

Abstract
Mitochondrial replacement therapy, a procedure to generate embryos with the nuclear genome of a donor mother and the healthy mitochondria of a recipient egg, has recently emerged as a promising strategy to prevent transmission of devastating mitochondrial DNA diseases and infertility. The procedure may produce an embryo that is free of diseased mitochondria. A recent study addresses important fundamental questions about the mechanisms underlying maternal inheritance and translational questions regarding the transgenerational effectiveness of this promising therapeutic strategy. This review considers recent advances in our understanding of maternal inheritance of mitochondria, implications for fertility and mitochondrial disease, and potential roles for the Balbiani body, an ancient oocyte structure, in mitochondrial selection in oocytes, with emphasis on therapies to remedy mitochondrial disorders.
Maternal transmission of mitochondria
Modern eukaryotic cells coevolved in a symbiotic relationship that began between microorganisms, archaebacteria and ancestral mitochondria two billion years ago. Along the way, much of the original mitochondrial genome was lost, with just over a few dozen genes among more than 1000 retained in the mitochondrial DNA [1, 2]. The mitochondrial genome of humans is comprised of 37 genes that encode the tRNAs and ribosomal RNA required to translate the proteins of the electron transport chain as well as proteins that are necessary for oxidative phosphorylation. The other gene functions that are required by mitochondria are supplied by the nuclear genome of the cell. The mitochondrial genome is organized into nucleoids composed of single or multiple copies of mitochondrial DNA due to fusion and fission cycles that ensure each daughter cell receives highly functional mitochondria during mitotic cell divisions [3]. These fusion and fission cycles can also generate mitochondria with distinct morphologies and even interconnected networks with presumably unique functions. Unlike the nuclear genome, the mitochondrial genome is not transmitted by sexual reproduction, but instead in most animals mitochondria are strictly inherited from one parent, usually the mother. Either before or after fertilization, mixing of maternal and paternal mitochondria is averted by elimination of any contaminating paternal mitochondria by seemingly diverse mechanisms, including ubiquitination modification and proteosomal degradation and mitophagy [4–11]. The involvement of conserved components (e.g. autophagy machinery), and the conserved reliance on redundant mechanisms to eliminate paternal mitochondria underscore the evolutionary importance of uniparental transmission of mitochondria, but the rationale behind this investment remains mysterious.
Despite a single parent of origin, variations in mitochondrial DNA between a mother and her progeny and between siblings exceeding levels expected in a single generation have been observed. This variation can be extensive and has been observed in Holstein cows [12], mouse models [13] and patients [14, 15]. For example, a heteroplasmic mother, an individual with more than one type of mitochondria due to accumulation of mutations, can produce progeny with only the mutant mitochondrial DNA (homoplasmic). Such enrichment of mutant mitochondria indicates the existence of a mitochondrial bottleneck. Bottlenecks enforce mechanisms that omit cells or individuals, in this context mitochondria, with extensive mutational burden in mitochondrial DNA, ideally such that only the healthiest mitochondria are transmitted. In all animals, mitochondria and other organelles are asymmetrically enriched within oocytes (reviewed in [16–18]). In animals that use maternal inheritance of germ plasm (the substance that contains determinants of the germline of the next generation) to specify the germline, mitochondria are enriched in areas where the germ plasm (the components that will specify the germ cells of the next generation) localizes [19] and reviewed in [17, 20]. Thus, subcellular enrichment of mitochondria has led to models whereby the oocyte selects the healthiest mitochondria for the next generation, by mechanisms that remain to be understood. Mitochondrial bottlenecks are thought to occur in the developing germline — both during oogenesis and later in embryonic primordial germ cells (PGCs) in some animals. Several models of mitochondrial inheritance have been proposed and include random segregation or stochastic mechanisms as well as more direct selection mechanisms that will be discussed briefly herein.
Heteroplasmy, Mitochondrial segregation and disease
In an individual with a mitochondrial disease, different cells or populations may have differing amounts of the mutant mitochondria, ranging from complete homplasmy —individuals with all mutant mitochondrial DNA as occurs in Leigh’s Syndrome [21–23] to more tissue specific accumulation and manifestation such as the blindness that occurs in patients with Leber hereditary optic neuropathy (Wallace 1988)-organs and systems with high bioenergetic demand, such as the brain and heart, are most sensitive to mitochondria associated pathologies (reviewed in [2]). Stochastic and tissue-specific selection of mitochondria are mechanisms that have been proposed to explain variation in accumulation of mutant mitochondria and mitochondrial disease in somatic cells. Unlike nuclear DNA, mitochondrial DNA replication is not limited by the cell cycle [24]. Consequently, according to the stochastic or “mitotic segregation” mechanism, a heteroplasmic cell can generate progeny with distinct mitochondrial DNA composition over a short time, depending on the extent to which mitochondrial DNA replication occurs before or after the cell divides. According to this model, most cells in a heteroplasmic individual would be expected to have a random distribution of normal and potentially pathogenic mitochodria. Such heteroplasmic individuals are healthy if the cells have sufficient numbers or activity of normal mitochondria to provide adequate respiratory and other functions. In contrast, cells with an unfavorable ratio of normal to mutant mitochondria show signs of disease. This stochastic pattern of inheritance can explain why mitochondrial mutations do not affect all cells in an individual equally. An alternative possibility, based on examination of heteroplasmic mice, indicates that variation from cell to cell in a heteroplasmic individual may not simply be stochastic, but is influenced by tissue specific mechanisms or bottlenecks that drive mitochondria selection [25].
Why is mitochondria homogeneity beneficial?
The reasons why heteroplasmy is to be avoided are not immediately intuitive since heterogeneity is favorable in other contexts. For example, heterozygosity and variation generated by sexual reproduction and recombination of the nuclear genome is generally regarded as beneficial both in terms of evolution and in serving to alleviate mutational burden and demise due to Muller’s rachet. Given the symbiotic evolution of the eukaryotic cell, this begs the question of whether mitochondrial bottlenecks and mechanisms to ensure that embryos are comprised of maternal but not paternal mitochondria are good for the oocyte, good for the mitochondria or both? Is this simply a relic of a time before the symbiotic relationship between the eukaryotes and mitochondria began, or are there benefits or biological constraints that render selection of mitochondria essential for certain cells? In terms of propagation of genetic information, it is easy to imagine how developmental bottlenecks coupled with purifying selection would be of evolutionary benefit to the mitochondria due to the shift in reliance on the nuclear genome to produce many products that are necessary to fulfill mitochondrial functions. In light of the exciting potential of mitochondrial replacement strategies to provide healthy mitochondria and cure diseases associated with mitochondrial dysfunction, and to prevent transmission of mitochondrial diseases including infertility [2, 26–29] it is increasingly important to understand mechanistically how these bottlenecks work, when they occur, why they are in place in terms of developmental functions, what benefits they confer, and the extent of, and importance of, pairing between mitochondrial and nuclear genomes. Several possibilities have been imagined, and a few are considered briefly below.
