

Abstract
Time series of single-cell transcriptome measurements can reveal dynamic features of cell differentiation pathways. From measurements of whole frog embryos spanning zygotic genome activation through early organogenesis, we derived a detailed catalog of cell states in vertebrate development and a map of differentiation across all lineages over time. The inferred map recapitulates most if not all developmental relationships and associates new regulators and marker genes with each cell state. We find that many embryonic cell states appear earlier than previously appreciated. We also assess conflicting models of neural crest development. Incorporating a matched time series of zebrafish development from a companion paper, we reveal conserved and divergent features of vertebrate early developmental gene expression programs.
Metazoan development represents a big jump in complexity compared to unicellular life in two aspects: cell type differentiation and cell spatial organization. The fertilized egg was once itself thought to be very complex, concealing the spatial and compositional differentiation of the adult. However, it is now clear that the egg is a rather simple cell and the complexity of the embryo and adult arises by bootstrapping: repeatedly exploiting spatial asymmetries to build the complex embryo by stages. In vertebrate embryos, as well as in other phyla, a provisional body plan organization arises before overt cell type specific differentiation and this provides a scaffold for the emerging complexity.
The process of increasing spatial and cell type heterogeneity has been difficult to follow comprehensively at the molecular level. Many insights have emerged from genetic studies and experimental embryology, but those analyses generally focus on a few transcription factors and/or secreted signaling molecules. Instead development involves alterations in many different intracellular and intercellular circuits. Recent technology advances have enabled the mapping of mRNA content in single cells over time, which could in principle systematically reveal differentiation pathways across entire developing embryos (Fig. 1A).
Fig. 1. Dissection of early Xenopus tropicalis development by scRNA-seq.
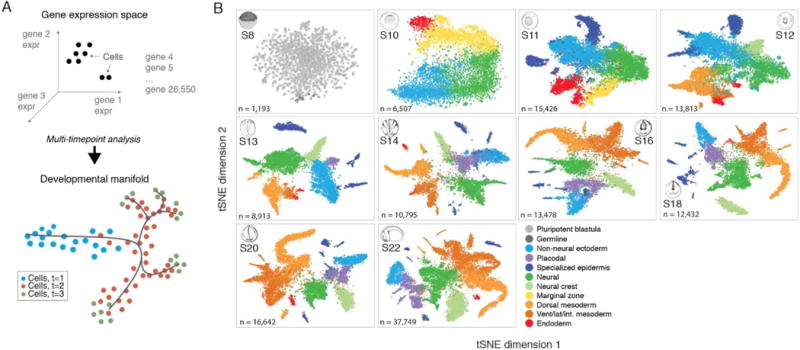
(A) Single-cell transcriptomes represent points in a high-dimensional gene expression space. By collecting single-cell transcriptomes over time of embryo development, it is possible to infer a continuum gene expression manifold connecting cell states across all lineages. (B) Summary of scRNA-seq developmental timecourse including 136,966 single-cell transcriptomes sampled over ten embryonic stages (S8-22). tSNE plots show increasing cell population structure over time. Colors indicate major tissues grouped by germ layer. Further details on subclustering shown in (figs. S3, S5, and S6), and at: tinyurl.com/scXen2018.
Two years ago we developed a versatile single-cell transcriptomic method based on droplet microfluidics that accurately measures genome-ide RNA levels in individual cells at high throughput (1). We applied this method to analyze all of the cells in the embryos of the Western claw-toed frog, Xenopus tropicalis over the first day of life post-fertilization. Along with its larger relative X. laevis, the frog embryo serves as one of the best-studied model systems of early vertebrate development. The egg undergoes 12 rapid cleavage divisions, during which time zygotic transcription is suppressed. The resulting four thousand cells are initially pluripotent. However, within 2 hours of activating zygotic transcription at the midblastula transition (2, 3), embryos go through gastrulation to establish the major germ layers, followed by fate commitment and progressive differentiation (4). We profiled embryos prior to the onset of zygotic transcription, through fate commitment and into the early tailbud stage - a point at which dozens of distinct cell types have formed encompassing the early progenitors of most major organs. In the course of this work we developed general computational tools that can relate cell states over time, providing a global view of gene expression diversification in the early embryo. Finally, we compared the results of this analysis to our recently completed time series of the developing zebrafish, Danio rerio. Through systematic analyses across lineages, time, and species we note generalities and differences in embryonic cell differentiation in the two vertebrate clades, which separated about 400 million years ago.
Single-cell RNA-seq of whole developing embryos
Xenopus blastomeres are large and fragile, with cell diameters up to 50 μm (X. tropicalis, compared with 100 μm for X. laevis) at the onset of zygotic transcription (stage 8.5). To preserve the integrity of cells through tissue dissociation and microfluidic handling, we optimized the inDrop platform to accommodate large cells. We also formulated a dissociation buffer that uses alkaline pH to promote dissociation, and handling procedures that minimize shear stresses placed on cells. The new protocol enables rapid and complete dissociation of whole embryos to single cells within 25 min with minimal mechanical agitation and cell handling, preservation of >95% cell viability, and background scRNA-seq signal (indicative of cell lysis) of <1-4% of total cellular RNA from stage 10 onward (fig. S1).
The samples for scRNA-seq were taken at ten time-points from zygotic genome activation (stage 8, 5 hpf) through early organogenesis (stage 22, 22 hpf), profiling a total of 136,966 single cells in two replicate experiments (table S1 and movies S1 to 3). The first replicate consisted of 42 k cells sequenced to a depth of 5.4 k UMIs per cell on average (99% of genes from bulk RNA-seq observed in pools of >100 cells), whereas the second replicate of 95 k cells was sequenced to an average depth of 1.4 k UMIs per cell. The number of measured cells allows detection of rare sub-populations such as germ cells. Overall, we expect to observe transcriptional states as rare as 0.1% of the embryo represented by at least 10 cells with 95% confidence (fig. S2). There may be rare or transient sub-populations that are missed given the total number of cells, time sampling, and the depth of sequencing we used.
Low-dimensional visualization of the scRNA-seq timeseries shows a pattern of increasing complexity over time, with cells fragmenting from a continuum of gene expression states in the early gastrula into distinct clusters at later time-points (Fig. 1B and figs. S3 and S4). To relate the scRNA-seq data to known embryonic cell types, we first classified gene expression states at each timepoint by a hierarchical clustering approach (Figs 1B and fig. S3). These cluster assignments were robust to different clustering algorithms (figs. S5 and S6). We then annotated each cluster by matching cluster-specific genes to >2000 in situ measurements of marker genes for embryonic cell types documented on the Xenopus Bioinformatics database (Xenbase) (5) (Data S1). We additionally shared our annotations with experts in the Xenopus research community, convened in part for that purpose at a recent Jamboree meeting, who helped to standardize our cell state nomenclature and choice of annotations. In total, we identified 87 distinct cell type annotations, many of which persisted in clusters across multiple time-points (average of 2.9 timepoints each) (Fig. 2C). Of these 87 states, 69 corresponded unambiguously to a specific Xenbase anatomical term, whereas the remaining 18 states correspond to finer structure (i.e., cell subtypes), or unidentified cell states within these anatomically defined tissues. Further substructure may be resolved by manual inspection of marker genes employing prior expert knowledge.
