM (Lonza) + 10% HI-FBS. CD19 CAR, GFP CAR, and TGF-β CAR Jurkat cell lines were generated with lentiviral transduction followed by fluorescence-activated sorting (FACS) using the FACSAria (II) at the UCLA Flow Cytometry Core Facility. Lentivirus was produced using HEK293T cells as previously described50.
Generation of CAR-expressing primary human T cells.
CD4+, CD8+, or mixed CD4+ and CD8+ T cells were isolated with the RosetteSep CD4+ or CD8+ Human T-cell Enrichment Cocktail (Stemcell Technologies) from healthy donor whole blood obtained from the UCLA Blood and Platelet Center. T-cells stimulated with CD3/CD28 Dynabeads (Thermo Fisher Scientific) at a 1:1 cell:bead ratio for 2 days were transduced with lentivirus at a multiplicity of infection of 1.5–3. T cells were cultured in complete RPMI and fed 50 U/ml IL-2 (Thermo Fisher Scientific) and 1 ng/ml IL-15 (Miltenyi Biotec) every 2 to 3 days. Dynabeads were removed after 9 days of culture. Transduced cells were enriched by magnetic bead-based sorting (Miltenyi Biotec) and continued to be cultured with IL-2/IL-15 supplementation every 2 to 3 days.
Production of anti-TGF-β scFvs, CD19ecto, and GFP ligands.
To produce solutions containing the anti-TGF-β scFvs or CD19ecto, HEK293T cells were transfected with plasmids encoding the desired protein preceded by the murine IgG κ light chain signal peptide (for the TGF-β scFvs) or its native signal peptide (CD19ecto). Sixteen hours post-transfection, cells were washed with 1X phosphate buffered saline (PBS) and replaced with fresh DMEM culture media (without serum). For the anti-TGF-β scFvs, supernatant was harvested 20 hours after media change; solutions were concentrated and washed with PBS using 10 kDa cutoff Amicon Ultra-15 centrifugal units (EMD Millipore). For CD19ecto, supernatant was harvested at 24 hours and 48 hours after media change and pooled; solutions were concentrated and washed with serum-free RPMI using 10 kDa cutoff Amicon Ultra-15 centrifugal units.
Monomeric superfolder GFP and covalently-linked dimeric superfolder GFP (D117C) ligands were a gift from Todd Yeates (University of California, Los Angeles) and previously described17. EGFP was expressed from the pET-28α vector with an N-terminal cleavable His6 tag in BL21-DE3 Escherichia coli. Cultures were grown until an OD600 of 0.6, then induced with 1 mM IPTG at room temperature overnight. Cells were harvested at 5000 x g for 30 min at 4°C, then stored at -80°C until purification. Protein purification was performed as previously described, using Ni-nitrilotriacetic acid agarose beads to bind His6-tagged proteins, then tobacco etch virus protease to remove the tag17. Proteins were subsequently analyzed at the UCLA Protein Expression Technology Center with an EnRich 650 sec size-exclusion column (Bio-Rad) on a Bio-Rad DuoFlow system with an injection volume of 350 μL and a flow rate of 0.75 mL/min. Areas under the size-exclusion chromatography traces were calculated numerically using 0.2 s steps.
Western blots.
