

Abstract
Host recognition of intracellular viral RNA and subsequent induction of cytokine signaling are tightly regulated at the cellular level and are a target for manipulation by viruses and therapeutics alike. Here, we characterize chromosome 6 ORF 106 (C6orf106) as an evolutionarily conserved inhibitor of the innate antiviral response. C6orf106 suppresses the synthesis of interferon (IFN)-α/β and proinflammatory tumor necrosis factor (TNF) α in response to the dsRNA mimic poly(I:C) and to Sendai virus infection. Unlike canonical inhibitors of antiviral signaling, C6orf106 blocks interferon-regulatory factor 3 (IRF3) and, to a lesser extent, NF-κB activity without modulating their activation, nuclear translocation, cellular expression, or degradation. Instead, C6orf106 interacts with IRF3 and inhibits IRF3 recruitment to type I IFN promoter sequences while also reducing the nuclear levels of the coactivator proteins p300 and CREB-binding protein (CBP). In summary, we have defined C6orf106 as a negative regulator of antiviral immunity that blocks IRF3-dependent cytokine production via a noncanonical and poorly defined mechanism. This work presents intriguing implications for antiviral immunity, autoimmune disorders, and cancer.
Keywords: interferon-regulatory factor (IRF), innate immunity, cytokine, interferon, NF-κB
Introduction
The innate immune response is a crucial frontline defense mechanism against invading pathogens. Intracellular detection of pathogen-associated molecular patterns (PAMPs)2 is mediated by membrane-bound Toll-like receptors (TLRs) or cytoplasmic retinoic acid–inducible gene I (RIG-I)–like receptors (RLRs) and nucleotide-binding oligomerization domain–containing (NOD)-like receptors. Engagement of these receptors with their agonists results in the activation of complex signaling pathways, culminating in the production of cytokines and antimicrobial compounds. A critical component of this response is the type I interferon (IFN) system, which induces a local antiviral state upon detection of viruses, intracellular bacteria, or their replicative intermediates (1). Production of type I IFN is also vital for the recruitment and priming of cells involved in both innate and adaptive immune responses (2–5).
Viral replication is typically detected by TLRs 3 and 7/8 in endosomal compartments (6, 7), whereas RIG-I and/or melanoma differentiation–associated gene 5 (MDA5) recognize short or long viral dsRNA intermediates in the cytosol (8, 9). TLR3 then activates TIR domain–containing adapter inducing IFN-β (TRIF) (10), whereas RIG-I/MDA5 interact via their caspase recruitment domains with mitochondrial activated signaling proteins (MAVS) (11). Activation of TRIF or MAVS then promotes recruitment of multiple cytosolic effectors, resulting in the phosphorylation and dimerization of interferon-regulatory factor (IRF) 3 or liberation of NF-κB from its inhibitory complex, respectively. These transcription factors are then imported into the nucleus with activating transcription factor 2 (ATF2) and c-Jun and bind to the promoter region of the IFNB gene along with resident transcription factor high mobility group I(Y) (12). Formation of this multiprotein complex, termed the IFN-β enhanceosome, then recruits transcriptional activators general control of amino acid synthesis protein 5 (GCN5), p300, and/or cAMP-response element–binding protein–binding protein (CBP) to unmask the downstream TATA box and initiate transcription (13). Activated NF-κB and ATF2/c-Jun can also initiate expression of various proinflammatory cytokines such as interleukins and tumor necrosis factor (TNF) α (14–16).
Although the rapid detection of viral infection and production of type I IFN are vital for the inhibition of virus replication and the clearance of infected cells, excessive or prolonged signaling through this pathway is detrimental. As such, numerous negative regulators of the type I IFN induction pathway have been characterized. Modifications of cytosolic and nuclear signaling effectors by phosphorylation, de-, or polyubiquitination are important regulatory mechanisms. Examples of this include ubiquitin-specific peptidase 21 (Usp21), which deubiquitinates RIG-I (17), and serine phosphorylation of the caspase recruitment domain (18) by negative regulator proteins prevents further initiation of signaling. The majority of characterized inhibitors of IRF3 activity target IRF3 for ubiquitination and degradation, for example Forkhead box protein O1 (FOXO1) (19), RTA-associated ubiquitin ligase (RAUL) (20), and tripartite motif–containing 26 (TRIM26) (21). Fas-associated factor 1 has been shown to prevent interactions of IRF3 with importins (22), thus inhibiting nuclear trafficking in response to viral stimuli. An additional inhibitor of IRF3, cellular FLICE-like inhibitory protein (cFLIPL) inhibits IFN-β transcription by binding to IRF3 and preventing its association with CBP within the nucleus (23). Many viruses themselves also target the TLR/RLR pathways as a method of immune evasion (24–26).
To study host molecules required for virus infections in an expansive and unbiased manner, we recently performed a genome-wide analysis of host genes required for infection of human cells by Hendra virus (HeV), a negative-strand RNA virus belonging to the family Paramyxoviridae (27). This screen identified a protein of unknown function encoded by chromosome 6 ORF 106 (C6orf106) as being required for HeV infection. Several studies highlight the importance of the type I IFN pathway in the context of henipavirus infection and pathogenesis. (i) The highly pathogenic henipaviruses HeV and Nipah virus encode immune-evading V proteins that antagonize type I IFN signaling pathways (28, 29). (ii) The nonpathogenic henipavirus Cedar virus initiates a robust IFN-β response to virus infection and lacks V protein coding capacity (30). (iii) HeV infection in vitro is abrogated by recombinant IFN-β stimulation (30). We therefore hypothesized that C6orf106 modulates the type I interferon signaling pathway in response to viral-like stimuli. In the present study, we demonstrate that C6orf106 is an evolutionarily conserved inhibitor of IRF3-dependent antiviral cytokine production that targets IRF3 activity in the nucleus.
Results
C6orf106 suppresses antiviral cytokine synthesis
Our previous study showed that transfecting cells with siRNAs targeting C6orf106 significantly impaired both HeV and Nipah virus infection (27). Our bioinformatics analyses have also shown that C6orf106 is highly evolutionarily conserved with homologs in many animal species (Table S1). Based upon this and the rationale presented above, we hypothesized that C6orf106 antagonizes antiviral signaling. siRNA reagents targeting C6orf106 resulted in a >90% decrease in C6orf106 expression at both the mRNA and protein levels compared with cells transfected with siNEG, a negative control siRNA (Fig. 1, a and b, respectively). In addition, cells were transfected with a C6orf106 overexpression vector (pCAGGs-C6orf106-FLAG) as well as a nonspecific GFP overexpression control (pCAGGs-GFP) (Fig. 1c). Modulating C6orf106 expression did not negatively impact cell viability compared with control cells (Fig. 1d). As a positive control for reduced cell viability, cells were transfected with an siRNA targeting polo-like kinase 1, a gene associated with apoptosis (31).
