

Abstract
Hereditary hemochromatosis (HH), a disease marked by chronic iron overload from insufficient expression of the hormone hepcidin, is one of the most common genetic diseases. One form of HH (Type III) results from mutations in the transferrin receptor-2 (TfR2). TfR2 is postulated to be a part of signaling system that is capable of modulating hepcidin expression. The molecular details of TfR2’s role in this system remain unclear, however. TfR2 is predicted to bind the iron carrier transferrin (Tf) when the iron-saturation of Tf is high. To better understand the nature of these TfR-Tf interactions, a binding study with the full-length receptors was conducted. In agreement with previous studies with truncated forms of the receptors, holo-Tf binds to the homolog, TfR1, significantly stronger than TfR2. However, the binding constant for Tf-TfR2 is still far above that of physiological holo-Tf levels, inconsistent with the hypothetical model and suggests that other factors mediate the interaction. One possible factor, Apo-Tf, only weakly binds TfR2 at serum pH, thus will not be able to effectively compete with holo-Tf. Tf-binding to a TfR2 chimera containing the TfR1-helical domain indicates that the differences in the helical domain account for differences in on-rate of Tf, and non-conserved inter-receptor interactions are necessary for the stabilization of the complex. Conserved residues at one possible site of stabilization, the apical arm junction, are not important for TfR1-Tf binding, but are critical for the TfR2-Tf interaction. Our results highlight the differences in Tf interactions with the two TfRs.
Keywords: Iron homeostasis, hereditary hemochromatosis, hepcidin, transferrin, transferrin receptor, TfR1, TfR2
FOR TABLE OF CONTENTS USE ONLY
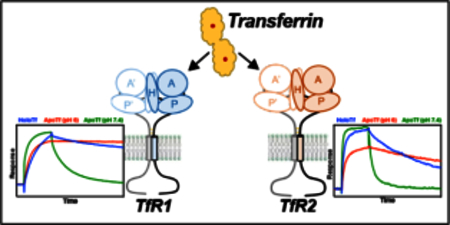
INTRODUCTION
Iron is an essential nutrient and the most abundant transition metal in the body. Homeostasis of iron is a key process that ensures that both tissues meet their demand and that iron is not able to generate excess reactive oxygen species or serve as a growth platform for pathogens. The serum iron carrier transferrin (Tf), is a central component of the iron homeostasis process. Tf is a bi- lobal protein that binds one ferric ion per lobe with exceptionally high affinity 1, and serves to solubilize the ferric ion for transport in blood. In addition, iron-loaded Tf (holo-Tf) serves as the main source of iron for most tissues. Circulating, holo-Tf binds to the cell-surface transferrin receptor-1 (TfR1) and is endocytosed. Through an endocytic process known as the Tf cycle, the iron is released from Tf and is taken up into the cell. Iron-depleted Tf (apo-Tf) remains bound to TfR1 and recycles to the cell surface, where it is released from TfR1 at the neutral pH of the extracellular environment 2 Thus the pH sensitivity of the interaction of holo-Tf and apo-Tf with TfR1 is critical to the Tf cycle. Underlining the critical nature of this process, a TfR1-null mouse is embryonic lethal, demonstrating the importance of this cycle in growth and development 3.
A homolog to TfR1, TfR2, is also able to bind circulating Tf. The physiological role of this interaction is through the maintenance of iron levels within the body rather than cellular iron uptake 45. Mutations in TfR2 have been identified to be responsible for a form of hereditary hemochromatosis (HH, type III; 4), a disorder marked by chronic iron overload. In HH, the iron overload is a result of insufficient expression of the hormone, hepcidin. Hepcidin is expressed and secreted primarily in hepatocytes and binds to the sole iron export protein, ferroportin, inducing internalization and degradation 6 Thus, hepcidin secretion results in increased sequestration of iron within cells, particularly in cell types of high iron efflux (e.g. intestinal epithelial cells, macrophages, and hepatocytes), and a decrease in circulating iron. In iron overload diseases such as HH, increased iron transport due to the failure to regulate ferroportin levels properly, leads to iron deposition in tissues, which includes the liver and heart, leading to significant tissue damage from oxidative stress.
How, precisely, the interaction of Tf and TfR2 leads to increased hepcidin expression is unresolved. Treatment of TfR2-expressing cells with holo-Tf, but not apo-Tf, leads to increased receptor half-life 7. Both TfRs are capable of forming complexes with HFE 8–10, another plasma membrane protein that, when mutated or absent, causes the most common form of HH, Type I. HFE has been shown to compete with holo-Tf for TfR1 binding 9 Disrupting the TfR1-HFE interaction through point mutation results in inappropriately high hepcidin expression 11. Thus, a proposed mechanism of iron-sensing is that HFE is in complex with TfR1 at low Tf-saturation, but upon increased systemic iron, HFE is displaced by holo-Tf and binds TfR2 11. Whether HFE-TfR2 complex or HFE in complex with another protein initiates a signaling cascade to induce hepcidin expression is controversial. Signaling then decreases both passively with falling Tf iron saturation and actively through cleavage from the protease matriptase-2 12. These hypothetical iron-sensing mechanisms are supported by multiple in vitro and in vivo experiments, but mechanisms remain unknown. Though multiple crystal or cryo-EM structures exist for TfR1 13,17, little structure-function data has been shown for TfR2. As such, the structural differences between TfR1 and TfR2 that account for the differences in receptor function are unresolved.