For CAR expression analysis, harvested cells were treated with 1 mg/mL tunicamycin (MP Biomedicals) overnight. For assessment of TGF-β–induced SMAD phosphorylation, cells were treated with indicated levels of human TGF-β1 (referred to as TGF-β throughout manuscript, PeproTech) for 30 min at 37°C. Two million cells were lysed in 35 μL of lysis buffer containing RIPA buffer (1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate, with protease and phosphatase inhibitor) supplemented with 0.5 mM phenylmethanesulfonylfluoride (PMSF) for 45 min on ice. Centrifuged cell lysate or concentrated cell culture supernatant was collected and mixed with Bolt LDS sample buffer (Thermo Fisher Scientific) containing 5% β-mercaptoethanol (Sigma-Aldrich) and heated at 70°C for 10 min. The SuperSignal Molecular Weight Protein Ladder (Thermo Fisher Scientific) was loaded with samples to assess protein sizes. Protein separation was performed by SDS-PAGE with pre-cast 4–12% Bis-Tris gels (Thermo Fisher Scientific). Gels were transferred to nitrocellulose membranes using the iBlot gel transfer system (Invitrogen). Membranes were either blocked with 1% bovine serum albumin in TBS-T (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) for 1 hour and then probed with antibodies using the SNAP i.d. 2.0 Protein Detection system (Millipore), or blocked with 5% bovine serum albumin in TBS-T for 1 hour and then probed with antibodies overnight. Primary antibodies for CD3ζ (#551034, BD Biosciences), phosphorylated SMAD2/3 (clone D27F4, Cell Signaling Technology), CD19 (#AP1494a, Abgent), and HRP-conjugated GAPDH (clone GAPDH 71.1, Sigma-Aldrich), as well as HRP-conjugated anti-mouse or anti-rabbit IgG (H+L) secondary antibodies (Jackson ImmunoResearch) were used. Membranes were treated with SuperSignal West Dura Extended Substrate (Thermo Fisher Scientific) and imaged using the ChemiDoc XRS+ System (Bio-Rad). Gel images were quantified using Fiji (“Fiji is just ImageJ”).
Flow cytometry and antibody staining.
Unless otherwise specified, flow cytometry in this study was performed with a MACSQuant VYB cytometer (Miltenyi Biotec). T-cells were assessed for surface presentation of epitopes using fluorescently labeled monoclonal antibodies for CD69 (clone FN50, BioLegend), or DYKDDDDK (clone L5, BioLegend; or #130–101-571, Miltenyi Biotec). EGFRt expression was measured with Erbitux (Bristol-Myers Squibb) biotinylated in-house (EZ-link Sulfo-NHS-Biotin, Pierce) followed by PE-conjugated streptavidin (Jackson ImmunoResearch). Flow data were analyzed and gated in FlowJo (TreeStar) (see Supplementary Fig. 8 for an example of gating strategy). Unless otherwise noted, data shown are drawn from biological triplicates (i.e., three distinct samples).
T-cell expansion assays.
T cells (unstained or stained with 0.5 μM CellTrace Violet (CTV; Thermo Fisher Scientific)) were seeded at 5 x 104 cells/2 mL/well in 24-well plates with 3.5 × 105 γ-irradiated (80 Gy) TM-LCLs stained with 0.2 μM CellTrace CFSE (Thermo Fisher Scientific). Cultures were supplemented every 2 days with 50 U/ml IL-2 and 1 ng/ml IL-15, with or without 5 ng/mL TGF-β. Every 3 or 4 days, wells were supplemented with 50 μL to 180 μL of complete media to compensate for evaporation, and 50 μL aliquots of resuspended cultures were removed for propidium iodide (Thermo Fisher Scientific) staining and viable T-cell quantification by flow cytometry. A note on TGF-β concentration used in this study: To our knowledge, there has been no report of typical TGF-β concentrations found in tumor tissue. Instead, published studies have relied on ELISA or receptor binding assays performed on blood plasma, with widely varying results (0.5 to 25 ng/mL in human plasma)55. It has been shown that TGF-β concentrations are significantly higher in plasma samples from cancer patients compared to healthy controls56, but the precise level of active TGF- β at tumor local environments remains unknown. Active TGF-β concentrations are expected to be higher at tumor sites than in plasma, as solid tumors are known to secrete TGF-β, and latent forms of TGF-β (produced by either tumor or other cell types) are known to be activated by tumor-associated metalloproteases57. Based on available information, we have chosen to use 5 ng/mL as the standard TGF-β input concentration for experiments described in this study because it is within the reported range of physiological TGF- β concentrations and is consistent with the conditions used by various research groups studying TGF- β signaling in T cells58,59.
