

Abstract
In eukaryotes, the synthesis and uptake of sterols undergo stringent multivalent regulation. Both individual enzymes and transcriptional networks are controlled to meet changing needs of the many sterol pathway products. Regulation is tailored by evolution to match regulatory constraints, which can be very different in distinct species. Nevertheless, a broadly conserved feature of many aspects of sterol regulation is employment of proteostasis mechanisms to bring about control of individual proteins. Proteostasis is the set of processes that maintain homeostasis of a dynamic proteome. Proteostasis includes protein quality control pathways for the detection, and then the correction or destruction, of the many misfolded proteins that arise as an unavoidable feature of protein-based life. Protein quality control displays not only the remarkable breadth needed to manage the wide variety of client molecules, but also extreme specificity toward the misfolded variants of a given protein. These features are amenable to evolutionary usurpation as a means to regulate proteins, and this approach has been used in sterol regulation. We describe both well-trod and less familiar versions of the interface between proteostasis and sterol regulation and suggest some underlying ideas with broad biological and clinical applicability.
Keywords: ERAD, SCAP, INSIG, SSD, HMG-CoA reductase, Hrd1
INTRODUCTION
The continuous requirement for sterols and related molecules, either produced by the sterol-synthesizing mevalonate pathway or taken up from the extracellular world, raises a biochemical double-edged sword. The sterol synthetic pathway is metabolically costly, and later stages are heavily oxygen dependent. Furthermore, pathway intermediates can cause toxicity in a variety of biophysical and pharmacological ways. Sterols and related molecules also act as ligands in signaling and development. As a result, concentrations of sterol pathway molecules must be tightly regulated in space and time. Regulation involves simultaneous control of synthesis, uptake, and cellular efflux of a variety of molecules that are all made by this highly branched metabolic pathway.
A surprising number of sterol regulatory mechanisms pertain to protein quality control, which is the set of cellular processes that detect and allow remediation of misfolded proteins. Proteostasis pathways have been around since the dawn of proteins, and evolution has apparently capitalized on the specificity and alacrity of these mechanisms in many facets of sterol regulation. Our review is not comprehensive, but fortunately many current examples exist (Brown & Goldstein 2008, Jiang & Song 2014, Jo & DeBose-Boyd 2010, Raychaudhuri et al. 2012, Sharpe et al. 2014, Ye & DeBose-Boyd 2011, Zhang et al. 2012). This review tells a particular set of stories about using protein quality control to regulate metabolism. We hope to provide new ways of thinking about both sterol regulation and proteostasis. We unabashedly own our bias, being involved in and intrigued by this interface between protein folding and physiological control. So in one sense the old saying “all is yellow to the jaundiced eye” may apply. Alternatively, we think we are providing a new pair of glasses, rose tinted by protein folding, to view the pervasive, highly conserved, and eminently translatable processes of sterol biology. We describe the best-studied routes of sterol pathway regulation to demonstrate the prevalence of proteostatic tactics employed to effect regulation. In addition, we highlight newer examples as avenues for ongoing inquiry into two of the oldest cellular professions: protein quality control and sterol synthesis.
BACKGROUND
The sterol pathway produces a dazzling variety of lipid molecules (Figure 1). The breadth of products includes not only cholesterol and other sterols, but also a massive collection of isoprenoids (based on the 2-methyl butyl carbon skeleton) and derivatives used in all walks of organismal life. This highly branched sequence of reactions—also termed the mevalonate pathway—includes essential molecules in nearly every cellular process. Approximations for the number of distinct natural isoprenoids top 30,000 (Lange et al. 2000). Accordingly, in a sense the sterol/mevalonate pathway can be viewed as a Darwinian combinatoric library. Regulation of sterol synthesis occurs at several levels, including coordinated feedback regulation of multiple gene groups. In addition, several key enzymes are controlled. The prevailing theme in all these layers is modification of protein levels rather than activity per se, through the use of protein synthesis, trafficking, quality control, and degradation.
Figure 1.
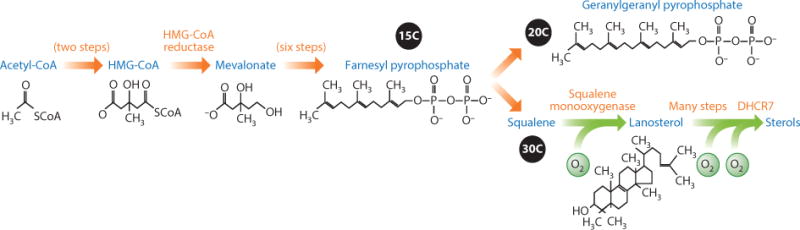
The sterol or mevalonate pathway. Only the salient features of this widely conserved and essential biosynthetic pathway are shown. The carbon number for some intermediates is shown in black circles (because carbon is black). Orange arrows indicate the early, oxygen-independent section of the pathway. Green arrows indicate the later, sterol-synthesizing, oxygen-dependent section of the pathway.
Sterol synthesis in eukaryotes occurs by the conversion of acetate in the greater-than-30-step mevalonate pathway (Figure 1). The pathway proceeds by sequential addition of 2-carbon acetate groups from acetyl-CoA to produce 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA). The reduction of HMG-CoA to mevalonic acid by HMG-CoA reductase (HMGR) is considered a rate-controlling step in many circumstances (Geelen et al. 1986). Mevalonate then undergoes phosphorylation and loss of CO2 to produce isopentanoid building blocks, which are used to assemble the remarkable variety of molecules seen in the biosphere. The sterol pathway condenses these first molecules into the 15-carbon farnesyl pyrophosphate and then the 30-carbon squalene, which is then oxidized and rearranged to produce the first sterol shape, lanosterol. Next, a variety of reactions produce the main sterol of a given organism (mammals: cholesterol; yeast: ergosterol; plants: phytosterol) used to create both biologically amenable membranes and a variety of high-potency sterol-based mediators and hormones (Bloch 1965). Numerous enzymes of the mainstream sterol pathway show highly conserved regulation. They are introduced as the examples relevant to our story arise below, along with the pertinent sterol-regulatory proteins. A motif parts list emerges that will be useful to frame our destructive and “misfolded” view of sterol regulation.
Mammalian sterol uptake is a whole-organism affair that starts with dietary sterols and ends with cellular uptake and distribution of sterols and other lipids. Sterols are given safe and soluble passage through the bloodstream as passengers in a variety of lipoprotein particles.
The most famous and best-studied example is the receptor-mediated uptake of plasma low-density lipoprotein (LDL) by the LDL receptor (LDLR). More recent additions to the uptake story include elucidation of the Niemann-Pick cholesterol intracellular transport proteins (Infante et al. 2008a,b; Ohgami et al. 2004; Vanier 2015) and the intestinal transport factor NPC1L1 (Niemann-Pick C1-Like 1), which allows for movement of dietary sterol molecules from the digestive tract to the intestinal cells that package them into chylomicrons (Abumrad & Davidson 2012, Altmann et al. 2004, Davies et al. 2000, Hussain 2014, Wang & Song 2012). Finally, sterol removal and excretion are parts of the entire balance. It is now clear that LDLRs mediate an important component of sterol removal due to catabolism and excretion after uptake (Favari et al. 2015, Jakulj et al. 2016). Furthermore, the reactions that break down sterols to foster their exit from the gut as bile acids compose another regulated component of sterol flux through mammals.
This review connects a number of sterol regulatory mechanisms to the seemingly separate field of proteostasis. Proteostasis includes the production, folding, delivery, and destruction of proteins and occurs constantly in all cells. A critical branch of proteostasis involves management of misfolded proteins. The folded integrity of the proteome is under constant challenge by the conditions of life. It has been eloquently noted that the typical cell operates in almost diametric opposition to the best practices of a biochemist (Goldberg 2003). A good biochemist keeps proteins isolated, at approximately 1 mg/ml concentrations, in the cold and away from reactive chemicals. A good cell keeps its proteins in complex mixtures; at approximately 200–300 mg/ml concentrations; often at 37°C; and in the presence of oxygen, reactive oxygen species, light of many wavelengths, and numerous reactive metabolic intermediates. So protein-based life is a compromise between the good kinetics of action and the unfortunate kinetics of misfolding. The ongoing struggle to maintain a functional, folded proteome requires a network of corrective processes collectively termed protein quality control. Protein quality control is a beautiful example of evolution’s ability to adapt cellular specificity—in this case the high selectivity that protein quality control pathways display for misfolded versions of their normally folded clients—to effect regulation.
GLOBAL REGULATION OF STEROL-RELATED PROTEIN EXPRESSION BY THE SREBP PATHWAY
Study of the molecular mechanisms of sterol regulation has a rich history. A key turning point came with a focus on mechanisms of LDL uptake more than 40 years ago by the Brown and Goldstein group (Brown & Goldstein 1974, Goldstein & Brown 1973, Schneider et al. 1982). These researchers’ discovery of, and studies on, the LDLR launched a multidecade endeavor that brought the physiological significance and the biochemical beauty of sterol regulation into wide view (Brown & Goldstein 2008; Goldstein & Brown 2009, 2015). In the course of unraveling familial hypercholesterolemia, they discovered that normal receptor-mediated LDL (and thus sterol) uptake causes marked downregulation of both sterol synthesis and LDL uptake. Feedback inhibition of sterol synthesis was profound, showing a more-than-100-fold decrease in the activity of the rate-limiting enzyme HMGR in some cases (Brown et al. 1973). These observations set the stage for deep mechanistic studies, leading to rich lines of inquiry and new biological fields that continue today. The basic model can be stated as global coordinated transcriptional control of synthesis and uptake, overlaid by posttranslational control of individual proteins. Many of the underlying mechanisms involve surprising adaptations of both synthetic and degradative aspects of proteostasis. Although the global transcriptional control is both very important and beautifully efficient, it has been reviewed numerous times in great detail (Horton et al. 2002, Osborne & Espenshade 2009, Ye & DeBose-Boyd 2011). We cover this topic here more briefly because the involved molecules define a useful parts list in exploration of the sterol-proteostasis interface.