Fig. 2. Inference of developmental cell state transitions from gene expression similarity.
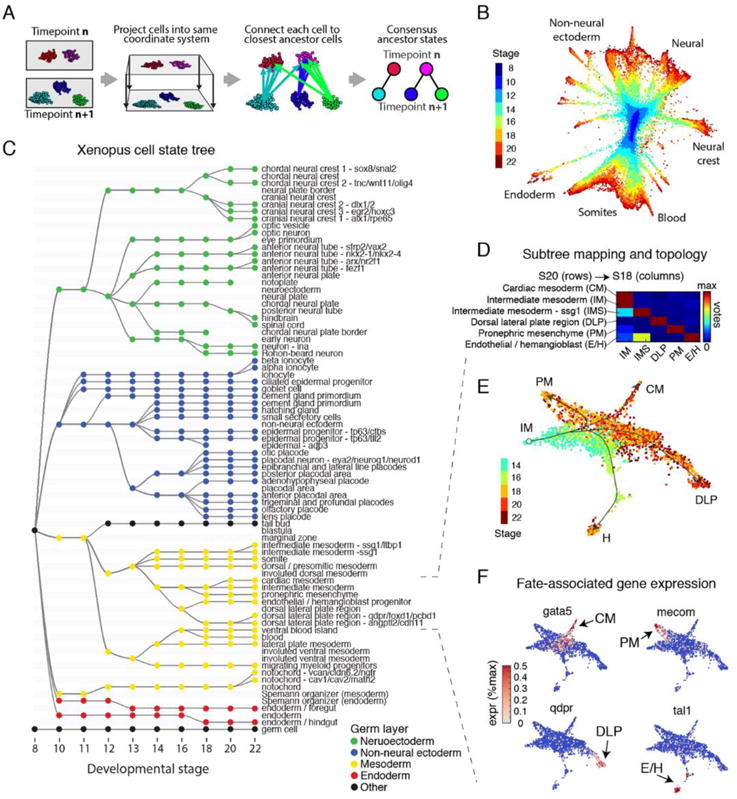
(A) Schematic of the mapping algorithm used to make similarity connections between clusters across time. (B) Global visualization of single cells profiled in the Xenopus developmental timecourse using a kNN-graph (Wagner et al.). (C) Cell state tree showing all inferred developmental transitions. Generated by applying the mapping algorithm in (A). (D) Representative cell voting outcomes between time points, generated during state tree construction. (E) Single-cell visualization of a representative sub-tree, showing lateral and intermediate mesoderm fates. Lines indicate corresponding topology of the cell state tree. (F) Marker gene expression associated with the formation of each intermediate mesoderm cell state.
Reconstructing developmental cell state transitions
Fate mapping studies have previously established a set of spatial lineage relationships in the Xenopus embryo; these generally support the notion of a hierarchical pattern of differentiation, albeit with some exceptions (6). We investigated the extent to which these known hierarchical lineage relationships are reflected in our map of expression states. A close match was not assured since there are several reasons cell lineage hierarchies might not be reflected in molecular expression. First, cellular states are defined not only by tissue but by spatial position and cell cycle state, which cross lineage boundaries. Second, asymmetric cell divisions could lead to discontinuities in cell state as a mother cell partitions into daughter cells with distinct molecular compositions. Third, although the lineage history of individual cells consist entirely of discrete bifurcation (mitosis) events, cell states could be continuous or show complex branching structures or even loops. Yet, counteracting this, in the lineage data of the early embryo, new gene expression generally arises in single contiguous domains.
To ask whether the single-cell gene expression states conform to a branching pattern, and if so, whether the pattern overlaps with the previously deduced lineage patterns, we devised a simple algorithm that connects each cell to its most likely ancestors (i.e., nearest neighbors) in the previous timepoint, and uses the consensus of these connections to assign an ancestor to each cell state (Fig. 2A). By applying this algorithm to all 259 cell states, comprised of 136,966 Xenopus cells spread over ten timepoints (Fig. 2B), we generated a detailed map of putative cell state transitions during early development (Fig. 2C and fig. S7). The map recapitulates known expression domains of master regulators within each germ layer over time (fig. S8). Significantly, only 17% of all votes fell outside of consensus ancestor clusters, showing that cell states over development can generally be approximated by a tree-like structure (Fig. 2D and fig. S7). The majority of votes cast outside of consensus ancestor states occurred at early timepoints, due to their continuum structure. The branching nature of cell states can also be appreciated by inspecting individual lineages at single-cell resolution (Fig. 2, E and F). The inferred cell state tree structure was consistent with ancestor assignments generated using an alternative state graph coarse-graining algorithm reported in our companion paper (7) (fig. S9).
The ancestor assignments largely agree with the known lineage relationships. Of all cross-time-point edges inferred by the algorithm (Fig. 2C), we could confirm 234 (91%) by comparison to Xenbase anatomy ontology (XAO) (8, 9) and the literature (Data S2). Of the remainder, 22 could not be ruled in or out, and only 2 (1%) were identifiably incorrect. The two errors, which occurred in somites and erythroid lineages, reflected a specific limitation of the tree-building approach: in both lineages, mature states initially arise from progenitors, but then form a parallel branch, rather than continuously arising from progenitors anew at each time point. Visualization of the raw scRNA-Seq data reveals the underlying asynchronous differentiation process (fig. S10). In addition to these errors, the cell state tree does not consistently resolve spatial localizations, e.g., we do not resolve individual somites. The tree-building approach could additionally generate errors when applied to other data sets lacking the dense time-sampling carried out here. When inferring cell state transitions from scRNA-seq snapshots separated by large gaps in time, intermediate progenitor states may be overlooked. Overall these results support a growing body of single-cell bioinformatics methods that seek to infer developmental cell trajectories on the basis of continuity (10, 11) and distance minimization (12) in gene expression space, but also illuminate where this principle can be misleading.
Among the developmental relationships inferred on the tree, there is also a question about the timing at which known cell types first appear. Of the 69 cell types with an unambiguous match to Xenbase, 60 appeared in our data at the developmental stage indicated in XAO (8, 9) or earlier. Several appeared much earlier than previously recognized (Fig. 3A), including an endothelial/hemangioblast progenitor, which appeared from the dorsal lateral plate region at Stage 18, as compared to Stage 26 / 31 (respectively) as previously thought (13,14) (Fig. 3B). This afforded an opportunity to explore the earliest transcriptional events associated with the specification of these fates (Fig. 3C). Notably, the tail bud and multiple epidermal cell types also appeared much earlier than indicated by XAO (Fig. 3A and Data S2) and revealed previously unappreciated early transcriptional dynamics.
Fig. 3. scRNA-seq detects early transcriptional events during specification of embryonic cell states.