In the present study, we identify key differences in the binding of Tf to the TfRs that are likely to contribute to their varying roles in iron homeostasis. Like previous studies have shown, TfR1 binds holo-Tf with a significantly stronger affinity than to TfR2. For the first time, however, we show that TfR1 and TfR2 have distinct mechanisms for stabilizing a complex with holo-Tf. Interestingly, apo-Tf is also able to bind to each receptor at serum pH, forming short-lived complexes that likely inhibit the interaction of holo-Tf with TfR2 at low systemic levels of iron. Together, our results support a dynamic model of Tf-TfR interactions in the liver that serve to directly link circulating iron levels to hepcidin expression.
MATERIALS & METHODS
Transferrin Receptor Cloning
To generate an EGFP fusion with the receptors, human TfR1 or TfR2 genes were cloned into the pEGFP-N1 expression plasmid (Stratagene; primers listed in Supplemental Table 1). To generate the TfR2-TfR1 helical domain chimera, residues 1–636 of TfR2 were inserted to the 5’ end of residues 605–760 of TfR1 in pEGFP-N1 using Gibson Assembly cloning (New England Biolabs; primers listed in Supplemental Table 1) according to manufacturer’s procedure. Point-mutations were introduced with the Quikchange Lightning site-directed mutagenesis kit (Agilent). All plasmids were verified by Sanger sequencing.
Partial Purification of Transferrin Receptors
HEK-293S GnTI- cells were transiently transfected with EGFP-receptor fusion plasmid through transient transfection with PEI MAX 40K reagent (Polysciences, Inc.) according to manufacturer’s procedure and were harvested 48 hours post-transfection. (We note that the GnTI- subtype does not produce complex glycosylation modifications and previous studies have shown Tf-binding to the glycoproteins, TfR1 and TfR2, is not glycosylation dependent 18–20). Cells were lysed and membrane proteins solubilized by addition of 20 mM HEPES, pH 7.4, 150 mM NaCl, 1% laurylmaltose neopentyl glycol (LMNG), and protease inhibitor cocktail (Biotools). Solubilization of the EGFP-TfR2-TfR1HD chimera required the addition of 10 μΜ holo- Tf. Lysates were rotated for one hour at 4 °C. Insoluble material was removed by ultracentrifugation at 70,000 xG for 40 minutes at 4 °C. Solubilized protein was applied to a AKTAFPLC (G.E. Healthcare) with in-line Superose 6 Increase gel filtration column and Waters 474 scanning fluorescence detector (excitation: 488 nm, emission: 520 nm). The running buffer was 20 mM HEPES, pH 7.4, 150 mM NaCl, 0.01% LMNG. The fraction containing maximal GFP fluorescence was collected for use in SPR binding studies.
Transferrin- Transferrin Receptor Binding Analysis
Binding studies were performed with a Biacore X100 surface plasmon resonance (SPR) instrument. GFP-Trap nanobody (Chromatek) was covalently coupled to ~2000 response units (RU’s) on both flow-cells of a CM5 chip with the Biacore Amine Coupling Kit. Partially- purified EGFP-receptor was then immobilized on the antibody surface of one flow-cell to a density of ~200 RU’s. Running Buffer for neutral-pH experiments was 20 mM HEPES, 150 mM NaCl, 0.01% LMNG. Running Buffer for endosomal pH experiments was 20 mM PIPES, pH 6.0, 150 mM NaCl, 0.01% LMNG. Various concentrations of either apo- or holo-Tf were then injected over the chip surface at a flow rate of 30 μL/min for 180 seconds followed by Running Buffer for 480 seconds. Apo-Tf solutions were supplemented with 2-fold molar excess of desferrioxamine to chelate any trace iron. At the end of each dissociation phase, running buffer supplemented with 0.5 M MgCl2 was injected for 12 seconds to remove any remaining bound Tf from the receptor. A reference cell without receptor immobilized was used to subtract background response. Buffer contributions were eliminated by subtracting the Tf response data with buffer-only injection data. Kinetic and equilibrium binding analysis was performed with the BiaEvaluation software. Data was best fit to a heterogenous model that describes two independent binding sites on the immobilized receptor dimer, producing two sets of kinetic constants.
RESULTS
Full-length TfR1 binds holo-Tf with higher affinity than TfR2.
A binding study of the isolated full-length receptor was undertaken to elucidate the differences in Tf-binding of the two TfRs. Multiple surface plasmon resonance (SPR) binding studies have previously been undertaken for the TfR1-Tf interaction 11, 19, 21–26, but only a single study of TfR2 has been performed 22. Additionally, all previous TfR SPR studies have been performed with the truncated extracellular domains of the receptors, which could lack structural elements that alter binding behavior. The full-length receptors, solubilized from cell membranes with detergent (1% LMNG), displayed primarily monodisperse fluorescent chromatograms (Fig. 1A) and and in agreeance with previous studies (e.g. 27), the receptors are primarily in the dimeric state (Fig. S1A). The eluent from the peak fractions were used for SPR analysis. Similar to previous studies, the SPR binding data for both receptors fit a heterogenous model that describes two independent ligand binding sites (Fig. S1B). Under this model, TfR1 is the preferred binding partner for holo-Tf, with a KD1, KD2 of <0.1 nM, 3.8 nM versus TfR2’s KD1, KD2 of 19 nM, 49 nM (Fig. 1B and 1C; Table 1). The serum concentration of Tf is ~25 μM. In humans, Tf ironloading is 20–50% of capacity in the absence of anemia or iron overload 28. The binding affinity of holo-Tf for TfR2 suggests that Tf binding to TfR2 should be saturated even in low iron conditions, which runs counter to the model that TfR2 serves as a sensor for Tf saturation.
Figure 1.