Figure 1.

Validation of C6orf106 knockdown and overexpression in HeLa cells. a and b, HeLa cells were transfected with siRNAs targeting C6orf106 (siC6orf106) or a nontargeting control (siNEG) and assayed by qRT-PCR (a) or Western blotting (b) for C6orf106 expression levels. c, validation of C6orf106-FLAG expression by Western blotting. d, cell viability in HeLa cells treated with the indicated siRNAs or cDNAs. Values are normalized against cells treated with transfection reagent only (mock). Error bars for all graphs indicate ±1 S.D. of a minimum of three independent experiments; asterisks show significant differences as assessed by one-way ANOVA with Bonferroni post-test (*, p < 0.05; **, p < 0.01).
The synthetic dsRNA analog poly(I:C) is an established stimulator of type I IFN activation (8) and was utilized to mimic viral RNA replication. HeLa cells transfected with poly(I:C) for 6 h showed a robust up-regulation of IFN-α/β, interleukin-6 (IL-6), and TNFα (Fig. 2a). Cells depleted of C6orf106 showed significantly enhanced IFN-α/β and TNFα transcription in response to poly(I:C) compared with siNEG cells (Fig. 2b). Interestingly, the induction of IL-6 was not strongly altered by C6orf106 knockdown, suggesting that C6orf106 differentially regulates poly(I:C)-induced cytokines.
Figure 2.

C6orf106 suppresses antiviral cytokine synthesis. a, HeLa cells were treated with transfected poly(I:C) for 6 h, and the cells were collected and analyzed for mRNA expression of the listed cytokines by qRT-PCR. b, relative cytokine mRNA levels in HeLa cells stimulated with poly(I:C) 48 h post-transfection with siRNAs. c, relative mRNA levels in HeLa cells stimulated with poly(I:C) 24 h post-transfection with cDNAs. d, cell culture supernatants from c were assayed for IFN-β using ELISA. Relative cytokine mRNA levels in HeLa cells infected with SeV (400 hemagglutination units/well) post-transfection with siRNAs (e) or cDNA plasmids (f) are shown. g, HeLa cells were stimulated with 2000 enzyme units/ml IFN-α for 6 h, and ISG15 mRNA expression was determined by qRT-PCR. h, HeLa cells were stimulated with 20 ng/ml TNFα for 6 h, and IκBα mRNA expression was determined by qRT-PCR. i and j, HeLa cells were transfected with siRNAs (i) or cDNA plasmids (j) and then stimulated with poly(I:C) for the times shown. Relative mRNA levels of IFN-β over time were measured by qRT-PCR. k, mock cells were stimulated with poly(I:C) for the times shown, and endogenous C6orf106 and IFN-β mRNA levels were measured by qRT-PCR. Error bars indicate ±1 S.D. of three independent experiments, and asterisks show significant changes compared with controls as measured by one- or two-way ANOVA with Bonferroni post-test (***, p < 0.001; **, p < 0.01; *, p < 0.05 compared with 4 h).
In agreement with results shown in Fig. 2b, C6orf106 overexpression significantly reduced IFN-α and IFN-β mRNA expression in response to poly(I:C) (Fig. 2c). Conversely, induction of IL-6 and TNFα was not significantly affected. Furthermore, secretion of IFN-β protein was decreased in cells overexpressing C6orf106 (Fig. 2d). Given that the relative reduction in IFN-β mRNA and protein levels was similar, we attributed the decrease in IFN-β secretion to reduced mRNA synthesis.
To confirm C6orf106 as a negative regulator of antiviral cytokine transcription, cells were infected with Sendai virus (SeV), an RNA virus (genus Respirovirus, family Paramyxoviridae) that induces type I IFN cytokine transcription in HeLa cells (47). In agreement with poly(I:C) stimulation studies, C6orf106 inhibited IFN-α, IFN-β, and TNFα transcription induced by Sendai virus infection as demonstrated by both siRNA (Fig. 2e) and overexpression (Fig. 2f) experiments. Interestingly, although C6orf106 blocked type I IFN production in response to poly(I:C), type I IFN signaling itself was not affected as the overexpression of C6orf106 did not impact the induction of interferon-stimulated gene (ISG) 15 or IκBα in response to recombinant IFN-α (Fig. 2g) or TNFα (Fig. 2h), respectively.
The impact of C6orf106 on type I IFN production, but not signaling pathways downstream of IFN-α/β receptor activation, led us to postulate that C6orf106 inhibits IFN transcription in a negative feedback mechanism. To test this hypothesis, cells depleted of C6orf106, or those overexpressing C6orf106, were stimulated with intracellular poly(I:C) for increasing amounts of time. In control cells, IFN-β mRNA was rapidly induced and plateaued at 6 h of poly(I:C) treatment, whereas in C6orf106-knockdown cells, IFN-β mRNA synthesis continued to increase past 8–10 h (Fig. 2i). However, the overexpression of C6orf106 reduced IFN-β mRNA levels at all time points assayed (Fig. 2j). Intriguingly, levels of endogenous C6orf106 mRNA significantly increased over the poly(I:C) time course, peaking at 10 h (Fig. 2k). These data suggested that C6orf106 is induced by poly(I:C) treatment as part of a negative feedback loop to prevent excessive cytokine production.
C6orf106 differentially modulates the antiviral transcription factors IRF3 and NF-κB
IFN-α/β and TNFα transcription is primarily controlled by IRFs and NF-κB, respectively, that are activated and translocated to the nucleus in response to TLR and RLR ligands (15, 32, 33). As C6orf106 most significantly affects IFN-α/β transcription, we speculated that it would preferentially regulate IRF activity. To assess this, we transfected luciferase vectors containing IRF- or NF-κB–binding sites in cells depleted of C6orf106. In response to intracellular poly(I:C), C6orf106 depletion resulted in a ∼300% increase in ISRE-luciferase activity compared with cells transfected with siNEG (Fig. 3a). By comparison, depletion of C6orf106 caused a smaller (∼140%) but significant increase in NF-κB activity (Fig. 3b). Similarly, C6orf106 overexpression resulted in an ∼80% reduction of ISRE-luciferase activity (Fig. 3c) and a small (∼20%) but significant reduction in NF-κB activity (Fig. 3d). These results support our observations of differential cytokine regulation and suggest that C6orf106 impairs cytokine transcription primarily via an IRF-dependent mechanism.
Figure 3.