Cytokine production quantification.
To assess T-cell cytokine production, T cells were incubated at 105 cells/100 μL media/well in 96-well U-bottom plates in the presence or absence of 5 ng/mL TGF-β for 24 hours. Cytokine production in culture supernatant was measured with the BD Cytometric Bead Array Human Th1/Th2 Cytokine Kit II (BD Biosciences) according to manufacturer’s instructions. Results were collected by flow cytometry and analyzed using the FCAP Array v3.0.1 software (BD Biosciences). For intracellular cytokine staining, T cells were incubated at 105 cells/100 μL media/well in 96-well plates in the presence of 5 μg/mL Brefeldin A (BioLegend) and indicated levels of TGF-β for 24 hours. Cells were fixed with 1.5% formaldehyde; permeabilized with ice-cold methanol; stained with antibody binding to TNF-α (clone Mab11), IFN-γ (clone 4S.B3), and IL-2 (clone MQ1–17H12) (all from BioLegend); and assessed by flow cytometry.
NFAT and NFκB reporter assays.
NFκB EGFP reporter Jurkat cells with indicated constructs were seeded at 2 x 104 cells/100 μL/well, with or without 5 ng/mL TGF-β, in triplicate in 96-well U-bottom plates. Reporter induction was assessed by flow cytometry after 18 hours at 37°C. NFAT EGFP reporter Jurkats with indicated constructs were seeded at 5–10 x 104 cells/100 μL/well in triplicate in 96-well plates, with indicated levels of TGF-β. Reporter induction was assessed by flow cytometry after 8–18 hours at 37°C. For the variable cell-density assay, serially diluted cell mixtures were prepared and seeded in 100 μL in 96-well flat-bottom plates, with or without 5 ng/mL TGF-β. Additional wells at each dilution were counted by flow cytometry immediately after seeding to measure the exact cell densities. After 8 hours at 37°C, reporter induction was assessed by flow cytometry. Because of low cell numbers at the lower cell density conditions, wells were combined to accumulate a minimum event count of 1500/sample to quantify reporter induction metrics. Each replicate was a combined 16 wells for seeding at 5.9 x 102, 1.6 x 103, and 2.7 x 103 cells/cm2; 8 wells for seeding at 6.2 x 103 cells/cm2; 4 wells for seeding at 1.3 x 104 cells/cm2; and 1 well for seeding at 3.5 x 104 and 9.1 x 104 cells/cm2. Brightfield snapshots of cells seeded at each density were acquired with a Zeiss Axio Observer Z1 microscope and a 10x objective.
T-cell cytotoxicity assay.
TM-LCLs were labeled with 0.2 μM CellTrace CFSE and OKT3+ TM-LCLs were labeled with 0.5 μM with CellTracker Orange CMTMR (Thermo Fisher Scientific) following manufacturer’s protocols. Unlabeled CD8+ T cells carrying the indicated construct were seeded with 5 x 103 off-target TM-LCLs and 5 x 103 on-target OKT3+ TM-LCLs at various effector-to-target (E:T) ratios in 200 μL/well in triplicate, with or without 5 ng/mL TGF-β, in 96-well U-bottom plates. After a 4-hour incubation at 37°C, viable target cells were counted by flow cytometry. Co-culture with mock-transduced T cells served as the negative control. For off-target TM-LCLs, % specific lysis = 100 x [1 – (# TM-LCLs in sample)/(# TM-LCLs in negative control)]. For on-target OKT3+ TM-LCLs, % specific lysis = 100 x [1 – (# OKT3+ TM-LCLs in sample x N)/(# TM-LCLs in negative control)], where N = (# TM-LCLs in target-only well)/(# OKT3+ TM-LCLs in target-only well) is a normalization factor to account for any intrinsic differences in viability between the TM-LCL and OKT3+ TM-LCL cell lines.
Confocal microscopy.
