

Abstract
In addition to the intrinsic toxicity associated with the chemical composition of nanoparticles (NP) and their ligands, biofunctionalized NP can perturb specific cellular processes through NP-cell interactions and induce programmed cell death (apoptosis). In the case of the epidermal growth factor (EGF), nanoconjugation has been shown to enhance the apoptotic efficacy of the ligand, but critical aspects of the underlying mechanism and its dependence on the NP morphology remain unclear. In this manuscript we characterize the apoptotic efficacy of nanoconjugated EGF as function of NP size (with sphere diameters in the range 20–80 nm), aspect ratio (A.R., in the range of 4.5 to 8.6), and EGF surface loading in EGFR overexpressing MDA-MB-468 cells. We demonstrate a significant size- and morphology-dependence in this relatively narrow parameter space with spherical NP with a diameter of approx. 80 nm being much more efficient in inducing apoptosis than smaller spherical NP or rod-shaped NP with comparable EGF loading. The nanoconjugated EGF is found to trigger an EGFR-dependent increase in cytoplasmic reactive oxygen species (ROS) levels but no indications of increased mitochondrial ROS levels or mitochondrial membrane damage are detected at early time points of the apoptosis induction. The increase in cytoplasmic ROS is accompanied by a perturbation of the intracellular glutathione homeostasis, which represents an important check-point for NP-EGF mediated apoptosis. Abrogation of the oxidative stress through inhibition of EGFR signaling through the EGFR inhibitor AG1478 or addition of antioxidants N-acetyl cysteine (NAC) or tempol, but not trolox, successfully suppressed the apoptotic effect of nanoconjugated EGF. A model to account for the observed morphology dependence of EGF nanoconjugation enhanced apoptosis and the underlying NP-cell interactions is discussed.
Keywords: Reactive Oxygen Species, Mitochondria, Nanoparticle Uptake, Glutathione, Nanotoxicity, Redox Homeostasis
Introduction
The epidermal growth factor (EGF) receptor (EGFR) is a transmembrane receptor and a model receptor tyrosine kinase (RTK). EGFR is rapidly endocytosed after activation through ligand binding and subsequent dimerization.1 EGFR signaling is key for healthy cell growth and differentiation, but EGFR overexpression or dysregulation is associated with uncontrolled cell growth and cancer.2–3 The medical relevance of EGFR is, however, not limited to cancer. The receptor is also important for regulating neural plasticity4–5 and intestinal barrier function,6 and represents a therapeutic target in Altzheimer’s disease7. Intriguingly, EGFR signaling can, under specific conditions, also block proliferation and initiate a programmed cell death (apoptosis) pathway.8–10 There is now mounting evidence that this apparent contradiction results from a perturbation of the spatial regulation of EGFR signaling. Although EGFR is activated through ligand binding at the cell surface, activated EGFR signaling continues after uptake until the EGFR containing endosome containing the phosphorylated EGFR tail exposed in the cytoplasm is enclosed in a multivesicular body, or the EGF-EGFR complex is degraded. In the case of free EGF it was hypothesized that the signaling outcome can vary with the cellular location of signaling.11 Indeed, accumulation of activated EGFR in the limiting membrane of early endosomes was shown to trigger apoptosis.12–15
EGF-EGFR recognition is utilized to target cancer cells for diagnostic and therapeutic purposes with NP.16–19 The rational development of optimized targeting strategies requires a detailed understanding of NP-cell interactions during and after uptake.20–24 A particularly important question for a ligand-receptor pair, such as EGF-EGFR, whose signaling is potentially spatially regulated, is whether nanoconjugation of the ligand and its consequences on uptake and intracellular trafficking impacts the signaling outcome and whether this effect by itself has therapeutic potential. Indeed, recent studies have found that conjugation of EGF to gold NP results in apoptosis, as evidenced by increased caspase-3 levels, nucleus condensation, and annexin V/propidium ion staining patterns, at much lower concentration than observed for the free ligand.25 The gain in apoptotic efficacy due to nanoconjugation of EGF was confirmed in both EGFR overexpressors (A431, MDA-MB-468) as well as in cells with physiological EGFR expression levels (HeLa).25–26 The mechanism by which nanoconjugation of EGF “switches” the signaling outcome from proliferation to apoptosis and how the effect depends on the physico-chemical properties of the NP core remain, however, unclear. As an important first step towards an improved understanding of the underlying NP-cell interactions that modulate EGF signaling, we characterize in this work the dependence of NP-EGF induced apoptosis on the intrinsic NP parameters size, shape, and EGF surface loading and elucidate the role of NP-EGF induced oxidative stress in inducing apoptosis.
Experimental
NP Functionalization with EGF
100 μL of 10 mM PEG1 (HS–(CH2)11–(C2H4O)6–COOH) and 10 μL of 10 mM PEG2 (HS–CH2CH2–(C2H4O)77–N3) solutions were added to 20 mL of gold nanosphere colloid and incubated overnight at room temperature. The particles were then washed twice by centrifugation and resuspension in Millipore water. Subsequently, 2 μL of a 100 mg mL−1 propargyl-N-hydroxysuccinimidyl ester (C10H11NO5) solution in DMSO were added to 100 μL of a 1 mg mL−1 solution of EGF in 1× PBS, pH 7.4, and incubated on ice for 6 h. This mixture was then dialyzed against 0.5× PBS for 72 h. 5 μL of the obtained propargyl-PEG-EGF (concentration 10 nM) were then incubated overnight at 4 °C with 1 mL of the PEGylated gold nanospheres in the presence of 500 μM ascorbic acid and 100 μM CuSO4 (catalyst for the cycloaddition reaction). The resulting EGF-functionalized particles (NP-EGF) were washed by centrifugation (2×) and resuspended in 1 mL of DMEM.
NR Pegylation and Functionalization with EGF
2.5 μL of 10 mM solutions of PEG1 and PEG2 were added to 1 mL of Au NR colloid (concentration = 100 pM) in the presence of 3% v/v of Tween 20 and subsequently incubated under soft stirring overnight at room temperature. The PEGylated Au NR were washed twice by centrifugation (7,500 – 15,000 rpm, 15 min – 40 min depending on the NP dimensions) and resuspended in Millipore water. 2 μL of a 100 mg mL−1 propargyl-PEG-NHS ester solution in DMSO were added to 100 μL of a 1 mg mL−1 solution of EGF in 1× PBS, pH 7.4, and incubated in an ice bath for 6 h. This mixture was then dialyzed against 0.5× PBS for 72 h with a 3.5 kDa molecular weight membrane. 10 μL of the obtained propargyl-PEG-EGF were incubated overnight at 4 °C with 1 mL of the PEGylated Au NR colloid containing 500 μM ascorbic acid and 100 μM CuSO4 as catalyzer for the cycloaddition reaction). The resulting EGF-functionalized rods (NR-EGF) were washed 2× by centrifugation (7,500 – 15,000 rpm, 15 min – 40 min depending on the NR size). After that, NR-EGF were resuspended in 1 mL DMEM.
Caspase-3 Activity and Quantification of Apoptosis Enhancement
MDA-MB-468 cells were plated in 6-well dishes. After ≈ 80 % cell confluency was reached the growth medium was replaced by fresh medium containing NP or free EGF ligand controls. The cells were incubated with the NP preparations for 4 h and then washed three times with Hank’s buffer. After that the cells were further incubated in growth media for 20h. Finally, the particles are trypsinized with 0.5 mL of a 0.25% trypsin-EDTA solution. The trypsinization was quenched after 5 min by adding 0.5 mL of complete medium in each well. The cells were collected by centrifugation (300g, 5 min) and washed once with 1× PBS. Subsequently, the cells were resuspended in 1× PBS (800 μL). The EnzChek® Caspase-3 assay was applied to determine apoptosis activity according to the manufacturer’s protocol in a 96-well plate. Fluorescence was quantified with a Perkin Elmer 1420 Victor-3 multilabel counter using excitation and emission wavelengths of λexc = 485 nm and λem = 535 nm, respectively. The apoptosis enhancement was calculated from the measured Caspase-3 levels as the change in apoptosis relative to the no treatment control. All Caspase-3 activities were normalized by the total protein concentration determined by a BCA assay to account for differences in sample size.