C6orf106 predominantly targets IRF-driven transcription. HeLa cells were transfected with siRNAs for 24 h, then transfected with ISRE- (a) or NF-κB-firefly luciferase (luc) (b) and Renilla luciferase vectors for 20 h, and then stimulated with poly(I:C) for 6 h. Alternatively, HeLa cells were transfected with GFP or C6orf106-FLAG and ISRE- (c) or NF-κB-firefly luciferase (d) and Renilla luciferase vectors for 20 h and then stimulated with poly(I:C) for 6 h. All cell lysates were assayed for luciferase activity, normalized to the transfection control Renilla luciferase. Error bars indicate ±1 S.D. of triplicate experiments; asterisks indicate significant differences as determined by Student's t test (*, p < 0.05; **, p < 0.01).
C6orf106 does not impair activation or nuclear translocation of IRF3 and NF-κB
Following RLR/TLR activation, a cascade of signaling effectors results in IRF3 phosphorylation and dimerization, which then allow its association with importins and nuclear trafficking. To test whether C6orf106 affects IRF3 phosphorylation upon recognition of viral RNA-like stimuli, cytoplasmic and nuclear cell fractions were isolated and probed for phosphorylated IRF3 (Ser-396). Poly(I:C) stimulation increased levels of phosphorylated IRF3 in the nuclear fraction of both control and C6orf106-overexpressing cells (Fig. 4a) with equal ratios of phospho-IRF3:total IRF3 observed in both groups. Additionally, nuclear accumulation of p65, phospho-IRF3, and total IRF3 was observed to equal extents in control and C6orf106-overexpressing cells.
Figure 4.

C6orf106 does not impair activation or nuclear translocation of IRF3 and NF-κB. a, HeLa cells transfected with GFP or C6orf106-FLAG for 20 h were stimulated with poly(I:C) for 6 h and then lysed, and cellular proteins were separated into cytosolic and nuclear fractions. Fractions were probed for the transcription factors IRF3 and p65 as well as C6orf106 and the loading controls GAPDH (cytosol) and fibrillarin (nucleus). b, HeLa cells as in a were fixed and labeled for C6orf106-FLAG (green), IRF3 (left panel) or p65 (right panel), and nuclei (blue). White scale bars, 10 μm. c, Fn/c ratios for treatment groups shown in b. Error bars indicate ±1 S.D. of a typical experiment from duplicate experiments; asterisks indicate significant differences as determined by one-way ANOVA with Dunn's multiple comparison test (***, p < 0.001).
To confirm this, mock- and poly(I:C)-stimulated cells were fixed and probed for C6orf106 and IRF3 or p65, and the ratio of nuclear to cytoplasmic staining (Fn/c) was calculated using confocal microscopy analysis. In agreement with previous studies (34, 35), C6orf106 subcellular staining was observed diffusely both in the cytoplasm and nucleus but was more concentrated at the periphery of the cytoplasm (Fig. 4, b and c, panels 3 and 4). In GFP control cells, stimulation with poly(I:C) caused increased nuclear staining of both IRF3 and p65 (Fig. 4b). Nuclear staining of IRF3 and p65 was also observed in cells overexpressing C6orf106 (Fig. 4b). A significant increase in both IRF3 and p65 Fn/c values was observed upon poly(I:C) stimulation in both control and C6orf106-overexpressing cells (Fig. 4c), confirming that C6orf106 does not impair nuclear trafficking of IRF3 and/or p65. Intriguingly, we also observed a small but significant increase in IRF3 and p65 Fn/c values and observed modest nuclear staining in unstimulated cells overexpressing C6orf106 compared with GFP (Fig. 4c).
C6orf106 does not induce IRF3 degradation
Several inhibitors of nuclear IRF3 and IFN transcription target IRF3 for ubiquitination and degradation (19, 20, 36). Although we did not observe any gross changes in IRF3 levels in C6orf106-expressing cells (Fig. 4a), we wanted to exclude this as a possible mechanism of action. Cells expressing C6orf106 were treated with cycloheximide (CHX; an inhibitor of mammalian protein translation), and IRF3 protein levels were assessed by Western blotting (Fig. 5a). Cells were stimulated with poly(I:C) to determine whether upstream antiviral signaling was also required for potential IRF3 degradation. No discernible differences in IRF3 or p65 protein labeling were observed. As an added confirmation, C6orf106-expressing cells were preincubated with the proteasomal inhibitor MG132 before poly(I:C) stimulation with no increase in IRF3/p65 levels observed (Fig. 5b). We therefore concluded that C6orf106 inhibits IRF3 activity without impacting IRF3 protein expression levels. As a positive control to demonstrate the activities of CHX and MG132 during this experiment, expression levels of cyclin B1, a protein with a known high turnover (48), were also measured. Compared with mock cells, cyclin B1 expression was reduced by CHX treatment and increased by MG132 (Fig. 5c).
Figure 5.
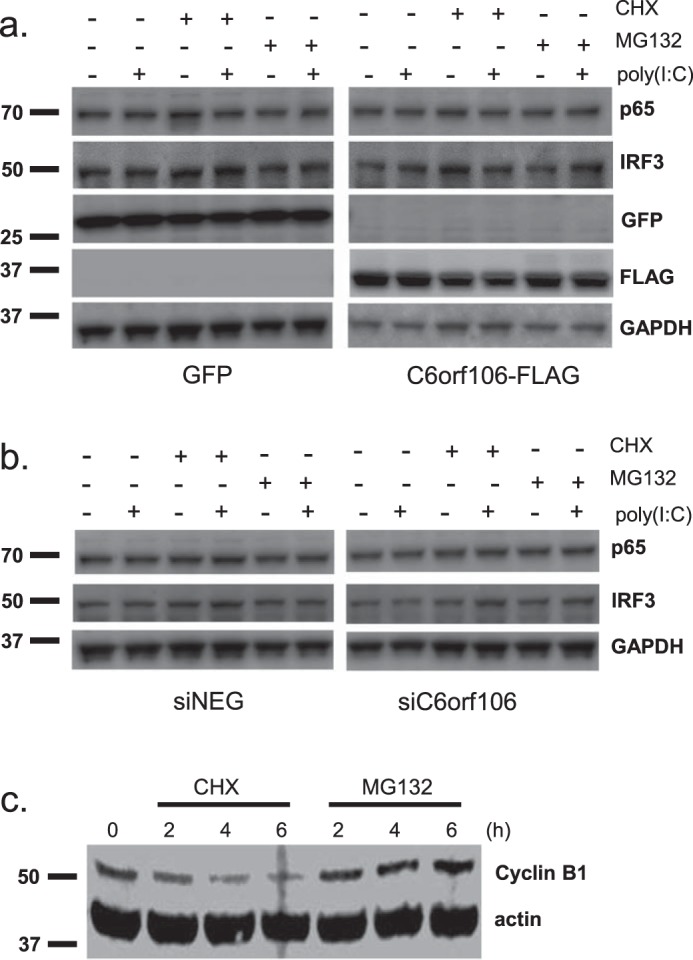
C6orf106 does not alter cellular protein levels of IRF3 and NF-κB. HeLa cells were transfected with cDNAs for 20 h (a) or siRNAs for 40 h (b). Cells were then pretreated for 1 h with CHX (20 μg/ml) or MG132 (10 μm) and then stimulated with poly(I:C) for 6 h. Cells were lysed, and cellular levels of IRF3, p65, and C6orf106 (endogenous or FLAG-tagged) were measured by Western blotting. c, immunoblot showing expression levels of cyclin B1 and actin in HeLa cells treated for the indicated times with CHX (20 μg/ml) or MG132 (10 μm).