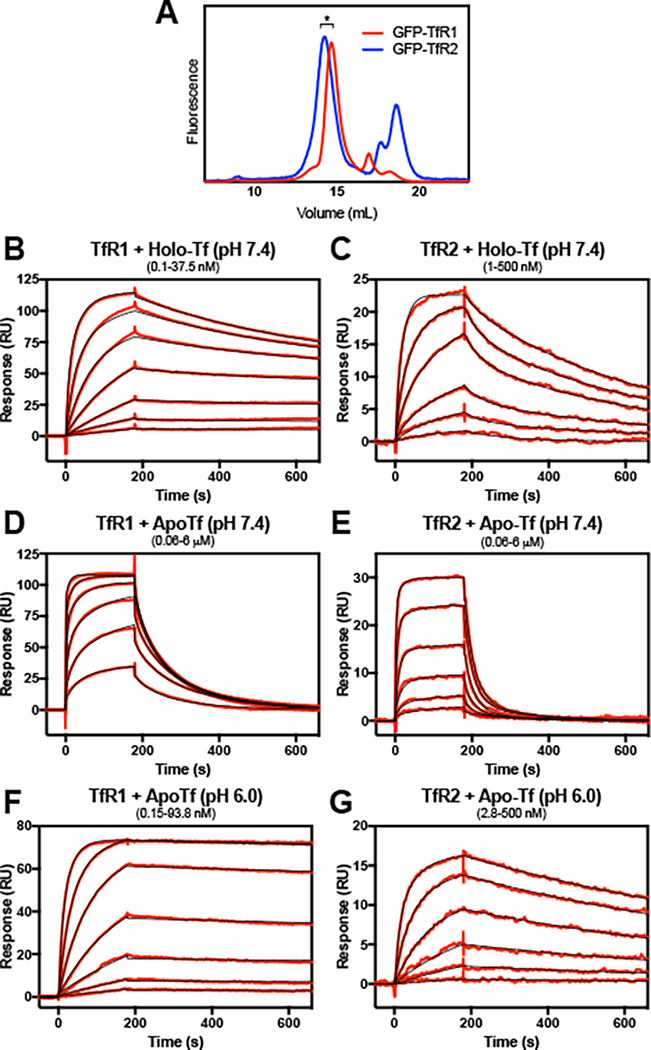
The interaction of full-length TfRs with holo- and apo-Tf. For surface plasmon resonance analysis, full-length EGFP-TfR1 or EGFP-TfR2 were solubilized from cell membranes with 1% LMNG and post-ultracentrifugation supernatant was applied to an FPLC with in-line Superose 6 size exclusion column and fluorometer (A). Shorter forms of EGFPRTfR2 represent EGFP cleaved from the full-length protein. The receptor from peak fractions (*) were immobilized to the surface of an a-GFP-CM5 chip to a density of ~200 RU’s and treated with varying concentrations of holo- or apo-Tf. Concentrations of Tf tested are the following: (B) 0.15 – 37.5 nM, with a 2.5-fold dilution series. (C) 1 – 500 nM, with a 2.8-fold dilution series; (D and E) 0.06 – 6 μM, with a 2.5-fold dilution series; (F) 0.15 – 93.8 nM, with a 2.5-fold dilution series; (G) 2.8 – 500 nM, with a 2.8-fold dilution series. Reference cell-subtracted binding response data is indicated in red, and heterogenous model fits are shown in black. Extrapolated kinetic constants from the model fits are shown in Table 1.
Table 1.
Comparison of binding affinities of Tf receptor constructs. The indicated construct was immobilized to the GFP-Trap-CM5 sensor chip surface and treated with various concentrations of the indicated analyte. Sensorgram data was fit with a heterogenous ligand model and kinetic data extrapolated. Denoted values (*) are below the sensitivity of the instrument, thus KD’s are represented as <0.01 nM in the text.
| Construct | Analyte | ka1 (1/μM*s) |
kd1 (1/ms) |
Kd1 (nM) |
ka2 (1^M*s) |
kd2 (1/ms) |
Kd2 (nM) |
|---|---|---|---|---|---|---|---|
| EGFP-TfR1 | Holo-Tf | 2.80 | 0.0006* | 0.0002* | 0.636 | 2.43 | 3.83 |
| EGFP-TfR1 | Apo-Tf (pH 7.4) | 0.45 | 22 | 49 | 0.019 | 6.44 | 344 |
| EGFP-TfR1 | Apo-Tf (pH 6.0) | 1.01 | 0.0012 | 0.0011 | 3.01 | 0.127 | 0.042 |
| EGFP-TfR1 G647A | Holo-Tf | 4.74 | 1.1 | 0.23 | 2.58 | 72.3 | 28.1 |
| EGFP-TfR1 H318Q | Holo-Tf | 3.01 | 0.020 | 0.0067 | 0.694 | 3.37 | 4.86 |
| EGFP-TfR1 H318Q | Apo-Tf (pH 6.0) | 1.22 | 0.0002* | 0.0002* | 4.31 | 2.88 | 0.668 |
| EGFP-TfR1 E759N | Holo-Tf | 4.06 | 0.21 | 0.053 | 0.857 | 3.56 | 4.16 |
| EGFP-TfR1 E759N | Apo-Tf (pH 6.0) | 1.45 | 0.0003* | 0.0002* | 5.73 | 3.65 | 0.637 |
| EGFP-TfR2 | Holo-Tf | 0.11 | 2.0 | 19 | 0.523 | 25.5 | 48.6 |
| EGFP-TfR2 | Apo-Tf (pH 7.4) | 0.066 | 44 | 670 | 0.0063 | 8.16 | 1300 |
| EGFP-TfR2 | Apo-Tf (pH 6.0) | 0.15 | 1.1 | 7.0 | 0.034 | 0.643 | 18.8 |
| EGFP-TfR2 Q340H | Holo-Tf | 0.010 | 0.0008 | 0.080 | 0.097 | 31.7 | 325 |
| EGFP-TfR2 N800E | Holo-Tf | 0.012 | 2.7 | 220 | 0.071 | 34.9 | 496 |
| EGFP-TfR2-TfR1HD | Holo-Tf | 0.56 | 3.2 | 5.7 | 0.052 | 31.3 | 606 |
| EGFP-TfR2-TfR1HD | Apo-Tf (pH 7.4) | 0.51 | 2.5 | 4.8 | 0.163 | 78.3 | 480 |
| EGFP-TfR2-TfR1HD | Apo-Tf (pH 6.0) | 0.16 | 0.35 | 2.3 | 1.03 | 4.95 | 4.81 |
TfR1 and TfR2 are able to weakly bind apo-Tf at serum pH.