Mitochondrial and nuclear genome compatibility
The “genome compatibility” hypothesis proposes that optimal matching of mitochondrial and nuclear genomes is important for efficient cellular respiration. This hypothesis suggests that purifying selection may be necessary to ensure optimal component pairing for proper mitochondrial function. Accordingly, if mitochondrial and nuclear genomes were matched then the native mitochondria would be expected to have selective advantage over those acquired (by mutation or by transfer during assisted reproduction and mitochondrial replacement described above) because “native” mitochondria would be more compatible with components encoded by the nuclear genome. Evidence from studies in which two homoplasmic metaphase II oocyte halves (one with spindle and one without) were fused demonstrated that mitochondrial segregation occurs during preimplantation primate development, but did not appear to be influenced by nuclear-mitochondrial bias, as native mitochondria were not preferentially selected over acquired mitochondria in somatic lineages [28]. Moreover, Analysis of mt-DNA mutator mice, mutant for a mitochondrial polymerase, with potentially pathogenic mtDNA mutations revealed bias against mutations within coding genes, but did not detect selection against pathogenic mitochondrial mutations in noncoding or tRNA genes prior to preantral stages [30, 31]. Recently, Yamada and colleagues provided additional evidence arguing against a strict nuclear-mitochondria pairing model in human embryos and cell lines, at least with respect to mitochondrial function. In their study, combining maternal genomes with different mitochondrial haplotypes did not compromise mitochondrial activity based on several indicators of mitochondrial function, including respiratory chain activity, oxygen consumption, coupling, respiratory, and capacity among other parameters [32].
Notably, despite attaining functional compatibility, heteroplasmic environments eventually led to instability of the mitochondrial genome by a mechanism that has not been determined [32]. In other studies of rodent and primate systems where heteroplasmy was high, genetic drift toward homoplasmy due to mitochondrial segregation was observed. For example, segregation was observed in primate embryos produced from oocytes with two mitochondrial DNA haplotypes [28]. In other studies using various wild strains of mice, Burgstaller and colleagues generated animals with variable degrees of heteroplasmy, and found that segregation was greatest in the haplotypes with the largest genetic distance [33]. This would seem to suggest that more closely related mitochondria and nuclear genomes might have a selective advantage given enough divergence.
Mitochondrial replacement therapy
Because of the uniparental, and usually maternal mode of mitochondrial inheritance diseases caused by mitochondria dysfunction are propagated from mother to offspring in a “dominant” manner. Mitochondrial replacement therapy, which involves transfer of a maternal nucleus from a mother with abnormal mitochondria (hereafter donor) to the enucleated oocyte or egg of a woman with healthy mitochondria (hereafter recipient oocyte or egg), recently emerged as a promising strategy to prevent transmission of devastating mitochondrial DNA diseases and infertility (Figure 1). The procedure, when successful, results in embryos, referred to as three parent embryos, with the healthy mitochondria of the recipient egg and the nuclear genome of the donor mother, that are ideally free of diseased mitochondria. However, evidence shows that contaminating donor mitochondria accompany the nucleus during the transfer procedure [27, 28, 32], raising questions about whether bottlenecks or selection of mitochondria in subsequent generations will lead to biased enrichment of the donor mitochondria over time. This is a potential concern for individuals generated by mitochondrial replacement therapy if critical tissues become diseased due to enrichment for the “contaminating” mitochondria. As discussed below, this potential drift toward diseased donor mitochondria is a potentially more significant issue for female embryos that are generated by mitochondrial replacement therapy because their mitochondria will be transmitted to the next generation.
Figure. 1. MitoDNA transfer procedure/timing relative to oocyte bottlenecks.
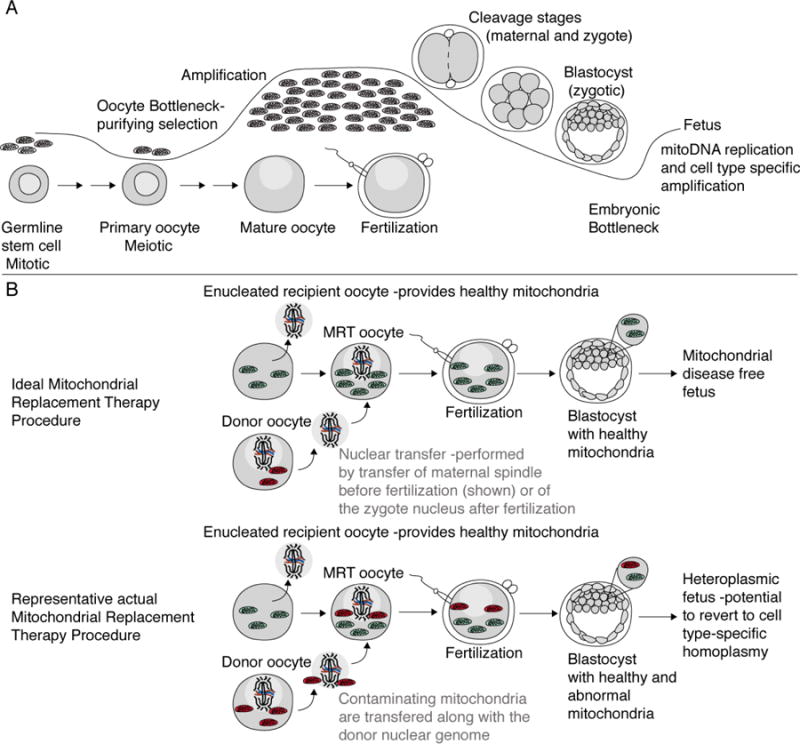
A) Schematic depicting the stages of oogenesis and the timing of mitochondrial bottlenecks and amplification in oocytes and early embryos. B) Schematics depict ideal mitochondrial replacement procedure to generate a healthy embryo free of diseased mitochondria, and the observed mitochondrial contamination during the transfer, which can lead to heteroplasmy and cell type specific disease or reversion to homoplasmy.
Maternal-effect genes are those genes expressed by the mother that are transcribed in the oocyte to produce all of the RNAs and proteins that will be needed throughout oogenesis and into early embryogenesis. Prior to the meiotic divisions, oocytes become transcriptionally silenced so the late stage oocytes and early embryos must utilize the maternally provided RNAs and proteins for their development. Like these maternal-effect genes, mitochondria are maternally inherited so the products of the mitochondrial genome at this stage can be considered within the scope of maternal-effect processes. If the bottleneck mechanism involves matching nuclear and mitochondrial genomes, then presumably this selection would be carried out in early oocytes by components encoded by the nuclear and mitochondrial genomes. Because the transfer procedure to generate a three parent embryo is performed relatively late in oogenesis, at stages after potential mitochondrial bottleneck selection and amplification during oocyte growth that will be discussed later in this review, mitochondria transfered at this late stage would have already passed the earlier selection and be identified as maternal, whether marked or unmarked, as so far ubiquitination or similar marks have only been reported for paternal mitochondria. Therefore, no biased elimination would be expected. Thereafter, the early embryo environment might be receptive to the mitochondria of both mothers despite the apparent mismatch between the mitochondrial and nuclear genomes. This is plausible because although the embryo has the nucleus of another oocyte, the embryo for a period of time expresses the products of its original nuclear genome — the persisting maternal RNA and protein products that were produced at stages prior to the transfer, and upon genome activation the RNA and protein products produced by the new donor nuclear genome. Moreover, in human embryos paternal mitochondria linger until around the time of genome activation, acquisition of pluripotency and the first fate decisions[34], indicating that prior to this time the mechanisms to clear “foreign” mitochondria might not be as robust.