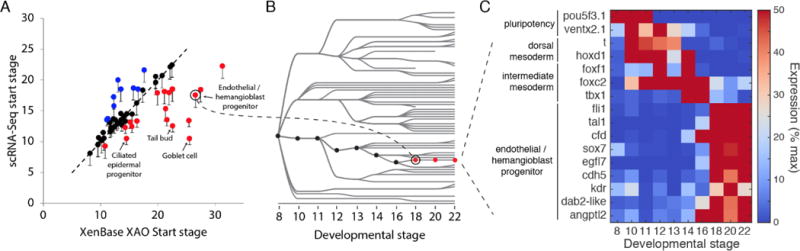
(A) Time of first appearance for each cell state in the cell state tree as compared to documented appearance times in the Xenopus anatomy ontology (XAO). Red/blue points are detected early/late in scRNA-seq as compared to XAO. 60 of 69 states appear as early or earlier than documented. Error bars represent time interval of scRNA-seq experiment. (B and C) scRNA-seq reveals an early endothelial / hemangioblast progenitor that appears at stage 18 (red lineage), as compared to stage 26 for hemangioblasts and stage 31 for endothelial cells [XAO; (12, 13)], with recognizable activation of the endothelial/hemangioblast gene expression program (C).
Overall, the cell state tree provides a resource for probing gene expression in the early Xenopus embryo, incorporating annotations of cell states, linking related states across time, and discovering the earliest transcriptional record of the specification of each cell type. It is now possible to identify genes that are differentially expressed between bifurcating cell states, to assess gene expression changes along the developmental history of each lineage, and to automatically identify specific marker genes for each cell state (fig. S11). We have tabulated genes differentially expressed at every embryonic cell fate choice (Data S3), indicating potential fate regulators, as well as marker genes for every cell state (Data S4). The complete data set, which is available through an interactive online browser (tinyurl.com/scXen2018) supports visualization of gene expression across the cell state tree, identification of enriched genes in specific states, differential expression analysis between cell states, co-expression analysis of gene pairs, and visualization of the dynamics of gene expression along particular cell state tree lineages. Interactive plots of single cells from each stage are also available through this browser.
Divergence of developmental gene expression between frog (amphibians-anurans) and fish (teleosts)
There are numerous cladograms of gene sequence that confirm the basic paleontological record of vertebrates, reflected in clear differences as well as similarities between Xenopus and zebrafish in their sequenced genomes. But whenever there is a change in anatomy, cell type, and physiology there must be an underlying alteration in the developmental program. Time-series single-cell gene expression measurements offer an opportunity to look deeply into these developmental gene expression programs and give us clues about the most consequential evolutionary changes across species. We first asked whether the same (i.e., orthologous) genes are conserved in developing tissues in frog and fish using data from our companion paper on zebrafish development (7), which spans a similar developmental period to the frog. We processed the zebrafish time series through the same tree building algorithm over annotated cell states. We manually aligned cell states between species (Fig. 4A, red shading) on the basis of tissue name, marker gene expression, developmental stage, and lineage relationships. In some cases, nomenclature of homologous tissues differed slightly between species. Thirty tissues could be matched with high confidence, jointly covering 66 of 87 Xenopus states (79% cells) and 83 of 122 zebrafish states (83% cells). The cell state tree alignment indicated broad conservation of lineage topologies between species, although the observed abundances of cell types differed markedly between species, and the proportion of matched states and cells decreased over time as species-specific features accumulated-such as specialized epidermal cell types in both species (fig. S12). Notably, the zebrafish neural ectoderm grew to 60% of sequenced cells in the embryo at 24 hpf, compared to 31% in the Stage 22 frog (fig. S13). Differences in cell survival rates during InDrops processing in either species could affect estimates of cell state abundances.
Fig. 4. Similarities and differences in developmental cell state hierarchies and gene expression between frog and fish.
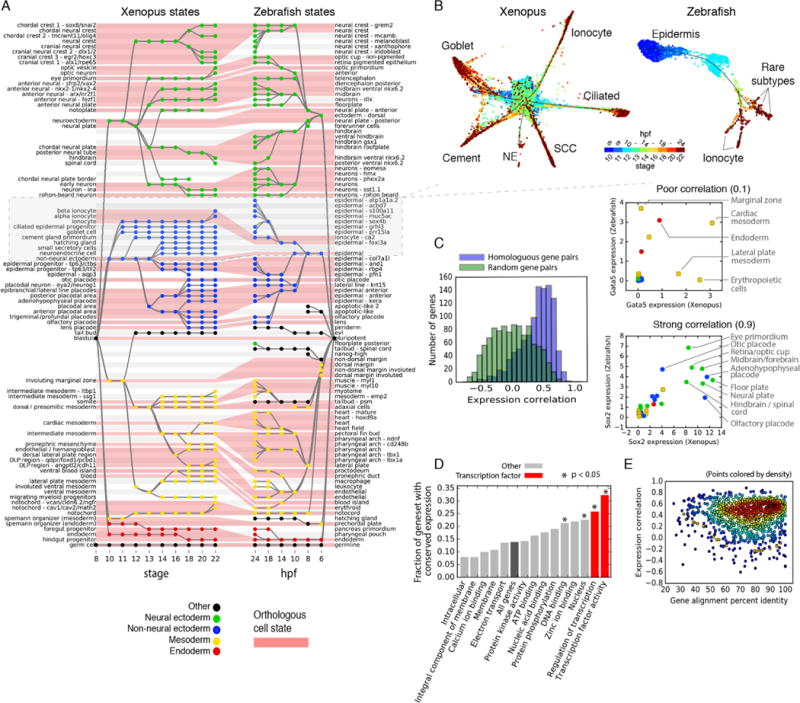
(A) Xenopus and Zebrafish cell state trees aligned by orthologous cell states (red shading). Gray/white stripes provide a visual guide. (B) Single-cell visualization of matched epidermal subtrees in frog and fish showcase similarities and differences in developmental hierarchy. SCC, small secretory cell; NE, neuroendocrine cell. Unidentified zebrafish cell types are labeled by marker genes. (C) Ortholog genes across species have variable conservation of cell state specific expression. Just 30% of self-similar orthologs are conserved at a 95% FDR compared to random gene pairs. Right panels: examples of highly (Sox2) and poorly (Gata5) correlated TFs across species. (D and E) Function, not sequence, predicts gene expression conservation: (D) Orthologs with highly conserved expression patterns across species are enriched in TF-associated GO terms. P-values show Bonferroni-corrected binomial test results. (E) Protein sequence conservation is not correlated with gene expression conservation (r = 0.01; P = 0.6).