TfR2 is stabilized by its interaction with holo-Tf, but not apo-Tf 7. We asked whether apo-Tf in the serum could compete with the holo-form, preventing the stabilization of TfR2 under low iron conditions when apo-Tf is predominant. Earlier studies with the truncated forms of TfR1 indicated that at neutral pH apo-Tf had a sufficiently low affinity for TfR1 that it effectively did not compete with holo-Tf over the physiological range of Tf concentration in the blood 21. In the present study, we find that TfR1 is able to bind apo-Tf at pH 7.4 (KD1, KD2 of 49 nM, 344 nM; Fig. 1D and Table 1), but apo-Tf dissociates much more rapidly than holo-Tf (Fig. 1B), which is consistent with previous studies 2, 29. TfR2 is able to form weaker, short-lived complexes with apo-Tf as compared to holo-Tf (KD1, KD2 of 0.67 μM, 1.3 μM; Fig. 1E and Table 1). In the context of TfR2’s putative role in hepcidin signaling, this interaction alone is unlikely prevent the binding of holo-Tf when serum Tf-iron saturation levels are low. Other components, either in the serum (e.g. monoferric Tf) or in the membrane (e.g. HFE) may prevent binding of holo-Tf at low saturation levels.
TfR1 and TfR2 form stable complexes with apo-Tf at endosomal pH.
In the Tf cycle, the Tf- TfR1-containing endosome undergoes a decrease in pH to ~6 as the result of V-type ATPase. This change in pH induces conformational changes within the Tf-TfR1 complex to favor the release of Fe3+ from Tf and to stabilize the apo-Tf-TfR1 complex 2, 14. The extent to which TfR2 undergoes a similar transition is unknown. In the case of full-length TfR1, apo-Tf at pH 6.0 shows near irreversible binding (Fig. 1F), while TfR2 at pH 6 shows an over 2-fold increase in affinity as compared to holo-Tf (KD1, KD2 of 7.0 nM, 19 nM; Fig. 1G and Table 1). The data for TfR1 is in agreement with the canonical model of the TfR1-Tf cycle, in that TfR1 forms a stable complex with apo-Tf after endosomal acidification and subsequent iron release.
A TfR2-TfR1 chimera containing the helical domain of TfR1 is unable to form stable complexes with holo-Tf.
The ectodomain sequences of the two receptors share 47% identity (67% similarity) and it is not obvious by inspection what sequence differences account for the altered affinities. To test whether TfR1’s higher affinity for holo-Tf is due to a difference in binding site residues, we created a TfR2/TfR1 chimera that replaces the helical domain of TfR2 (residues 637–801) with that of TfR1 (residues 605–760). The chimera displayed a polydisperse fluorescence chromatogram (Fig. 2A) indicating that it had decreased stability from the wild- type receptors (Fig. 1A). Addition of 10 μM holo-Tf to the solubilization buffer greatly increased the presence of a primary receptor species that was used for SPR characterization. The chimera is able to bind holo-Tf with high affinity, but has a much faster off-rate than TfR1 (KD1, KD2 of 5.4 nM, 261 nM; Fig. 2B). This suggests that the receptor relies upon interactions with structural elements beyond the helical domain to stabilize the complex. Figs 2C & D). Further, the stabilizing interactions are not precisely conserved between TfR1 and TfR2.
Figure 2.
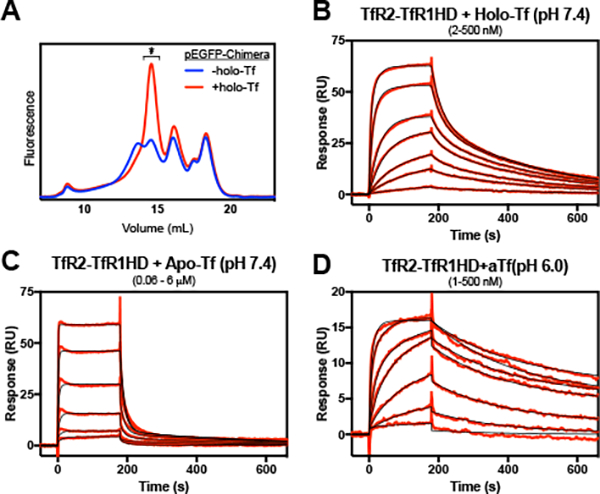
The interaction of a TfR2-TfR1 helical domain chimera with holo- and apo-Tf. The chimera receptor was solubilized in the absence or presence of 10 μM holo-Tf and applied to a Superose 6 column with in-line fluorometer (A). The receptor peak fraction from the +holo-Tf run (*) was immobilized to the SPR chip surface to a density of ~200 RU’s and treated with various concentrations of Tf. Concentrations of holo- or apo-Tf injected were the following: (B) 2 – 500 nM, with a 2.5-fold dilution series; (C) 0.06 – 6 μΜ, with a 2.5-fold dilution series; (D) 1 – 500 nM, with a 2.8-fold dilution series. Reference cell-subtracted binding response data is indicated in red, and heterogenous model fits are shown in black. Extrapolated kinetic constants from the model fits are shown in Table 1.