ROS Quantification
Flow cytometry was used to quantify ROS generation. Cytoplasmic ROS generation was quantified using the CellROX deep red flow cytometry assay kit (Invitrogen, C10422). 1 mM N-acetylcysteine (NAC) was used as antioxidant to suppress ROS generation and 200 μM Tert-butyl hydroperoxide (TBHP) was used as positive control to induce ROS. Mitochondrial superoxide levels were quantified with MitoSOX Red Mitochondrial Superoxide Indicator (Invitrogen, M36008). This compound is a hydroethidine derivate that selectively localizes to the mitochondria.
GSH Quantification
Cellular GSH levels were measured with the GSH/GSSG ratio detection assay kit (Fluorimetric-Green) following the manufacturer’s protocol (Abcam, ab138881). GSH levels were evaluated after incubation for 4h with NP78.9-EGF.
Mitochondrial Membrane Potential Assay
Cells were cultured in 6-well dishes. 20 μM tetraethylbenzimidazolylcarbocyanine (JC1) was added to control and sample wells and incubated for 10 min at 37°C. Wells were washed with Hank’s buffer (HBSS) twice before NP78.9-EGF were added and incubated for 4 h. 4 μl of carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) was included as positive control. Cells were detached, washed with PBS and then analyzed through flow cytometry with an emission wavelength of 590 nm.
ICP-MS Quantification of Cellular NP Uptake
MDA-MB-468 cells were plated in 6-well dishes. After 24 h, the old growth medium was replaced with a fresh medium containing nanoconjugated EGF, with NP-EGF concentrations varying depending on the NP core size between 8 – 128 pM, or PEGylated NP at a concentration of 100 pM for 4 h and then washed three times with Hank’s buffer. After that the cells were further incubated in growth media for 20h before they were trypsinized with 1 mL of 0.25% trypsin-EDTA solution and subsequent quenching of the enzyme by the addition of 1 mL of complete medium in each well. Cells were collected by centrifugation (200g, 5 min) and subsequently washed twice with 1× PBS. The cell densities of the samples were determined through a hemacytometer (no less than 200 cells). Aqua regia (1 mL) was added to the cells in a total volume of 10 μL to dissolve the Au NP. The mixture was then dried overnight at 65 °C and re-dissolved in HCl solution (1 mL, 2%). After an additional 4-fold dilution through Millipore water, the samples were inserted into a VG Plasma Quad ExCell ICP-MS. For etching control experiments an additional step was performed before trypsinization. 1 mL of the I2/KI aqueous solution (0.34 mM I2, 2.04 mM KI in PBS) was added to the culture well and incubated at room temperature for 2 min and then washed 3 times with pre-warmed Hank’s buffer.
Quantification of EGFR Phosphorylation
NP-EGF were incubated with MDA-MB-468 cells in DMEM for defined time durations. The cells were washed in Hank’s buffer to remove the particles and then detached and lysed. The lysate was diluted 10× before phosphorylation levels were measured using a phosphorylated human EGFR (pY1068) Elisa kit following the manufacturer’s directions.
For additional Methods and Materials, please refer to the ESI.
Results
Apoptosis Enhancement through Spherical NP-EGF and the Effect of NP Size
The design of the NP-EGF used in this study is schematically depicted in Figure 1a. The NP surface is passivated with a self-assembled monolayer of (HS–(CH2)11–(C2H4O)6–COOH, PEG1) interspersed with (HS–CH2CH2–(C2H4O)77–N3, PEG2) for the covalent attachment of alkyne functionalized EGF through the CuI catalyzed 1,3-dipolar cycloaddition reaction.27–28 We included three spherical NP sizes in the commonly as nanoparticle defined size range between 1–100 nm: 21.5 ± 0.9 nm (NP21.5), 40.4 ± 1.0 nm (NP40.4), 78.9 ± 1.3 nm (NP78.9). In initial test experiments we also tested 98.1 ± 0.8 (NP98.1) particles, but these larger NP lacked sufficient stability when incubated with cells and resulted in unacceptable levels of agglomeration (Figure S1).
Figure 1.

a) Schematic drawing of the NP-EGF used in this work. PEG1 = HS–(CH2)11–(C2H4O)6–COOH; PEG2 = HS–CH2CH2–(C2H4O)77–N3. The inset shows SEM images of the different NP cores used. The average NP size (± standard deviation) of the NP is (clockwise): 21.5 ± 0.9 nm; 40.4 ± 1.0 nm; 78.9 ± 1.3 nm. b)–d) Hydrodynamic diameter (intensity statistics) as determined by DLS of citrate capped b) NP21.5, c) NP40.4, and d) NP78.9. e) UV-Vis for NP40.4 before and after functionalization with EGF. f) Number of EGF bound per NP for (left to right): NP78.9-EGF, NP40.4-EGF, NP21.5-EGF. Inset shows ELISA calibration standard obtained with free EGF. g) Zeta-Potentials of NP21.5 before (NP21.5-PEG) and after (NP21.5-EGF) functionalization with EGF.
SEM images of the NP cores and size distributions as determined by dynamic light scattering (DLS, intensity statistics) for NP and NP-EGF are summarized in Figure 1a–d. Figure 1e contains UV-Vis spectra for NP40.4 before and after functionalization with EGF. A small systematic red-shift of about 5 nm indicates a change in the local refractive index due to a successful loading of the NP with EGF.29–30 The number of EGF ligands per NP was determined by ELISA (Figure 1f). We found that on average 14, 30, 150 EGF were bound to the different NP diameters (in order of increasing diameter), corresponding to an average EGF surface density of (8.17±2.64)×103 μm−2. The zeta-potential increase obtained after cross-linking (Figure 1g) further confirms a successful functionalization of the NP cores with EGF.
Previous studies have shown that EGF nanoconjugation triggers apoptosis in different cancer cell lines (A431, MDA-MB-468, HeLa).25–26 In this study, we focus on the EGFR-overexpressing MDA-MB-468 breast cancer cell line, which is an established model system for investigating the apoptotic efficacy of free EGF.14 We chose a relatively short incubation time of NP-EGF with cells of 4 h in our studies to provide a stringent test of the efficacy of NP-EGF with different NP core diameters and shapes and to minimize detrimental effects of long colloidal NP incubation times (agglomeration, NP settlement, non-specific binding, etc.). The NP were incubated in serum-free DMEM to minimize corona21, 31–33 formation around the NP. After 4 h, the NP-EGF containing DMEM was removed and exchanged with fresh culture medium. The NP were then incubated for another 20h before apoptosis was quantified by measuring the activity of the activated death protease caspase-3,34–35 which plays a central role in the execution of both extrinsic and intrinsic apoptosis.36 The apoptosis enhancement in % was calculated from the measured caspase-3 levels ([Casp]) of NP-EGF treated cells and no treatment control as 100*([Casp(treated)]-[Casp(control)])/[Casp(control)].
When characterizing the impact of NP size on NP-EGF induced apoptosis, one needs to account for the differences in surface area, and thus total EGF concentration, for the investigated NP diameters. In our experiments we adjusted the NP concentration to keep the effective EGF concentration constant at approximately 1.0 nM. We chose this concentration as it lies significantly below the threshold required for apoptosis induction by free EGF ligand. Unless otherwise noted, we used the following NP concentrations: 128.0 pM (NP21.5), 32.0 pM (NP40.4), and 8.0 pM (NP78.9). The stability of the NP-EGF under the chosen experimental conditions was assayed by UV-Vis and dynamic light scattering (Figures S2 and S3). Only NP21.5-EGF, showed some minor self-association, presumably due to the higher particle concentration in this case. However, as the contribution from agglomerates was low even in the case of NP21.5-EGF, all NP preparations were used as prepared.