The early cleavages of the blastula are reductive, generating smaller cells with each nuclear replication and di vision cycle. During this period of rapid di vision, no replication of mitochondrial DNA has been detected in several vertebrates examined [28, 35, 36]; thus, despite differences in onset between species each cleavage prior to zygotic genome activation and acquisition of pluripotency would generate cells with less mitochondrial DNA per cell. This period without mitochondrial replication has been identified as a genetic bottleneck in primates based on examination of heteroplasmic embryos [28]. In that work, prior to 8-cell stage, cells were heteroplasmic, thereafter, specific tissues of the fetuses at later stages showed evidence of homoplasmy attributed to tissue- specific differences in mitochondrial copy number at stages after DNA replication resumes [28]. This genetic bottleneck in embryogenesis corresponds to the period when the maternal products are degraded and are replaced by the products of the zygotic genome. The progeny of mitochondrial replacement therapy (MRT) would be subject to this bottleneck, and if cell-type specific selection favors the contaminating mitochondria then the individual will suffer from mitochondrial disease in those tissues. In the recent work from Yamada and colleagues, such enrichment of contaminating, albeit healthy mitochondria in their case, was observed in cardiac and connective tissues of embryos and human stem cell lines generated by mitochondrial replacement therapy [32].
Despite the potential for tissue specific accumulation of dysfunctional mitochondria, MRT understandably is of considerable appeal to couples coping with a devastating mitochondrial disease as such intervention provides the only hope for a healthy child. Depending on the tissues and proportion of diseased mitochondria, this drift toward homoplasmy might not be problematic in first generation males or females. However, because of the uniparental inheritance and mitochondrial bottlenecks within the prospective female germline, selection bias in favor of the “contaminating mitochondria” as they pass through the bottleneck owing to their shared maternal origin with the nuclear genome would be a significant limitation to MRT and potentially detrimental, as it could conceivably lead to reversion to the diseased donor mitochondria population. Thus, MRT females even with an otherwise healthy life would face the same mitochondrial disease and infertility as their mothers. A recent study that examined the fate of healthy donor mitochondria in recipient human oocytes indicates that in the majority of cases the donor mitochondria are eliminated over time; however, their data also point to persistence and even complete reversion or drift toward the mutant donor mitochondria in a significant number of cases [32]. This study both sheds light on and raises important basic research questions about the still poorly understood mechanisms underlying maternal inheritance of mitochondria, as well as translational implications and caution regarding the transgenerational effectiveness of this promising therapeutic strategy.
Mechanisms for organelle level sorting of mitochondria in oocytes
The existence of a mitochondrial bottleneck during early oogenesis is widely recognized, but remains incompletely understood, both in terms of timing and mechanism. So far, emphasis has been on the population of mitochondria in late stage oocytes and fertilized embryos. Like the potential advantages and reasons why uniparental inheritance of mitochondria might be important, how and when individual mitochondria are selected by the oocyte, and what features of oocyte cell biology might render it uniquely suited for transgenerational transmission of mitochondria remain open questions. It is possible that this property is intrinsic to differences in meiosis between males and femalesߝalthough meiosis in both sexes generates four haploid cells, all of which become sperm in males, whereas only one oocyte is produced in females. Bottlenecks are characterized by selection of only a limited population by a strong purifying mechanism. As oogenesis progresses, oocytes undergo considerable growth as they actively produce stores, including RNAs, proteins, and mitochondria in preparation for the later stages of meiosis and the developmental events of early embryogenesis prior to activation of the zygotic genome. Early differentiated or primary oocytes are small and have fewer mitochondria, which will undergo amplification to produce the plentiful mitochondria of the significantly larger fertilization-competent late oocytes; therefore, the primary oocyte stage fulfills the bottleneck requirement for a limited population. That a bottleneck exists is clear, as quantitative assessment of the mitochondrial numbers in mouse provided evidence for reduction of mitochondrial numbers in oocytes [37], as well as for alternative mechanisms that would involve selection and amplification rather than strictly elimination of mitochondria in mouse [38, 39]. In addition to containing comparably fewer mitochondria, primary oocytes also form unique transient subcellular structures that are not present in later stage oocytes, specifically the Balbiani body. The Balbiani body, also known as the mitochondrial cloud because of the enrichment of these organelles in this nonmembrane bound subcellular compartment, is an ancient and conserved asymmetric feature of oocytes (reviewed in [16, 40]. Little is known about how this structure forms or is regulated because it is only transiently present in primary oocytes and thus has been difficult to access, but it has been long appreciated as a potential sorting or amplification depot for oocyte mitochondria.
Owing to the colocalization between mitochondria and the germ plasm of some animals, models whereby the Balbiani body selects the healthiest mitochondria for the next generation have been proposed [41, 42]. Early as well as more recent studies thus examined the distribution of mitochondria in oocytes using reporters of mitochondrial respiration to identify more active mitochondria [43–45]. Consistent with a role in selecting the “most fit” mitochondria, these studies revealed an enrichment of the most active mitochondria within the Balbiani body [43–45], but how individual mitochondria are selected remains an open question. It is possible that sequestration of mitochondria within the Balbiani could lead to differential marking of the mitochondria. For example, this could occur by promoting postranslational modification of a mitochondrial protein, facilitating novel associations with proteins that are spatially restricted to the Balbiani body, or by preventing the sequestered mitochondria from receiving a destruction mark. The question of whether the mitochondria are presorted and then selected based on their activity, or only become activated once localized within the Balbiani body remains to be determined. Moreover, whether the proteins of mitochondria within the Balbiani body are otherwise differentially marked from those outside the Balbiani body has not been demonstrated.
Single cell sequencing approaches allow the variation in individual oocytes to be determined and thus provide a picture of population level selection. To capture selection at the level of the organelle in oocytes one would need to pinpoint precisely when the bottleneck occurs and analyze mitochondrial content at stages just before and after the bottleneck. A recent study by Otten and colleagues measured mitochondrial copy number to identify the timing of mitochondrial DNA bottlenecks in zebrafish [35]. This work revealed distinct bottleneck events in germline and nongermline cells, and provided evidence that in the female germline a bottleneck occurs in stage I oocytes [35]. In zebrafish, stage I encompasses oocytes that overlap with the period prior to and when the Balbiani body is present (Figure 2). In the Otten study, these early stage oocytes were not subdivided according to meiotic stage and oogonia were mixed with meiotic stages from zygotene to diplotene; therefore, it was not possible to pinpoint the precise stage when the bottleneck occurs. If the bottleneck mechanism includes selection of mitochondria by sequestration or exclusion from the Balbiani-body localized, then future analysis and comparison of mitochondrial DNA copy number and variation in oocytes sorted based on meiotic stage and phase of Balbiani body development (e.g. prior to formation, after formation, during translocation and after reaching the cortex) may prove to be informative and should provide suffiicient resolution to pinpoint the timing of the oocyte bottleneck. In addition, isolation and proteomic analysis of mitochondrial proteins of the Balbiani body associated mitochondria and cytoplasmic mitochondria should provide insight into whether distinct posttranslational modifications are associated with these mitochondrial populations.