The “apical arm junction” is critical for the Tf-TfR2 interaction, but not the Tf-TfR1 interaction.
The crystal structure of the ectodomain of TfR1 revealed multiple interactions between helical domain and adjacent apical and protease-like domains that could explain the instability found with the chimera described above. A comparison of the crystal structures of the TfR1 ectodomain and the TfR1 ectodomain bound to Tf (Fig. 3A) showed a region conformational change at the junction of the TfR1 dimerization and Tf-binding interfaces. At the junction is a disordered loop of the apical domain. This “arm” is positioned away from the Tf binding site when no ligand is present, but rotates towards the Tf binding site with the ligand bound (14, Fig. 3B). The arm has a histidine residue at the position nearest to the interface (His- 318) and is proximal to not only the bound Tf, but also the carboxyl terminus of the other subunit of the dimer. The apical domain arm is well-conserved between TfR1 and TfR2, except at the position of the histidine, which, in TfR2, is instead a glutamine (Gln-340, Fig. 3B). Similarly, the carboxyl terminus is well-conserved between the two receptors, except at the penultimate residue (Glu-759 in TfR1, Asn-800 in TfR2; Fig. 3B and 3C). Other residues at the C-terminus participate in accelerating Fe3+-release in TfR1 26. Thus, the C-terminus, together with the apical domain arm, may form a basis for the different Tf binding behavior between receptors.
Figure 3.
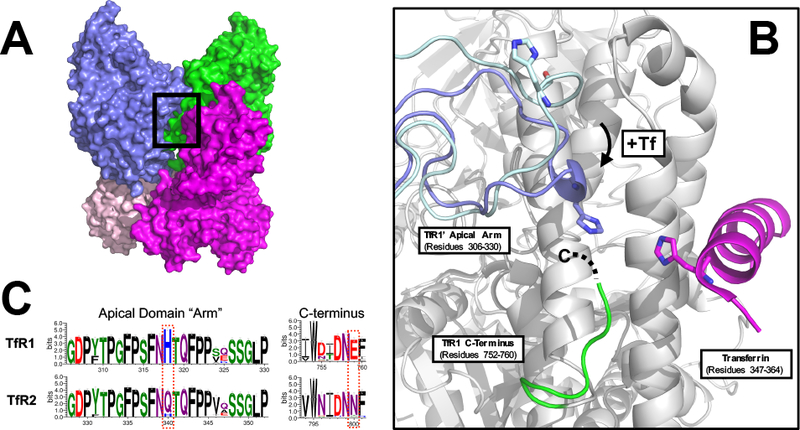
The apical arm junction of the transferrin receptor-transferrin complex. (A) The crystal structure of the Tf-TfRl ectodomain complex determined by Eckenroth, et. al (PDB ID 3S9N;14) with region of apical arm junction outlined by black box. The TfRl ectodomain subunits are colored in blue and green and the Tf molecules are colored in magenta and salmon. (B) Isolated view of the apical arm junction of the Tf-TfRl ectodomain complex. For clarity, the ectodomain residues for the Tf-binding TfRl subunit are shown as grey with the exception of the C-terminal residues 752–758 (green), and the Tf molecule has been removed with the exception of residues 347–364 (magenta). The apical domain residues 306–330 are shown in two conformations: without Tf bound (light blue; PDB ID 1CX8; 16) and with Tf bound (blue; PDB ID 3S9N). Note that the structure lacked sufficient electron density to assign residues 759–760 of the TfRl C-terminus, represented as a dashed line. (C) Sequence conservation of the apical domain arm and the C-terminus between TfR1 and TfR2. Multiple sequence alignments were generated from vertebrate TfR1 and TfR2 sequences obtained from the NCBI nr database through ClustalW. Alignments were then processed by the Weblogo 3 server to generate consensus sequences for the respective receptors. Residue positions are indicated on the x-axis, with numbering according to the human sequences.
To test if incompatibility in this site accounts for the instability of the TfR2-TfR1HD chimera, point mutants were created at non-conserved positions in the apical arm and the carboxyl terminus. The H318Q mutation in TfR1 had no significant effect on holo-Tf binding (Fig. 4A) and the E759N mutation only had a small impact on binding (Fig. 4B). Similarly, neither mutation had altered binding to apo-Tf at endosomal pH (Fig. 4C and 4D). Together, the data suggests that those specific residues have little to no effect as a primary stabilizing mechanism in TfR1 that is lacking in the chimera. Contrary to expectations, both mutations (Q340H and N800E) severely impaired holo-Tf binding affinity to the TfR2 and reduced maximal binding responses (Fig. 4E and 4F; Table 1). Though a crystal structure is lacking for TfR2, and as a result we cannot be certain they position similar to TfR1, they are each clearly key to the binding of holo-Tf. For the chimera construct, introduction of the mutations (Q340H or E759N) does not provide a significant increase in affinity, while reducing the maximal binding levels (Fig. 4G and 4H). Together this data indicates that the TfR1 and TfR2 rely upon different interactions to stabilize a complex with Tf.
Figure 4.