C6orf106 interacts with IRF3 and inhibits binding to its DNA consensus sequence
Following activation and nuclear translocation, IRF3 binds to the promoter regions of target genes to initiate transcription. To assess whether C6orf106 modulates IRF3–DNA binding, we isolated cell nuclei and probed the DNA binding capabilities of nuclear IRF3 to its consensus sequence. Poly(I:C) treatment of control cells resulted in an ∼5-fold increase in nuclear IRF3–DNA binding compared with unstimulated cells (Fig. 6a). A significant decrease in IRF3–DNA binding was observed in cells overexpressing C6orf106. When the reciprocal experiment was performed, IRF3–DNA binding induced by poly(I:C) was significantly enhanced in cells lacking C6orf106 (Fig. 6b). To assess whether this inhibition was specific, the impact of C6orf106 on p65–DNA binding was also assessed. Unlike IRF3, the overexpression of C6orf106 did not impair the binding of p65 to its consensus sequence in response to poly(I:C) (Fig. 6c). In fact, depleting C6orf106 led to a small but significant decrease in p65–DNA binding upon poly(I:C) stimulation (Fig. 6d). We next assessed whether C6orf106 binds IRF3 to prevent its association with the promoter. As shown in Fig. 6e, C6orf106 could be coimmunoprecipitated with immobilized anti-IRF3 antibody but not with an irrelevant IgG isotype, suggesting that C6orf106 interacts with IRF3 to modulate DNA binding.
Figure 6.

C6orf106 interacts with IRF3 and inhibits binding to its DNA consensus sequence. HeLa cells were transfected with cDNAs and stimulated with poly(I:C), and the nuclear proteins were extracted using a hypotonic lysis method. Nuclear proteins (10 μg) were analyzed for IRF3–DNA (a) or p65–DNA (c) binding. Alternatively, HeLa cells depleted of C6orf106 were stimulated with poly(I:C), and nuclear proteins were analyzed for IRF3–DNA (b) or p65–DNA (d) binding. e, HEK293T cells transfected with IRF3 alone or in combination with C6orf106 were stimulated with poly(I:C), lysed, and subjected to indirect immunoprecipitation with an anti-IRF3 antibody. Immunoprecipitated (IP) samples and input controls were probed with anti-FLAG antibody for Western blotting. An IgG isotype was used as a negative control for the immunoprecipitation experiment. Error bars indicate ±1 S.D. of triplicate experiments; asterisks indicate significant differences as determined by two-way ANOVA with Bonferroni post-test (***, p < 0.001; **, p < 0.01; ns, not significant).
C6orf106 expression is associated with decreased levels of the transactivator proteins p300 and CBP
The DNA binding activity of IRF3 requires phosphorylation by cytosolic effectors as described above in addition to association with the nuclear transactivators p300/CBP (37). As we only observed a modest interaction of C6orf106 with IRF3 (Fig. 6e) and no effect on IRF3 phosphorylation (Fig. 4a), we investigated whether C6orf106 interferes with other components of the IFN-β enhanceosome complex. Nuclear protein isolates used for the DNA binding assay (Fig. 6a) were probed for the enhanceosome proteins IRF3, p65, c-Jun, CBP, and p300 in the presence of either C6orf106 or GFP. We observed a modest decrease in p300/CBP protein levels in unstimulated C6orf106-expressing cells compared with the control (Fig. 7a). This decrease was even more pronounced upon poly(I:C) stimulation (compare column 4 with columns 3 and 1). Again, there were no discernible changes in nuclear p65 or IRF3 levels, suggesting that this effect was specific to the nucleus-resident p300/CBP proteins. In cells depleted of C6orf106, poly(I:C) stimulation induced a modest increase in nuclear p300 levels compared with controls cells (Fig. 7b). CBP and p300 were detected at extremely low levels in the cytoplasmic fraction, demonstrating that C6orf106 had no observable impact on CBP or p300 localization (Fig. 7c).
Figure 7.

C6orf106 reduces nuclear levels of the transactivator proteins p300 and CBP. Nuclear and cytosolic fractions were isolated from HeLa cells expressing C6orf106-FLAG (a) or knocked down with siRNAs targeting C6orf106 (b) and stimulated with poly(I:C) as shown. Equal amounts of nuclear lysates were probed for members of the enhanceosome complex as shown. c, cytosolic fractions were also probed for the nuclear transactivators CBP/p300.
Discussion
We present here the first characterization of a highly conserved novel protein, C6orf106, in the regulation of IRF3-dependent cytokine transcription. The regulation of cytokine transcription is of great importance in both infection and steady-state environments. Insufficient or delayed production of antimicrobial agents leads to enhanced pathogenicity and chronic infection, whereas excessive or prolonged responses can result in the development of autoimmune disorders or systemic inflammatory effects such as septic shock. We show that C6orf106 does not interfere with cytoplasmic effectors and the activation/nuclear translocation of IRF3 or NF-κB (Fig. 4) nor does it promote degradation of these transcription factors in the nucleus (Fig. 5). Instead, it interacts with IRF3 and impairs its association with DNA promoter regions by enhancing the degradation of the nuclear transactivators p300/CBP (Figs. 6 and 7) required for DNA binding.
Thus, we propose that C6orf106 is a new member of an emerging class of proteins that act to regulate antiviral cytokine transcription from within the nucleus. A working model for C6orf106 modulation of IFN-β transcription is presented in Fig. 8. In cells with low levels of C6orf106 (left panel), dsRNA initiates a signaling cascade resulting in the activation and nuclear translocation of IRF3 and NF-κB. These then associate with p300/CBP and other components of the enhanceosome complex (e.g. ATF2/c-Jun) to bind promoter regions and drive IFNβ gene expression. However, when C6orf106 levels are high (induced by dsRNA activation), nuclear C6orf106 interacts with IRF3 and impairs association with promoter regions by decreasing p300/CBP levels, thus reducing the extent of IFN-β transcription. We tentatively suggest that C6orf106 may utilize activated IRF3 binding to target p300/CBP and are currently investigating how C6orf106 is able to mediate their degradation in the context of virus infection.
Figure 8.