Transcriptional control of sterol uptake and synthesis involves the transcription factor SREBP (sterol response element–binding protein). This classic bHLH-zip transcription factor is the soluble N terminus of a larger two-spanning membrane protein that resides in the endoplasmic reticulum (ER). The membrane anchor allows the SREBP to be regulated by the classic ER vesicle trafficking pathway used to process and deliver approximately 30% of all cellular proteins to various destinations in the secretory pathway. Cleavage and hence activation of SREBP rely on a protein termed SCAP (SREBP cleavage-activating protein), which is required for liberating the soluble N-terminal transcription factor from its membrane anchor. SCAP normally traffics between the ER and the Golgi complex by binding to one of the coat proteins from an ER transport vesicle. SCAP also binds to the C terminus of SREBP, and SCAP thus serves as an Uber driver for SREBP, taking it to the Golgi complex to be sequentially cleaved by two separate Golgi-resident proteases that liberate the active, soluble SREBP N terminus. Cholesterol regulates SREBP activation through this SCAP-based transport (Figure 2). When cellular cholesterol levels are high, SCAP binds to an ER-localized multispanning membrane protein termed INSIG (insulin-induced gene 1; Insig in mammals) that retains it in the ER, thus preventing activation by prohibiting SREBP trafficking to the Golgi complex. Put simply, high cholesterol blocks SREBP trafficking to its cleavage site, and low cholesterol allows SREBP trafficking to its cleavage site. Because active SREBP brings about expression of sterol synthetic enzymes and the LDLR, this scenario makes homeostatic sense. When cholesterol is low, both sterol synthesis and uptake are increased, and when cholesterol is abundant, both are curtailed. It is worth remembering that the ER-to-Golgi trafficking pathway is a major conduit of protein processing and transport, and so the theme of proteostasis looms large in the mechanism of regulation that evolution has chosen for this multigene portion of sterol regulation. Furthermore, the SCAP regulatory mechanism reveals a number of cis and trans components that are employed in a variety of other sterol regulation events. Accordingly, there is a revealing unity in several of the independent processes of sterol regulation and an interesting consistency to the idea that proteostasis has been harnessed to effect highly specific regulation.
Figure 2.
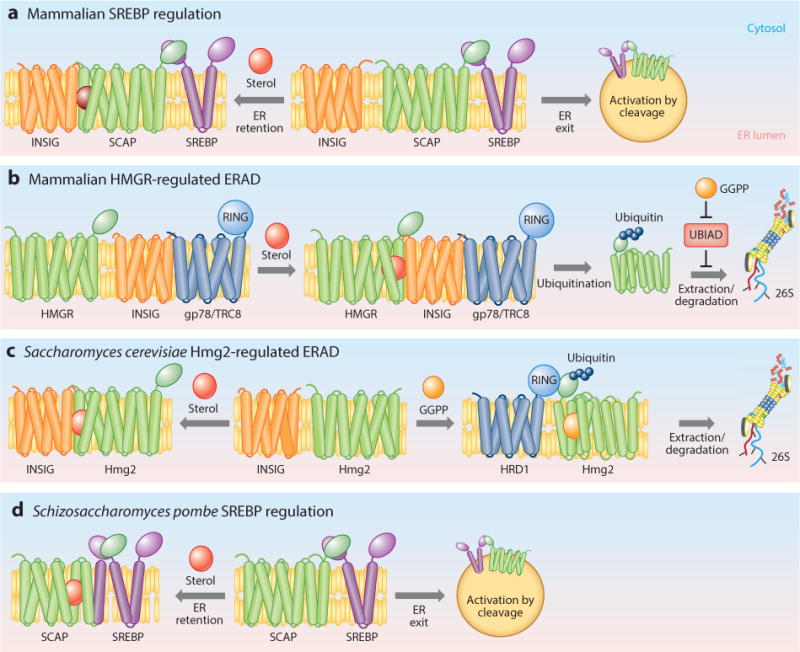
Eukaryotic tactics of sterol pathway regulation. A highly stylized cartoon showing the identities and basic features of the proteins involved in control of the sterol pathway. (a) Mammalian regulated ER retention of SCAP (and bound SREBP). (b) Mammalian regulated ER-associated degradation (ERAD) of HMG-CoA reductase (HMGR). (c) Saccharomyces cerevisiae regulated ERAD of the Hmg2 HMGR isozyme. (d) Schizosaccharomyces pombe regulated ER retention of SCAP (and bound SREBP). Sterol-sensing domain proteins are shown in green, INSIGs in orange, SREBP cargo in purple, and the ER membrane in yellow (the lumen is below, and the cytosol is above). Ligands are ball shaped. GGPP denotes geranylgeranyl pyrophosphate.
THE STEROL-SENSING DOMAIN, INSIG, AND LIPID SIGNALS: THE CENTRAL DOGMA OF STEROL REGULATION
SCAP is an eight-spanning membrane protein. Embedded in the SCAP sequence is an intriguing motif termed the sterol-sensing domain (SSD). The SSD was first noted by aligning the sequences of a variety of integral membrane proteins that have something to do with sterols (Kuwabara & Labouesse 2002, Nohturfft et al. 1998, Osborne & Rosenfeld 1998). Although this description may sound glib, the breadth of SSD protein actions renders it fairly accurate. SSD is found in proteins with diverse functions in sterol regulation, including enzymes, trafficking components, signaling devices, and degradation factors. Thus, an ongoing goal of sterol regulation is to decide whether there is an underlying common function of the SSD. The SSD consists of highly conserved residues within or near transmembrane spans 2–6 of SCAP. The SSD was originally speculated to bind cholesterol and to thus allow an overlay of regulation or sensing (Goldstein et al. 2006, Radhakrishnan et al. 2004). However, further studies on SCAP indicate that SSD function is distinct from sterol binding (Motamed et al. 2011). Nevertheless, the extreme conservation of this motif over two billion years of evolution demands focus on its function. The highly conserved residues of the SSD are consistently demonstrable as important in a variety of regulatory actions, indicating the phenotypic importance of the SSD. Through the lens of proteostasis, an interesting hypothesis about an SSD function emerges. This view also depends on another key player in the sterol regulatory arena: INSIG proteins.
The INSIG proteins (Insig1 and -2 in mammals) reside in the ER, where they anchor SCAP in a cholesterol-dependent manner (Yang et al. 2002). The INSIG proteins are highly conserved in both sequence and function from yeast to mammals (Burg et al. 2008, Flury et al. 2005). The ability of cholesterol and other sterols to regulate the trafficking of SCAP—and thus the processing of SREBP—is due to a cholesterol-dependent change in the conformation of SCAP that allows binding to Insig and thus precludes trafficking (Adams et al. 2003, Brown et al. 2002). Direct biochemical assays on purified SCAP demonstrated that cholesterol binds to SCAP with a nanomolar Kd, causing a structural change in the pure protein. The original idea that the SSD was responsible for sterol binding was supplanted by studies showing that the first luminal loop of the protein (outside the SSD) was necessary and sufficient for high-affinity binding of sterols (Motamed et al. 2011). The model from a variety of studies is that the SSD is responsible for engaging INSIG; it has even been proposed that the SSD designation be changed to IBD (INSIG binding domain) to emphasize this function (Motamed et al. 2011). However, in the context of SSD action outside of the mammalian arena, the SSD has autonomous on-pathway functions that suggest that the IBD moniker is too narrow.
The story of these parts is further complicated by the observation that INSIGs also have affinity for a distinct class of sterols, and INSIG binding of sterols can similarly restrict SCAP to the ER. The structure activity of INSIG-sterol binding is distinct from that of SCAP, as far as has been measured, with the strongest ligands being oxidized species (Adams et al. 2004, Radhakrishnan et al. 2007). Thus, there may be in this regulatory axis an ability to monitor a number of facets of sterol anabolism, catabolism, and transport. A variety of studies show that both the SSD and INSIGs have long and separate evolutionary histories that came together in eukaryotes (Hausmann et al. 2009, Kuwabara & Labouesse 2002, Ren et al. 2015). What is clear is that the functional relationship between SSD and INSIG proteins is often but not always intertwined; with this parts list now in hand, we turn to examples that employ these components in revealing ways in mammals and other eukaryotes.
REGULATED DEGRADATION OF HMG-COA REDUCTASE: ERADICATION OF EXCESS ACTIVITY
HMGR undergoes feedback-regulated degradation as a part of pathway control. In cell culture, the activity of HMGR varies up to 200 fold, depending on sterol abundance and availability. Similarly, in vivo in animals, liver levels of the enzyme can oscillate more than two orders of magnitude during a typical diurnal cycle (Hwang et al. 2016). This impressive range appears to be entirely due to altering the amount of HMGR protein. Although the cytosolic HMGR catalytic domain undergoes control by phosphorylation (Beg et al. 1985, Omkumar et al. 1994), this modification is reserved for switching between catabolic and anabolic states. Accordingly, HMGR production and degradation are used to regulate the activity of this enzyme. Synthesis is controlled en bloc by the SREBP pathway described above, and this mechanism accounts for approximately 10–20-fold variation. The remainder of the variation is due to regulated degradation of the HMGR enzyme, which responds in the expected way to flux through the sterol pathway: When levels of sterol pathway products are abundant, HMGR undergoes rapid degradation (Edwards et al. 1983). When pathway flux is slowed, HMGR becomes stable, and the difference in half-life is on the order of 30 min in the rapidly degraded state to more than 6 h when stable.