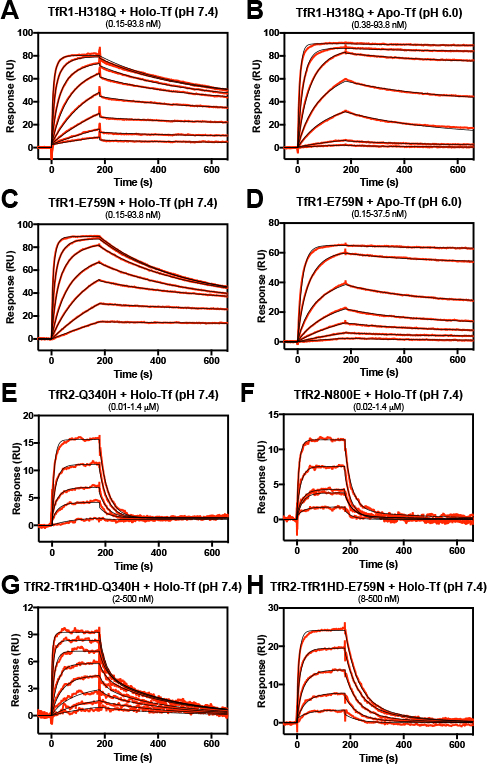
The role of the TfR apical arm junction in Tf binding. Mutant receptors were immobilized to the SPR chip surface to a density of ~200 RU’s and treated with increasing concentrations of Tf. Concentrations of holo- or apo-Tf used in the experiment were the following: (A, C) 0.15 – 93.8 nM, with a -fold dilution series of 2.5; (B) 0.38 – 93.8 nM, with a -fold dilution series of 2.5; (D) 0.15 – 37.5 nM, with a -fold dilution series of 2.5; (E) 0.01 – 1.4 μΜ, with a 2.8-fold dilution series (excluding 23 nM); (F) 0.02 nM - 1.4 μM, with a 2.8-fold dilution series; (G) 2 – 500 nM, with a 2.8-fold dilution series; (H) 8 – 500 nM, with a 2.8-fold dilution series. Reference cell-subtracted binding response data is indicated in red, and heterogenous model fits are shown in black. Extrapolated kinetic constants from the model fits are shown in Table 1.
DISCUSSION
In hepatocytes, Tf-mediated hepcidin expression is likely occurring through the balance of several factors: the serum concentrations of the different forms of Tf (diferric, monoferric, apo- Tf), the affinities for the various Tf’s between the TfR’s, the affinities for HFE between the TfR’s, and the cell surface levels of the receptors (e.g. Fe3+-dependent expression levels of TfR1 via the IRP/IRE system). As a result, it is critical to understand the ligand binding behavior of the receptors to better understand the system.
In agreement with previous studies, we show holo-Tf will preferentially bind TfR1 to TfR2 at serum pH. The binding behavior indicates the receptors not only bind Tf with different affinities, but stabilize the binding complex in unique ways. In the full-length receptor system used here, the KD’s for both receptors are lower than those previously described. Mutation of a critical Tf- binding residue in TfR1 (Gly-647) to alanine significantly reduces binding (Fig. S1C), thus, the increased affinity is likely not due to non-specific interactions. Further, TfR1, unlike TfR2, is selective for human holo-Tf and does not bind to ovoTf (Fig. S2A and S2B), as has been previously found 30, suggesting the detergent-solubilized receptors are still in a functional state. Rather, the orientation of the N-lobes of the two Tf molecules in the crystal structure with TfR1 suggests that the ligand may interact with the stalk region of the receptor, thereby providing additional interactions that may be responsible for the increased affinities that we measured.
The interaction of diferric Tf and TfR2 is considered to be critical for its role in increasing hepcidin gene expression. The interaction re-routes internalized receptor from a degradation to a endosomal recycling pathways 31, thereby increasing the half-life of the protein. Although the binding of holo-Tf to TfR2 is weaker than to TfR1, in the context of physiological concentrations of serum Tf, the determined KD’s suggests it should bind efficiently even at low saturation levels. This suggested insensitivity of TfR2 to Tf saturation opposes in vivo data 5 and is likely a result of the simplified two-component system used here. Apo-Tf, which does not induce stabilization of the receptor, interacts with TfR2 at serum pH, but at a ~30-fold reduced affinity. As a result, it is not likely to effectively compete with holo-Tf for TfR2 binding. Whether monoferric Tf is capable of binding TfR2 at a higher affinity than apo-Tf or promoting stabilization of the receptor is unknown. Future studies will assess if that population could prohibit stabilization of TfR2 at low Tf saturation. Other proteins that have been found to interact with TfR2, such as HFE8 , hemojuvelin32 , or CD8133 may also influence Tf binding behavior at the cell surface.
At endosomal pH, TfR2 forms a stable complex with apo-Tf (Fig. 1G). This stability is analogous to that observed for TfR1 as part of the Tf cycle. Though iron uptake is not thought to be the primary function of TfR2, TfR2 has been found to promote Tf-Fe3+ uptake in cultured cells 27, 34. Furthermore, recent evidence shows that TfR2 traffics holo-Tf to lysosomes erythroid progenitors for iron uptake 35. As with TfR1, stabilization of the apo-Tf-TfR2 complex may be a key aspect of an uptake process.
The Tf-binding ectodomains of the TfR’s are quite similar, with 46% sequence identity. The helical domain specifically, which comprises nearly all of the binding site, has 50% sequence identity (69% similarity). The TfR1 Tf-binding site consists of 20 amino acids forming 30 interactions with Tf 14. Of those interactions, 8 are not conserved in TfR2, from 5 of the 20 amino acids. The TfR2 chimera that exchanges the helical domain for TfRl’s restores all but one of the non-conserved interactions. With those changes, the maximal binding and on-rate is enhanced from WT-TfR2, but still below that observed for TfR1. Further, the off-rate of the complex is increased from with wild-type TfR2 receptor. These data suggest that the difference in the association of TfR2 with Tf may be primarily through binding site residues, but the stability of the complex is due to unconserved mechanisms. This may include conformational changes upon binding that initiate stabilizing interactions of the helical domain with either the apical or protease-like domains.