Working model of C6orf106 (C6)-mediated inhibition of IRF3-dependent cytokine transcription.
This mechanism differs from most canonical cellular inhibitors of IRF3 and IFN-β transcription. For example, IRF3/7 inhibitors such as RAUL and FOXO1 possess E3 ligase activity that results in IRF3 ubiquitination and degradation via the proteasome (19, 20). Similarly, RBCC protein interacting with PKC1 (RBCK1) binds and ubiquitinates IRF3 (36). Selective targeting of phosphorylated IRF3 has also been shown for TRIM26 (21), a protein with E3 ligase activity, which degrades nuclear IRF3 to regulate IFN-β transcription. C6orf106 does not contain any identifiable E3 ligase domains, although it does potentially encode a ubiquitin binding–like motif in its N-terminal region. To our knowledge, only one other cellular protein has been demonstrated to inhibit IRF3 activity without inducing degradation. cFLIPL binds IRF3 and prevents interactions with CBP and the IFN-β promoter; this inhibitory activity was mapped to a nuclear localization domain in the C terminus (23). We also observed C6orf106 nuclear localization as well as an intriguing nuclear retention of inactive IRF3 in cells expressing C6orf106, similar to cFLIPL (23). We propose that this may be a result of inadvertent interactions of nuclear C6orf106 or cFLIPL with inactive IRF3, which has been shown to shuttle between the cytosol and nucleus in unstimulated cells (38). Most recently, cFLIPL was also shown to modulate IRF7 activity by preferentially interacting with the activating kinase IKKα, preventing downstream IFN-α transcription (39). Thus, it is possible that C6orf106 may also have other roles in the regulation of cytosolic effectors in response to other PAMPs (such as CpG DNA), and we are currently investigating the extent of PAMP-induced signaling affected by C6orf106.
C6orf106 was reported in a genome-wide screen of host factors required for HeV infection (27). Based upon the data we present here we suggest that C6orf106 facilitates HeV infection by its modulation of antiviral cytokine transcription and/or modulation of p300/CBP. Many DNA viruses utilize p300/CBP to modulate host gene transcription and promote replication (40–42). In the context of antiviral signaling, herpes simplex virus-1 ICP0 sequesters activated IRF3–CBP–p300 complexes in distinct foci in the nucleus to prevent IFN-β induction (43). Like C6orf106, ICP0 does not impact IRF3 activation or nuclear trafficking. The coronavirus porcine epidemic diarrhea virus encodes nonstructural protein 1, which suppresses IFN-β and ISG expression by selectively degrading CBP and therefore preventing enhanceosome formation. Our current work suggests that C6orf106 promotes infection by several negative-sense viruses (data not shown) and thus may be involved in a common mechanism for promoting viral replication via modulation of host transcription. Future studies will further investigate putative roles for C6orf106 and CBP/p300 in paramyxovirus replication.
In summary, our study represents the first functional characterization of C6orf106 in the context of antiviral immunity and provides evidence that transcription factors are regulated via a multitude of different proteins and mechanisms. This study reveals a previously unappreciated role for C6orf106 in type I IFN and proinflammatory cytokine production as well as the regulation of nucleus-resident transactivation proteins p300/CBP. Intriguingly, increased levels of C6orf106 have been observed in both lung and breast cancer cells where C6orf106 promotes cell proliferation and migration (34, 35). Degradation of p300 has also been associated with lung cancer cell proliferation and expression of metastasis genes (44, 45), supporting an additional role for C6orf106 in cancer progression. Future work will be directed toward defining the roles of C6orf106 in virus infection and inflammation and their potential convergence with concepts in cancer biology.
Experimental procedures
Cells
HeLa cells (ATCC CCL-2), human embryonic kidney (HEK) 293T (ATCC CRL-3216), and Vero cells (ATCC CRL-81) were maintained in growth medium (Eagle's modified Eagle medium supplemented with 10% (v/v) fetal bovine serum (FBS), 10 mm HEPES, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). All cells were kept at 37 °C in a humidified incubator (5% CO2).
C6orf106 plasmid generation
The full-length coding sequence of human C6orf106 (accession number NP_077270.1) was synthesized by GenScript with SacI and XhoI restriction sites at the 5′- and 3′-ends respectively. This was ligated into the mammalian expression vector pCAGGs, transformed, and amplified in chemically competent Escherichia coli (MAX Efficiency DH5α competent cells, Life Technologies). Plasmids were amplified and extracted using a Qiagen EndoFree Plasmid Maxi kit as specified by the manufacturer.
Antibodies
Antibodies were sourced from manufacturers as follows: Abcam, p65 (ab16502), GFP (ab183734), fibrillarin (ab5821), p300 (ab14984), and CBP (ab2832); Thermo Scientific, c-Jun (MA5-15881); Cell Signaling Technology, IRF3 (11904S), phospho-IRF3 (Ser-396) (4947S), cyclin B1 (V152) (4135S), GAPDH (2118S); Invitrogen, actin (MA5-11869). M2 anti-FLAG mAb was a kind gift from G. Lovrecz (CSIRO Manufacturing).
Transfections and drug treatments
HeLa cells were reverse transfected with 40 nm siRNA (GE Healthcare) using 0.5 μl of Dharmafect-1 (GE Healthcare) in Opti-MEM (Life Technologies). For DNA transfections, 300 ng of DNA was incubated with 1 μl of Lipofectamine 2000 in Opti-MEM and used to reverse transfect HeLa cells. Cells were stimulated with transfected high-molecular-weight poly(I:C) (Invivogen) (5 μg/ml with 1.5 μl of Lipofectamine 2000) for 6 h. For nucleus isolation studies, media and DNA:Lipofectamine amounts were scaled up accordingly (10-fold for 6-cm dish; 30-fold for 10-cm dish). Cycloheximide and MG132 were purchased from Sigma-Aldrich and diluted in DMSO as specified by the manufacturer.
RNA purification, reverse transcription, and quantitative real-time PCR
Cells were lysed in TRIzol (Life Technologies), and RNA was extracted according to the manufacturer's protocols. Following DNase treatment (RQ1 DNase, Promega), 500 ng of RNA was reverse transcribed to DNA using Superscript III reverse transcriptase (Life Technologies) or SensiFast reverse transcriptase (Bioline) first-strand cDNA synthesis protocols. qRT-PCR was performed using SYBR Green (Applied Biosystems, Foster City, CA) on a StepOne Plus PCR cycler (Applied Biosystems). PCR cycling for gene detection was at 95 °C for 10 min followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min. A melting curve analysis was performed to eliminate primer–dimer artifacts and to verify the specificity of the assay. Cytokine expression and virus RNA transcription data were analyzed using the ΔΔCT method and were normalized to GAPDH. Primers used in qRT-PCR analyses are shown in Table S2.