Regulated degradation of HMGR is a highly selective process, targeting only this protein. The high specificity makes this branch of sterol regulation particularly interesting as a novel clinical target and has engendered much research (Sever et al. 2003a,b; Ravid et al. 2000; Roitelman & Simoni 1992). HMGR is an ER-resident integral membrane protein, with an N-terminal eight-spanning membrane anchor followed by a linker and a highly conserved cytoplasmic catalytic domain that catalyzes the rate-limiting production of mevalonic acid. The large transmembrane anchor is necessary and sufficient for regulated degradation by ubiquitination. The ubiquitin pathway involves a cascade of enzymatic reactions that result in construction of a multiubiquitin chain on a substrate, which targets that protein for destruction by the 26S proteasome. Ubiquitination is brought about by transfer of ubiquitin from one of a small number (10 in yeast, ∼50 in mammals) of E2 ubiquitin-conjugating enzymes (UBCs) to the substrate or substrate-bound growing multiubiquitin chain (Amm et al. 2014, Kleiger & Mayor 2014). Transfer of ubiquitin from the UBC to the substrate is catalyzed by an E3 ubiquitin ligase, which is responsible for the specificity of ubiquitination. HMGR degradation is accomplished by regulated ubiquitination, in which the enzyme is tagged with multiple copies of the ubiquitin, followed by its extraction from the ER membrane and proteolysis by the cytosolic 26S proteasome. Our understanding of mammalian HMGR ubiquitination owes much to the elegant work of the DeBose-Boyd group (Jo et al. 2011; Song et al. 2005a,b). The initial model involved a single E3 ubiquitin ligase and some of the familiar components from the SREBP story: Like SCAP, the HMGR transmembrane anchor has an SSD, and this domain is required for regulated degradation. When sterol levels are high, HMGR associates with Insig. Insig binding brings an ER-resident E3 ubiquitin ligase known as gp78 into close proximity with HMGR, causing its ubiquitination and eventual degradation by the cytosolic proteasome. gp78 is one of a number of E3 ubiquitin ligases involved in ER-associated protein degradation (ERAD), by which misfolded proteins of the ER are selectively detected and destroyed (Brodsky 2012, Hirsch et al. 2009, Needham & Brodsky 2013). A variety of highly conserved pathways participate in ERAD. Because many of the maladies of aging involve pathologies associated with misfolded proteins, degradative quality control is an active field of endeavor. Although the regulated recruitment of gp78 to HMGR is highly specific, the range of substrates that are removed by ERAD pathways is very broad. Thus, in this aspect of sterol regulation, again a highly general proteostatic process is adapted to bring about a highly specific regulatory outcome. For SREBP, the high-throughput conveyor belt of ER-to-Golgi transport is involved, and in this context, one of the broad-specificity ERAD quality control pathways operates in conjunction with protein synthesis and folding to ensure a well-folded proteome.
Since those initial studies, it has become clear that mammalian HMGR degradation is more complex than this initial understanding. Another multispanning ER-resident E3 ubiquitin ligase termed TRC8 is also capable of supporting regulated degradation of HMGR (Jo et al. 2011). Moreover, gp78 can apparently degrade TRC8. Furthermore, TRC8 has its own SSD and can interact with INSIGs, allowing for a fairly complex interplay of these factors (Jo et al. 2011, Lee et al. 2010). Significantly, even this more nuanced two-E3 view has been challenged using gp78−/− null cells (Tsai et al. 2012); evidence from those studies indicates that the model of E3 participation in HMGR degradation may be incomplete. Finally, the individual regulatory proteins that mediate HMGR and SREBP regulation clearly undergo ERAD by a variety of mechanisms. For example, TRC8 is a substrate of gp78, and the Insig proteins are also subject to degradation when they are not bound to a client (Lee et al. 2006, Lee et al. 2010). The final understanding of the true complexity of these intertwined systems may be resolvable only by newer gene editing methods, perhaps using systems biology approaches.
The principal sterol that brokers the gp78-INSIG association with HMGR is derived from lanosterol, rather than cholesterol (Lange et al. 2008; Song et al. 2005a,b). Lanosterol is produced only by the sterol pathway. Thus, regulated degradation of HMGR is keyed to sterol synthesis rather than simple sterol abundance, appropriate for HMGR’s rate-limiting status in this pathway. The HMGR presumably has a binding site for 24,25-dihydrolanosterol, although this has not been demonstrated. HMGR stability is also controlled by the 20-carbon isoprenoid geranylgeranyl pyrophosphate (GGPP). The original observation was that addition of the 20-carbon isoprene geranylgeraniol (GGOH, the unphosphorylated form of GGPP) accelerated sterol-mediated degradation of HMGR (Sever et al. 2003a,b). Recent studies have implicated the multispanning integral membrane prenyltransferase UBIAD1 (UbiA prenyltransferase domain–containing 1) by the following mechanism: UBIAD1 binds to HMGR during its sterol-dependent ubiquitination and prohibits the extraction of ubiquitinated HMGR from the ER membrane for degradation. Added GGOH is converted into the pathway intermediate GGPP, which then binds to UBIAD1, promoting its liberation from HMGR and thus allowing the steps downstream of its degradation to proceed (Schumacher et al. 2015, 2016). Thus, mammalian HMGR stability is keyed to two biosynthetic signals: GGPP and lanosterol (Figure 1).
The highly conserved sterol/mevalonate pathway is found in all eukaryotes. We next discuss two examples of sterol pathway regulation separated from mammals by one billion years of evolution: regulated degradation of HMGR in Saccharomyces cerevisiae (which we term yeast) and the SREBP pathway in Schizosaccharomyces pombe. Study of common regulatory components such as INSIGs and SSDs in these model citizens of the biosphere reveals core functions and novel insights applicable to all examples.
REGULATED DEGRADATION OF HMG-COA REDUCTASE IN SACCHAROMYCES CEREVISIAE: HRD BEHAVIOR
Regulated ubiquitination of HMGR was first discovered and studied in S. cerevisiae (Hampton & Bhakta 1997, Hampton et al. 1996), and its study continues to provide insights into both sterol regulation and ERAD (Neal et al. 2017, Sato et al. 2009, Vashistha et al. 2016). HMGR is essential for life in yeast, allowing synthesis of many isoprenoids and ergosterol, which is the principal sterol. Yeast HMGR has a body plan very similar to that of mammalian HMGR: an eight-spanning N-terminal ER membrane anchor and a C-terminal cytosolic catalytic domain connected by a linker to the membrane anchor. The mammalian catalytic domain, which is ~60% identical to the yeast catalytic region, can provide the needed HMGR activity to yeast. In contrast, the eight-spanning N-terminal transmembrane region has far more limited homology but nevertheless has a clear SSD that is critical for regulated degradation of HMGR. There are two isozymes of HMGR in yeast: Hmg1 and Hmg2. Either isozyme can supply the essential enzyme activity, but only Hmg2 undergoes regulated degradation.
Regulated degradation of Hmg2 follows the dictates of feedback regulation: High flux through the mevalonate pathway promotes degradation, whereas lowered flux causes stabilization. In this way, Hmg2 half-life can vary between 15 min and 6 h (Gardner et al. 1998, Shearer & Hampton 2004). Genetic analysis of Hmg2 degradation led to the discovery of the HRD genes (which are responsible for Hmg-CoA reductase degradation), along with many other substrates. The HRD1 and HRD3 genes encode a pair of ER-resident proteins that form the core subunits of a highly conserved E3 ubiquitin ligase, which is responsible for ubiquitination of Hmg2, leading to proteasome-mediated degradation (Bays et al. 2001, Gardner et al. 2000, Hampton & Garza 2009, Hampton et al. 1996, Vashistha et al. 2016). The Hrd1 protein is a multispanning E3 ubiquitin ligase with an N-terminal transmembrane region and a C-terminal E3 ubiquitin ligase domain. A canonical RING-H2 domain in this E3 domain is responsible for brokering the transfer of ubiquitin, primarily from the E2 Ubc7, to Hmg2. Hrd3 (Sel1L in mammals) has a large luminal domain and is found in stoichiometric association with Hrd1. Loss of either protein causes complete removal of the HRD pathway and full stabilization of Hmg2 and all other HRD degradation substrates. Surprisingly, in addition to a role in Hmg2 degradation, the HRD pathway is central to eukaryotic ERAD. As mentioned above, ERAD is conserved in eukaryotes and is instrumental in alleviating potentially lethal ER stress (Brodsky & Skach 2011, Koenig & Ploegh 2014, Travers et al. 2000, Walter & Ron 2011). Hrd1 is a clear homolog of gp78, the main E3 ubiquitin ligase used in mammalian HMGR degradation, with a similar layout, sequence identities throughout the protein, and similar E2 requirements (Fang et al. 2001; Song et al. 2005a,b). Thus, the theme of regulation by use of protein quality control is extant across the impressive evolutionary gap that separates yeast from hepatocytes. However, unlike the complex involvement of other E3 ubiquitin ligases in addition to gp78 in mammals, Hrd1 appears to be the sole E3 ubiquitin ligase required for HMGR degradation.