The alignment of the cell state trees also highlighted significant changes in developmental patterning between the two species, which could further be appreciated by deeper inspection of the underlying single-cell data. In the epidermis, for example, the two species show a radiation of common and distinct cell types from gata2-expressing non-neural ectoderm (Fig. 4B and fig. S14). Trajectories of differentiating ionocytes can be seen in both species, whereas ciliated cells expressing foxj1, goblet cells expressing itln1, and small secretory cells expressing met (fig. S14), each of which is an important component of the Xenopus epidermis, are absent in zebrafish. These different epidermal cell types likely reflect the demands of development in different environments, potentially requiring an early immune barrier in the frog but not the fish. Rearrangements of lineage topology can also be appreciated: Xenopus and zebrafish both produce specialized hatching glands (HG) that secrete shared hatching enzymes and that are specified by the conserved transcription factor klf17, seen by scKNA-Seq (fig. S15). Single-cell trajectories link the HG to non-neural ectoderm in Xenopus, but to the organizer mesoderm in zebrafish (Fig. 4A and fig. S15), reflecting a known difference in HG germ layer origin between Amphibians and Teoleosts (15).
Other more subtle changes in lineage topology involved the addition or loss of specialized progenitor cell states within a partially shared developmental history. For example, xanthoblasts appear at 18 hpf in zebrafish from an early neural crest population, but do not appear until stage 46 in Xenopus (16), after the shared ancestral neural crest population has progressed through multiple additional specialized neural crest intermediate states. Similarly, myeloid cells appear in Xenopus at S14 from early involuted ventral mesoderm, whereas they (macrophages and leukocytes) appear only at 18-24 hpf in zebrafish, after the ventral mesoderm has differentiated through specialized cardiovasculature and lateral plate intermediate states. Terminal differentiation of myeloid cells in both cases involves activation of several conserved master regulators including cybb, cyba, spib, and cebpa. These observations support a degree of independence between the differentiation path characterizing a lineage and the activation of particular conserved terminal cell phenotypes (17). Variation in differentiation paths can reflect not only drift or flexibility in developmental programs, but also selected features of embryonic cells including roles in interacting with and instructing other differentiation and morphogenetic mechanisms.
To obtain a systematic view of similarities and differences in developmental gene expression across species, we used the aligned cell state trees to investigate whether orthologous genes show correlation in expression values across matched embryonic tissues (Fig. 4C). Focusing on the subset of genes that were robustly expressed and dynamically varying in both species, we observed that orthologous genes correlated in their expression between species (Fig. 4C), seemingly supporting the notion that cell state gene expression is largely conserved. However, only a minority (30%) of orthologous pairs were correlated with high confidence (5% FDR) compared to random pairs. For example, in contrast to the well conserved (r = 0.9) neuroectodermal marker sox2, we identified many examples of poorly correlated genes such as the transcription factor gata5 (r = 0.1) (Fig. 4C). Despite being a conserved endoderm and cardiac master regulator, gata5 is also expressed in the erythropoietic tissue only of Xenopus (18), and in the marginal zone only of zebrafish (19) at a distinct spatiotemporal location of endoderm specification compared to the vegetal expression seen in Xenopus. Accordingly, we were surprised to find that the genes best marking cell states within one species did not generally perform well at marking the same cell states within the other (fig. S16).
We tried to understand what properties predict whether a gene shows conserved expression patterns across tissues across species, i.e., whether specific functional categories of genes are more likely to be conserved in expression dynamics. Analysis of gene ontology annotations spanning diverse cellular processes revealed a striking enrichment of gene expression conservation among transcription factors, identifying three statistically significant (P < 0.05; binomial-test, Bonferroni corrected) functional annotations that include “nucleus”, “regulation of transcription” and “transcription factor activity” (Fig. 4D). By contrast, protein sequence conservation was uncorrelated to gene expression conservation (r = 0.01; P = 0.6). Orthologous genes with 20-40% sequence identity had the same expression conservation as those with 95-100% sequence identity (average expression conservation of 0.41 versus 0.38, P = 0.5; t test). A gene’s function is therefore more strongly predictive of its conservation in developmental gene expression programs across species than conservation of its protein sequence, decoupling two fundamental processes underlying evolutionary changes in embryonic development.
Reuse of developmental transcription factors
Given the close association and conservation of transcription factors with differentiation and hence cell type, we asked two questions. How are transcription factors deployed across cell types and across time during development? And how at the level of genes, do new cell identities emerge at branch points in development? These questions can be examined here in dozens of cell states and in the unperturbed setting of the embryo, rather than in cell culture.
Classical experiments have described differentiation in terms of the activation of single transcription factors, called master regulators, which established cell identity and after which other transcription factors progressively refined the phenotype (20) (Fig. 5A). Alternative views, however ascribe the initial specification of cell identity to interactions among multiple transcription factors, a ‘combinatorial code’, capable of defining a larger number of identities with different combinations from a small set of TFs (21, 22) (Fig. 5B). We asked which of these views best describes early development. This is not just of theoretical interest since knowing which transcription factor(s) could specify a given cell type could aid in the formulation of combinations that could be used therapeutically (23). Our experiments identify some cases where candidate TFs are activated just once in early development and after which they persist and may define a lineage, suggesting a master regulator model. But we also see TFs that are expressed more than once de novo in distinct and unrelated cell states, which may support a model of combinatorial deployment.
Fig. 5. TF reuse is pervasive in vertebrate development.
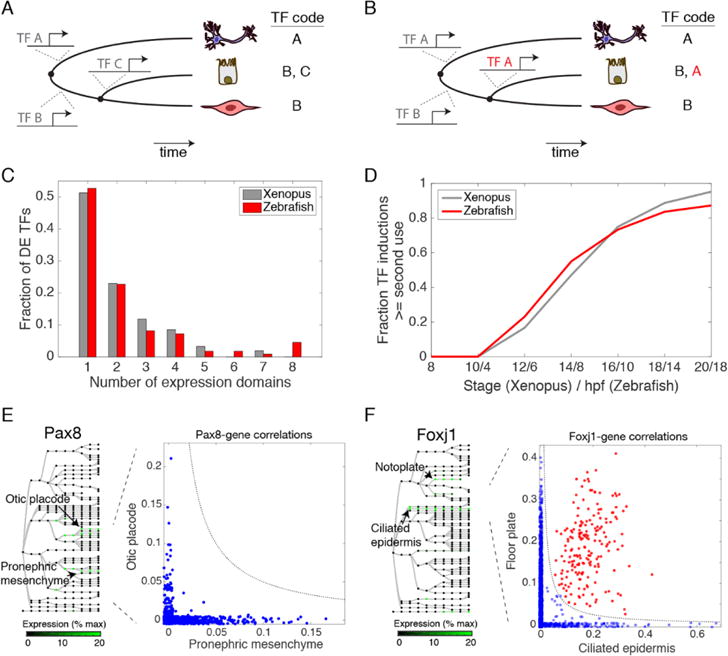
(A) Progressive programming of cell identity through sequential TF activation. (B) Programming of cell identity through combinatorial reuse of TFs. TF A is reused (red) in combination with TF B to generate a new fate. (C) Half of differentially expressed (DE) TFs are induced more than once in early frog and fish development. (D) Reused TFs increasingly dominate new TF expression during fate choices over time. (E and F) Reused TFs correlate with context-dependent (blue; off-diagonal) or conserved (red; on-diagonal) gene expression modules: (E) Pax8 correlates with different genes in the otic placode and the pronephric mesenchyme. (F) Foxj1 correlates with ciliogenesis genes in both the floor plate and in ciliated epidermal cells.