Scanning confocal imaging was acquired with a Nikon Ti-E equipped with C2 laser LED excitation and a Nikon Apo 60x 1.4 NA oil objective to observe cells in 50 μL volumes in 48-well glass-bottom plates. To assess Ca2+ flux, cells were loaded with 10 μM Fluo-4 AM (Thermo Fisher Scientific) in PBS for 20 min at room temperature and images were acquired with a 488-nm laser and a 525/50 emission filter. The subcellular distribution of mCherry-tagged CARs was imaged with a 561-nm laser and a 600/50 emission filter. For time-lapse experiments, 0.5 μL 5 ng/μL TGF-β was applied to the edge of the well after cells had settled, and images were acquired near the center of the well every 10 or 20 seconds. For single time-point experiments, images were acquired 15 min after applying 50 ng/mL TGF-β. When applicable, latrunculin A (EMD Millipore) or cytochalasin D (Fisher Scientific) were applied at 10 μM 15 min before image acquisition. For microscopy of ZAP70, cells were incubated with or without 50 ng/mL TGF-β for 15 min, fixed and permeabilized with 4% paraformaldehyde and 0.1% Triton X-100, stained with Alexa Fluor 488-conjugated ZAP70 antibody (clone IE7.2, Thermo Fisher Scientific), and imaged with a 488-nm laser and a 525/50 emission filter. In all cases, 16-bit images were acquired using NIS Elements 4.2 and maximum cutoffs and colorization were set using Fiji.
Biochemical modeling
Receptors were considered to be in one of three states: R, RL, and R2L, representing the unbound, singly bound, and dually bound species. We defined activation to be the number of activated receptors over the total number of receptors to arrive at three possible expressions for activation:
Two reactions were considered:
Applying the law of mass action and taking the total receptor number of receptors as generally constant (CR), we arrive at three independent relations:
With three relations and four variables, we can simplify the three activation expressions to functions of one variable, the ligand concentration:
Plots of each of these expressions are generated with R 3.3.2, and shown in Fig. 4a, where constants are set at order-of-magnitude estimates. We take CR ~0.01 nM·cm, which is equivalent to ~20 molecules/μm3. This value is derived from the estimates that (i) the number of CARs is about half the number of CD3 molecules (densitometry analysis of Supplementary Fig. 3b), (ii) there are ~104 CD3 molecules expressed60, and (iii) the radius of a Jurkat T cell is ~5 μm. We take K1 ~0.01 nM-1, a value within the range of typical binding constants for monoclonal antibodies. To estimate K2, we set the dissociation constant K2-1 to be approximately a tenth of the total concentration of receptors, CR, giving an estimate of K2 ~1000 nM-1·cm-1. Conceptually, the receptor term R can be expanded to include monomeric CARs or dimeric CARs. Of note, our model does not include the special case where two TGF-β scFvs in a dimeric CAR can bind to a single homodimeric TGF-β molecule. The two scFvs in a TGF-β CAR dimer are covalently connected via disulfide bonds in the IgG4 hinge domain, with the first disulfide bond being only 8 amino acids away from the scFv, thus providing little flexibility in scFv movement within the dimeric receptor. Furthermore, the TGF-β homodimer (25 kDa) is smaller than each bulky glycosylated scFv domain (27 kDa + glycosylation mass). As a result, steric hindrance will most likely preclude the possibility of both subunits of a pre-dimerized CAR binding to a single homodimeric TGF-β molecule.
Statistical analyses.