A disordered apical domain loop, or “arm” as described here (residues 306–330), was identified as possibly participating in the Tf interaction from the crystal structure of TfR1 bound to Tf. The arm, and residue His-318, in particular, from one TfR1 monomer is drawn away from the binding site in the crystal structure lacking Tf 16. With Tf-bound, however, the arm is in close proximity to the Tf bound to the other monomer, as well as the C-terminus of that monomer 14. Both the arm and the C-terminus are highly conserved between TfR1 and TfR2 sequences, with the exception of His-318 (Gln-340 in TfR2) and the penultimate residues, Glu-759 in TfR1 and Asn-800 in TfR2 (Fig. 3C). These observations led to the hypothesis that, upon Tf binding, the conformational change of the apical domain arm and concurrent interaction between His-318, the C-terminus, and Tf is a key stabilizing mechanism for the interaction of Tf with TfR1 and that an altered (or missing) version of it in TfR2 accounts for the weaker complex stability.
Though highly conserved in TfR1 sequences across different species (Fig. 3C), both His-318 and Glu-759, however, are not critical for the interaction with holo- or apo-Tf (Fig. 4). Thus, in the context of the Tf cycle, they are not important for holo-Tf complex formation or stabilization of the apo-Tf-TfR1 complex after endosomal acidification and iron loss. There are a number of histidine residues in TfR1 that have been found to be involved in the conformational changes that drive iron release from Tf due to the protonation during endosomal acidification. Further, the Tf residue nearest the junction, His-349, has previously been shown to be essential for release of iron from the C-lobe 36. His-318 of TfR1 may similarly be required for the transition from a holo-complex to an apo- one, but the stable end states. The tolerance for substitution with the amide group-containing sidechains of the equivalent residues in TfR2 (Gln-340 and Asn-800, respectively) suggests that only the weak nucleophilicity is required, if they are involved in binding at all.
The inverse mutations in TfR2, however, do have a significant negative impact on holo-Tf binding. Lacking a crystal structure for TfR2, we cannot be certain the apical domain arm undergoes the same conformational change, forming a similar convergence of structural elements. The near-complete conservation of the elements, coupled with the substantially weaker Tf-binding with either TfR1-residue substitution, suggests that they are at least both involved in complex formation. Introduction of the mutations into the chimera protein do not significantly alter the kinetics, but do significantly impair maximal binding responses, particularly for the Q340H mutant, further illustrating the importance of that position in TfR2-Tf complex formation. The fact that neither mutation improves binding for the chimera demonstrates the role of inter-domain interactions beyond the apical arm junction are critical for stabilizing the complex, especially for TfR1’s high affinity interaction with Tf.
The differences in TfR1 and TfR2 binding behavior with Tf are likely central to the differing functions they perform. During the Tf cycle, TfR1 is not a passive receptor only to permit holo- Tf entry, rather the receptor serves to drive Fe3+ release from each lobe of the protein after acidification of the endosome through an assembly of conformational changes. Thus the interaction may be high affinity as the result of the extensive contacts required to alter the Tf structure prior to iron release. As TfR2 does not participate in the canonical Tf cycle, some specific interactions necessary for TfR1 function have likely been lost through evolution, with those favoring a specific conformational change that transduces a signal across the membrane retained.
Alongside a putative role for monoferric Tf in modulating the holo-Tf-TfR2 association, future studies should address if, and how, HFE influences TfR2’s interaction with holo-Tf. Soluble TfR2 ectodomain did not exhibit binding to HFE 22, but subsequent immunoprecipitation (IP) experiments have identified HFE-TfR2 complexes 8 Further, IP experiments have indicated HFE binding requires the stalk domain of TfR2 10, which is lacking in the ectodomain-only constructs. As TfR2 and HFE mutations each represent similar hereditary hemochromatosis sub-types, a better molecular understanding of the TfR2-HFE-Tf interaction(s) and their direct impact on modulating hepcidin expression is needed.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the OHSU Molecular Microbiology & Immunology department for use of the Biacore X100 instrument.
Funding Sources
NIH T-32 Training Grant GMMBI0231C (M.D.K.); NIH R01DK072166 (C.A.E.).
ABBREVIATIONS
- HH
hereditary hemochromatosis
- Tf
transferrin
- TfR
transferrin receptor
- HFE
high Fe protein
- SPR
surface plasmon resonance
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- [1].Aisen P, Leibman A, and Zweier J (1978) Stoichiometric and site characteristics of the binding of iron to human transferrin, J Biol Chem 253, 1930–1937. [PubMed] [Google Scholar]
- [2].Klausner RD, Ashwell G, van Renswoude J, Harford JB, and Bridges KR (1983) Binding of apotransferrin to K562 cells: explanation of the transferrin cycle, Proc Natl Acad Sci U S A 80, 2263–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levy JE, Jin O, Fujiwara Y, Kuo F, and Andrews NC (1999) Transferrin receptor is necessary for development of erythrocytes and the nervous system, Nat Genet 21, 396–399. [DOI] [PubMed] [Google Scholar]
- [4].Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, and Gasparini P (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22, Nat Genet 25, 14–15. [DOI] [PubMed] [Google Scholar]
- [5].Robb A, and Wessling-Resnick M (2004) Regulation of transferrin receptor 2 protein levels by transferrin, Blood 104, 4294–4299. [DOI] [PubMed] [Google Scholar]
- [6].Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, and Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization, Science 306, 2090–2093. [DOI] [PubMed] [Google Scholar]
- [7].Johnson MB, and Enns CA (2004) Diferric transferrin regulates transferrin receptor 2 protein stability, Blood 104, 4287–4293. [DOI] [PubMed] [Google Scholar]