In Figure 2a we summarize the measured apoptosis enhancement (relative to the no treatment control) for spherical NP-EGF with the investigated core diameters and for the following controls: 1 nM and 40 nM free EGF and the supernatant of the last wash for NP78.9-EGF. We also performed apoptosis measurements for pegylated NP (NP21.5-PEG, NP40.4-PEG, NP78.9-PEG) that did not contain any EGF (Figure 2b). None of the different NP-PEG samples, the free EGF samples, or the last wash show any significant apoptosis enhancement. This is different for nanoconjugated EGF. NP78.9-EGF achieves a significant (p < 0.0001) increase in the average apoptosis enhancement (up to approx. 40%), whereas conjugates with smaller NP core (NP21.5-EGF and NP40.4-EGF) failed to induce apoptosis (no significant change) if the NP exposure was limited to 4h. When comparing the apoptosis levels measured for one effective EGF concentration with different NP sizes, the peptide concentration bound to a NP is not necessarily equivalent to that of the free peptide, as all of the peptides bound to one NP effectively interact with one (or a few) receptors to which the NP is bound. We emphasize, however, that this effect further increases the “effective” EGF concentration of the smaller NP when compared with NP78.9-EGF, making the apoptosis enhancement for NP78.9 even more significant. In fact, even greatly increased concentrations of smaller NP-EGF with diameters < 78.9 nm did not achieve comparable apoptosis levels as observed for 8.0 pM NP78.9-EGF (vide infra).
Figure 2.
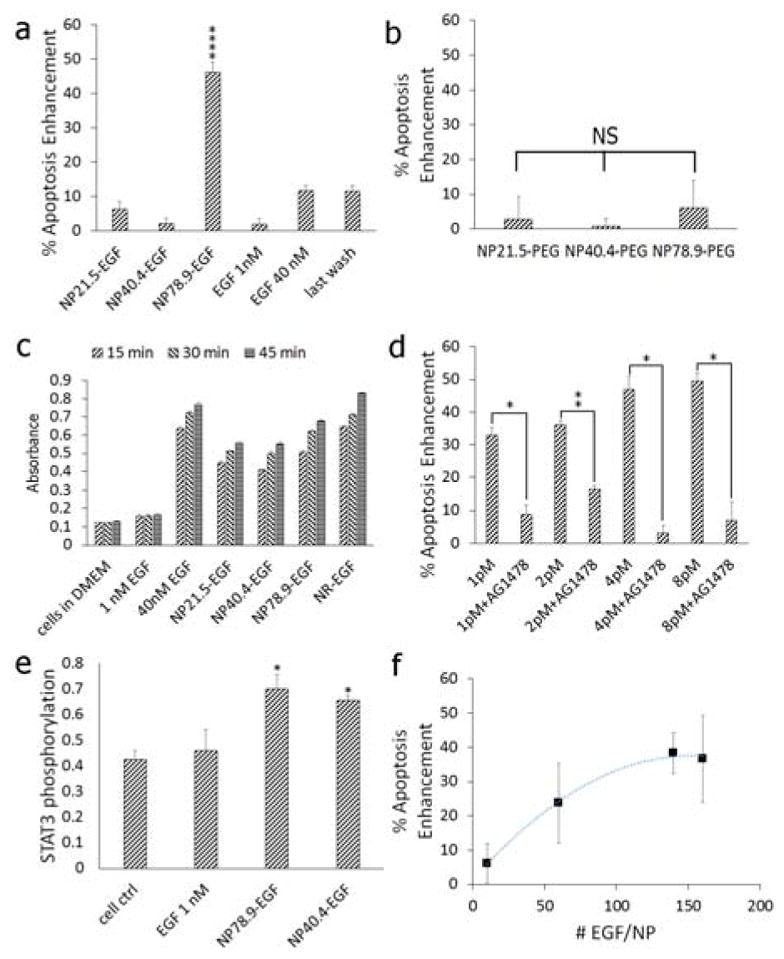
a) Caspase-3 activity (relative to the no treatment control) measured after 4h of exposure to (from left to right) NP21.5-EGF, NP40.4-EGF, NP78.9-EGF, NP98.1-EGF,1 nM free EGF, 40 nM free EGF, and the supernatant of the last wash of NP98.1-EGF. The effective EGF concentration for the different NP-EGF concentrations was 1.0 nM (see text). The plotted data were collected in at least six independent experiments. Error bars show standard errors of the mean. Only NP78.9-EGF show a significant increase at a significance level of ****p < 0.0001. b) Caspase-3 activity (relative to the no treatment control) for pegylated NPs (no EGF) as function of NP core size. NS = not significant. c) ELISA absorbance signal of phosphorylated EGFR after 15, 30, and 45 min of incubation with (from left to right): no treatment control, 1 nM free EGF, 40 nM free EGF, NP21.5-EGF, NP40.4-EGF, NP78.9-EGF, NR-EGF (AR = 8.6). d) Apoptosis enhancement as function of NP78.9-EGF concentration in the absence and presence of the RTK inhibitor AG1478 (250 nM). The effective EGF concentrations are 0.15 nM, 0.30 nM, 0.60 nM, 1.20 nM. *p < 0.05, **p < 0.01. e) STAT3 phosphorylation for no treatment control, 1 nM free EGF, and NP78.9-EGF and NP40.4-EGF with an effective EGF concentration of 1 nM (*p < 0.05 relative to the cell control). f) Apoptosis enhancement as function of number of EGF peptides bound to NP78.9-EGF. Data in (b) and (f) were collected in three independent experiments; data in (c) – (e) were collected from two independent experiments. Error bars show standard deviations.
We measured the average EGFR activation as global phosphorylation for NP21.5-EGF, NP40.4-EGF, NP78.9-EGF, EGF-conjugated nanorods (NR-EGF) with an aspect ratio of AR = 8.6, as well as for 1 nM and 40 nM free EGF, and the no treatment control 15, 30, and 45 min after addition of EGF or its nanoconjugate. The measured phosphorylation levels (Figure 2c) confirm that all EGF nanoconjugates retain the ability to activate EGFR. For NP78.9-EGF, we also evaluated EGFR phosphorylation in the presence and absence of EGFR-selective RTK inhibitor (AG1478). In the presence of the inhibitor we observed, as expected, a suppression of phosphorylation (Figure S4).
For NP78.9-EGF we evaluated the apoptosis enhancement for different concentrations in the absence and presence of the EGFR-selective RTK inhibitor AG1478 (Figure 2d). In the absence of the inhibitor we observed a systematic increase of apoptosis enhancement as function of increasing concentrations, while the RTK inhibitor suppressed apoptosis for all NP78.9-EGF concentrations. The significant difference between the experimental conditions +/− AG1478 confirms that the observed apoptosis is EGFR-dependent.
The signal transducer and activator of transcription 3 (STAT3) has been indicated to play a major role in initiating apoptosis in response to free EGF.37 Indeed, we found significantly enhanced STAT3 phosphorylation levels for nanoconjugated EGF but not for free EGF (Figure 2e) after 4h of NP-EGF exposure, which is consistent with an overall higher apoptotic efficacy of NP-EGF when compared with free EGF. We did, however, not detect any significant difference in STAT3 activation between NP40.4-EGF and NP78.9-EGF although according to Figure 2a the larger NP core has a higher apoptotic efficacy. This finding implies that STAT3 activation alone is insufficient to account for the differences observed for nanoconjugated EGF with different core sizes, confirming a NP-dependent modulation of the apoptotic efficacy of EGF.