Figure. 2. Germline development and asymmetries in early oogenesis and potential mitochondrial bottleneck in zebrafish.
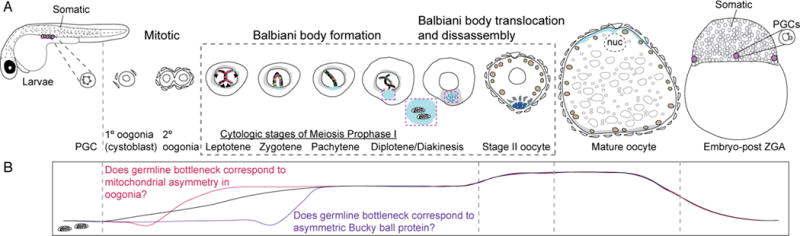
A) Cartoons depicting the stages of germ cell development beginning with the primordial germ cells (PGCs) in larvae. Cellular asymetries have been observed in mitotic oogonia of female zebrafish. Asymmetric Bucky ball protein (light blue) is detected at zygotene stage, and is required for Balbiani body formation during prophase I (this period corresponds to stage I oocytes in zebrafish). The Balbiani body translocated to the prospective vegetal cortex and then disassembles in stage II ooctyes. B) Schematic graph depicting potential mitochondrial bottleneck and purifying selection stages in zebrafish. Grey dashed lines indicate stages/periods of oogenesis analyzed by Boke and colleagues. In their study, they observed a dramatic increase in mitochondrial DNA copy number between PGC stages and stage I oocytes, a smaller increase between stage I and stage II oocytes and mature oocytes, and a decline in embryos (black lines). Because stage I oocytes are diverse in terms of cytological stage of prophase I and phase of Balbiani body development the precise stage when the bottleneck and purifying selection, and how it relates to polarity in mitochondrial distribution in oogonia (red line), and asymmetric enrichment of Bucky ball protein (purple line) and Balbiani body formation and enrichment of mitochondria there is not known.
Zebrafish mutants and transgenic oocytes that are mutant for or overexpress Bucky ball protein, a vertebrate specific protein with uncharacterized protein domains, disrupt Balbiani body formation and fail to localize mitochondria at diplotene stage [46–48]. We have previously demonstrated that Bucky ball protein is asymmetrically localized at zygotene stage before the Balbiani body forms [48], raising the possibility that oocyte polarization was initiated earlier than had been previously thought. We, and others recently provided evidence that this is indeed the case [49, 50]. These studies used electron microscopy to examine and quantify mitochondria distribution in the female germline during this period, and found that mitochondria are asymmetrically distributed in stages prior to Balbiani body formation [49, 50]. This is one of the stages represented within the early ooctye bottleneck identified by Otten and colleagues, raising the possibility that a mechanism to enrich mitochondria, and thus a potential purifying selection could occur prior to or at zygotene stage, when asymmetric Bucky ball protein is first detected [48](Figure 2). Alternatively, it is possible that an earlier mitochondrial bottleneck could function in a Bucky ball and Balbiani body independent manner because mitochondria are initially asymmetrically distributed at earlier stages in bucky ball mutants [48, 49] although mitochondria are not sequestered in bucky ball mutants at later stages [46–48]. If selection occurs prior to or independent of Balbiani body formation then the mitochondria composition of bucky ball mutants would be expected to resemble the variation in wild-type reported by Otten and colleagues. In contrast, if the Balbiani body is required for purifying selection then mitochondria of bucky ball mutants might show more variation compared to wild-type.
The Xenopus homolog of Bucky ball is known as XVelo (Vegetally localized -1), first identified based on the vegetal pole localization of xvelo RNA [51]. A recent study identified XVelo as a Balbiani body enriched protein, and identified prion-like sequences within the amino terminal region of Xvelo [52] (Figure 3). In that work, the authors showed that the prion-like domain (PLD) of XVelo was necessary and sufficient for Balbiani body localization of overexpressed XVelo fusion proteins in ovo, and to drive formation of amyloid-like meshworks in in vitro and cell free reconstitution assays [52]. Notably, with respect to a potential role for the Balbiani body as an oocyte mitochondrial bottleneck discussed above, Boke and colleagues showed that these XVelo meshworks could entrap mitochondria by a mechanism that requires the prion-like domain [52]. Although the amino acid level conservation between XVelo and zebrafish Bucky ball is poor over much of the sequence, the proteins are most similar in their amino terminal region [47, 52], which includes the PLD. Evidence for a conserved activity includes the ability of both proteins to drive assembly of structures that can recruit mitochondria and RNAs [46–48], and experiments in which versions of XVelo and Bucky ball with swapped PLD domains supported stable matrix formation in photobleaching and recovery assays [52]. Interestingly, versions of XVelo containing the PLD, but lacking the C-terminal region could still form a matrix in overexpression assays, but could not recruit mitochondria to the matrix [52]. Therefore, the PLD is sufficient for matrix formation, but the C-term is required to coordinate mitochondrial recruitment either directly or indirectly. Notably, both of the previously characterized zebrafish bucky ball mutant alleles that encode for truncated Bucky ball proteins (if any protein is produced) lacking the entire C-terminus or only the last few dozen amino acids of the protein fail to support Balbiani body formation and recruitment of mitochondria and RNA [46, 47]. Moreover, consistent with an important role for the C-terminus in recruiting mitochondria and Balbiani body proteins, the overexpression phenotypes of transgenic animals expressing Bucky ball, and rescue of the mitochondria localization and Balbiani body formation defects of bucky ball mutants require the Bucky ball C-terminus in transgenic [48] and transient rescue assays [47].
Figure. 3. Comparison of Bucky ball/XVelo/Oskar functional domains.