The HRD pathway is responsible for the degradation of many misfolded ER proteins, including both luminal and integral membrane substrates (Bordallo et al. 1998, Hampton et al. 1996, Vashist & Ng 2004). All misfolded model substrates examined are constitutively degraded by the HRD pathway. By contrast, pathway signals regulate the entry of wild-type Hmg2 into the always operating HRD pathway (Hampton & Rine 1994). Use of drugs and molecular biological approaches indicated that the 15-carbon isoprenoid farnesyl pyrophosphate (FPP) was the source of a signal for enhanced degradation (Figure 1) (Gardner & Hampton 1999, Shearer & Hampton 2005). Subsequent work revealed that the de facto signal is in fact GGPP—the same molecule that enhances degradation of mammalian HMGR. However, unlike the mechanism proposed in mammals, in which GGPP enhances sterol-regulated ERAD of HMGR, in yeast GGPP is the primary signal for HRD-dependent Hmg2 degradation (Garza et al. 2009). Our ongoing studies suggest a simple model for GGPP action with the possibility of broad utility and possible clinical application.
The conundrum of normally folded Hmg2 undergoing regulated degradation by the constitutive HRD quality control pathway is resolved with a misfolding step as part of the regulatory circuit. It appears that GGPP causes reversible misfolding of Hmg2, allowing for enhanced degradation by the HRD machinery. This idea emerged from our initial studies with the chemical chaperone glycerol, in which Hmg2 undergoing rapid regulated degradation was immediately and strongly stabilized by the addition of glycerol, indicating that Hmg2 undergoing regulated degradation behaved like a misfolded protein (Gardner et al. 2001). An in vitro limited proteolysis assay was developed to explore this idea and is depicted in Figure 3. Hmg2 with a protected epitope tag in isolated ER microsomes undergoes a characteristic time-dependent proteolysis pattern when treated with trypsin. In vitro Hmg2 trypsinolysis is greatly slowed by the direct addition of glycerol or other chemical chaperones (Shearer & Hampton 2004) and is accelerated by the direct addition of GGPP (Garza et al. 2009). Our first studies of this in vitro structural regulation were with the neutral isoprenoid farnesol (FOH; FPP without phosphates), which showed a similar specific alteration in Hmg2 structure (Shearer & Hampton 2005). However, the effect of FOH was seen only at millimolar concentrations of FOH. By contrast, the effects of GGPP in vitro were observed with an EC50 in the low-nanomolar (∼50 nM) range (Figure 3). This finding is consistent with the clear role of GGPP as the bona fide physiological regulator of Hmg2 stability in vivo (Garza et al. 2009). The effect of GGPP on Hmg2 structure is fully reversible, and GGPP does not result in wholesale misfolding of Hmg2, because the thermal denaturation curve is unaffected at any concentration (Wangeline & Hampton 2017). Importantly, the effects of GGPP on Hmg2 stability in vivo or on Hmg2 structure in vitro are reversed by the addition of chemical chaperones such as glycerol, indicating that GGPP causes Hmg2 misfolding. The microsomal limited trypsinolysis assay has provided a simple, powerful approach to understanding GGPP-mediated regulated misfolding and the Hmg2 SSD.
Figure 3.
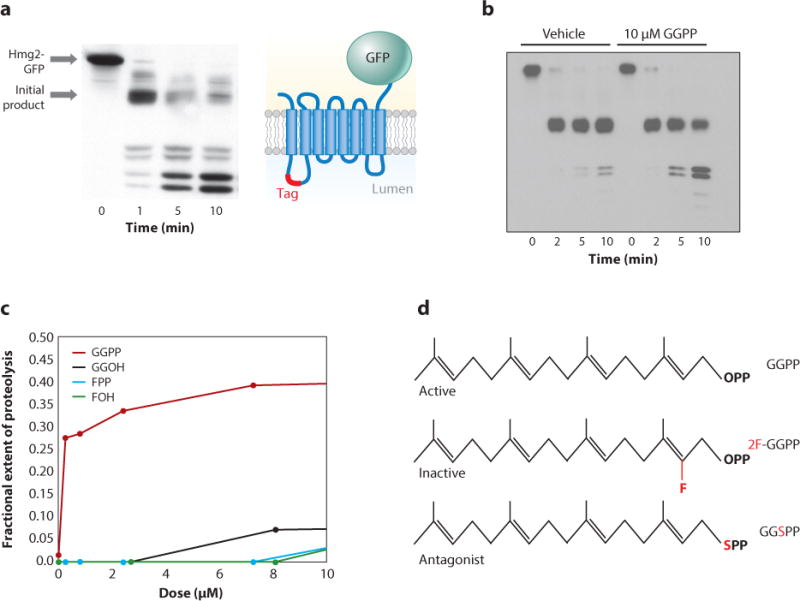
Geranylgeranyl pyrophosphate (GGPP)-mediated reversible misfolding of Hmg2. (a) An example of the trypsinolysis assay used to explore Hmg2-GFP structure. The time course of the trypsinolysis using a high concentration of trypsin, measured by SDS-PAGE and epitope tag blotting. The dark fragment in the 1-min lane is the initial product generated by rapid removal of GFP at the start of the assay. Hmg2-GFP with a luminal epitope is also depicted. (b) Effect of added GGPP on the rate of Hmg2 cleavage in the limiting trypsinolysis assay. (c) Concentration dependence of GGPP in altering Hmg2-GFP structure in the trypsinolysis assay, along with several other isoprenoids. GGPP EC50 ∼ 50 nM. Extent of proteolysis is defined as the fraction of the major fragment degraded. Abbreviations: FOH, farnesol; FPP, farnesyl pyrophosphate; GGOH, geranylgeraniol. Adapted from Wangeline & Hampton (2017). (d) The GGPP effect is highly specific; 2F-GGPP has no effect on Hmg2 trypsinolysis, whereas GGSPP is an antagonist of GGPP in vitro and in vivo.
THE NECESSARY AND AUTONOMOUS STEROL-SENSING DOMAIN
The ongoing work on Hmg2 has allowed us to study a critical and autonomous function for the SSD. The Hmg2 N-terminal transmembrane domain has a clearly defined SSD, with residues found in transmembrane spans 2–6 (Kuwabara & Labouesse 2002, Theesfeld et al. 2011). The simplicity of the in vitro Hmg2 regulatory readout provides a unique opportunity to move toward understanding SSD function. Deeply conserved residues are shared in SSDs spread over billions of years of evolution. We found in our initial studies of the Hmg2 SSD that many of these highly conserved residues are critical for regulated degradation of Hmg2 (Theesfeld et al. 2011). A typical example is serine 215 (S215). The S215A mutant is completely stable in vivo at all levels of GGPP, whether added exogenously or by forced production with enzymes. The limited proteolysis assay revealed that the S215A mutant of Hm2 no longer undergoes the characteristic GGPP-stimulated structural transition. An alanine scan of all Hmg2 conserved SSD residues resulted in a simple binning of residue functions: Either alanine substitution had no effect on GGPP-stimulated degradation, or a replacement caused strong stabilization of Hmg2. In no case did replacement of a highly conserved SSD residue cause destabilization of Hmg2. Thus, the SSD apparently allows for GGPP-regulated misfolding. Taken alone, this observation leads to the conclusion that SSD mediates either GGPP recognition or misfolding mediated by GGPP bound to a different site. We are currently involved in resolving these two scenarios but favor the transmission role for the reasons described below.
Whatever the molecular function of the SSD, a key general feature emerges from these studies: The SSD has a bona fide regulatory function independent of the INSIG proteins known to be intimately involved in SSD protein action in mammals. Importantly, yeast has two INSIG proteins, Nsg1 and Nsg2; they interact with Hmg2 and participate in sterol regulation. However, removal of both Nsg1 and Nsg2 has no effect on GGPP-stimulated Hmg2 degradation. Thus, the Hmg2 SSD is autonomously required for permission to enter the HRD pathway, and alteration of many of the highly conserved residues—often to amino acids that are more suited to be within a lipid bilayer—results in a version of Hmg2 that can no longer undergo feedback regulation.
TWO SIGNALS: THE LOGIC OF HMG2 REGULATION
The independence of Hmg2 SSD–dependent function from INSIGs is not due to an absence of these proteins in yeast sterol regulation. In fact, yeast INSIGs provide an additional layer of regulation to regulated degradation of Hmg2. In striking contrast to the mammalian situation, Nsg1 inhibits Hmg2 degradation, so sufficient levels of Nsg1 will block the GGPP-dependent degradation of Hmg2. Nsg-mediated stabilization of Hmg2 apparently occurs at the level of regulated misfolding as well: The presence of stoichiometric levels of Nsg1 slows the in vitro limited proteolysis of Hmg2 in a manner very similar to that of other stabilizing influences such as chemical chaperones (Flury et al. 2005).