Effect of EGF Surface Density on Apoptosis Enhancement
Another effect that modifies the apoptotic efficacy of nanoconjugated EGF is multivalency. Multivalent presentation of a ligand on a NP leads to increased binding avidities and can potentially impact signaling outcomes. Indeed, we observed that the apoptosis enhancement increases with growing EGF density on the NP. This is exemplified for NP78.9-EGF with different EGF densities in Figure 2f. The measured apoptosis initially increases with the number of bound EGF/NP but then converges. For the two highest EGF densities, 140 EGF/NP (≈7.0×103 μm−2) and 160 EGF/NP (≈8.0×103 μm−2) no difference in apoptosis enhancement is detected. Unless otherwise noted, we used an average EGF surface density of (7.8±2.3)×103 μm−2 with EGF surface densities > 6000 μm−2 for all NP sizes throughout this manuscript. These NP-EGF lie in the “high” EGF loading regime where the apoptosis enhancement is independent of the EGF surface concentration. To account for potential differences in the NP uptake due to different binding affinities related to variations in the number of available EGF per particle as well as intrinsic size differences in the uptake mechanism itself, we next quantified the uptake of NP-EGF as function of NP core size.
Size versus Concentration Effect
We first validated that the EGF bound to the NP is available and that the NP-EGF uptake is EGF-specific. Figure 3a shows darkfield scattering images of MDA-MB-468 cells after incubation with NP78.9 –EGF (left) and NP78.9–PEG (right). Only the EGF-functionalized NP show a systematic binding, indicating that the observed binding is EGF-mediated. We quantified the uptake for EGF- and PEG-functionalized NP of all core sizes by ICP-MS (Figure 3b). For the 1 nM EGF preparations the number of NP per cell increases in the following order NP78.9-EGF < NP40.4-EGF < NP21.5 -EGF. For all investigated NP sizes the uptake of NP-EGF was higher than that of NP-PEG, confirming a receptor-mediated uptake of the nanoconjugated EGF for all NP sizes.26 Selective removal of surface bound NP with mild KI/I2 etching38 under conditions that removed all solvent-accessible NP (Figure S5) revealed that even for the largest NP size at least 50% of the NP were protected from the etchant through internalization (labeled NP78.9-EGF-etched in Figure 3b).
Figure 3.
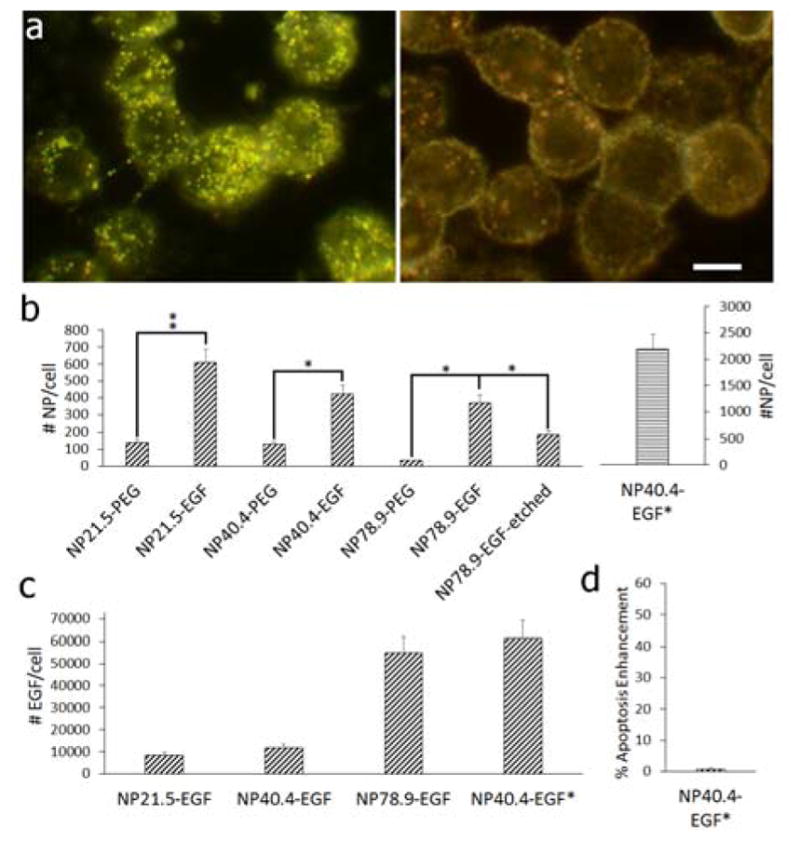
a) Darkfield image of MDA-MB-468 cells after incubation with NP78.9-EGF (left) and NP78.9-PEG (right) under otherwise identical conditions. Scale bar is 10 μm. b) Average number of particles uptake into MDA-MB-468 cells determined by ICP-MS for NP-PEG and NP-EGF with core diameters (left to right) of 21.5 nm, 40.4 nm, 78.9 nm. For the NP diameter of 78.9 nm we also included the data obtained after mild KI/I2 etching that preferentially removes surface bound NPs (“etched”). The effective EGF concentration for all conditions was 1 nM, except for NP40.4-EGF*, which contained an effective EGF concentration of 1.2 μM. *p < 0.05; **p < 0.01. c) Average number of EGF molecules delivered per cell for (left to right): NP21.5-EGF, NP40.4-EGF, NP78.9-EGF, NP40.4-EGF*. d) Apoptosis enhancement obtained for NP40.4-EGF*. Data in (b) – (d) were collected from at least two independent experiments.
Figure 3c summarizes the average number of EGF peptides delivered per cell for all NP-EGF conjugates with a constant effective EGF concentration of 1 nM. The numbers were generated by multiplying the number of delivered NP by the number of bound EGF. Although the number of uptaken NP decrease with growing diameter, the number of delivered EGF molecules increases for the different NP cores in the order NP21.5 < NP40.4 < NP78.9 due to the large difference in bound ligands per particle.
The stark differences in the number of EGF molecules delivered between different NP sizes raises the question whether the high apoptotic efficacy of NP78.9-EGF is related to the physical size of the NP78.9 core or whether the higher intracellular concentration of EGF is the dominating factor. To address this important question we measured the apoptosis enhancement for a smaller NP core at a greatly increased concentration. We used 400 nM NP40.4-EGF (effective EGF concentration 1.2 μM) under otherwise identical conditions as before and obtained a significantly higher NP uptake (included as NP40.4-EGF* in Figure 3b). The average number of EGF delivered per cell under these conditions (6.1×104 EGF/cell) was even higher than for NP78.9-EGF (Figure 3c). However, despite the strongly increased intracellular EGF concentration, NP40.4-EGF still failed to result in a measurable apoptosis enhancement (Figure 3d). This observation contradicts a simple EGF (or NP) concentration effect as cause for the strong apoptosis of NP78.9 and, instead, suggests an enhancement mechanism that is more sensitive to details of the nanoconjugation (size of the NP, ligand density, etc.) than to the number of delivered EGF. For instance, it is conceivable that the nanoconjugation has a crucial effect on the temporospatial regulation of EGFR signalling. Intriguingly, in cells exposed to free EGF, NP21.5-EGF, or NP78.9-EGF (effective EGF concentration was 1 nM in all cases) for 15 min and subsequently maintained in serum-free medium for another 60 min before immunolabelling the early endosome marker EEA1, optical microscopy revealed a higher early endosome concentration for NP78.9 than for NP21.5 or free EGF (Figure S6). Considering that NP21.5-EGF achieved comparable or higher intracellular NP concentrations as NP78.9-EGF (Figure 3b), this observation suggests that the NP78.9 core triggers an accumulation of EGF in early endosomes, which is consistent with previous tracking studies that showed longer dwell times.26
NR-EGF Fail to Enhance Apoptosis
In addition to the size, the shape is another physical NP property with potential relevance for determining the apoptotic efficacy of nanoconjugated EGF. To address the role of the AR in EGF-mediated apoptosis enhancement we synthesized NR with average lengths (AR given in parenthesis) of 45.8±5.5 nm (2.5), 60.9±7.3 nm (3.6), 71.6±9.6 nm (5.4), and 88.9±11.0 nm (8.6) using the seed-mediated method described by Vigderman et al39. The anisotropic growth of NR was achieved with cetyltrimethylammonium bromide (CTAB) as surface ligand. As this ligand is cytotoxic, we exchanged it against benign PEG in a postsynthetic step before EGF was introduced through a CuI catalyzed 1,3-dipolar cycloaddition reaction (Figure 4a–c). SEM images and UV-Vis spectra of the NR used in this work are provided in Figure 4d–g. Peak extinction wavelengths and zeta-potentials before/after PEGylation as well as the volumes and surface areas of the NR used in this work are summarized in Table S1.