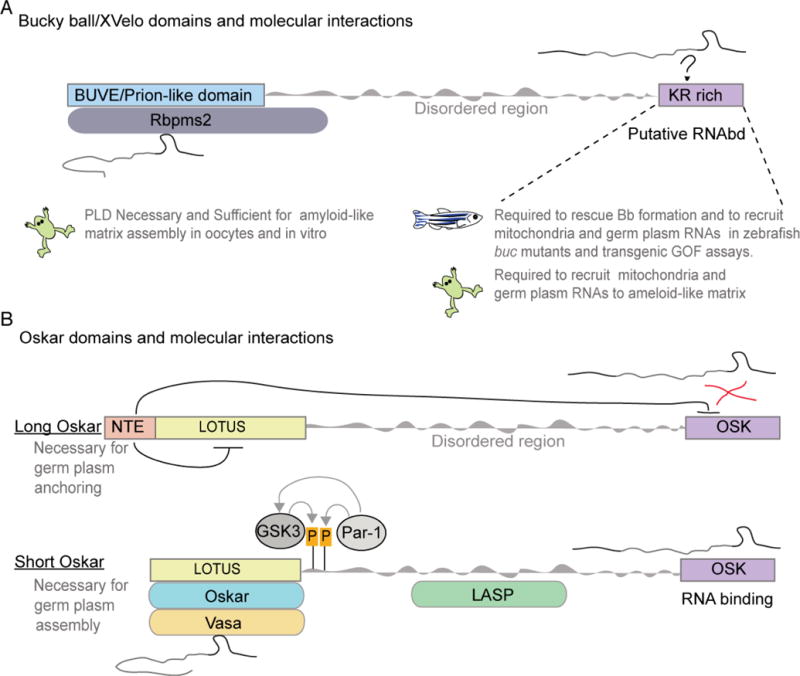
A) Schematic depicting the protein domains of Xenopus Velo and zebrafish Bucky ball. The amino terminus harbors a prion-like domain (PLD), also known as the BUVE (Bucky ball Velo domain) that is essential for assembly of an amyloid-like matrix. The Carboxy terminus is K/R rich and has properties suggestive of RNA binding function. The C-terminus is required for Balbiani body assembly in zebrafish, including recruiting RNAs and mitochondria, and for entrapment of mitochndria and RNAs in XVelo assembled amyloid-like matrix in Xenopus oocytes and reconstitution assays. The PLD and KR rich region are separated by an intrinsically disorded region. B) Schematic depicting long and short forms of Drosophila Oskar proteins. The Amino termimal extension (NTE) of the long isoform is essential for nucleating actin filaments and anchoring of germ plasm components to the posterior cortex in oocytes. The LOTUS domain mediates Oskar dimerization and interactions with protein binding partners, like Vasa. The actin binding protein Lasp binds to the disordered region. The OSK domain is composed of basic and hydrophilic amino acids and is thought to mediate interactions with the RNA effectors of Oskar. Short Oskar activity and stabiity are regulated by posttranslational modifications, including phosphorylation and ubiquitination (not shown). Although long Oskar has the LOTUS and OSK domains it does not bind to Vasa nor does it bind to RNAs. The NTE is thought to inhibit these activities.
Interestingly, Boke and colleagues identified residues in the C-terminus of Xvelo that lack a known RNA recognition consensus sequence, but have characteristics that might confer RNA binding properties similar to reports for Drosophila Oskar, another intrinsically disordered protein with essential roles in germ cell formation [53]. Consistent with XVelo meshworks supporting RNA recruitment, which is a previously described function of zebrafish Bucky ball [46–48], versions of Xvelo with the prion-like domain but lacking the rest of the protein formed meshworks, but did not recruit a labeled nanos/xcat-2 RNA there [52]. Both XVelo and Bucky ball, like Oskar, have previously been shown to bind RNA binding proteins [48, 52, 53]. Thus, it appears that a potential shared feature of the germ plasm assemblers may be the capacity to recruit RNAs by two mechanisms — direct interaction via their putative RNA binding domains and through their interactions with RNA binding proteins. However, in the case of XVelo it remains to be determined whether recruitment of nanos/xcat-2 to the network occurs via direct or indirect RNA interactions, as both XVelo and Bucky ball have been shown to interact with the conserved RNA binding protein, Rbpms2 (RNA binding protein with multiple splice isoforms), which binds to Balbiani body localized germ plasm RNAs such as bucky ball and nanos/xcat-2 [48, 54–56]. Notably, XVelo/Bucky ball interactions with Rbpms2 protein are mediated via the XVelo/Bucky ball N-terminus [48, 54], which is also the region harboring the prion-like domain[52]; however, whether interaction with Rbpms2 promotes or represses Buc/XVelo functions in Balbiani body and matrix formation, is involved in bucky ball RNA regulation, or only facilitates recruitment of RNAs to the assembled structure remains to be determined.
The seemingly shared ability of independently evolved [57, 58] maternal germ plasm regulators to engage multiple proteins and RNA partners provide new insight and testable models for mechanisms by which germ plasm assembly, mitochondrial recruitment and Balbiani body formation are regulated, including additional support for the possibility that a key aspect of regulation could include differential association with a unique repertoires of RNAs, directly via RNA binding domain interactions and either directly or indirectly through interactions with RNA bindi ng proteins [48, 52, 53]. Consistent with the notion of temporally distinct regulation and activities, Boke and colleagues found that XVelo could only promote assembly of amyloid-like networks in oocytes, but not in eggs. It is intriguing that the ability to recruit mitochondria and RNA to the matrix based on the evidence so far appear to be linked in these vertebrate oocytes, suggesting a coordinated mechanism, or that recruitment of one is prerequisite for recruitment of the other, for example one could imagine that an RNA encoding a protein that is required to select or anchor mitochondria might be specifically translated or localized within the Balbiani body. In Drosophila milton mutants recruitment of mitochondria to the Balbiani body is uncoupled from RNA localization there [59]. In Drosophila RNA labeling and tracking experiments indicates that the germ plasm effector RNAs become entrapped and concentrated in the localized germ plasm owing to their affinity for germ plasm proteins [60, 61]. In Drosophila, tudor mutants that disrupt an RNA binding protein that acts downsteam of oskar, mitochondria associate with pole granules; however, recruitment or transfer of mitochondrial ribosomal RNAs to the surface of polar granules (germ plasm) is disrupted [62–65]. Animals that don’t use maternal germ plasm, including humans, have Balbiani bodies and genes like oskar or bucky ballXvelo. This raises the possibility that nonmembrane bound compartments formed by self assembling proteins like XVelo, Bucky ball and Oskar originated for mitochondrial entrapment and selection, and were coopted or coupled to amass, chaperone and coordinately regulate cohorts of RNA, like the germ plasm through their interactions with RNA binding proteins, which have been shown to undergo dynamic phase transitions through concentration dependent interactions with their RNA and protein partners (Figure 4) [66–69]. Compared to Velo or Bucky Ball, much more is understood about the complex regulation of Oskar, which includes regulation by post-transcriptional and posttranslational mechanisms, such as phosphorylation and ubiquitination (Figure 3) (reviewed in [70]). Further analysis of XVelo/Bucky ball functional domains and the genes regulating Balbiani body development will be required to resolve these questions. Important lines of investigation ongoing and for the future include further defining the binding partners of the vertebrate specific Velo and Bucky ball, and applying genetic and biochemical approaches to tease out the mechanisms that regulate, both spatially and temporally, the various RNA and protein associations of these proteins in the context of their functional consequences to germ cell development and fertility.
Figure. 4. Mitochondria asymmetry, Balbiani body formation, and germ plasm assembly as potential oocyte bottlenecks.