This action of Nsg1 occurs at native levels of both Hmg2 and Nsg1 and was initially perplexing. Why would Hmg2 have elaborate stability regulation only to be blocked by Nsg1? The logic of this extra layer of regulation came from the realization that, as in the mammal, the Hmg2-Nsg1 interaction is sterol dependent. Nsg1 inhibits Hmg2 degradation by interacting with the Hmg2 transmembrane region, and this interaction requires lanosterol. When lanosterol is present, Nsg1 binds to Hmg2, prohibiting GGPP-mediated regulated degradation (Theesfeld & Hampton 2013). Why have this extra layer of regulation? We think the action of Nsg1 allows an oxygen contingency to Hmg2-regulated degradation. The first part of the sterol pathway up to the 30-carbon squalene is oxygen independent. Later steps are strongly oxygen dependent (Figure 1). Accordingly, GGPP can be made in the absence of oxygen, whereas production of the first bona fide sterol, lanosterol, requires oxygen. Because of the interplay of GGPP-stimulated Hmg2 degradation and lanosterol/Nsg1-mediated stabilization, Hmg2 will undergo regulated degradation only when GGPP is abundant and lanosterol is not. These are exactly the biochemical conditions of anaerobiosis, and this model is detailed in Theesfeld & Hampton (2013). Whatever the purpose of this two-signal control, it is interesting that both yeast and mammals measure GGPP and lanosterol to regulate HMGR activity. Mammalian HMGR is degraded when lanosterol is abundant, and degradation is further enhanced when GGPP is also abundant (Sever et al. 2003a,b). Yeast has evolved a distinct response to the same two signals, using the same core molecular devices. Although our model pertains to anaerobiosis, Hmg2-regulated degradation occurs in all growth conditions, allowing for easy study in any circumstance in which Hmg2 levels surpass those of Nsg1 (Flury et al. 2005).
GGPP-AND LIGAND-REGULATED MISFOLDING: THE DARK SIDE OF ALLOSTERY
The key observation from Hmg2-regulated degradation is that GGPP-mediated Hmg2 degradation is dependent on the SSD, and this dependence represents an autonomous function of this motif. INSIG-mediated, sterol-dependent regulation of Hmg2 stability inhibits the action of GGPP, but GGPP-regulated Hmg2 degradation is completely INSIG independent. Accordingly, Hmg2 regulation presents an unparalleled opportunity to learn about the conserved function of the SSD motif and its involvement in many independent signaling axes.
We have more recently focused on the nature of the GGPP- and SSD-dependent misfolding of Hmg2. Our recent work indicates that GGPP-stimulated ERAD of Hmg2 may be a form of allosteric regulation, with many of the hallmarks of this classic mode of enzyme control but with ligand-dependent misfolding as the structural readout. Using both the in vitro limited proteolysis assay (Shearer & Hampton 2004, 2005) and in vivo Hmg2 degradation, a variety of experiments point to an allosteric model of induced Hmg2 misfolding (Wangeline & Hampton 2017).
The allosteric misfolding model arose from in vitro studies of the GGPP regulator, which by every criterion behaves like an allosteric ligand. The in vitro structural effects of GGPP are rapid and fully reversible. Washing Hmg2 microsomes treated with even 100 times the GGPP needed for the maximal change in Hmg2 structure restores Hmg2 to its untreated state. As mentioned above, GGPP is potent in this assay, with an EC50 in the 50 nM range—a concentration that is below the typical Km for yeast enzymes that process this normal isoprenoid. The most revealing data emerge from structure-activity studies. Because GGPP is a substrate of the prenyltransferases needed to activate the RAS oncogene and other signaling proteins, many analogs have been produced in the study of prenylation. We found that subtle changes in the GGPP structure render the analogs unable to cause any changes in the limited proteolysis assay or any change in Hmg2 stability in vivo (Wangeline & Hampton 2017). GGPP consists of the 20-carbon alcohol gernanylgeraniol in phosphoester linkage with a pyrophosphate group (Figure 1). Substitution of the alcohol O with an S, or replacement of a 2C methylene H with F, completely removes the ability of these analogs to affect Hmg2 structure in vitro. Furthermore, the GGSPP analog, but not the 2F version, is a GGPP antagonist of GGPP, reversing the GGPP-mediated changes in Hmg2 structure in vitro and GGPP-stimulated degradation of Hmg2 in vivo (Figure 3). The existence of an antagonist is strong, if indirect, evidence for a specific GGPP binding site required for reversible misfolding of Hmg2.
The action of GGPP sounds similar to that of an allosteric regulator, which is a generally reversible binding ligand for the regulated protein. We further explored this idea by examining features of the Hmg2 protein itself. We found that the Hmg2 transmembrane domain forms multimers, as is often the case for allosteric proteins. Neither the interaction with GGPP, nor the presence of conservative SSD mutants that remove the effects of GGPP, affects Hmg2 multimerization. Importantly, these highly ligand-specific effects of GGPP are reversed by a variety of chemical chaperones in vitro, indicating that folding state is the susceptible feature of Hmg2 being controlled by the ligand (Wangeline & Hampton 2017). Our current view is that reversible high-affinity binding of GGPP to a specific binding site on the Hmg2 quaternary structure causes a transition to a misfolded state that is more susceptible to ERAD-based quality control.
The idea that a small-molecule ligand can trigger the entry of a specific, normal protein to enter a degradative quality control pathway has interesting implications. Because quality control is highly specific to subtle changes in folding and is widely used in all kingdoms of life, the possibility exists for the inherent specificity of quality control to be similarly controlled by a small molecule in cases of natural protein regulation. Two examples include ligand-triggered degradation of the ER-bound IP3 receptor in mammals (Wojcikiewicz et al. 1994, 2009) and regulated destruction of an early enzyme of the LPS biosynthetic pathway in Escherichia coli (Fuhrer et al. 2006). Whatever the extent of such regulation in nature, developing molecules that can bring about ligand-regulated misfolding might allow for selective pharmaceutical manipulation of previously undruggable targets. This novel approach to drug development is being actively explored in a variety of ways (Lai & Crews 2016), and harnessing the power of allosteric misfolding could be a distinct approach that mimics natural modes of regulation. There are many known cases of pharmacological chaperones, that is, ligands that improve the folding and stability of their protein binding partners, allowing more of the protein to be accumulated by skirting destruction by quality control (Bernier et al. 2004, Leidenheimer & Ryder 2014). In contrast, we posit that GGPP is a so-called pharmacological chaotrope, causing selective misfolding of its binding partner. Because GGPP-regulated degradation of Hmg2 has many parallels to allosteric regulation but results in detectable misfolding, we suggest the term mallostery to refer to this potentially pervasive ligand-mediated alteration of a protein’s folding state (Figure 4).
Figure 4.
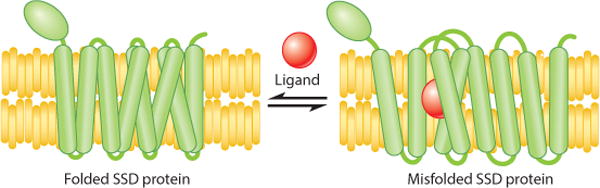
Mallostery, or a reversible, ligand-mediated misfolding model for sterol-sensing domain (SSD) proteins, in which the SSD motif imparts the ability to undergo reversible misfolding. We posit that the SSD protein functions as a multimer and effects a variant of allosteric regulation, allowing for ligand-mediated misfolding as a regulatory tactic.
GENERALIZING STEROL-SENSING DOMAIN AUTONOMY: STEROL-REGULATED SCAP IN SCHIZOSACCHAROMYCES POMBE
Our studies on Hmg2 reveal an example in which the SSD is absolutely required for a regulatory function but operates autonomously. This is in contrast to the mammalian examples, in which the INSIGs are intimately involved in the sterol-mediated regulation of SSD-containing proteins HMGR and SCAP. How often is SSD function divorced from INSIGs? The autonomy of the SSD is also evident in another fruitful nonmammal, S. pombe, as revealed by the beautiful work from the Espenshade group on sterol-regulated SREBP cleavage (Hughes et al. 2005, Porter et al. 2010, Raychaudhuri et al. 2012). This example is particularly intriguing; the main function of SREBP (Sre1 in S. pombe) regulation is apparently to employ the sterol pathway as a biochemical indicator of oxygen status (Hughes et al. 2005, Todd et al. 2006). Because sterol synthesis is strongly dependent on oxygen (Figure 1), biosynthesis of sterols is a hallmark of oxygen abundance. As in the mammal, when sterol levels drop in S. pombe, SREBP processing by cleavage to an active transcription factor increases, and transcripts are produced. But in S. pombe, the majority of these transcripts encode proteins needed for life at low oxygen, and removal of SREBP results in poor survival following hypoxia. The regulation of SREBP is similar to that in mammals: SCAP (Scp1 in S. pombe) mediates the trafficking of SREBP from the ER to the Golgi complex, where SREBP is cleaved into its soluble, transcriptionally active form. As in the mammal, in S. pombe the presence of sterol regulates SREBP processing: High sterols preclude SCAP exit from the ER, and thus SREBP processing ceases. Low sterols allow for exit and processing of SCAP and bound SREBP. However, an important difference is in the complete lack of a role for INSIG (Ins1 in S. pombe) proteins in this process. Indeed, they are present and mediate regulatory phosphorylation of HMGR (Burg et al. 2008), but the sterol-dependent retention of SCAP (with bound SREBP) is completely INSIG independent (Hughes et al. 2005). As with Hmg2, the ability of S. pombe SCAP to respond to its sterol pathway signal requires a number of conserved residues of the SSD (Hughes et al. 2008). The SSD point mutants that alter SCAP function do so by allowing SCAP to cycle constitutively at all levels of sterol. Thus, as in Hmg2, the SSD is needed for a sterol pathway molecule to alter the regulated protein. Significantly, the cleavage mechanism of S. pombe SREBP is quite different from that in mammals, revealing an intriguing new mechanism of protein processing in the Golgi compartment (Lloyd et al. 2013; Stewart et al. 2011, 2012). Nevertheless, the use of SCAP to ferry SREBP out of the ER in a sterol-regulated manner is very similar to what is seen in mammals and S. pombe and is thus revealing in comparison.