To understand the prevalence of these modes of TF use, we scored the number of times a TF was expressed independently, i.e., in states that share a non-expressing common ancestor. From this analysis, we found that the expression of half of developmentally-variable TFs is initiated only once during early development (52% in frog, 54% in zebrafish) (Fig. 5C). The detected single-use TF induction events occurred in just 35% of annotated cell states, seemingly inconsistent with the view that distinct master TFs define each fate. Reused-TFs, by contrast, cover 58% of tissue annotations in a complex combinatorial code (Data S5). In the remaining 7% of cell states, a specific TF induction was not detected. Over time, the average fraction of transcription factors being reused in a second circumstance climbs, reaching ~90% of new TF expression initiation events by stage 20 (18.5 hpf) in Xenopus and by 20 hpf in zebrafish (Fig. 5D). Together these results underscore the importance of combinatorial TF reuse in developmental gene expression programs.
Consistent with the notion that combinatorial interactions could lead to complex gene expression responses, the expression of the same TF in different tissues did not generally correlate with the same gene sets, as seen in the example of Pax8, which is independently expressed in the otic placode and pronephric mesenchyme (Fig. 5E). Some putative downstream targets of reused TFs could however be identified in more than one tissue, as in the case of Foxj1, which is expressed in both the ciliated epidermis and the floor plate, and orchestrates motile cilia formation. Many of the genes correlating with foxj1 transcript abundance within the floor plate also correlated with foxj1 in the epidermis, and were highly enriched for ciliogenesis GO terms (Fig. 5F). Interestingly, we found that TFs deployed just once over the observed time series were more conserved in their expression pattern across the two-species compared to reused TFs (median correlation 0.72 vs. 0.48 in frog; 0.64 vs. 0.36 in fish). This may indicate that reused TFs are more commonly rearranged across tissues during evolution to generate novelties, whereas TFs whose field of expression is established just once might have more conserved regulatory functions.
Multilineage gene expression priming during fate choices
It has been shown in several specific cell types - including hematopoietic stem cells (24) and mouse ES cells (25) - that uncommitted cells may coexpress competing fate regulators associated with more than one terminal cell fate. Such gene expression overlap, followed by refinement as cells differentiate, is known as multilineage priming (MLP) (Fig. 6A). MLP has been argued to be a part of the process of cell type decision making, via competition between fate regulators, and also to play a role in specifying an undifferentiated state by blocking lineage commitment (25–28).
Fig. 6. Refinement of promiscuous multilineage gene expression during early embryonic fate choices.
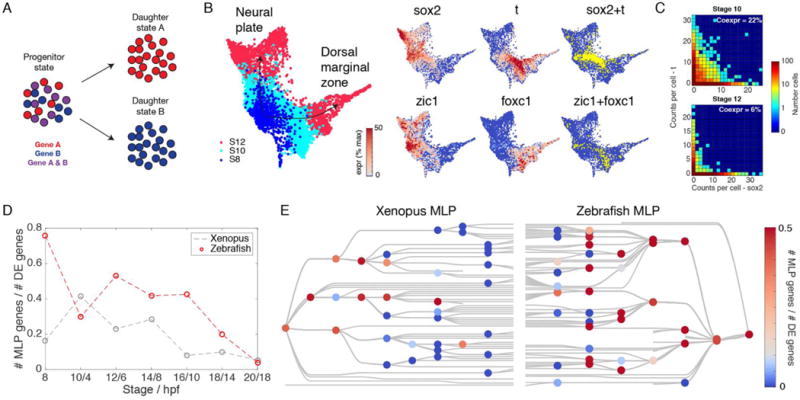
(A) Illustration of multilineage priming (MLP). Two genes, specific to daughter states A and B respectively, transiently overlap in the ancestor progenitor state as a fate decision is being made. (B and C) MLP during the fate choice between neural plate and dorsal marginal zone in Xenopus. Sox2 and T, as well as Zic1 and Foxc1, overlap in progenitor cells before becoming specific. (D and E) Global patterns of MLP in early development: (D) Multilineage primed genes are initially pervasive among differentially expressed (DE) genes at fate branch points, but become progressively rarer. (E) MLP frequency shown for each cell fate choice on the cell state trees indicates sporadic MLP at later time points.
The significance of MLP for cell fate decisions is clearly supported by these specific examples, but its importance would be further strengthened if it were generally associated with branch points in differentiation. Our scRNA-seq data offered an opportunity to assess this over entire embryos. Wardle and Smith had previously (29) noted transient overlap and then refinement between the ectoderm-specific TF Sox2, and the mesoderm-specific TF Brachyury (T) during gastrula stages in Xenopus. This was recapitulated in our data, which shows co-expression of these markers in 26% of cells expressing either gene at stage 10, but refinement to just 3% overlap by stage 14 (Fig. 6, B and C). Our data also show numerous other examples, including similar MLP between early ectodermal TF Zic1, and the mesodermal TF Foxc1, which overlap in expression at stage 10 (7% coexpression) before refinement by stage 12 (<3% coexpression) (Fig. 6B). Across all fate choices in the embryo, we identify 412 MLP genes in Xenopus (Data S6). We find that MLP is initially widespread, encompassing a considerable fraction of highly-lineage-specific genes at each branch point (>4-fold differentially expressed), but reduces over time (Fig. 6, D and E). For lineage splitting events that occur before or up until the end of gastrulation, over 70% of highly-lineage-specific genes overlapped in their progenitor state in fish and ~50% in frog. By contrast, for subsequent lineage splits, MLP ultimately drops to <10% in both species (Fig. 6D). All lineages showed multilineage overlap before gastrulation, whereas only selected cell fate choices showed frequent MLP after gastrulation (Fig. 6E).
Where MLP did occur, we wondered if it reflected generally noisy gene expression, or more specifically co-expression of regulators of cell fate. Suggestive of the latter possibility, MLP genes were significantly enriched for TF-associated GO terms including ‘transcription factor activity’, ‘regulation of transcription’, ‘nucleus’, and ‘DNA binding’ as compared to all DE genes (fig. S17) (2-fold enriched; P < 0.001 binomial- test, Bonferroni corrected).
Assessing the retention of pluripotency during neural crest development
The utility of single-cell gene expression maps extends from studying general features of development to clarifying specific developmental relationships. Among embryonic lineages, the neural crest is unique in its broad fate potential, contributing to multiple tissues spanning germ layer boundaries. It remains an open question how this unique fate potential arises. As the neural crest forms (S13/14), it expresses at least eight pluripotency genes - foxd3, c-myc, id3, tfap2, ventx2, ets1, snail, and oct25 - which are also expressed in the early blastula (S8/9) (30). Functional assays showed that several of these genes are required for multipotency of both blastula and neural crest cells (30). This raised the intriguing possibility that neural crest may retain multipotency from the blastula stage (a “retention model”), by contrast to the classical view that a field of ectodermal tissue reacquires multipotency during early neurulation (Fig. 7A). However, this model remains a hypothesis because a multipotent intermediate state between blastula and neural crest stages has not been directly identified.