IFN-β ELISA
Cell culture supernatants were analyzed for IFN-β secretion using a sandwich ELISA kit from Elisakit according to the manufacturer's protocols. IFN-β concentrations were calculated by comparison with standards using a polynomial regression method.
Sendai virus infections
HeLa cells were infected with 400 hemagglutination units of SeV in 250 μl of Eagle's modified Eagle medium with no FCS and incubated at 37 °C for 1 h (rocking every 20 min to facilitate infection). After 1 h, infectious medium was replaced with growth medium, and incubation continued at 37 °C for 24 h. Cell lysates were collected in 500 μl of TRIzol, and RNA was purified and analyzed for cytokine expression as described above.
Dual-Luciferase assays
HeLa cells were reverse transfected with 100 ng of GFP/C6orf106-FLAG plasmids in conjunction with 100 ng of either ISRE-luciferase or NF-κB-luciferase plasmids (firefly) and 50 ng of a Renilla luciferase plasmid. At 20 h post-transfection, cells were stimulated with poly(I:C) for 6 h, then lysed, and assayed for successive firefly and Renilla luciferase activities using the Dual-Luciferase kit (Promega).
Cell fractionation and IRF3/p65 DNA binding assays
HeLa cells were reverse transfected with pCAGGS-GFP or pCAGGs-C6orf106-FLAG for 20 h or with siNEG/siC6orf106 for 48 h and then stimulated with 5 μg/ml transfected poly(I:C) at 37 °C for 5 h. Cells were detached from dishes using trypsin-EDTA (Life Technologies), pelleted, and washed in ice-cold PBS containing phosphatase/protease inhibitors. Fractions were isolated using the Pierce NE-PER nuclear and cytoplasmic protein extraction kit (Life Technologies). For the IRF3 DNA binding assay, cells were incubated on ice in hypotonic buffer (20 mm HEPES, 5 mm NaF, 10 μm Na2MoO4, 0.1 mm EDTA) for 15 min followed by cytoplasmic membrane disruption with 0.1% Nonidet P-40. Nuclei were pelleted at 12,000 × g for 30 s, washed in hypotonic buffer, and lysed in complete lysis buffer (Abcam) containing phosphatase and protease inhibitors and DTT on ice for 45 min. Debris was removed by centrifugation, and nuclear protein extracts were quantified by bicinchoninic acid assay (Life Technologies). 10 μg of nuclear protein (in duplicate) was applied to an IRF3 transcription factor assay plate (ab207210, Abcam) or NF-κB transcription factor assay plate (ab210613, Abcam), and DNA binding was assayed as indicated by the manufacturer. Briefly, nuclear lysates were incubated in 96-well plates containing short DNA sequences corresponding to the consensus binding sequence for IRF3 or p65 for 1 h. Following washes, wells were probed with anti-IRF3 or anti-p65 antibodies (±control oligos to confirm specific binding) and then an HRP-conjugated secondary antibody. Bound transcription factors were detected using a colorimetric HRP reaction blanked against a complete lysis buffer–only control.
Immunoprecipitation
HEK293T cells were transfected with 1 μg of pCAGGS-IRF3-FLAG or in combination with C6orf106 in 6-well plates using FuGENE 6 transfection reagent (Promega). Poly(I:C) stimulation was performed 24 h post-transfection for 6 h via FuGENE 6 reagent after growth medium replacement. Cells were harvested and lysed using 500 μl of Nonidet P-40 buffer (PBS, 1% Nonidet P-40 (v/v), 1 mm phenylmethylsulfonyl fluoride) supplemented with protease inhibitor (Roche Applied Science) and 1 mg/ml bovine serum albumin (BSA) on ice for 20 min. Soluble fractions of the cell lysates were recovered by centrifugation at 16,000 × g for 30 min at 4 °C. For immunoprecipitation, 400 μl of the cell lysates was incubated with 40-μl aliquots of recombinant Protein A-Sepharose 4B (Zymed Laboratories Inc.) and 8 μl of anti-IRF3 mAb (Cell Signaling Technology) or IgG isotype at 4 °C for 16 h. The slurries were washed three times with 600 μl of Nonidet P-40 buffer to remove unbound proteins and resuspended in 2× reducing SDS loading buffer for Western blot analysis using anti-FLAG M2 antibody.
Western blotting
Cells were lysed in SDS lysis buffer (50 mm Tris-HCl, pH 8.0, 2 mm EDTA, pH 8.0, 150 mm NaCl, 0.5% (w/v) SDS, 10% glycerol) or lithium dodecyl sulfate sample buffer (Life Technologies) supplemented with a protease/phosphatase inhibitor mixture (Life Technologies). A BCA assay (Life Technologies) was used to quantify total protein concentration, and equal amounts were loaded on a 4–12 or 3–8% gradient NuPAGE polyacrylamide gel in lithium dodecyl sulfate sample buffer. Protein lysates were separated at 120 V and then transferred to a nitrocellulose membrane using the TransBlot system (Bio-Rad). Following blocking in 3% skim milk powder/Tris-buffered saline (+0.05% Tween 20) or 5% BSA (Fraction V, Sigma-Aldrich), membranes were incubated with primary antibodies in blocking solution at 4 °C overnight. Alexa Fluor 647– or HRP-conjugated secondary antibodies were diluted 1:2000 in blocking solution and incubated with the blot for 3 h at room temperature. Membranes were washed and rinsed in Tris-buffered saline with Tween 20 and incubated with enhanced chemiluminescence developing solution (Bio-Rad) and detected on a Chemi-Doc (Bio-Rad) using both luminescence and fluorescence detectors.
Immunofluorescence
Cells were fixed in 4% (w/v) paraformaldehyde in PBS at room temperature for 20 min followed by permeabilization with 0.1% (v/v) Triton X-100 and quenching with 0.2 m glycine for 10 min each at room temperature. Fixed cells were blocked in 1% BSA in PBS for 30 min, then incubated with primary antibodies in blocking solution for 1 h at room temperature, washed three times in 0.2% BSA in PBS, and incubated with secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 568 (Life Technologies) in 1% BSA in PBS at room temperature for 1 h. Cells were washed in PBS, then counterstained with the nuclear dye DAPI (0.5 μg/ml), and viewed on a Leica SP5 confocal microscope. Fn/c ratios were calculated using ImageJ from at least 50 representative cells per sample as described previously (46).
Author contributions
R. L. A., T. E. A., A. G. D. B., and C. R. S. conceptualization; R. L. A. data curation; R. L. A. and T. E. A. formal analysis; R. L. A. validation; R. L. A. and C. R. S. investigation; R. L. A. visualization; R. L. A. and Y. C. L. methodology; R. L. A. writing-original draft; R. L. A., T. E. A., and C. R. S. project administration; R. L. A., Y. C. L., T. E. A., A. G. D. B., and C. R. S. writing-review and editing; T. E. A., A. G. D. B., and C. R. S. supervision; T. E. A. and C. R. S. funding acquisition; A. G. D. B. and C. R. S. resources.