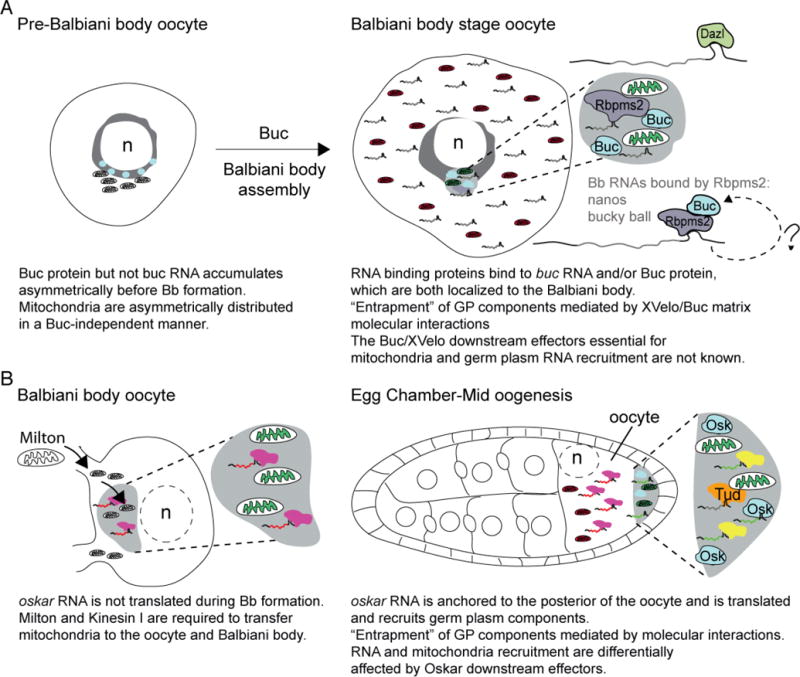
A) Schematic depicts an early zebrafish oocyte prior to Balbiani body formation. Buc protein is asymmetric next to the nucleus, and mitochondria are asymmetrically distributed in oocytes in a Buc-independent manner at this stage. Buc is required for Balbiani body assembly and recruitment of the most active mitochondria (green) there. RNAs and proteins, including RNA binding proteins (like Dazl and Rbpms2) are also recruited to the Balbiani body at this time. Although the mechanism of recruitment is not understood, a mechanism whereby Buc forms a self-assembling network that can recruit germ plasm (GP) components directly, and through conserved interactions with RNA binding proteins has been proposed for Balbiani body assembly in zebrafish and Xenopus oocytes. Analysis of zebrafish maternal-effect mutants and overexpression assays indicates that Buc/XVelo coordinate recruitment, possibly by entrapment, of mitochondria and RNAs to the nonmembrane bound Balbiani body. B) In Drosophila, one of 16 cyst cells is specified as the oocyte. Mitochondria are transferred to the oocyte in a Kinesin and Milton dependent manner to form the Balbiani body. At this stage, oskar RNA is present, but is translationally repressed; therefore, if recruitment of mitochondria at this stage requires oskar this would be an RNA function rather than a coding function of oskar. In mid-oogenesis, oskar RNA is anchored at the posterior pole and is translated there. Once translated, short Oskar mediates germ plasm assembly components, including mitochondria via self-assembly and interactions with its RNA and protein effectors, including RNA binding proteins that lead to entrapment of components within the germ plasm. Tudor (Tud) binds to mitochondria and RNA, but is not required to localize mitochondria to the posterior pole. Therefore, in Drosophila recruitment of mitochondria and germ plasm RNAs to nonmembrane bound compartments can be uncoupled by Oskar downstream effectors. Balbiani body formation is a conserved feature of oocyte development and analysis of mitochondria indicates bottlenecks within these stages; however, it remains to be determined if selection at either of these stages influences mitochondrial content of oocytes or the embryonic germline.
The prior observations that zebrafish bucky ball mutants and transgenics have ample mitochondria and, at least in terms of oocyte growth are indistinguishable from normal oocytes provides evidence that sorting to the Balbiani body is not required for mitochondria amplification in zebrafish [46, 48]. However, whether or not the mitochondria of zebrafish bucky ball mutants are sufficient for growth but are less fit, or if bucky ball mutant oocytes are more prone to heteroplasmy in the absence of Balbiani body enrichment, which might be expected if the Balbiani body serves to select the mitochondria as part of an ancient oocyte bottleneck, has yet to be determined. Moreover, the question of whether the mitochondria that colocalize with the germ plasm in the Balbiani body are later selectively sorted to the primoridial germ cells of the embryo remains to be addressed, but should be feasible with modern genome editing technologies.
Looking forward
Stem cell and replacement therapies have been extremely effective for blood diseases, including mitochondrial diseases with single or limited organ involvement such as mitochondrial neurogastrointestinal encephalomyopathy [71]; however, the maternal inheritance pattern and potential to revert to the diseased mitochondria in oocytes motivates caution for treatment of infertility and other maternally inherited mitochondrial diseases. Because these replacement therapies offer significant therapeutic potential as they are potentially curative, and are the only hope for maternally-inherited mitochondrial diseases, further research into the extent of reversion and potential approaches to minimize or alleviate reversion altogether. The relationship between genetic distance and segregation patterns observed by Burgstaller and colleagues indicating that more closely related mitochondria might be less subject to germline segregation, raises the possibility that mitochondria matching, akin to blood type matching of donors and recipients -in this case using a donor and recipient with a common maternal lineage might prove to be an effective strategy for mitochondrial therapy to replace de novo mitochondrial mutations, but further testing is required to determine if such matching is effective. In addition, developing mitochondrial replacement therapies that eliminate contaminating mitochondria or to screen for their presence so that only nuclear genomes devoid of mitochondria are used for the transfer procedure. Alternatively, a better understanding of the basic mechanisms and genes involved in mitochondria selection in the germline and oocyte bottlenecks will potentially offer new strategies not only to screen for persisting diseased mitochondria, but also may facilitate development of treatments or genetic approaches to mark diseased mitochondria for destruction or to endow healthy mitochondria with features that will confer selective advantage.
Highlights.
This review considers recent advances in our understanding of maternal inheritance of mitochondria, implications for fertility and mitochondrial disease, and potential roles for the Balbiani body, an ancient oocyte structure, in mitochondrial selection in oocytes, with emphasis on therapies to remedy mitochondrial disorders.