Statistical tests were performed in Excel (Microsoft Office) or R 3.3.2. Student’s t tests with unequal variances were used to compare continuous variables between two groups, with the Sidak correction for multiple comparisons. When pairwise comparisons were performed between a single control group and multiple treatment groups, Dunnett’s test was performed. One-way repeated measures ANOVA was performed when comparing repeated measures of means with respect to a single variable. All tests were two-tailed with a hypothesis-specific family alpha level of 0.05.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (DP5OD012133, grant to Y. Chen; F30CA183528, fellowship to Z. Chang). We thank Drs. M. Jensen (Seattle Children’s Research Institute), S. Forman (City of Hope National Medical Center), D. Kohn (University of California, Los Angeles), X. Lin (University of Texas MD Anderson Cancer Center), A. Weiss (University of California, San Francisco), and T. Yeates (University of California, Los Angeles) for materials used in this work. We also thank Y. Choi, H. Ho, and R. Smolkin for assistance and support in the lab.
Footnotes
Author Contributions
Z.L.C. and Y.Y.C. designed the project, participated in data analysis throughout, and wrote the manuscript. Z.L.C., M.H.L, and Y.Y.C edited and revised the manuscript. Z.L.C. developed the TGF-β CAR system and performed and analyzed microscopy, western blot, and computational modeling experiments. Z.L.C., M.H.L., and U.T. performed and analyzed TGF-β CAR flow cytometry, cytokine production, and cell expansion experiments. X.C. performed the CD19 CAR experiments. Z.L.C. and N.J.B. developed and tested the GFP CAR system.
Competing Financial Interests Statement
We declare competing financial interests in the form of a pending patent application whose value may be affected by the publication of this manuscript.
Data availability
All data generated during this study are available from the corresponding author upon request.
References
- 1.Sadelain M, Rivière I & Riddell S Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang ZL & Chen YY CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol. Med. 23, 430–450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown CE et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali A et al. HIV-1-Specific Chimeric Antigen Receptors Based on Broadly Neutralizing Antibodies. J. Virol. 90, 6999–7006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellebrecht CT et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hombach A et al. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res. 58, 1116–9 (1998). [PubMed] [Google Scholar]
- 7.Lanitis E et al. Redirected Antitumor Activity of Primary Human Lymphocytes Transduced With a Fully Human Anti-mesothelin Chimeric Receptor. Mol. Ther. 20, 633–643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan KF et al. Bypassing immunization: optimized design of ‘designer T cells’ ag ainst carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin. Cancer Res. 5, 3928–41 (1999). [PubMed] [Google Scholar]
- 9.Westwood JA et al. The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J. Immunother. 32, 292–301 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Ma Q, DeMarte L, Wang Y, Stanners CP & Junghans RP Carcinoembryonic antigen-immunoglobulin Fc fusion protein (CEA-Fc) for identification and activation of anti-CEA immunoglobulin-T-cell receptor-modified T cells, representative of a new class of Ig fusion proteins. Cancer Gene Ther. 11, 297–306 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Carpenter RO et al. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 19, 2048–2060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGuinness RP et al. Anti-tumor activity of human T cells expressing the CC49-zeta chimeric immune receptor. Hum. Gene Ther. 10, 165–73 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Chmielewski M et al. T Cells That Target Carcinoembryonic Antigen Eradicate Orthotopic Pancreatic Carcinomas Without Inducing Autoimmune Colitis in Mice. Gastroenterology 143, 1095–1107.e2 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Irving BA & Weiss A The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 64, 891–901 (1991). [DOI] [PubMed] [Google Scholar]
- 15.Letourneur F & Klausner RD T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc. Natl. Acad. Sci. U. S. A. 88, 8905–9 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romeo C & Seed B Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 64, 1037–46 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Leibly DJ et al. A Suite of Engineered GFP Molecules for Oligomeric Scaffolding. Structure 23, 1754–1768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirchhofer A et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17, 133–138 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Tang JCY et al. A nanobody-based system using fluorescent proteins as scaffolds for cell-specific gene manipulation. Cell 154, 928–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mack M, Riethmüller G & Kufer P A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 92, 7021–5 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbanska K et al. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 72, 1844–1852 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamada K et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 18, 6436–45 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Rabinovich GA, Gabrilovich D & Sotomayor EM Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 25, 267–296 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flavell RA, Sanjabi S, Wrzesinski SH & Licona-Limón P The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 10, 554–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koehler H, Kofler D, Hombach A & Abken H CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 67, 2265–73 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Bunnell SC et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 158, 1263–75 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campi G, Varma R & Dustin ML Actin and agonist MHC–peptide complex–dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 202, 1031–1036 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim ST et al. The alphabeta T Cell Receptor Is an Anisotropic Mechanosensor. J. Biol. Chem. 284, 31028–31037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y-C et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol. 184, 5959–63 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Chen W, Evavold BD & Zhu C Accumulation of Dynamic Catch Bonds between TCR and Agonist Peptide-MHC Triggers T Cell Signaling. Cell 157, 357–368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y et al. A Flow Cytometry Method to Quantitate Internalized Immunotoxins Shows that Taxol Synergistically Increases Cellular Immunotoxins Uptake. Cancer Res. 70, 1082–1089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hargreaves PG & Al-Shamkhani A Soluble CD30 binds to CD153 with high affinity and blocks transmembrane signaling by CD30. Eur. J. Immunol. 32, 163–73 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Schwesinger F et al. Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. U. S. A. 97, 9972–7 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu KH & Butte MJ T cell activation requires force generation. J. Cell Biol. 213, 535–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Cordoba S-P, Dushek O & Anton van der Merwe, P. Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling. Proc. Natl. Acad. Sci. U. S. A. 108, 19323–19328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobbins J et al. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci. Signal. 9, ra75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Merwe PA & Dushek O Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Yokosuka T et al. Spatiotemporal Regulation of T Cell Costimulation by TCR-CD28 Microclusters and Protein Kinase C θ Translocation. Immunity 29, 589–601 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadelain M Chimeric antigen receptors: driving immunology towards synthetic biology. Curr. Opin. Immunol. 41, 68–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalos M & June CH Adoptive T Cell Transfer for Cancer Immunotherapy in the Era of Synthetic Biology. Immunity 39, 49–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuda M, Koga M, Nishida E & Ebisuya M Synthetic signal propagation through direct cell-cell interaction. Sci. Signal. 5, ra31 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Gordon WR et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev. Cell 33, 729–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morsut L et al. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwarz KA, Daringer NM, Dolberg TB & Leonard JN Rewiring human cellular input–output using modular extracellular sensors. Nat. Chem. Biol. 13, 202–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanash SM, Pitteri SJ & Faca VM Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Hu S, Loo JA & Wong DT Human body fluid proteome analysis. Proteomics 6, 6326–6353 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedorov VD, Themeli M & Sadelain M PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 5, 215ra172–215ra172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloss CC, Condomines M, Cartellieri M, Bachmann M & Sadelain M Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grada Z et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2, e105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC & Chen YY T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Online methods references
- 51.Jonnalagadda M et al. Chimeric Antigen Receptors With Mutated IgG4 Fc Spacer Avoid Fc Receptor Binding and Improve T Cell Persistence and Antitumor Efficacy. Mol. Ther. 23, 757–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen P, Moisini I & Geiger TL Identification of a murine CD28 dileucine motif that suppresses single-chain chimeric T-cell receptor expression and function. Blood 102, 4320–4325 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Moeller M et al. A functional role for CD28 costimulation in tumor recognition by single-chain receptor-modified T cells. Cancer Gene Ther. 11, 371–379 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 183, 5563–74 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gressner AM, Weiskirchen R, Breitkopf K & Dooley S Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 7, d793–807 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Junker U et al. Transforming growth factor beta 1 is significantly elevated in plasma of patients suffering from renal cell carcinoma. Cytokine 8, 794–8 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Yu Q & Stamenkovic I Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–76 (2000). [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Q et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res. 65, 1761–9 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Foster AE et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 31, 500–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.El Hentati F-Z, Gruy F, Iobagiu C & Lambert C Variability of CD3 membrane expression and T cell activation capacity. Cytometry B. Clin. Cytom. 78, 105–14 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.