Supplementary Material
Acknowledgments
We thank Dr. Gregory Moseley (Monash University, Australia) for the kind gift of the IRF3-FLAG expression plasmid. We also thank the Pathology and Pathogenesis Group at Australian Animal Health Laboratory, CSIRO for microscopy assistance. We acknowledge the Australian Microscopy and Microanalysis Research Facility for support with equipment within the microscopy facility.
This work was supported by the CSIRO and CSIRO Office of the Chief Executive postdoctoral fellowships (to R. L. A. and Y. C. L.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2.
- PAMP
- pathogen-associated molecular pattern
- RIG-I
- retinoic acid–inducible gene I
- TLR
- Toll-like receptor
- RLR
- RIG-I–like receptor
- IRF
- interferon-regulatory factor
- CBP
- CREB-binding protein
- CREB
- cAMP-response element–binding protein
- IFN
- interferon
- MDA5
- melanoma differentiation–associated gene 5
- TRIF
- TIR domain–containing adapter inducing IFN-β
- MAV
- mitochondrial activated signaling protein
- ATF2
- activating transcription factor 2
- TNF
- tumor necrosis factor
- FOXO1
- Forkhead box protein O1
- RAUL
- RTA-associated ubiquitin ligase
- TRIM26
- tripartite motif–containing 26
- cFLIPL
- cellular FLICE-like inhibitory protein
- HeV
- Hendra virus
- C6orf106
- chromosome 6 ORF 106
- IL
- interleukin
- SeV
- Sendai virus
- ISRE
- interferon-sensitive response element
- Fn/c
- ratio of nuclear to cytoplasmic staining
- CHX
- cycloheximide
- ISG
- interferon-stimulated gene
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- qRT-PCR
- quantitative RT-PCR
- HRP
- horseradish peroxidase
- ANOVA
- analysis of variance
- IκBα
- inhibitor κBα.
References
- 1. Schoggins J. W., and Rice C. M. (2011) Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 10.1016/j.coviro.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee C. K., Rao D. T., Gertner R., Gimeno R., Frey A. B., and Levy D. E. (2000) Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 165, 3571–3577 10.4049/jimmunol.165.7.3571 [DOI] [PubMed] [Google Scholar]
- 3. Ito T., Amakawa R., Inaba M., Ikehara S., Inaba K., and Fukuhara S. (2001) Differential regulation of human blood dendritic cell subsets by IFNs. J. Immunol. 166, 2961–2969 10.4049/jimmunol.166.5.2961 [DOI] [PubMed] [Google Scholar]
- 4. Le Bon A., Thompson C., Kamphuis E., Durand V., Rossmann C., Kalinke U., and Tough D. F. (2006) Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 176, 2074–2078 10.4049/jimmunol.176.4.2074 [DOI] [PubMed] [Google Scholar]
- 5. Tough D. F. (2012) Modulation of T-cell function by type I interferon. Immunol. Cell Biol. 90, 492–497 10.1038/icb.2012.7 [DOI] [PubMed] [Google Scholar]
- 6. Alexopoulou L., Holt A. C., Medzhitov R., and Flavell R. A. (2001) Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 10.1038/35099560 [DOI] [PubMed] [Google Scholar]
- 7. Lund J. M., Alexopoulou L., Sato A., Karow M., Adams N. C., Gale N. W., Iwasaki A., and Flavell R. A. (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 101, 5598–5603 10.1073/pnas.0400937101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., and Fujita T. (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 9. Triantafilou K., Vakakis E., Kar S., Richer E., Evans G. L., and Triantafilou M. (2012) Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 125, 4761–4769 10.1242/jcs.103887 [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto M., Oshiumi H., and Seya T. (2011) Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. 21, 67–77 10.1002/rmv.680 [DOI] [PubMed] [Google Scholar]
- 11. Seth R. B., Sun L., Ea C. K., and Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 12. Honda K., and Taniguchi T. (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- 13. Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., and Thanos D. (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell 103, 667–678 10.1016/S0092-8674(00)00169-0 [DOI] [PubMed] [Google Scholar]
- 14. Libermann T. A., and Baltimore D. (1990) Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol. Cell. Biol. 10, 2327–2334 10.1128/MCB.10.5.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collart M. A., Baeuerle P., and Vassalli P. (1990) Regulation of tumor necrosis factor α transcription in macrophages: involvement of four κB-like motifs and of constitutive and inducible forms of NF-κB. Mol. Cell. Biol. 10, 1498–1506 10.1128/MCB.10.4.1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zagariya A., Mungre S., Lovis R., Birrer M., Ness S., Thimmapaya B., and Pope R. (1998) Tumor necrosis factor α gene regulation: enhancement of C/EBPβ-induced activation by c-Jun. Mol. Cell. Biol. 18, 2815–2824 10.1128/MCB.18.5.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fan Y., Mao R., Yu Y., Liu S., Shi Z., Cheng J., Zhang H., An L., Zhao Y., Xu X., Chen Z., Kogiso M., Zhang D., Zhang H., Zhang P., et al. (2014) USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 211, 313–328 10.1084/jem.20122844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nistal-Villán E., Gack M. U., Martínez-Delgado G., Maharaj N. P., Inn K. S., Yang H., Wang R., Aggarwal A. K., Jung J. U., and García-Sastre A. (2010) Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-β production. J. Biol. Chem. 285, 20252–20261 10.1074/jbc.M109.089912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lei C. Q., Zhang Y., Xia T., Jiang L. Q., Zhong B., and Shu H. B. (2013) FoxO1 negatively regulates cellular antiviral response by promoting degradation of IRF3. J. Biol. Chem. 288, 12596–12604 10.1074/jbc.M112.444794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu Y., and Hayward G. S. (2010) The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 10.1016/j.immuni.2010.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang P., Zhao W., Zhao K., Zhang L., and Gao C. (2015) TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 11, e1004726 10.1371/journal.ppat.1004726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song S., Lee J. J., Kim H. J., Lee J. Y., Chang J., and Lee K. J. (2016) Fas-associated factor 1 negatively regulates the antiviral immune response by inhibiting translocation of interferon regulatory factor 3 to the nucleus. Mol. Cell. Biol. 36, 1136–1151 10.1128/MCB.00744-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gates L. T., and Shisler J. L. (2016) cFLIPL interrupts IRF3-CBP-DNA interactions to inhibit IRF3-driven transcription. J. Immunol. 197, 923–933 10.4049/jimmunol.1502611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowie A. G., and Unterholzner L. (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 10.1038/nri2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grandvaux N., tenOever B. R., Servant M. J., and Hiscott J. (2002) The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis. 15, 259–267 10.1097/00001432-200206000-00008 [DOI] [PubMed] [Google Scholar]