Acknowledgments
I am grateful to my lab members for discussions and comments on the review. I apologize for neglecting to cite all relevant literature due to space constraints. Work in the FLM lab is supported by NIH R01/GM089979.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Boussau B, Karlberg EO, Frank AC, Legault BA, Andersson SG. Computational inference of scenarios for alpha-proteobacterial genome evolution. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9722–9727. doi: 10.1073/pnas.0400975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harbor perspectives in biology. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annual review of genetics. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 4.Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 5.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Q, Li H, Xue D. Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans. Cell research. 2011;21:1662–1669. doi: 10.1038/cr.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLuca SZ, O’Farrell PH. Barriers to male transmission of mitochondrial DNA in sperm development. Developmental cell. 2012;22:660–668. doi: 10.1016/j.devcel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 1999;402:371–372. doi: 10.1038/46466. [DOI] [PubMed] [Google Scholar]
- 9.Luo SM, Ge ZJ, Wang ZW, Jiang ZZ, Wang ZB, Ouyang YC, Hou Y, Schatten H, Sun QY. Unique insights into maternal mitochondrial inheritance in mice. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13038–13043. doi: 10.1073/pnas.1303231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura Y, Yoshinari T, Naruse K, Yamada T, Sumi K, Mitani H, Higashiyama T, Kuroiwa T. Active digestion of sperm mitochondrial DNA in single living sperm revealed by optical tweezers. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1382–1387. doi: 10.1073/pnas.0506911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q, Li H, Li H, Nakagawa A, Lin JL, Lee ES, Harry BL, Skeen-Gaar RR, Suehiro Y, William D, Mitani S, Yuan HS, Kang BH, Xue D. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science. 2016;353:394–399. doi: 10.1126/science.aaf4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koehler CM, Lindberg GL, Brown DR, Beitz DC, Freeman AE, Mayfield JE, Myers AM. Replacement of bovine mitochondrial DNA by a sequence variant within one generation. Genetics. 1991;129:247–255. doi: 10.1093/genetics/129.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nature genetics. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh SS, Fahy E, Bodis-Wollner I, Sherman J, Howell N. Longitudinal study of a heteroplasmic 3460 Leber hereditary optic neuropathy family by multiplexed primer-extension analysis and nucleotide sequencing. American journal of human genetics. 1996;58:325–334. [PMC free article] [PubMed] [Google Scholar]
- 15.Larsson NG, Tulinius MH, Holme E, Oldfors A, Andersen O, Wahlstrom J, Aasly J. Segregation and manifestations of the mtDNA tRNA(Lys) A—>G(8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. American journal of human genetics. 1992;51:1201–1212. [PMC free article] [PubMed] [Google Scholar]
- 16.Marlow FL. Maternal Control of Development in Vertebrates: My Mother Made Me Do It! San Rafael (CA): 2010. [PubMed] [Google Scholar]
- 17.Kloc M, Jedrzejowska I, Tworzydlo W, Bilinski SM. Balbiani body, nuage and sponge bodies — The germ plasm pathway players. Arthropod structure & development. 2014 doi: 10.1016/j.asd.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 18.de Smedt V, Szollosi D, Kloc M. The balbiani body: asymmetry in the mammalian oocyte. Genesis. 2000;26:208–212. doi: 10.1002/(sici)1526-968x(200003)26:3<208::aid-gene6>3.3.co;2-e. [DOI] [PubMed] [Google Scholar]
- 19.Heasman J, Quarmby J, Wylie CC. The mitochondrial cloud of Xenopus oocytes: the source of germinal granule material. Developmental biology. 1984;105:458–469. doi: 10.1016/0012-1606(84)90303-8. [DOI] [PubMed] [Google Scholar]
- 20.Marlow F. Primordial Germ Cell Specification and Migration. F1000Res. 2015;4 doi: 10.12688/f1000research.6995.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakase H. [Leigh’s syndrome and mitochondrial myopathy] Nihon Rinsho. 1993;51:2403–2408. [PubMed] [Google Scholar]
- 22.Sakuta R, Goto Y, Horai S, Ogino T, Yoshinaga H, Ohtahara S, Nonaka I. Mitochondrial DNA mutation and Leigh’s syndrome. Annals of neurology. 1992;32:597–598. doi: 10.1002/ana.410320428. [DOI] [PubMed] [Google Scholar]
- 23.Lombes A, Nakase H, Tritschler HJ, Kadenbach B, Bonilla E, DeVivo DC, Schon EA, DiMauro S. Biochemical and molecular analysis of cytochrome c oxidase deficiency in Leigh’s syndrome. Neurology. 1991;41:491–498. doi: 10.1212/wnl.41.4.491. [DOI] [PubMed] [Google Scholar]
- 24.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 25.Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nature genetics. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 26.Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, Zhu J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157:1591–1604. doi: 10.1016/j.cell.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 27.Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, Ramsey C, Masterson K, Battaglia D, Lee D, Wu D, Jensen J, Patton P, Gokhale S, Stouffer R, Mitalipov S. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HS, Ma H, Juanes RC, Tachibana M, Sparman M, Woodward J, Ramsey C, Xu J, Kang EJ, Amato P, Mair G, Steinborn R, Mitalipov S. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell reports. 2012;1:506–515. doi: 10.1016/j.celrep.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuels DC, Wonnapinij P, Chinnery PF. Preventing the transmission of pathogenic mitochondrial DNA mutations: Can we achieve long-term benefits from germ-line gene transfer? Hum Reprod. 2013;28:554–559. doi: 10.1093/humrep/des439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freyer C, Cree LM, Mourier A, Stewart JB, Koolmeister C, Milenkovic D, Wai T, Floros VI, Hagstrom E, Chatzidaki EE, Wiesner RJ, Samuels DC, Larsson NG, Chinnery PF. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nature genetics. 2012;44:1282–1285. doi: 10.1038/ng.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS biology. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada M, Emmanuele V, Sanchez-Quintero MJ, Sun B, Lallos G, Paull D, Zimmer M, Pagett S, Prosser RW, Sauer MV, Hirano M, Egli D. Genetic Drift Can Compromise Mitochondrial Replacement by Nuclear Transfer in Human Oocytes. Cell Stem Cell. 2016;18:749–754. doi: 10.1016/j.stem.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgstaller JP, Johnston IG, Jones NS, Albrechtova J, Kolbe T, Vogl C, Futschik A, Mayrhofer C, Klein D, Sabitzer S, Blattner M, Gully C, Poulton J, Rulicke T, Pialek J, Steinborn R, Brem G. MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell reports. 2014;7:2031–2041. doi: 10.1016/j.celrep.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ankel-Simons F, Cummins JM. Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13859–13863. doi: 10.1073/pnas.93.24.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otten AB, Theunissen TE, Derhaag JG, Lambrichs EH, Boesten IB, Winandy M, van Montfoort AP, Tarbashevich K, Raz E, Gerards M, Vanoevelen JM, van den Bosch BJ, Muller M, Smeets HJ. Differences in Strength and Timing of the mtDNA Bottleneck between Zebrafish Germline and Non-germline Cells. Cell reports. 2016;16:622–630. doi: 10.1016/j.celrep.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 36.Piko L, Matsumoto L. Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Developmental biology. 1976;49:1–10. doi: 10.1016/0012-1606(76)90253-0. [DOI] [PubMed] [Google Scholar]