Figure 4.

a) Gold NR were functionalized with PEG1 and PEG2 in the presence of 3% v/v of Tween 20 to stabilize the NPs and to introduce binding sites for EGF (see text). b) Human EGF was modified with a terminal propargyl residue. c) The modified EGF was tethered to the binding sites of the PEGylated NR through the Cu(I) catalyzed 1,3-dipolar cycloaddition reaction. d) – g) UV-Vis spectra and SEM images of the nanorods used in this work with AR = 2.5, 3.6, 5.4, 8.6. h) Apoptosis enhancement measured for NR-EGF as function of AR under the same experimental conditions as for NP-EGF (1 nM effective EGF concentration, 4h incubation with rods). i) Average number of NR-EGF (AR = 8.6) per cell after (left) and before (right) KI/I2 etching. j) Relative uptake of (left to right): NP78.9-EGF, NP78.9-EGF + amantadine, NP78.9-EGF + nystatin into MDA-MB-468 determined by ICP-MS. k) Cellular uptake of (left to right): NR-EGF (AR=8.6), NR-EGF + amantadine, NR-EGF + nystatin. Data in (h)–(k) were collected from at least two independent experiments. Error bars show standard deviations. **p < 0.01; *p < 0.05; NS = not significant.
The EGF loadings of the NR were in order of increasing AR: 23, 58, 101, 160 EGF/NR. The average EGF density for all NR was (2.7±2.0)×104 μm−2. Like before for the spherical NP, the NR were incubated with NR-EGF corresponding to an effective EGF concentration of 1 nM. All experimental conditions were identical to the spherical NP-EGF studies (4h incubation with NR, followed by 20h incubation in serum containing medium), but none of the investigated AR led to a measurable enhancement in apoptosis (Figure 4h). This is remarkable, considering i.) that the length of the two longest NR is comparable to the diameter of NP78.9 and ii.) that the number of bound EGF for the highest AR even slightly exceeds that of NP78.9-EGF. Differences in the uptake behavior between NR and NP have recently attracted a lot of interest. It was observed that high AR NRs are taken up much more slowly than spherical NP.40–41 We confirmed that the high AR NR-EGF used in this work were still uptaken by comparing the cellular gold content before and after removing solvent-accessible gold NR with KI/I2 etchant through ICP-MS (Figure 4i). The large fraction of protected NR observed in these experiments confirms their intracellular uptake. The NR uptake, especially for high AR NR does, however, not necessarily occur along the same pathway as that of spherical NP.42–43 This is relevant as shape-dependent differences in the uptake mechanisms can lead to different intracellular NP-EGF distributions.44–46
To verify differences in the uptake of spherical NP78.9-EGF and high AR NR-EGF (AR = 8.6), we performed uptake inhibition assays for clathrin- and caveolae-mediated endocytosis (Figure 4j,k). We measured the cellular gold content though ICP-MS for cells treated with nystatin as inhibitor for caveolae-mediated endocytosis or amantadine as inhibitor for clathrin-mediated endocytosis. The inhibition studies reveal that NP78.9-EGF uptake (Figure 4j) is reduced through amantadine but not by nystatin, as expected for clathrin-mediated uptake. In contrast, for NR-EGF amantadine does not affect uptake (Figure 4k). Instead, we observe a reduction in uptake after nystatin treatment, indicative of caveolae-mediated endocytosis. Positive controls for the inhibitors are provided in Figure S7.
NP78.9-EGF Induced Oxidative Stress
Oxidative stress plays a central role in apoptosis, with relevance for both induction as well as execution. Although oxidative stress is often indicated in NP-induced apoptosis,47–49 there are different mechanisms of ROS formation and the generated ROS can have different functions. Depending on the chemical composition of the NP and its ligands, NP and ligands may create ROS through redox-reactions. Inert NP can trigger cellular responses that result in ROS formation. For gold50–51 and silver NP52–53, as well as for many metal-oxide NP54–55, ROS generation has been observed as consequence of mitochondrial damage that results in apoptosis.56 For biofunctionalized NP, as for the NP investigated in this work, the mechanism of ROS generation is furthermore expected to depend on the interplay between ligand, NP, and cellular receptor. Relevant in this context is that EGFR activation triggers ROS formation as signaling molecules, and H2O2, in particular, has been indicated in receptor transactivation.57–60
As a first step towards a quantitative understanding of the relationship between EGF nanoconjugation and oxidative stress, we measured both cytoplasmic ROS and mitochondrial superoxide levels after addition of NP78.9-EGF. Figure 5a summarizes the cytoplasmic ROS levels after NP78.9-EGF incubation with MDA-MB-468 cells for 4h, and Figure 5b shows cytoplasmic ROS levels after maintaining the cells for an additional 20h after removal of NP78.9-EGF. Several controls were included: no treatment, treatment with tert-Butyl hydroperoxide (TBHP), and NP-EGF in the presence of NAC. Importantly, for both investigated time points, NP78.9-EGF was found to induce a strong increase in cytoplasmic ROS concentration that was almost identical to that obtained with the positive control, TBHP. The ROS levels for NP78.9-EGF significantly exceeded those of free EGF at the same effective concentration (1 nM ligand). In Figure 5b we also included the ROS levels for NP78.9-PEG obtained under otherwise identical conditions. The ROS levels for the latter were even lower than those of free EGF. The increased ROS levels for NP78.9-EGF result from the synergistic interactions between NP core and conjugated EGF.
Figure 5.

a) Quantification of ROS levels through fluorescence flow cytometry using a CellRox assay. Fluorescence histogram for no treatment control, TBHP positive control, NP78.9-EGF + NAC (NAC), and NP78.9-EGF are included. b) Same data as in (a) but after additional culturing for 20h after removal of NP78.9-EGF. NP78.9-PEG was included as additional control. c) Quantification of mitochondrial ROS under the same conditions as in (a). d) JC1 mitochondrial membrane potential assay. NP78.9-EGF does not result in a measurable reduction in fluorescence intensity relative to the no treatment control, indicating intact mitochondria membranes. FCCP = carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone is a positive control that reduces the mitochondria potential.
We monitored the mitochondrial superoxide (O2−) concentration as the primary ROS generated in the mitochondria. Figure 5c shows the mitochondrial superoxide concentrations for cells incubated for 4h with NP78.9-EGF and maintained an additional 20h in the incubator after removal of NP78.9-EGF as well as for positive and negative controls. No significant increase in mitochondrial superoxide concentration was detected after NP78.9-EGF treatment. We also did not detect any decrease in mitochondrial membrane potential (Figure 5d). Overall, the data in Figure 5a–d show that NP78.9-EGF result in a significant increase in cytoplasmic ROS, but that the effect on mitochondrial ROS and membrane integrity is negligible.