VIEWING STEROL-SENSING DOMAIN FUNCTION WITH PROTEOSTASIS-TINTED GLASSES
Is there an underlying consistency to the varied actions of this widely conserved motif? The two principal perplexing features are (a) the varied number of sterol pathway ligands or signals that seem to promote regulation through SSD-bearing clients and (b) the varied role of INSIG proteins in SSD-dependent regulation. We posit that the unifying function of the SSD in these cases is to allow ligand-mediated misfolding as a path to regulation. In the simplest case, S. pombe SCAP normally cycles between the Golgi complex and the ER with the bound SREBP cargo. When ergosterol (the S. pombe main sterol) binds to SCAP, SCAP is retained in the ER and no longer assists in trafficking of SREBP to its activation site. One of the primary responses to misfolded ER proteins is retention in the ER, as happens when ergosterol is elevated and presumably binds to S. pombe SCAP. Consistent with this idea, mutations of many of the highly conserved SSD residues in S. pombe SCAP produce a version that still exits the ER and transports SREBP but no longer shows sterol-dependent ER retention. This scenario sounds very similar to that of Hmg2, which undergoes ligand-dependent misfolding in the presence of GGPP, a totally distinct signal.
The role of INSIGs in Hmg2 regulation may fit a model of regulated quality control as well. The function of yeast INSIGs is to thwart Hrd1-dependent degradation of Hmg2. We have posited that the INSIGs represent a family of sterol-dependent chaperones for the misfolded SSD state (Flury et al. 2005, Theesfeld & Hampton 2013). Thus, GGPP-engaged Hmg2 is susceptible to INSIG binding, which stabilizes it or protects it from Hrd1-mediated ubiquitination. Stabilization of misfolded proteins is certainly one function of traditional chaperones and may be how INSIGs stabilize Hmg2 in the presence of abundant GGPP. Similarly, mammalian SCAP undergoes a structural transition in the presence of a third signal, cholesterol, and is retained in the ER by virtue of a now-permitted interaction with INSIG. If the response to sterol by mammalian SCAP were similar to that in S. pombe, then this response would be consistent with INSIG stabilizing the misfolded state and causing retention, also a function of traditional chaperones (Araki & Nagata 2011, Gidalevitz et al. 2013, Kim et al. 2013). Finally, mammalian HMGR responds to a fourth signal, lanosterol or a related metabolite (Lange et al. 2008; Song et al. 2005a,b), to undergo degradation by an ERAD quality control E3 ubiquitin ligase, gp78, that is a Hrd1 homolog. This interaction requires INSIG engagement of HMGR when sterols are abundant. Because there are numerous cases of more traditional chaperones or protein trafficking factors recruiting ubiquitin E3 ligases to degrade misfolded clients (Chen et al. 2011, Guerriero & Brodsky 2012), this embellishment would again be consistent with INSIGs serving as SSD-specific chaperones. Although this view is probably too simplistic, it creates an interesting framework for useful hypothesis generation, which would include a plethora of powerful tools and models not normally accessed in sterol biology.
If the SSD is required for programmed misfolding, it could have at least two roles: binding of the mallosteric ligand and a more structural role that allows ligand-mediated misfolding to occur. The number of distinct ligands in the four examples above implies that the SSD may be a motif that permits alteration of the protein’s folding state rather than binding per se. Consistent with this idea, the recent work on mammalian SCAP has clearly implicated a luminal loop, and not the SSD, of SCAP as a nanomolar-affinity sterol binding site (Motamed et al. 2011). This modularity would result in the SSD permitting regulated misfolding for a variety of distinct ligands. Whatever the model, it will be important and interesting to parse these two models with molecular biological and structural approaches. Recent structures of the NPC1 SSD region indicate that a new mechanistic era in understanding the SSD is now beginning (Li et al. 2016, Zhao et al. 2016).
An example of the utility of a quality control view of sterol regulation may be found in the complexities of regulated degradation of mammalian HMGR. As described above, there is some disagreement about the E3 ubiquitin ligases that are required for mammalian HMGR degradation. The explanation may lie in the nature of protein quality control. It is clear that quality control E3 ubiquitin ligases have broad and overlapping substrate ranges, as is also the case for the folding branches of proteostatic quality control (Gidalevitz et al. 2013, Vembar & Brodsky 2008). Accordingly, removal of one quality control factor will often unveil or induce another (Hirsch et al. 2009, Stolz et al. 2013). A telling example of ERAD flexibility for HMGR arose when hamster HMGR and its cognate INSIG were coexpressed in Drosophila cells. Indeed, sterol-regulated degradation of the mammalian HMGR occurred in this heterologous system. However, the E3 ubiquitin ligase required for regulated ubiquitination was none other than the endogenous Drosophila Hrd1 (dHrd1), the prototype ERAD quality control E3 ubiquitin ligase (Faulkner et al. 2013, Nguyen et al. 2009). This finding indicates that the transplanted mammalian HMGR can undergo sterol-regulated entry into a canonical ER quality control pathway in the correct circumstances. Another implication is that INSIG SSD–dependent sterol regulation of mammalian HMGR degradation may also involve regulated misfolding and may thus be susceptible to the known flexibility of ERAD E3 ubiquitin ligases. This feature could provide an explanation for, or at least a mechanistic framework with which to resolve, the varied roles of E3 ubiquitin ligases involved in mammalian HMGR ERAD.
Whether or not there is an underlying unitary explanation for SSD-mediated sterol regulation based on quality control degradation, this approach to modulation is widely employed in sterol regulation and in many other niches of biology. Below are some briefly described examples to illustrate this point and to hopefully inspire ideas and experiments.
MARCHING TO A DIFFERENT LIGASE: STEROL-REGULATED ENDOPLASMIC RETICULUM–ASSOCIATED DEGRADATION OF SQUALENE MONOOXYGENASE
Squalene monooxygenase (SM, or Erg1 in yeast) is the first oxygen-dependent step in the sterol pathway, creating an epoxide from the 30-carbon isoprenoid squalene, which is next rearranged to lanosterol, the first sterol of the pathway (Figure 1). SM is an integral ER membrane protein. It was first reported that SM undergoes cholesterol-regulated proteasome-mediated degradation in mammalian cells (Gill et al. 2011). High cholesterol levels cause increased degradation of SM, whereas low cholesterol levels decrease degradation rate. Subsequent proteomic studies in yeast indicated that SM regulation is highly conserved, and they implicated the yeast Doa10 RING-H2 E3 ubiquitin ligase (Foresti et al. 2013). This distinct ERAD pathway works alongside the HRD pathway in eukaryotes. The conserved involvement of this E3 ubiquitin ligase in SM regulation was confirmed by the finding that MARCH6 (a DOA10 homolog) is responsible for regulated SM degradation in mammals (Foresti et al. 2013, Zelcer et al. 2014). Curiously, yeast SM (Erg1) degradation is keyed to lanosterol rather than cholesterol (Foresti et al. 2013). Furthermore, the first 100 amino acids of mammalian SM is sufficient for cholesterol-regulated degradation, and although yeast lacks this region of the protein, it shows sterol-regulated degradation by the same ERAD pathway (Howe et al. 2015).
Cholesterol-regulated degradation of SM is thus another example of signal-regulated entry into an ERAD pathway. There is no SSD in SM, nor is its degradation dependent on INSIGs. However, the features of mammalian SM regulation bear many similarities to those of yeast Hmg2. SM is stabilized in vivo by application of the chemical chaperone glycerol, implying that cholesterol is causing misfolding. Cysteine-based scanning and a PEGylation cysteine modification assay confirm that SM undergoes a cholesterol-dependent conformational change (Howe et al. 2015). Because there is a lack of known motifs, further analysis of the first 100 amino acids of mammalian SM, and its interaction with cholesterol, will be important for understanding this version of regulated misfolding.
SMITH-LEMLI-OPITZ SYNDROME
Smith-Lemli-Opitz syndrome is a single-gene metabolic disease with challenging and complex clinical symptoms (Kelley & Hennekam 2000, Nowaczyk & Irons 2012). The disease is caused by hypomorphic mutations in the single late sterol pathway enzyme 7-dehydrocholesterol reductase (DHCR7) (Fitzky et al. 1998, Wassif et al. 1998). Thus, clinical approaches for this currently incurable disease include ways to elevate DHCR7 levels in patients. Recent work (e.g., by Prabhu et al. 2016) indicated that DHCR7 is subject to cholesterol-regulated degradation. Elevating the level of cholesterol in cultured mammalian cells causes selective degradation by the proteasome. DHCR7 is a multispanning ER membrane protein with a highly diverged SSD. Thus, it is presumably a substrate for one (or more) of the several mammalian ERAD ubiquitin ligases. So far, MARCH6 has been shown to be not involved, leaving open a variety of other pathways that will be interesting to test. Importantly, the mammalian Insigs appear to be uninvolved in degradation. Because we now know that SSDs are sufficient to promote ERAD and other ER-based quality control processes, it will be important to test the conserved residues for any role in DHCR7 stability or retention. Finally, DHCR7 loss of function causes buildup of intermediate sterols that are normally converted to cholesterol by the enzyme. Perhaps some of these serve as atypical degradation signals, and thus their buildup furthers disease severity by promoting degradation of the already compromised enzyme. Understanding the degradation of this critical enzyme will certainly reveal new aspects of regulated degradation and may pave the way to a novel proteostasis-based approach to this syndrome.