- [8].Goswami T, and Andrews NC (2006) Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing, J Biol Chem 281, 28494–28498. [DOI] [PubMed] [Google Scholar]
- [9].Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N, Tsuchihashi Z, Sigal E, Bjorkman PJ, and Schatzman RC (1998) The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding, Proc Natl Acad Sci U S A 95, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen J, Chloupkova M, Gao J, Chapman-Arvedson TL, and Enns CA (2007) HFE modulates transferrin receptor 2 levels in hepatoma cells via interactions that differ from transferrin receptor 1-HFE interactions, J Biol Chem 282, 36862–36870. [DOI] [PubMed] [Google Scholar]
- [11].Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, and Andrews NC (2008) The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression, Cell Metab 7, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wahedi M, Wortham AM, Kleven MD, Zhao N, Jue S, Enns CA, and Zhang AS (2017) Matriptase-2 suppresses hepcidin expression by cleaving multiple components of the hepcidin induction pathway, J Biol Chem 292, 18354–18371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Abraham J, Corbett KD, Farzan M, Choe H, and Harrison SC (2010) Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses, Nat Struct Mol Biol 17, 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, and Mason AB (2011) How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH, Proc Natl Acad Sci U S A 108, 13089–13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bennett MJ, Lebron JA, and Bjorkman PJ (2000) Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor, Nature 403, 46–53. [DOI] [PubMed] [Google Scholar]
- [16].Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, and Harrison SC (1999) Crystal structure of the ectodomain of human transferrin receptor, Science 286, 779–782. [DOI] [PubMed] [Google Scholar]
- [17].Cheng Y, Zak O, Aisen P, Harrison SC, and Walz T (2004) Structure of the human transferrin receptor-transferrin complex, Cell 116, 565–576. [DOI] [PubMed] [Google Scholar]
- [18].van Dijk JP, van der Zande FG, Kroos MJ, Starreveld JS, and van Eijk HG (1993) Number and affinity of transferrin-receptors at the placental microvillous plasma membrane of the guinea pig: influence of gestational age and degree of transferrin glycan chain complexity, J Dev Physiol 19, 221–226. [PubMed] [Google Scholar]
- [19].Lebron JA, West AP Jr., and Bjorkman PJ (1999) The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor, J Mol Biol 294, 239–245. [DOI] [PubMed] [Google Scholar]
- [20].Zhao N, Zhang AS, Worthen C, Knutson MD, and Enns CA (2014) An iron-regulated and glycosylation-dependent proteasomal degradation pathway for the plasma membrane metal transporter ZIP14, Proc Natl Acad Sci U S A 111, 9175–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, and Bjorkman PJ (1998) Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor, Cell 93, 111–123. [DOI] [PubMed] [Google Scholar]
- [22].West AP Jr., Bennett MJ, Sellers VM, Andrews NC, Enns CA, and Bjorkman PJ (2000) Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE, J Biol Chem 275, 38135–38138. [DOI] [PubMed] [Google Scholar]
- [23].West AP Jr., Giannetti AM, Herr AB, Bennett MJ, Nangiana JS, Pierce JR, Weiner LP, Snow PM, and Bjorkman PJ (2001) Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites, J Mol Biol 313, 385–397. [DOI] [PubMed] [Google Scholar]
- [24].Giannetti AM, Snow PM, Zak O, and Bjorkman PJ (2003) Mechanism for multiple ligand recognition by the human transferrin receptor, PLoS Biol 1, E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Giannetti AM, and Bjorkman PJ (2004) HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface, J Biol Chem 279, 25866–25875. [DOI] [PubMed] [Google Scholar]
- [26].Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, and Bjorkman PJ (2005) The molecular mechanism for receptor-stimulated iron release from the plasma iron transport protein transferrin, Structure 13, 1613–1623. [DOI] [PubMed] [Google Scholar]
- [27].Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, and Koeffler HP (1999) Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family, J Biol Chem 274, 20826–20832. [DOI] [PubMed] [Google Scholar]
- [28].Tsung SH, Rosenthal WA, and Milewski KA (1975) Immunological measurement of transferrin compared with chemical measurement of total iron-binding capacity, Clin Chem 21, 1063–1066. [PubMed] [Google Scholar]
- [29].Leverence R, Mason AB, and Kaltashov IA (2010) Noncanonical interactions between serum transferrin and transferrin receptor evaluated with electrospray ionization mass spectrometry, Proc Natl Acad Sci U S A 107, 8123–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shimo-Oka T, Hagiwara Y, and Ozawa E (1986) Class specificity of transferrin as a muscle trophic factor, J Cell Physiol 126, 341–351. [DOI] [PubMed] [Google Scholar]
- [31].Chen J, Wang J, Meyers KR, and Enns CA (2009) Transferrin-directed internalization and cycling of transferrin receptor 2, Traffic 10, 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].D’Alessio F, Hentze MW, and Muckenthaler MU (2012) The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation, J Hepatol 57, 1052–1060. [DOI] [PubMed] [Google Scholar]
- [33].Chen J, and Enns CA (2015) CD81 promotes both the degradation of transferrin receptor 2 (TfR2) and the Tfr2-mediated maintenance of hepcidin expression, J Biol Chem 290, 7841–7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Robb AD, Ericsson M, and Wessling-Resnick M (2004) Transferrin receptor 2 mediates a biphasic pattern of transferrin uptake associated with ligand delivery to multivesicular bodies, Am J Physiol Cell Physiol 287, C1769–1775. [DOI] [PubMed] [Google Scholar]
- [35].Khalil S, Holy M, Grado S, Fleming R, Kurita R, Nakamura Y, and Goldfarb A (2017) A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2, Blood Advances 1, 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Steere AN, Byrne SL, Chasteen ND, Smith VC, MacGillivray RT, and Mason AB (2010) Evidence that His349 acts as a pH-inducible switch to accelerate receptor- mediated iron release from the C-lobe of human transferrin, J Biol Inorg Chem 15, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.