NP78.9-EGF Impact on Intracellular Glutathione Homeostasis and Its Effect on Apoptosis
GSH is a major thiol-based redox buffer that in healthy cells is present in high concentrations in its reduced state and that is converted into its oxidized dimer state GSSG through reaction with ROS.61–62 Importantly, the cellular GSH content is indicated to play a direct role in the induction and regulation of apoptosis.63–64 Figure 6a shows the cellular GSH/GSSG concentration ratio for NP78.9-EGF, NP40.4-EGF, NP21.5-EGF, and for NP78.9-EGF in the presence of the EGFR-selective RTK inhibitor AG1478, the anti-oxidants NAC (N-actylcysteine), trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), or tempol (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl). In agreement with the size-dependence of the EGF nanoconjugation induced apoptosis, we found that the drop in GSH/GSSG ratio was specific to the NP78.9 core; smaller NP cores did not induce a significant reduction in the GSH/GSSG ratio under the chosen experimental conditions. Importantly, the presence of the EGFR-specific RTK inhibitor AG1478, or of the anti-oxidants NAC, trolox or tempol mitigated the oxidative stress induced by NP78.9-EGF and prevented a significant drop of the GSH/GSSG ratio.
Figure 6.
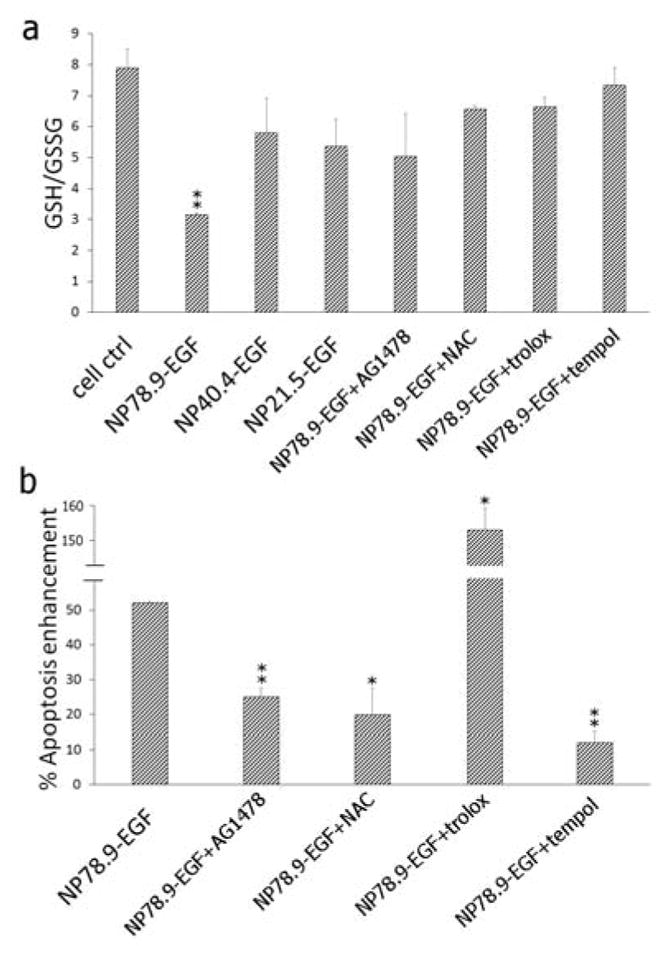
a) GSH/GSSG ratio for no treatment control, NP78.9-EGF, NP40.4-EGF NP21.5-EGF, NP78.9-EGF + AG1478, NP78.9-EGF + NAC, NP78.9-EGF + trolox, NP78.9-EGF + tempol. b) Apoptosis enhancement for NP78.9-EGF, NP78.9-EGF + Ag1478, NP78.9-EGF + trolox, NP78.9-EGF + tempol. All presented data were recorded from three independent experiments. Error bars show standard deviations. Significance was evaluated relative to the no treatment control (a) or NP78.9-EGF (b). **p < 0.01; *p < 0.05.
When we measured the apoptosis enhancement for NP78.9-EGF in the presence of the EGFR inhibitor or the three antioxidants (Figure 6b), we found that inhibition of EGFR phosphorylation through AG1478 and the reduction of oxidative stress through NAC or tempol very efficiently suppressed apoptosis. Surprisingly, the anti-oxidant trolox did not suppress the apoptotic efficacy of NP78.9-EGF, instead, an even higher apoptosis enhancement was detected for the combination of NP78.9-EGF and trolox. We verified in control experiments that trolox alone did not induce apoptosis under the chosen experimental conditions (Figure S8), confirming that the detected gain in apoptosis enhancement resulted from a synergistic effect between NP78.9-EGF and trolox. A potential explanation for the different effects of trolox and NAC or tempol could be related to the different chemical nature of the anti-oxidants. Different than the GSH precursor NAC that removes ROS by enhancing the glutathione response,65 or the stable radical tempol whose primary reaction product after reaction with ROS is an oxoammonium cation,66–67 trolox is converted into a reactive radical cation by reacting with ROS. The observed failure of trolox to inhibit apoptosis could, therefore, be caused by the radical nature of the reaction product. The trolox radical cation is a strong oxidant and its reactivity could result in cellular damage and other detrimental cell processes. The apoptotic effect may be further potentiated by the preferential localization of this vitamin D derivate to cellular membranes.
Discussion
The systematic analysis of NP-EGF induced apoptosis in Figure 2 confirms that neither EGF ligand nor NP by itself results in apoptosis under the chosen experimental conditions. The observed apoptosis must, therefore, be a result of the conjugation of EGF to NP and the accompanying changes in the cellular response to ligand and core. EGF surface density, NP size, and shape all interact to influence the apoptotic efficacy of nanoconjugated EGF. High EGF densities favor apoptosis, spherical NP are more effective than NR, and the largest NP investigated in this work with a diameter of 80 nm are particularly effective. NP-EGF with smaller core diameters can also induce apoptosis, but require longer co-incubation times than the 4 h applied in this work.25 All of the investigated EGF nanoconjugates activate EGFR. Even though the effective EGF concentration was constant in all cases, the measured phosphorylation differs between the nanoconjugates (Figure 2c). NP78.9-EGF and NR-EGF (AR = 8.6) show higher phosphorylation levels than NP21.5-EGF and NP40.4-EGF. In fact, NP78.9-EGF and NR-EGF achieve similar phosphorylation levels as 40 nM free EGF, although the effective EGF concentration of the nanoconjugates is only 1 nM. But despite similar phosphorylation levels for free EGF (40 nM), NP78.9-EGF, and NR-EGF, only NP78.9-EGF effectively induced apoptosis (Figure 2a). The observation that comparable EGFR phosphorylation levels yield different apoptosis enhancements, implies that the total EGFR activation alone is insufficient to account for the observed apoptosis differences under the chosen experimental conditions. Instead, differences in the activation process that are related to the structure and ligand presentation by EGF-presenting NP seem to play a key role for the induction of apoptosis. For one, the increase in apoptosis with increasing EGF surface density in Figure 2f confirms that multivalent EGF presentation enhances the apoptotic signaling. But the shape of the NP also plays an important role. For instance, NR-EGF (AR = 8.6) with comparable EGF surface loadings and total phosphorylation levels as NP78.9-EGF did not induce apoptosis (Figure 4h). We conclude that an accurate evaluation of apoptotic efficacy requires an explicit consideration of NP size, ligand density, and shape.
Based on the putative mechanism for free EGF induced apoptosis enhancement, which attributes apoptosis to an accumulation of activated EGFR in early endosomes,12–13, 15, 68 it is conceivable that it is not the global EGFR signaling but specifically the endosomal EGFR signaling that is the determining factor for NP-EGF induced apoptosis enhancement. Apoptosis could then be the result of EGF nanoconjugation induced changes in the temporospatial intracellular signaling patterns of bound EGFR. The fact that NP40.4-EGF failed to induce comparable apoptosis levels as NP78.9-EGF, even when the concentration of delivered EGF was much higher (Figure 3b–d) emphasizes a central role of the physical NP size in determining the apoptotic efficacy of nanoconjugated EGF. Shape and size of a stiff NP have direct implications for the trafficking of NP-EGF tethered EGFR as vesicle sorting and trafficking require a high degree of structural flexibility, or pleomorphism, of the participating vesicles in the dense matrix of the cytoplasm.69 Gold NP are hard, and under typical cellular conditions non-deformable objects. The structural flexibility of a vesicle of a given size (for instance formed through clathrin-mediated endocytosis) and, thus, its mobility in the cytoplasm are expected to decrease with increasing size of the contained gold NP cargo.70 Indeed, we observed higher early endosome concentrations for NP78.9-EGF than for NP21.5-EGF (Figure S6), which could result from an overall slower intracellular trafficking of the larger NP core.26 Confocal scans also indicated some differences in the spatial distribution of EEA1 for NP78.9-EGF, which motivate further detailed experimental investigation of the impact of NP size on the trafficking of nanoconjugated EGF.