FINAL THOUGHTS
The high selectivity of proteostatic processes has been adapted in many ways to effect regulation. Elucidating these mechanisms of control will hopefully lead to new and creative ways to modify the sterol pathway in a variety of clinical circumstances. Imitating nature’s tendency to employ ligand-mediated misfolding could usher in new approaches to protein targeting and new classes of drugs that work through proteostatic mechanisms.
Acknowledgments
This work was supported by NIH grants 5R37DK051996-18 (to R.Y.H.) and 5T32GM007240-40 (to CMG trainee M.A.W.). The authors wish to express their gratitude to members of the Hampton lab and numerous colleagues at the University of California, San Diego, for oft-tapped input and vibrant discussions. We wish to dedicate this work to the memory of Susan Lindquist, who recently passed, far too soon. She was a remarkable leader in many aspects of proteostasis and an energetic and inspiring colleague. Her work was always creative and cutting edge, and her presence at conferences was an optimal fusion of high-level science and fun interaction. She will be deeply missed.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abumrad NA, Davidson NO. Role of the gut in lipid homeostasis. Physiol Rev. 2012;92(3):1061–85. doi: 10.1152/physrev.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CM, Goldstein JL, Brown MS. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. PNAS. 2003;100(19):10647–52. doi: 10.1073/pnas.1534833100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279(50):52772–80. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- Altmann SW, Davis HR, Zhu L-J, Yao X, Hoos LM, et al. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303(5661):1201–4. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta Mol Cell Res. 2014;1843(1):182–96. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2011;3(11):a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3(1):24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Beg ZH, Stonik JA, Brewer HB. Phosphorylation of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase and modulation of its enzymic activity by calcium-activated and phospholipid-dependent protein kinase. J Biol Chem. 1985;260(3):1682–87. [PubMed] [Google Scholar]
- Bernier V, Lagacé M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15(5):222–28. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bloch K. The biological synthesis of cholesterol. Science. 1965;150(3692):19–28. doi: 10.1126/science.150.3692.19. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum–associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9(1):209–22. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151(6):1163–67. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23(4):464–75. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10(2):237–45. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. PNAS. 1973;70(7):2162–66. doi: 10.1073/pnas.70.7.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. PNAS. 1974;71(3):788–92. doi: 10.1073/pnas.71.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res. 2008;50(Suppl):15–27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JS, Powell DW, Chai R, Hughes AL, Link AJ, Espenshade PJ. Insig regulates HMG-CoA reductase by controlling enzyme phosphorylation in fission yeast. Cell Metab. 2008;8(6):522–31. doi: 10.1016/j.cmet.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3(8):a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JP, Levy B, Ioannou YA. Evidence for a Niemann-Pick C (NPC) gene family: identification and characterization of NPC1L1. Genomics. 2000;65(2):137–45. doi: 10.1006/geno.2000.6151. [DOI] [PubMed] [Google Scholar]
- Edwards PA, Lan SF, Fogelman AM. Alterations in the rates of synthesis and degradation of rat liver 3-hydroxy-3-methylglutaryl coenzyme A reductase produced by cholestyramine and mevinolin. J Biol Chem. 1983;258(17):10219–22. [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. PNAS. 2001;98(25):14422. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner RA, Nguyen AD, Jo Y, DeBose-Boyd RA. Lipid-regulated degradation of HMG-CoA reductase and Insig-1 through distinct mechanisms in insect cells. J Lipid Res. 2013;54(4):1011–22. doi: 10.1194/jlr.M033639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favari E, Chroni A, Tietge UJF, Zanotti I, Escolà-Gil JC, Bernini F. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. 2015;224:181–206. doi: 10.1007/978-3-319-09665-0_4. [DOI] [PubMed] [Google Scholar]
- Fitzky BU, Witsch-Baumgartner M, Erdel M, Lee JN, Paik YK, et al. Mutations in the Δ7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. PNAS. 1998;95(14):8181–86. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flury I, Garza R, Shearer A, Rosen J, Cronin S, Hampton RY. INSIG: a broadly conserved transmembrane chaperone for sterol-sensing domain proteins. EMBO J. 2005;24(22):3917–26. doi: 10.1038/sj.emboj.7600855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, Carvalho P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife. 2013;2013(2):e00953. doi: 10.7554/eLife.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer F, Langklotz S, Narberhaus F. The C-terminal end of LpxC is required for degradation by the FtsH protease. Mol Microbiol. 2006;59(3):1025–36. doi: 10.1111/j.1365-2958.2005.04994.x. [DOI] [PubMed] [Google Scholar]
- Gardner R, Cronin S, Leader B, Rine J, Hampton R, Leder B. Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1998;9(9):2611–26. doi: 10.1091/mbc.9.9.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY. A highly conserved signal controls degradation of 3-hydroxy-3methylglutaryl-coenzyme A (HMG-CoA) reductase in eukaryotes. J Biol Chem. 1999;274(44):31671–78. doi: 10.1074/jbc.274.44.31671. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Shearer AG, Hampton RY. In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol. 2001;21(13):4276–91. doi: 10.1128/MCB.21.13.4276-4291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151(1):69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Tran PN, Hampton RY. Geranylgeranyl pyrophosphate is a potent regulator of HRD-dependent 3-hydroxy-3-methylglutaryl-CoA reductase degradation in yeast. J Biol Chem. 2009;284(51):35368–80. doi: 10.1074/jbc.M109.023994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geelen MJ, Gibson DM, Rodwell VW. Hydroxymethylglutaryl-CoA reductase—the rate-limiting enzyme of cholesterol biosynthesis. FEBS Lett. 1986;201(2):183–86. doi: 10.1016/0014-5793(86)80604-4. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta. 2013;1833(11):2410–24. doi: 10.1016/j.bbamcr.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011;13(3):260–73. doi: 10.1016/j.cmet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–99. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. PNAS. 1973;70(10):2804–8. doi: 10.1073/pnas.70.10.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–38. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161–72. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum–associated degradation in human physiology. Physiol Rev. 2012;92(2):537–76. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Bhakta H. Ubiquitin-mediated regulation of 3-hydroxy-3-methylglutaryl-CoA reductase. PNAS. 1997;94(24):12944–48. doi: 10.1073/pnas.94.24.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7(12):2029–44. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109(4):1561–74. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol. 1994;125(2):299–312. doi: 10.1083/jcb.125.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann G, von Mering C, Basler K. The Hedgehog signaling pathway: Where did it come from? PLOS Biol. 2009;7(6):e1000146. doi: 10.1371/journal.pbio.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458(7237):453–60. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Investig. 2002;109(9):1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe V, Chua NK, Stevenson J, Brown AJ. The regulatory domain of squalene monooxygenase contains a re-entrant loop and senses cholesterol via a conformational change. J Biol Chem. 2015;290(46):27533–44. doi: 10.1074/jbc.M115.675181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Stewart EV, Espenshade PJ. Identification of twenty-three mutations in fission yeast Scap that constitutively activate SREBP. J Lipid Res. 2008;49(9):2001–12. doi: 10.1194/jlr.M800207-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Todd BL, Espenshade PJ. SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell. 2005;120(6):831–42. doi: 10.1016/j.cell.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol. 2014;25(3):200–6. doi: 10.1097/MOL.0000000000000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Hartman IZ, Calhoun LN, Garland K, Young GA, et al. Contribution of accelerated degradation to feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol metabolism in the liver. J Biol Chem. 2016;291(26):13479–94. doi: 10.1074/jbc.M116.728469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, Goldstein JL. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem. 2008a;283(2):1052–63. doi: 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. PNAS. 2008b;105(40):15287–92. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakulj L, van Dijk TH, de Boer JF, Kootte RS, Schonewille M, et al. Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab. 2016;24(6):783–94. doi: 10.1016/j.cmet.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Jiang W, Song B-L. Ubiquitin ligases in cholesterol metabolism. Diabetes Metab J. 2014;38(3):171–80. doi: 10.4093/dmj.2014.38.3.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, DeBose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45(3):185–98. doi: 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, Lee PCW, Sguigna PV, DeBose-Boyd RA. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. PNAS. 2011;108(51):20503–8. doi: 10.1073/pnas.1112831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37(5):321–35. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82(1):323–55. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014;24(6):352–59. doi: 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig P-A, Ploegh HL. Protein quality control in the endoplasmic reticulum. F1000Prime Rep. 2014;6:49. doi: 10.12703/P6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara PE, Labouesse M. The sterol-sensing domain: multiple families, a unique role? Trends Genet. 2002;18(4):193–201. doi: 10.1016/s0168-9525(02)02640-9. [DOI] [PubMed] [Google Scholar]
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2016;16(2):101–14. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange BM, Rujan T, Martin W, Croteau R. Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. PNAS. 2000;97(24):13172–77. doi: 10.1073/pnas.240454797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange Y, Ory DS, Ye J, Lanier MH, Hsu F-F, Steck TL. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem. 2008;283(3):1445–55. doi: 10.1074/jbc.M706967200. [DOI] [PubMed] [Google Scholar]
- Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol Chem. 2006;281(51):39308–15. doi: 10.1074/jbc.M608999200. [DOI] [PubMed] [Google Scholar]