- 26. Mulhern O., Harrington B., and Bowie A. G. (2009) Modulation of innate immune signalling pathways by viral proteins. Adv. Exp. Med. Biol. 666, 49–63 10.1007/978-1-4419-1601-3_4 [DOI] [PubMed] [Google Scholar]
- 27. Deffrasnes C., Marsh G. A., Foo C. H., Rootes C. L., Gould C. M., Grusovin J., Monaghan P., Lo M. K., Tompkins S. M., Adams T. E., Lowenthal J. W., Simpson K. J., Stewart C. R., Bean A. G., and Wang L. F. (2016) Genome-wide siRNA screening at biosafety level 4 reveals a crucial role for fibrillarin in henipavirus infection. PLoS Pathog. 12, e1005478 10.1371/journal.ppat.1005478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodriguez J. J., Wang L. F., and Horvath C. M. (2003) Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J. Virol. 77, 11842–11845 10.1128/JVI.77.21.11842-11845.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satterfield B. A., Cross R. W., Fenton K. A., Agans K. N., Basler C. F., Geisbert T. W., and Mire C. E. (2015) The immunomodulating V and W proteins of Nipah virus determine disease course. Nat. Commun. 6, 7483 10.1038/ncomms8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marsh G. A., de Jong C., Barr J. A., Tachedjian M., Smith C., Middleton D., Yu M., Todd S., Foord A. J., Haring V., Payne J., Robinson R., Broz I., Crameri G., Field H. E., et al. (2012) Cedar virus: a novel henipavirus isolated from Australian bats. PLoS Pathog. 8, e1002836 10.1371/journal.ppat.1002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X., and Erikson R. L. (2003) Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 5789–5794 10.1073/pnas.1031523100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao X. J., Dong Q., Bindas J., Piganelli J. D., Magill A., Reiser J., and Kolls J. K. (2008) TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J. Immunol. 181, 3049–3056 10.4049/jimmunol.181.5.3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reimer T., Brcic M., Schweizer M., and Jungi T. W. (2008) poly(I:C) and LPS induce distinct IRF3 and NF-κB signaling during type-I IFN and TNF responses in human macrophages. J. Leukoc. Biol. 83, 1249–1257 10.1189/jlb.0607412 [DOI] [PubMed] [Google Scholar]
- 34. Jiang G., Zhang X., Zhang Y., Wang L., Fan C., Xu H., Miao Y., and Wang E. (2015) A novel biomarker C6orf106 promotes the malignant progression of breast cancer. Tumour Biol. 36, 7881–7889 10.1007/s13277-015-3500-5 [DOI] [PubMed] [Google Scholar]
- 35. Zhang X., Miao Y., Yu X., Zhang Y., Jiang G., Liu Y., Yu J., Han Q., Zhao H., and Wang E. (2015) C6orf106 enhances NSCLC cell invasion by upregulating vimentin, and downregulating E-cadherin and P120ctn. Tumour Biol. 36, 5979–5985 10.1007/s13277-015-3274-9 [DOI] [PubMed] [Google Scholar]
- 36. Zhang M., Tian Y., Wang R. P., Gao D., Zhang Y., Diao F. C., Chen D. Y., Zhai Z. H., and Shu H. B. (2008) Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res. 18, 1096–1104 10.1038/cr.2008.277 [DOI] [PubMed] [Google Scholar]
- 37. Suhara W., Yoneyama M., Kitabayashi I., and Fujita T. (2002) Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J. Biol. Chem. 277, 22304–22313 10.1074/jbc.M200192200 [DOI] [PubMed] [Google Scholar]
- 38. Kumar K. P., McBride K. M., Weaver B. K., Dingwall C., and Reich N. C. (2000) Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol. 20, 4159–4168 10.1128/MCB.20.11.4159-4168.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gates-Tanzer L. T., and Shisler J. L. (2018) Cellular FLIP long isoform (cFLIPL)-IKKα interactions inhibit IRF7 activation, representing a new cellular strategy to inhibit IFNα expression. J. Biol. Chem. 293, 1745–1755 10.1074/jbc.RA117.000541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ferrari R., Gou D., Jawdekar G., Johnson S. A., Nava M., Su T., Yousef A. F., Zemke N. R., Pellegrini M., Kurdistani S. K., and Berk A. J. (2014) Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16, 663–676 10.1016/j.chom.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. François S., Sen N., Mitton B., Xiao X., Sakamoto K. M., and Arvin A. (2016) Varicella-zoster virus activates CREB, and inhibition of the pCREB-p300/CBP interaction inhibits viral replication in vitro and skin pathogenesis in vivo. J. Virol. 90, 8686–8697 10.1128/JVI.00920-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L., Grossman S. R., and Kieff E. (2000) Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. U.S.A. 97, 430–435 10.1073/pnas.97.1.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melroe G. T., Silva L., Schaffer P. A., and Knipe D. M. (2007) Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-β induction. Virology 360, 305–321 10.1016/j.virol.2006.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeong M. J., Kim E. J., Cho E. A., Ye S. K., Kang G. H., and Juhnn Y. S. (2013) cAMP signalling decreases p300 protein levels by promoting its ubiquitin/proteasome dependent degradation via Epac and p38 MAPK in lung cancer cells. FEBS Lett. 587, 1373–1378 10.1016/j.febslet.2013.03.010 [DOI] [PubMed] [Google Scholar]
- 45. Wang S. A., Hung C. Y., Chuang J. Y., Chang W. C., Hsu T. I., and Hung J. J. (2014) Phosphorylation of p300 increases its protein degradation to enhance the lung cancer progression. Biochim. Biophys. Acta 1843, 1135–1149 10.1016/j.bbamcr.2014.02.001 [DOI] [PubMed] [Google Scholar]
- 46. Oksayan S., Wiltzer L., Rowe C. L., Blondel D., Jans D. A., and Moseley G. W. (2012) A novel nuclear trafficking module regulates the nucleocytoplasmic localization of the rabies virus interferon antagonist, P protein. J. Biol. Chem. 287, 28112–28121 10.1074/jbc.M112.374694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Monson E. A., Crosse K. M., Das M., and Helbig K. J. (2018) Lipid droplet density alters the early innate immune response to viral infection. PLoS One 13, e0190597 10.1371/journal.pone.0190597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bae Y., Choi D., Rhim H., and Kang S. (2010) Hip2 interacts with cyclin B1 and promotes its degradation through the ubiquitin proteosome pathway. FEBS Lett. 584, 4505–4510 10.1016/j.febslet.2010.10.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.