EGFR activation is known to trigger ROS generation,57, 71 and the increase in cytoplasmic ROS and reduction of the cellular GSH/GSSG ratio observed in this work is consistent with a persistent activation of cytoplasmic EGFR. The associated change in the cellular milieu represents a key check-point in EGF nanoconjugation induced apoptosis. Suppression of a drop in GSH/GSSG ratio by NAC or tempol, or inhibition of EGFR signaling through AG1478 successfully prevented NP-EGF induced apoptosis (Figure 6). Intriguingly, the nanoconjugated EGF did not trigger increased mitochondrial ROS levels, which corroborates the hypothesis that mitochondria-independent redox signaling in the cytoplasm is the origin of EGF nanoconjugation induced apoptosis.
For ligand-receptor pairs whose signaling outcomes are spatially regulated, differences in the uptake mechanism can result in different signaling results. Importantly, for NR-EGF (AR = 8.6) we found indications of caveolae-mediated endocytosis, whereas NP78.9-EGF was primarily uptaken by clathrin-mediated endocytosis (Figure 4j,k). A switch in the uptake mechanism from clathrin- to caveolae-mediated endocytosis for nanorods with a length of around 80 nm is in good agreement with previous findings by Liu et al.42 A transition from clathrin- to caveolae-mediated uptake changes the temporospatial distribution of the nanoconjugated EGF (and the bound EGFR). In addition, caveolin-1, which is a component of caveolae, can downregulate EGFR signaling by dephosphorylating the receptor tyrosine kinase.72–74 We, consequently, attribute the observed differences in the apoptotic efficacy of high AR NR and larger NP to a reduction of oxidative stress due to shape-dependent differences in the uptake and intracellular signaling of EGFR. Additional studies into the cellular mechanisms underlying the shape-dependent differences in oxidative stress upon uptake of nanoconjugated EGF are warranted.
Conclusion
NP-cell interactions are complex and can result in unexpected and potentially harmful signaling outcomes for nanoconjugated ligands targeted at cell surface receptors. If these interactions are understood in detail, they provide, however, also new opportunities for manipulating and controlling cell signaling. In this work, we have demonstrated that nanoconjugation modulates the apoptotic efficacy of EGF and that the magnitude of the effect depends not only on the ligand density of the NP but, in particular, on the size and shape of the NP core. Under the chosen experimental conditions NP78.9-EGF was much more efficient in inducing apoptosis than smaller NP or high AR NR with comparable EGF loading. The apoptosis was shown to be related to NP78.9-EGF induced oxidative stress. Cytoplasmic ROS generation and perturbation of the glutathione homeostasis were key events for EGF nanoconjugation mediated apoptosis and their abrogation prevented apoptosis. The successful induction of apoptosis in cancer cells through nanoconjugated EGF is particularly important and has translational potential as apoptosis evasion is one of the hallmarks of cancer and established EGFR targeted therapeutics suffer from rapid development of resistances.75
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health through grant R01CA138509 (BMR). JCF acknowledges Fulbright-Ministry of Education Program for being recipient of a Fulbright fellowship.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: Figures S1–S8 and additional Methods and Materials.
References
- 1.Sorkin A. Internalization and Degradation of the EGF Receptor. Humana Press; New York: 2008. [Google Scholar]
- 2.Lemmon MA, Bu ZM, Ladbury JE, Zhou M, Pinchasi D, Lax I, Engelman DM, Schlessinger J. Embo Journal. 1997;16:281–294. doi: 10.1093/emboj/16.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raymond E, Faivre S, Armand JP. Drugs. 2000;60:15–23. doi: 10.2165/00003495-200060001-00002. [DOI] [PubMed] [Google Scholar]
- 4.Abe K, Saito H. Brain Res. 1992;587:102–108. doi: 10.1016/0006-8993(92)91433-f. [DOI] [PubMed] [Google Scholar]
- 5.Barrie A, Chieregatti E, Miloso M, Benfenati F, Valtorta F. Mol Pharmacol. 1996;49:399–403. [PubMed] [Google Scholar]
- 6.Rowland KJ, Choi PM, Warner BW. Semin Pediatr Surg. 2013;22:101–111. doi: 10.1053/j.sempedsurg.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Chiang HC, Wu W, Liang B, Xie Z, Yao X, Ma W, Du S, Zhong Y. Proc Natl Acad Sci USA. 2012;109:16743–16748. doi: 10.1073/pnas.1208011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong DK, Kaufmann SH, Ottaviano YL, Furuya Y, Buckley JA, Isaacs JT, Davidson NE. Cancer Research. 1994;54:5280–5283. [PubMed] [Google Scholar]
- 9.Gill GN, Lazar CS. Nature. 1981;293:305–307. doi: 10.1038/293305a0. [DOI] [PubMed] [Google Scholar]
- 10.Kottke TJ, Blajeski AL, Martins LM, Mesner PW, Jr, Davidson NE, Earnshaw WC, Armstrong DK, Kaufmann SH. J Biol Chem. 1999;274:15927–15936. doi: 10.1074/jbc.274.22.15927. [DOI] [PubMed] [Google Scholar]
- 11.Tomas A, Futter CE, Eden ER. Trends Cell Biol. 2014;24:26–34. doi: 10.1016/j.tcb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceresa BP. Int J Mol Sci. 2013;14:72–87. doi: 10.3390/ijms14010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. J Biol Chem. 2012;287:712–722. doi: 10.1074/jbc.M111.294470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyatt DC, Ceresa BP. Exp Cell Res. 2008;314:3415–3425. doi: 10.1016/j.yexcr.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ceresa BP, Schmid SL. Curr Opin in Cell Biol. 2000;12:204–210. doi: 10.1016/s0955-0674(99)00077-0. [DOI] [PubMed] [Google Scholar]
- 16.Master AM, Sen Gupta A. Nanomedicine. 2012;7:1895–1906. doi: 10.2217/nnm.12.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tseng CL, Wang TW, Dong GC, Yueh-Hsiu Wu S, Young TH, Shieh MJ, Lou PJ, Lin FH. Biomaterials. 2007;28:3996–4005. doi: 10.1016/j.biomaterials.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Gao M, Su H, Lin G, Li S, Yu X, Qin A, Zhao Z, Zhang Z, Tang BZ. Nanoscale. 2016;8:15027–15032. doi: 10.1039/c6nr04439e. [DOI] [PubMed] [Google Scholar]
- 19.Kuroda S, Tam J, Roth JA, Sokolov K, Ramesh R. Int J Nanomed. 2014;9:3825–3839. doi: 10.2147/IJN.S65990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma A, Stellacci F. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 21.Fleischer CC, Payne CK. Acc Chem Res. 2014;47:2651–2659. doi: 10.1021/ar500190q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P, Wang X, LW, Hou X, Liu W, Chen C. Sci Technol Adv Mater. 2015;16:034610. doi: 10.1088/1468-6996/16/3/034610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Y, Yang X, Song S, Huang M, He C, Cui M, Chen J. Nanotechnology. 2012;23:045103. doi: 10.1088/0957-4484/23/4/045103. [DOI] [PubMed] [Google Scholar]