Fig. 7. Assessing the retention of pluripotency during neural crest development.
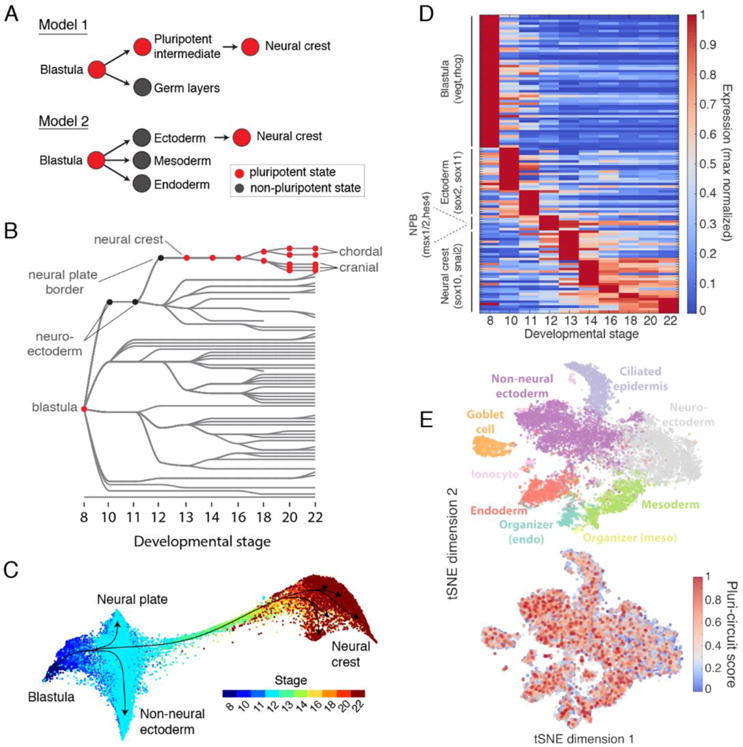
(A) Contrasting models of neural crest development. Model 1: neural crest emerges from an intermediate population that retains blastula pluripotency (29). Model 2: neural crest emerges from ectoderm and reactivates pluripotency. (B) Ancestors inferred from scRNA-seq support model 2, where neural crest derives from neural cells at the neural plate border. (C) Single-cell visualization (SPRING) of neuroectoderm, non-neural ectoderm, and neural crest also indicates that neural crest derives from the neural plate border. (D) Neural crest differentiation involves hundreds of >3- fold dynamic marker genes. (E) At stage 11, the shared pluripotency circuit proposed by Buitrago-Delgado et al. (30) - foxd3, c-myc (myca), id3, tfap2a, ventx2.1, ets1, and snail and pou3f5.2 - is expressed broadly in nonpluripotent cells. Score shows normalized aggregate expression; see fig. S18 for individual genes.
Our single-cell data offers an opportunity to re-examine these alternative hypotheses with a high-resolution view of the intermediate states between blastula- and neurula-stages in neural crest development. To support the “retention model”, we specifically searched for a sub-set of cells that maintain a blastula gene expression program, distinct from the remaining early ectoderm. To further increase the likelihood of finding such cells, we supplemented our whole embryo time series with additional scRNA-seq data (9308 cells) collected from tissue that we dissected from the neural plate border region - that is fated to become neural crest - of stage 11 embryos, immediately prior to the expression of neural crest genes (total of 15,426 stage 11 cells). These data in frog, and matching data in fish, failed to reveal a distinct cluster of cells defined by expression of the proposed pluripotency genes, nor by any other set of genes enriched in the blastula. Instead, the data supported a more conventional differentiation pathway through clearly identifiable neuroectodermal intermediates (Figs. 7B and 4A). The same results held when examining differentiation progression not at the cell cluster level, but at single-cell resolution (Fig. 7C). Far from showing a gene expression program that persists from blastula stage, we found that the inferred precursors of the neural crest showed large changes in gene expression with hundreds of dynamically varying genes (Fig. 7D). The proposed suite of eight pluripotency genes, in particular, were not limited to a single cluster of cells but were broadly expressed across most of the ectoderm, as well as in nonpluripotent states such as the endoderm and mesoderm at stage 11 (Fig. 7E) (see fig. S18 for individual genes). This broad expression pattern would have been more difficult to appreciate without single-cell data. The original experiments (30) could not weigh equally the expression of across all tissues because the whole mount in situ staining used emphasizes superficial tissues.
We conclude that, at least at the level of transcription, there is no evidence of a distinct expression program that persists from the blastula to give rise to the neural crest. We fail to see such cells both using unsupervised approaches (clustering) and by examining the set of eight shared genes previously proposed to maintain pluripotency. It is not possible to completely rule out the retention of a blastula-like pluripotency program in neural crest precursors from our data: if functional pluripotency was maintained by chromatin state or posttranslational modification it may be undetectable by scRNA-seq. Nevertheless, these new studies argue against a persistent transcriptional program preserved from an earlier stage uniquely in the neural crest. Rather, we argue in favor of a more conventional view of neural crest development proceeding from a well-defined ectodermal lineage.
Discussion
Embryonic development involves a carefully timed set of changes in cell behavior that drives the egg to a complex spatial and compositional pattern of cell types. A biochemical dissection of the underlying processes in development is complicated by limited material in the embryo and the heterogeneity of its composition, both of which must be considered together. Methods such as in situ hybridization have bridged these difficulties and can now be performed quantitatively and simultaneously over many genes (31, 32). However, registering such spatial information at single-cell resolution can be challenging, particularly in three-dimensional tissues. In an alternative approach, we and others have developed single-cell transcriptomic methods, which sacrifice spatial information and in return provide a universal modality of measurement that is easily adapted to diverse situations (1, 33). In a short time, these methods have been widely deployed and excelled in revealing cell population structures and dynamics (17, 34, 35). The resulting cell atlases can be computationally related to spatial structure using extensive in situ expression databases such as Xenbase (36, 37). For embryos, applying scRNA-Seq involves important considerations. The method should not preferentially select a single cell type. It should be highly efficient in yield of cells. The dissociation procedure adopted should be quick and complete. Background RNA released by lysed cells in the sample should be minimized. These problems are acute for Xenopus embryos, as it has large yolk-filled blastomeres that are sensitive to shear forces and handling. Yet these problems have been overcome with a method of efficient capture and very little cell lysis.
We also developed analytical methods for interpreting the time course measurements of single-cell gene expression in the embryo. Large temporal differences obscured temporal mappings between cell states using established tools. This challenge was solved in our analysis by using shared latent spaces to measure similarities between states across adjacent timepoints. Upon linking cell states over time based on their gene expression, we found a strong match to known lineages. There were some differences that could be explained by the heterochonic formation of the same cell type, such as in the differentiation of the somite tissue, and in some cases the transcriptional cell states did not cluster along spatial boundaries. Yet in other aspects the data showed high sensitivity and could detect the first appearance of numerous cell states far earlier than previously appreciated. As shown for the neural crest, the sensitivity and single-cell resolution of our gene expression resource can be used to test specific hypotheses about fundamental developmental processes.