- Lee JP, Brauweiler A, Rudolph M, Hooper JE, Drabkin HA, Gemmill RM. The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol Cancer Res. 2010;8(1):93–106. doi: 10.1158/1541-7786.MCR-08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidenheimer NJ, Ryder KG. Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol Res. 2014;83:10–19. doi: 10.1016/j.phrs.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. PNAS. 2016;113(29):8212–17. doi: 10.1073/pnas.1607795113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SJ-A, Raychaudhuri S, Espenshade PJ. Subunit architecture of the Golgi Dsc E3 ligase required for sterol regulatory element–binding protein (SREBP) cleavage in fission yeast. J Biol Chem. 2013;288(29):21043–54. doi: 10.1074/jbc.M113.468215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamed M, Zhang Y, Wang ML, Seemann J, Kwon HJ, et al. Identification of luminal loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J Biol Chem. 2011;286(20):18002–12. doi: 10.1074/jbc.M111.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Mak R, Bennett EJ, Hampton R. A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum–associated degradation (ERAD-M) substrates. J Biol Chem. 2017;292:3112–28. doi: 10.1074/jbc.M116.770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham PG, Brodsky JL. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim Biophys Acta Mol Cell Res. 2013;1833(11):2447–57. doi: 10.1016/j.bbamcr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AD, Lee SH, DeBose-Boyd RA. Insig-mediated, sterol-accelerated degradation of the membrane domain of hamster 3-hydroxy-3-methylglutaryl-coenzyme A reductase in insect cells. J Biol Chem. 2009;284(39):26778–88. doi: 10.1074/jbc.M109.032342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Brown MS, Goldstein JL. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J Biol Chem. 1998;273(27):17243–50. doi: 10.1074/jbc.273.27.17243. [DOI] [PubMed] [Google Scholar]
- Nowaczyk MJM, Irons MB. Smith-Lemli-Opitz syndrome: phenotype, natural history, and epidemiology. Am J Med Genet C Semin Med Genet. 2012;160C(4):250–62. doi: 10.1002/ajmg.c.31343. [DOI] [PubMed] [Google Scholar]
- Ohgami N, Ko DC, Thomas M, Scott MP, Chang CCY, Chang T-Y. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. PNAS. 2004;101(34):12473–78. doi: 10.1073/pnas.0405255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omkumar RV, Darnay BG, Rodwell VW. Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J Biol Chem. 1994;269(9):6810–14. [PubMed] [Google Scholar]
- Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: What a long, strange tRIP it’s been. Genes Dev. 2009;23(22):2578–91. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne TF, Rosenfeld JM. Related membrane domains in proteins of sterol sensing and cell signaling provide a glimpse of treasures still buried within the dynamic realm of intracellular metabolic regulation. Curr Opin Lipidol. 1998;9(2):137–40. doi: 10.1097/00041433-199804000-00010. [DOI] [PubMed] [Google Scholar]
- Porter JR, Burg JS, Espenshade PJ, Iglesias PA. Ergosterol regulates sterol regulatory element binding protein (SREBP) cleavage in fission yeast. J Biol Chem. 2010;285(52):41051–61. doi: 10.1074/jbc.M110.144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu AV, Luu W, Sharpe LJ, Brown AJ. Cholesterol-mediated degradation of 7-dehydrocholesterol reductase switches the balance from cholesterol to vitamin D synthesis. J Biol Chem. 2016;291(16):8363–73. doi: 10.1074/jbc.M115.699546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. PNAS. 2007;104(16):6511–18. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Sun L-P, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP. Mol Cell. 2004;15(2):259–68. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Ravid T, Doolman R, Avner R, Harats D, Roitelman J. The ubiquitin-proteasome pathway mediates the regulated degradation of mammalian 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2000;275(46):35840–47. doi: 10.1074/jbc.M004793200. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Young BP, Espenshade PJ, Loewen CJ. Regulation of lipid metabolism: a tale of two yeasts. Curr Opin Cell Biol. 2012;24(4):502–8. doi: 10.1016/j.ceb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R, Zhou X, He Y, Ke M, Wu J, et al. Crystal structure of a mycobacterial Insig homolog provides insight into how these sensors monitor sterol levels. Science. 2015;349(6244):187–91. doi: 10.1126/science.aab1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitelman J, Simoni RD. Distinct sterol and nonsterol signals for the regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem. 1992;267(35):25264–73. [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34(2):212–22. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WJ, Beisiegel U, Goldstein JL, Brown MS. Purification of the low density lipoprotein receptor, an acidic glycoprotein of 164,000 molecular weight. J Biol Chem. 1982;257(5):2664–73. [PubMed] [Google Scholar]
- Schumacher MM, Elsabrouty R, Seemann J, Jo Y, DeBose-Boyd RA. The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase. eLife. 2015;4:e05560. doi: 10.7554/eLife.05560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MM, Jun D-J, Jo Y, Seemann J, DeBose-Boyd RA. Geranylgeranyl-regulated transport of the prenyltransferase UBIAD1 between membranes of the ER and Golgi. J Lipid Res. 2016;57(7):1286–99. doi: 10.1194/jlr.M068759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever N, Song B-L, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-coA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003a;278(52):52479–90. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003b;11(1):25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- Sharpe LJ, Cook ECL, Zelcer N, Brown AJ. The UPS and downs of cholesterol homeostasis. Trends Biochem Sci. 2014;39(11):527–35. doi: 10.1016/j.tibs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY. Structural control of endoplasmic reticulum–associated degradation: effect of chemical chaperones on 3-hydroxy-3-methylglutaryl-coA reductase. J Biol Chem. 2004;279(1):188–96. doi: 10.1074/jbc.M307734200. [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY. Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J. 2005;24(1):149–59. doi: 10.1038/sj.emboj.7600498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B-L, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005a;1(3):179–89. doi: 10.1016/j.cmet.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Song B-L, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005b;19(6):829–40. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Stewart EV, Lloyd SJ-A, Burg JS, Nwosu CC, Lintner RE, et al. Yeast sterol regulatory element-binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain–containing subunit of the Golgi Dsc E3 ligase. J Biol Chem. 2012;287(1):672–81. doi: 10.1074/jbc.M111.317370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EV, Nwosu CC, Tong Z, Roguev A, Cummins TD, et al. Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol Cell. 2011;42(2):160–71. doi: 10.1016/j.molcel.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Besser S, Hottmann H, Wolf DH. Previously unknown role for the ubiquitin ligase Ubr1 in endoplasmic reticulum–associated protein degradation. PNAS. 2013;110(38):15271–76. doi: 10.1073/pnas.1304928110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld CL, Hampton RY. Insulin-induced gene protein (INSIG)-dependent sterol regulation of Hmg2 endoplasmic reticulum–associated degradation (ERAD) in yeast. J Biol Chem. 2013;288(12):8519–30. doi: 10.1074/jbc.M112.404517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld CL, Pourmand D, Davis T, Garza RM, Hampton RY. The sterol-sensing domain (SSD) directly mediates signal-regulated endoplasmic reticulum–associated degradation (ERAD) of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase isozyme Hmg2. J Biol Chem. 2011;286(30):26298–307. doi: 10.1074/jbc.M111.244798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd BL, Stewart EV, Burg JS, Hughes AL, Espenshade PJ. Sterol regulatory element binding protein is a principal regulator of anaerobic gene expression in fission yeast. Mol Cell Biol. 2006;26(7):2817–31. doi: 10.1128/MCB.26.7.2817-2831.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–58. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Leichner GS, Pearce MMP, Wilson GL, Wojcikiewicz RJH, et al. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol Biol Cell. 2012;23(23):4484–94. doi: 10.1091/mbc.E12-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT. Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis. 2015;38(1):187–99. doi: 10.1007/s10545-014-9794-4. [DOI] [PubMed] [Google Scholar]
- Vashist S, Ng DTW. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165(1):41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashistha N, Neal SE, Singh A, Carroll SM, Hampton RY. Direct and essential function for Hrd3 in ER-associated degradation. PNAS. 2016;113(21):5934–39. doi: 10.1073/pnas.1603079113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum–associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–86. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang L-J, Song B-L. Niemann-Pick C1-like 1 and cholesterol uptake. Biochim Biophys Acta Mol Cell Biol Lipids. 2012;1821(7):964–72. doi: 10.1016/j.bbalip.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Wangeline MA, Hampton RY. Ligand-regulated entry into the HRD ERAD pathway: the dark side of allostery. bioRxiv. 2017:177311. doi: 10.1101/177311. [DOI]
- Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, et al. Mutations in the human sterol Δ7-reductase gene at 11q12–13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63(1):55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz RJ, Furuichi T, Nakade S, Mikoshiba K, Nahorski SR. Muscarinic receptor activation down-regulates the type I inositol 1,4,5-trisphosphate receptor by accelerating its degradation. J Biol Chem. 1994;269(11):7963–69. [PubMed] [Google Scholar]
- Wojcikiewicz RJH, Pearce MMP, Sliter DA, Wang Y. When worlds collide: IP3 receptors and the ERAD pathway. Cell Calcium. 2009;46(3):147–53. doi: 10.1016/j.ceca.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, et al. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ye J, DeBose-Boyd RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 2011;3(7):a004754. doi: 10.1101/cshperspect.a004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Sharpe LJ, Loregger A, Kristiana I, Cook ECL, et al. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol Cell Biol. 2014;34(7):1262–70. doi: 10.1128/MCB.01140-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol. 2012;32(11):2541–46. doi: 10.1161/ATVBAHA.112.250571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ren J, Harlos K, Stuart DI, Lu G, et al. Structure of glycosylated NPC1 luminal domain C reveals insights into NPC2 and Ebola virus interactions. FEBS Lett. 2016;590(6):605–12. doi: 10.1002/1873-3468.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]