- 24.Grabinski C, Schaeublin N, Wijaya A, D’Couto H, Baxamusa SH, Hamad-Schifferli K, Hussain SM. ACS Nano. 2011;5:2870–2879. doi: 10.1021/nn103476x. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Reinhard BM. Biomater Sci. 2014;2:156–166. doi: 10.1039/C3BM60142K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, Xu F, Reinhard BM. Nanoscale. 2016;8:13755–13768. doi: 10.1039/c6nr02974d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolb HC, Finn MG, Sharpless KB. Angew Chem Internat Ed/ 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Boriskina SV, Wang H, Reinhard BM. ACS Nano. 2011;5:6619. doi: 10.1021/nn202055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat Mater. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 30.Pollitt MJ, Buckton G, Piper R, Brocchini S. Rsc Adv. 2015;5:24521–24527. [Google Scholar]
- 31.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Proc Natl Acad Sci USA. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Docter D, Westmeier D, Markiewicz M, Stolte S, Knauer SK, Stauber RH. Chem Soc Rev. 2015;44:6094–6121. doi: 10.1039/c5cs00217f. [DOI] [PubMed] [Google Scholar]
- 33.Hamad-Schifferli K. Nanomedicine. 2015;10:1663–16674. doi: 10.2217/nnm.15.6. [DOI] [PubMed] [Google Scholar]
- 34.Krajewska M, Wang H-G, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- 35.Cohen GM. Biochemical Journal. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stennicke HR, Salvesen GS. Biochim Biophys Acta. 1998;1387:17–31. doi: 10.1016/s0167-4838(98)00133-2. [DOI] [PubMed] [Google Scholar]
- 37.Jackson NM, Ceresa BP. Exp Cell Res. 2017;356:93–103. doi: 10.1016/j.yexcr.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho EC, Xie J, Wurm PA, Xia Y. Nano Lett. 2009;9:1080–1084. doi: 10.1021/nl803487r. [DOI] [PubMed] [Google Scholar]
- 39.Vigderman L, Zubarev ER. Chem Mater. 2013;25:1450–1457. [Google Scholar]
- 40.Huang C, Zhang Y, Yuan H, Gao H, Zhang S. Nano Lett. 2013;13:4546–4550. doi: 10.1021/nl402628n. [DOI] [PubMed] [Google Scholar]
- 41.Agarwal R, Singh V, Jurney P, Sreenivasan SV, Roy K. Proc Natl Acad Sci USA. 2013;110:17247–17252. doi: 10.1073/pnas.1305000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Wu F, Tian Y, Wu M, Zhou Q, Jiang S, Niu Z. Sci Rep. 2016;6:24567. doi: 10.1038/srep24567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Chen Z, Zhang L, Yung WY, Leung KC, Chan HY, Choi CH. Small. 2016;12:5178–5189. doi: 10.1002/smll.201601483. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Adv Mat. 2009;21:419–424. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Ji Y, Wu X, Xu H. ACS Appl Mater Interfaces. 2013;5:9856–9865. doi: 10.1021/am4033857. [DOI] [PubMed] [Google Scholar]
- 47.Manke A, Wang L, Rojanasakul Y. BioMed research international. 2013;2013:942916. doi: 10.1155/2013/942916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE. Nano Lett. 2006;6:1794–1807. doi: 10.1021/nl061025k. [DOI] [PubMed] [Google Scholar]
- 49.Lavado AS, Chauhan VM, Zen AA, Giuntini F, Jones DR, Boyle RW, Beeby A, Chan WC, Aylott JW. Nanoscale. 2015;7:14525–14531. doi: 10.1039/c5nr00795j. [DOI] [PubMed] [Google Scholar]
- 50.Butterworth KT, Coulter JA, Jain S, Forker J, McMahon SJ, Schettino G, Prise KM, Currell FJ, Hirst DG. Nanotechnology. 2010;21:295101. doi: 10.1088/0957-4484/21/29/295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan Y, Leifert A, Ruau D, Neuss S, Bornemann J, Schmid G, Brandau W, Simon U, Jahnen-Dechent W. Small. 2009;5:2067–2076. doi: 10.1002/smll.200900466. [DOI] [PubMed] [Google Scholar]
- 52.AshaRani PV, Low Kah Mun G, Hande MP, Valiyaveettil S. ACS Nano. 2009;3:279–290. doi: 10.1021/nn800596w. [DOI] [PubMed] [Google Scholar]
- 53.Chairuangkitti P, Lawanprasert S, Roytrakul S, Aueviriyavit S, Phummiratch D, Kulthong K, Chanvorachote P, Maniratanachote R. Toxicol in vitro. 2013;27:330–338. doi: 10.1016/j.tiv.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 54.Shi Y, Wang F, He J, Yadav S, Wang H. Toxicol Lett. 2010;196:21–27. doi: 10.1016/j.toxlet.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 55.Manna P, Ghosh M, Ghosh J, Das J, Sil PC. Nanotoxicology. 2012;6:1–21. doi: 10.3109/17435390.2011.552124. [DOI] [PubMed] [Google Scholar]
- 56.Wang X. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 57.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 58.DeYulia GJ, Jr, Carcamo JM, Borquez-Ojeda O, Shelton CC, Golde DW. Proc Natl Acad Sci USA. 2005;102:5044–5049. doi: 10.1073/pnas.0501154102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhuang S, Schnellmann RG. Am J Physiol Renal Physiol. 2004;286:F858–865. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 60.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Pias EK, Aw TY. Cell Death Differ. 2002;9:1007–1016. doi: 10.1038/sj.cdd.4401064. [DOI] [PubMed] [Google Scholar]
- 62.Pias EK, Aw TY. Faseb J. 2002;16:781–790. doi: 10.1096/fj.01-0784com. [DOI] [PubMed] [Google Scholar]
- 63.Circu ML, Aw TY. Biochim Biophys Acta. 2012;1823:1767–1777. doi: 10.1016/j.bbamcr.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han YH, Kim SH, Kim SZ, Park WH. J Cell Biochem. 2008;104:862–878. doi: 10.1002/jcb.21673. [DOI] [PubMed] [Google Scholar]
- 65.Sun SY. Cancer Biol Ther. 2010;9:109–110. doi: 10.4161/cbt.9.2.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krishna MC, Grahame DA, Samuni A, Mitchell JB, Russo A. Proc Natl Acad Sci USA. 1992;89:5537–5541. doi: 10.1073/pnas.89.12.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kudo W, Yamato M, Yamada K, Kinoshita Y, Shiba T, Watanabe T, Utsumi H. Free Radic Res. 2008;42:505–512. doi: 10.1080/10715760802112809. [DOI] [PubMed] [Google Scholar]
- 68.Ceresa BP. Endocytic Trafficking of the Epidermal Growth Factor Receptor in Transfomed Cells. In: Gunduz M, editor. Breast Cancer - Carcinogenesis, Cell Growth and Signalling Pathways. 2011. [Google Scholar]
- 69.Jovic M, Sharma M, Rahajeng J, Caplan S. Histol Histopathol. 2010;25:99–112. doi: 10.14670/hh-25.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartmann R, Weidenbach M, Neubauer M, Fery A, Parak WJ. Angew Chem Int Ed Engl. 2015;54:1365–1368. doi: 10.1002/anie.201409693. [DOI] [PubMed] [Google Scholar]
- 71.Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- 72.Couet J, Sargiacomo M, Lisanti MP. J Biol Chem. 1997;272:30429–30438. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 73.Agelaki S, Spiliotaki M, Markomanolaki H, Kallergi G, Mavroudis D, Georgoulias V, Stournaras C. Canc Biol Ther. 2009;8:1470–1477. doi: 10.4161/cbt.8.15.8939. [DOI] [PubMed] [Google Scholar]
- 74.Park WY, Park JS, Cho KA, Kim DI, Ko YG, Seo JS, Park SC. J Biol Chem. 2000;275:20847–20852. doi: 10.1074/jbc.M908162199. [DOI] [PubMed] [Google Scholar]
- 75.Chong CR, Janne PA. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.