In this study, we looked for general features of development that might be difficult to appreciate from examination of individual lineages. In both Xenopus and zebrafish, we observed a transition from pervasive multilineage gene expression during gastrulation, to more specific and combinatorial deployment of differentiation programs later. In particular, newly induced transcription factors increasingly demarcated multiple independent cell fates in the embryo after gastrulation, as transcriptional states within each germ layer are further specialized. The drop in the frequency of multilineage priming, and the increase in combinatorial TF deployment, co-occur with the formation of the first discrete embryonic cell states. In Xenopus, this transition appears to be particularly switch-like, occurring between stages 12 and 14, just 2.5 hrs apart in time. Curiously, the timing of appearance of distinct cells states correlates precisely with that of cell fate commitment in Xenopus (38, 39). The global nature of this transition is reminiscent of the midblastula transition, that occurs in stage 8 Xenopus embryos, when the gradual titration of DNA-to-histone ratios during blastula stage cleavage cycles abruptly drives transcriptional activation, a longer cell cycle, onset of asynchronous cell divisions, and the start of cell motility across the embryo (2, 3). We do not know whether any single mechanism coordinates the rapid appearance of multiple cell types in the post-gastrula embryo, however. A candidate process could be the formation of heterochromatin domains, which occurs on the appropriate timescale (40, 41), and could facilitate both the refinement of lineage program conflicts (42) and the combinatorial reuse of transcription factors by restricting their targets in a tissue- specific manner (43, 44).
In contrast to these seemingly conserved global features of transcriptional dynamics in embryos, we found that the tissue-specific expression of individual genes was relatively variable across species. Rather than preserving global transcriptional profiles, orthologous cell states maintained expression of just a subset of genes, which were most notably enriched for TFs, and among TFs, those that are used in a single tissue rather than those that are combinatorially reused across lineages. Developmental programs thus appear to preserve an underlying core set of transcriptional programs that define cellular hierarchies, while exploring significant variation in the way most genes are deployed across tissues to define cellular phenotypes. This finding supports previous arguments that cell identities and phenotypes may be decoupled across evolution (45). Indeed, new cell states and changes in cellular hierarchies across species could be associated with the deployment of TFs in novel locations or combinations, and concomitant acquisition of batteries of effector genes. We found that this expression plasticity is independent of variation in protein sequence itself, surprisingly decoupling a gene’s structure from its expression pattern in the embryo across evolution. Taken together, the approaches and analyses presented here establish first steps toward a data-driven dissection of developmental programs and how they change across species.
Supplementary Material
Acknowledgments
We thank A. Ratner for technical support, S. Wolock for assistance with data analysis, and the Bauer Core Facility at Harvard University for sequencing support.
Funding: A.M.K was supported by an Edward J. Mallinckrodt Foundation Grant, a Burroughs Wellcome Fund CASI Award, and NIH award 5R33CA212697-02. J.B. L.P. and M.W.K were supported by NIH award 2R01HD073104-06. M.W.K was also supported by R21HD087723.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/cgi/content/full/science.aar5780/DCl
Materials and Methods
Author contributions: J.A.B. designed and performed all single-cell experiments, processed and analyzed data, and generated figures. J.A.B., M.W.K, and A.M.K. conceived the study and wrote the manuscript. M.W.K, A.M.K., and L.P. assisted with experiments and data analysis. C.W. developed the web browser for viewing single-cell data and assisted with data analysis, generating figures, and writing the manuscript. D.E.W. and S.M. provided clustering analysis and annotations of zebrafish data.
Competing interests: A.M.K. and M.W.K are founders of 1Cell-Bio, Inc. All other authors declare no competing interests.
Data and materials availability: All data are available for interactive exploration at: tinyurl.com/scXen2018. The underlying scRN A-seq counts data can be downloaded from the same browser. Raw FASTQ files and scRNA-seq counts data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO), accession GSE113074.
REFERENCES AND NOTES
- 1.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newport J. Kirschner M. A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell. 1982;30:675–686. doi: 10.1016/0092-86748290272-0Medline. [DOI] [PubMed] [Google Scholar]
- 3.Newport J. Kirschner M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell. 1982;30:687–696. doi: 10.1016/0092-86748290273-2Medline. [DOI] [PubMed] [Google Scholar]
- 4.Heasman J. Patterning the early Xenopus embryo. Development. 2006;133:1205–1217. doi: 10.1242/dev.02304Medline. [DOI] [PubMed] [Google Scholar]
- 5.Karimi K, Fortriede JD, Lotay VS, Burns KA, Wang DZ, Fisher ME, Pells TJ, James-Zorn C, Wang Y, Ponferrada VG, Chu S, Chaturvedi P, Zorn AM, Vize PD. Xenbase: A genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Res. 2018;46:D861–D868. doi: 10.1093/nar/gkx936. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis RL, Kirschner MW. The fate of cells in the tailbud of Xenopus laevis. Development. 2000;127:255–267. doi: 10.1242/dev.127.2.255. Medline. [DOI] [PubMed] [Google Scholar]
- 7.Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018;360:aar4362. doi: 10.1126/science.aar4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segerdell E, Ponferrada VG, James-Zorn C, Burns KA, Fortriede JD, Dahdul WM, Vize PD, Zorn AM. Enhanced XAO: The ontology of Xenopus anatomy and development underpins more accurate annotation of gene expression and queries on Xenbase. J Biomed Semantics. 2013;4:31. doi: 10.1186/2041-1480-4-31Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segerdell E, Bowes JB, Pollet N, Vize PD. An ontology for Xenopus anatomy and development. BMC Dev Biol. 2008;8:92. doi: 10.1186/1471-213X-8-92Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinreb C, Wolock S, Tusi BK, Socolovsky M, Klein AM. Fundamental limits on dynamic inference from single-cell snapshots. Proc Natl Acad Sci U S A. 2018;115:E2467–E2476. doi: 10.1073/pnas.1714723115Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiebinger G, et al. Reconstruction of developmental landscapes by optimal-transport analysis of single-cell gene expression sheds light on cellular reprogramming. bioRxiv. 2017 Sep 27; doi: 10.1101/191056. 191056 [preprint] [DOI]
- 13.Walmsley M, Ciau-Uitz A, Patient R. Adult and embryonic blood and endothelium derive from distinct precursor populations which are differentially programmed by BMP in Xenopus. Development. 2002;129:5683–5695. doi: 10.1242/dev.00169Medline. [DOI] [PubMed] [Google Scholar]