- 37.Cree LM, Samuels DC, Lopes SC de Sousa, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nature genetics. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 38.Cao L, Shitara H, Sugimoto M, Hayashi J, Abe K, Yonekawa H. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS genetics. 2009;5:e1000756. doi: 10.1371/journal.pgen.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nature genetics. 2008;40:1484–1488. doi: 10.1038/ng.258. [DOI] [PubMed] [Google Scholar]
- 40.Kloc M, Bilinski S, Etkin LD. The Balbiani body and germ cell determinants: 150 years later. Current topics in developmental biology. 2004;59:1–36. doi: 10.1016/S0070-2153(04)59001-4. [DOI] [PubMed] [Google Scholar]
- 41.Cox RT, Spradling AC. A Balbiani body and the fusome mediate mitochondrial inheritance during Drosophila oogenesis. Development. 2003;130:1579–1590. doi: 10.1242/dev.00365. [DOI] [PubMed] [Google Scholar]
- 42.Pepling ME, Spradling AC. Female mouse germ cells form synchronously dividing cysts. Development. 1998;125:3323–3328. doi: 10.1242/dev.125.17.3323. [DOI] [PubMed] [Google Scholar]
- 43.Wilding M, Carotenuto R, Infante V, Dale B, Marino M, Di Matteo L, Campanella C. Confocal microscopy analysis of the activity of mitochondria contained within the ‘mitochondrial cloud’ during oogenesis in Xenopus laevis. Zygote. 2001;9:347–352. doi: 10.1017/s096719940100140x. [DOI] [PubMed] [Google Scholar]
- 44.Tworzydlo W, Kisiel E, Jankowska W, Witwicka A, Bilinski SM. Exclusion of dysfunctional mitochondria from Balbiani body during early oogenesis of Thermobia. Cell and tissue research. 2016 doi: 10.1007/s00441-016-2414-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YZ, Ouyang YC, Hou Y, Schatten H, Chen DY, Sun QY. Mitochondrial behavior during oogenesis in zebrafish: a confocal microscopy analysis. Development, growth & differentiation. 2008;50:189–201. doi: 10.1111/j.1440-169X.2008.00988.x. [DOI] [PubMed] [Google Scholar]
- 46.Marlow FL, Mullins MC. Bucky ball functions in Balbiani body assembly and animal-vegetal polarity in the oocyte and follicle cell layer in zebrafish. Developmental biology. 2008;321:40–50. doi: 10.1016/j.ydbio.2008.05.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bontems F, Stein A, Marlow F, Lyautey J, Gupta T, Mullins MC, Dosch R. Bucky ball organizes germ plasm assembly in zebrafish. Current biology, CB. 2009;19:414–422. doi: 10.1016/j.cub.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 48.Heim AE, Hartung O, Rothhamel S, Ferreira E, Jenny A, Marlow FL. Oocyte polarity requires a Bucky ball-dependent feedback amplification loop. Development. 2014;141:842–854. doi: 10.1242/dev.090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forbes MM, Draper BW, Marlow FL. The polarity factor Bucky ball associates with the centrosome and promotes microtubule rearrangements to establish the oocyte axis in zebrafish. Development. 2015 doi: 10.1242/dev.129023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elkouby YM, Jamieson-Lucy A, Mullins MC. Oocyte Polarization Is Coupled to the Chromosomal Bouquet, a Conserved Polarized Nuclear Configuration in Meiosis. PLoS biology. 2016;14:e1002335. doi: 10.1371/journal.pbio.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claussen M, Pieler T. Xvelo1 uses a novel 75-nucleotide signal sequence that drives vegetal localization along the late pathway in Xenopus oocytes. Developmental biology. 2004;266:270–284. doi: 10.1016/j.ydbio.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 52.Boke E, Ruer M, Wuhr M, Coughlin M, Lemaitre R, Gygi SP, Alberti S, Drechsel D, Hyman AA, Mitchison TJ. Amyloid-like Self-Assembly of a Cellular Compartment. Cell. 2016;166:637–650. doi: 10.1016/j.cell.2016.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeske M, Bordi M, Glatt S, Muller S, Rybin V, Muller CW, Ephrussi A. The Crystal Structure of the Drosophila Germline Inducer Oskar Identifies Two Domains with Distinct Vasa Helicase-and RNA-Binding Activities. Cell reports. 2015;12:587–598. doi: 10.1016/j.celrep.2015.06.055. [DOI] [PubMed] [Google Scholar]
- 54.Nijjar S, Woodland HR. Protein interactions in Xenopus germ plasm RNP particles. PloS one. 2013;8:e80077. doi: 10.1371/journal.pone.0080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song HW, Cauffman K, Chan AP, Zhou Y, King ML, Etkin LD, Kloc M. Hermes RNA-binding protein targets RNAs-encoding proteins involved in meiotic maturation, early cleavage, and germline development. Differentiation; research in biological diversity. 2007;75:519–528. doi: 10.1111/j.1432-0436.2006.00155.x. [DOI] [PubMed] [Google Scholar]
- 56.Aguero T, Zhou Y, Kloc M, Chang P, Houliston E, King ML. Hermes (Rbpms) is a Critical Component of RNP Complexes that Sequester Germline RNAs during Oogenesis. J Dev Biol. 2016;4 doi: 10.3390/jdb4010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Extavour CG, Akam M. Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development. 2003;130:5869–5884. doi: 10.1242/dev.00804. [DOI] [PubMed] [Google Scholar]
- 58.Ewen-Campen B, Schwager EE, Extavour CG. The molecular machinery of germ line specification. Molecular reproduction and development. 77:3–18. doi: 10.1002/mrd.21091. [DOI] [PubMed] [Google Scholar]
- 59.Cox RT, Spradling AC. Milton controls the early acquisition of mitochondria by Drosophila oocytes. Development. 2006;133:3371–3377. doi: 10.1242/dev.02514. [DOI] [PubMed] [Google Scholar]
- 60.Little SC, Sinsimer KS, Lee JJ, Wieschaus EF, Gavis ER. Independent and coordinate trafficking of single Drosophila germ plasm mRNAs. Nature cell biology. 2015;17:558–568. doi: 10.1038/ncb3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trcek T, Grosch M, York A, Shroff H, Lionnet T, Lehmann R. Drosophila germ granules are structured and contain homotypic mRNA clusters. Nature communications. 2015;6:7962. doi: 10.1038/ncomms8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding D, Whittaker KL, Lipshitz HD. Mitochondrially encoded 16S large ribosomal RNA is concentrated in the posterior polar plasm of early Drosophila embryos but is not required for pole cell formation. Developmental biology. 1994;163:503–515. doi: 10.1006/dbio.1994.1166. [DOI] [PubMed] [Google Scholar]
- 63.Kashikawa M, Amikura R, Nakamura A, Kobayashi S. Mitochondrial small ribosomal RNA is present on polar granules in early cleavage embryos of Drosophila melanogaster. Development, growth & differentiation. 1999;41:495–502. doi: 10.1046/j.1440-169x.1999.00451.x. [DOI] [PubMed] [Google Scholar]
- 64.Amikura R, Hanyu K, Kashikawa M, Kobayashi S. Tudor protein is essential for the localization of mitochondrial RNAs in polar granules of Drosophila embryos. Mechanisms of development. 2001;107:97–104. doi: 10.1016/s0925-4773(01)00455-5. [DOI] [PubMed] [Google Scholar]
- 65.Amikura R, Kashikawa M, Nakamura A, Kobayashi S. Presence of mitochondria-type ribosomes outside mitochondria in germ plasm of Drosophila embryos. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9133–9138. doi: 10.1073/pnas.171286998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 67.Brangwynne CP. Phase transitions and size scaling of membrane-less organelles. The Journal of cell biology. 2013;203:875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase -Separated Liquid Droplets by RNA-Binding Proteins. Molecular cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, Gladfelter AS. RNA Controls PolyQ Protein Phase Transitions. Molecular cell. 2015;60:220–230. doi: 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lehmann R. Germ Plasm Biogenesis—An Oskar-Centric Perspective. Current topics in developmental biology. 2016;116:679–707. doi: 10.1016/bs.ctdb.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Halter J, Schupbach WM, Casali C, Elhasid R, Fay K, Hammans S, Illa I, Kappeler L, Krahenbuhl S, Lehmann T, Mandel H, Marti R, Mattle H, Orchard K, Savage D, Sue CM, Valcarcel D, Gratwohl A, Hirano M. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330–337. doi: 10.1038/bmt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]