- 14.Liu F, Walmsley M, Rodaway A, Patient R. Fli1 acts at the top of the transcriptional network driving blood and endothelial development. Curr Biol. 2008;18:1234–1240. doi: 10.1016/j.cub.2008.07.048Medline. [DOI] [PubMed] [Google Scholar]
- 15.Nagasawa T, Kawaguchi M, Yano T, Sano K, Okabe M, Yasumasu S. Evolutionary Changes in the Developmental Origin of Hatching Gland Cells in Basal Ray-Finned Fishes. Zool Sci. 2016;33:272–281. doi: 10.2108/zs150183Medline. [DOI] [PubMed] [Google Scholar]
- 16.Yasutomi M, Hama T. Electron microscopic study on the xanthophore differentiation in Xenopus laevis, with special reference to their pterinosomes. J Ultrastruct Res. 1972;38:421–432. doi: 10.1016/0022-53207290080-9Medline. [DOI] [PubMed] [Google Scholar]
- 17.Briggs JA, Li VC, Lee S, Woolf CJ, Klein A, Kirschner MW. Mouse embryonic stem cells can differentiate via multiple paths to the same state. eLife. 2017;6:e26945. doi: 10.7554/eLife.26945Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber H, Symes CE, Walmsley ME, Rodaway AR, Patient RK. A role for GATA5 in Xenopus endoderm specification. Development. 2000;127:4345–4360. doi: 10.1242/dev.127.20.4345. Medline. [DOI] [PubMed] [Google Scholar]
- 19.Reiter JF, Alexander J, Rodaway A, Yelon D, Patient R, Holder N, Stainier DYR. Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 1999;13:2983–2995. doi: 10.1101/gad.13.22.2983Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weintraub H. The MyoD family and myogenesis: Redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-86749390610-3Medline. [DOI] [PubMed] [Google Scholar]
- 21.Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, Carninci P, Daub CO, Forrest ARR, Gough J, Grimmond S, Han JH, Hashimoto T, Hide W, Hofmann O, Kamburov A, Kaur M, Kawaji H, Kubosaki A, Lassmann T, van Nimwegen E, MacPherson CR, Ogawa C, Radovanovic A, Schwartz A, Teasdale RD, Tegnér J, Lenhard B, Teichmann SA, Arakawa T, Ninomiya N, Murakami K, Tagami M, Fukuda S, Imamura K, Kai C, Ishihara R, Kitazume Y, Kawai J, Hume DA, Ideker T, Hayashizaki Y. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–752. doi: 10.1016/j.cell.2010.01.044Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monod J, Jacob F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Hard Symp Quant Biol. 1961;26:389–401. doi: 10.1101/SQB.1961.026.01.048Medline. [DOI] [PubMed] [Google Scholar]
- 23.Cohen DE, Melton D. Turning straw into gold: Directing cell fate for regenerative medicine. Nat Rev Genet. 2011;12:243–252. doi: 10.1038/nrg2938Medline. [DOI] [PubMed] [Google Scholar]
- 24.Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, Enver T. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997;11:774–785. doi: 10.1101/gad.11.6.774Medline. [DOI] [PubMed] [Google Scholar]
- 25.Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, Ramanathan S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell. 2011;145:875–889. doi: 10.1016/j.cell.2011.05.017Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, Hirche C, Lutz C, Buss EC, Nowak D, Boch T, Hofmann WK, Ho AD, Huber W, Trumpp A, Essers MAG, Steinmetz LM. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19:271–281. doi: 10.1038/ncb3493Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shu J, Wu C, Wu Y, Li Z, Shao S, Zhao W, Tang X, Yang H, Shen L, Zuo X, Yang W, Shi Y, Chi X, Zhang H, Gao G, Shu Y, Yuan K, He W, Tang C, Zhao Y, Deng H. Induction of pluripotency in mouse somatic cells with lineage specifiers. Cell. 2013;153:963–975. doi: 10.1016/j.cell.2013.05.001Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052Medline. [DOI] [PubMed] [Google Scholar]
- 29.Wardle FC, Smith JC. Refinement of gene expression patterns in the early Xenopus embryo. Development. 2004;131:4687–4696. doi: 10.1242/dev.01340Medline. [DOI] [PubMed] [Google Scholar]
- 30.Buitrago-Delgado E, Nordin K, Rao A, Geary L, LaBonne C. Shared regulatory programs suggest retention of blastula-stage potential in neural crest cells. Science. 2015;348:1332–1335. doi: 10.1126/science.aaa3655Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11:360–361. doi: 10.1038/nmeth.2892Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090. doi: 10.1126/science.aaa6090Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karaiskos N, Wahle P, Alles J, Boltengagen A, Ayoub S, Kipar C, Kooks C, Rajewsky N, Zinzen RP. The Drosophila embryo at single-cell transcriptome resolution. Science. 2017;358:194–199. doi: 10.1126/science.aan3235Medline. [DOI] [PubMed] [Google Scholar]
- 35.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, Adey A, Waterston RH, Trapnell C, Shendure J. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–667. doi: 10.1126/science.aam8940Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol. 2015;33:503–509. doi: 10.1038/nbt.3209Medline. [DOI] [PubMed] [Google Scholar]
- 37.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forman D, Slack JM. Determination and cellular commitment in the embryonic amphibian mesoderm. Nature. 1980;286:492–494. doi: 10.1038/286492a0Medline. [DOI] [PubMed] [Google Scholar]
- 39.Heasman J, Wylie CC, Hausen P, Smith JC. Fates and states of determination of single vegetal pole blastomeres of X. laevis. Cell. 1984;37:185–194. doi: 10.1016/0092-86748490314-3Medline. [DOI] [PubMed] [Google Scholar]
- 40.Hontelez S, van Kruijsbergen I, Georgiou G, van Heeringen SJ, Bogdanovic O, Lister R, Veenstra GJC. Embryonic transcription is controlled by maternally defined chromatin state. Nat Commun. 2015;6:10148. doi: 10.1038/ncommsl0148Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Françoijs KJ, Stunnenberg HG, Veenstra GJC. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev Cell. 2009;17:425–434. doi: 10.1016/j.devcel.2009.08.005Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: Digging Waddington’s canal. Nat Rev Mol Cell Biol. 2009;10:526–537. doi: 10.1038/nrm2727Medline. [DOI] [PubMed] [Google Scholar]
- 43.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD, Wagner GP. The origin and evolution of cell types. Nat Rev Genet. 2016;17:744–757. doi: 10.1038/nrg.2016.127Medline. [DOI] [PubMed] [Google Scholar]
- 46.Sive HL, Grainger RM, Harland RM. Early Development of Xenopus laevis: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2000. [Google Scholar]
- 47.Savova V, Pearl EJ, Boke E, Nag A, Adzhubei I, Horb ME, Peshkin L. Transcriptomic insights into genetic diversity of protein-coding genes in X. laevis. Dev Biol. 2017;424:181–188. doi: 10.1016/j.ydbio.2017.02.019Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Maaten L, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 50.Weinreb C, Wolock S, Klein AM. SPRING: A kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics. 2018;34:1246–1248. doi: 10.1093/bioinformatics/btx792Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blitz IL, Paraiso KD, Patrushev I, Chiu WTY, Cho KWY, Gilchrist MJ. A catalog of Xenopus tropicalis transcription factors and their regional expression in the early gastrula stage embryo. Dev Biol. 2017;426:409–417. doi: 10.1016/j.ydbio.2016.07.002Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.