

Abstract
Th17 cells are a class of T helpers that secrete IL-17 and mediate pathogenic immunity responsible for autoimmunity including experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis. RORγt is the critical transcription factor that controls the differentiation of Th17 cells. However, little is known about the transcriptional co-factors for RORVγt in the regulation of Th17 differentiation. Here, we demonstrate that protein arginine N-methyltransferase 1 (PRMT1) associates with RORγt expanding Foxp3+ regulatory T cells. Consistently, pharmacological inhibition of PRMT1 impaired the generation of Th17 cells and prevented induction of EAE in mouse. Mechanistically, PRMT1-dependent modification of asymmetric histone 4 arginine 3 dimethylation (H4R3me2a), is required to stabilize the stimulatory STAT3 to displace the inhibitory STAT5 at IL-17 locus, resulting in the activation of IL-17 gene. Furthermore, PRMT1-facilitated recruitment of STAT3 overcame the inhibition of Th17 differentiation exerted by IL-2-induced STAT5 activation. PRMT1 thus regulates Th17 differentiation by controlling the reciprocal recruitment of STAT3 and STAT5. Our study thus reveals PRMT1 as a novel target for alleviating Th17-mediated autoimmunity by decreasing RORγt-dependent generation of pathogenic Th17 cells.
Introduction
T helper 17 (Th17) cells contribute to protective immunity against pathogens (1-3), as well as pathological immune reactions responsible for autoimmune diseases, including multiple sclerosis and psoriasis (4, 5). Retinoic acid-related orphan receptor gamma t (RORγt) is the transcription factor that control the generation of Th17 cells (6, 7). Activation of naïve T cells in the presence of TGFβ and IL-6 induces the up-regulation of RORγt and subsequent differentiation into Th17 cells (8, 9). Mutation of the RORγt gene causes severe immunodeficiency in both mice (6) and humans (10). Being a transcription factor, much of the previous studies have focused on RORγt-modulated target genes critical for the regulation of Th17 differentiation, which have demonstrated RORγt-mediated direct stimulation of the genes, including Il17a, Il17f, Ccr6, and Il23r (6, 7, 11, 12). However, less is known about the transcriptional co-factors for RORγt in the regulation of Th17 differentiation. Th17 cells produce the effector cytokines including IL-17A, IL-17F, IL-22, and GM-CSF to mediate pathological inflammation responsible for autoimmunity (13-15), hence targeting Th17 cells is considered a potential treatment for autoimmune diseases (16). Therefore, identification of the transcriptional co-factors critical for RORγt-dependent expression of inflammatory cytokines will not only reveal the basic mechanisms for regulating Th17 differentiation, but may also facilitate the development of novel therapeutics for the treatment of autoimmunity.
PRMTs are a family of nine protein arginine methyltransferases which catalyze the addition of methyl groups to an arginine residue, a form of post-translational modification (17). It is well established that PRMTs are involved in gene transcription, as they are recruited to promoters by transcription factors to epigenetically modify the histones. PRMT1 is the founding member of the PRMT family, and has been shown to function as a transcriptional co-activator by uniquely depositing dimethyl group on histone 4 arginine 3 (H4R3me2a), which facilitates the chromatin remodeling for gene activation (18, 19). PRMT1 has non-overlapping functions with other members of PRMTs, as indicated by the lethal phenotypes of mice deficient in PRMT1 (20, 21). PRMT1 was found to be overexpressed in breast cancers, prostate cancers and leukemia and responsible for oncogenesis (20, 22). PRMT1 inhibitors are thus developed for therapeutic treatment of cancers (23, 24). Much of the previous studies focus on PRMT1 function in cancers, whereas less is known about its role in T cells. With the development of popular immunotherapies, checkpoint inhibitors or CAR-T cells, it becomes increasingly important to understand the function of PRMT1 in T cells.
In this study, we demonstrate that PRMT1 is recruited to RORγt complex and regulates Th17 differentiation. WT PRMT1 increases, whereas enzymatically inactive PRMT1 or PRMT1 inhibitor decreases Th17 differentiation. Furthermore, we show that PRMT1-mediated deposition of H4R3me2a mark is required for recruiting stimulatory STAT3 and subsequently releasing inhibitory STAT5 at IL-17 gene locus, which leads to optimal activation of IL-17 gene expression. Finally, PRMT1 inhibitor-treated T cells also fail to induce EAE in vivo. Our study thus identifies the therapeutic candidature of PRMT1 and demonstrates the potential of PRMT1 inhibitors in the treatment of Th17-mediated autoimmune disorders.
Materials and Methods
Mice
C57BL/6 mice and Rag1tm1Mom (Rag1-/-) mice were purchased from the Jackson Laboratory. Mouse care and experimental procedures were performed under pathogen-free conditions following institutional guidance and approved protocols from the Institutional Animal Care and Use Committee at the Beckman Research Institute, City of Hope (no. 07023). Randomization was not used, and blinding was not performed.
Plasmids
A retroviral expression plasmid MIGR1 [murine stem cell virus (MSCV)-IRES-GFP] was used to clone mouse PRMT1 (GenBank: BC051953.1) cDNA. Specific point mutations of PRMT1 were introduced by using a mutagenesis kit (GeneArt® Site-Directed Mutagenesis System, Life Technologies Corporation, CA, USA). MSCV-LTRmiR30-PIG (LMP, Open Biosystems, GE Healthcare Dharmacon Inc), the control LMP has no insert, and vector-based retroviral PRMT1 short hairpin RNA (shRNA)- vectors (shPRMT1-1; shPRMT1-2; shPRMT1-3) were constructed by using following oligonucleotide sequences: shPRMT1: 5′TGCTGTTGACAGTGAGCGATGCCTGCAAGTGAAGAGGAACTAGTGAAGCCACAGATGTAGTTCCTCTTCACTTGCAGGCAGTGCCTACTGCCTCGGA; shPRMT1-2, 5′TGCTGTTGACAGTGAGCGCTGGGATTGAGTGTTCCAGTATTAGTGAAGCCACAGATGTAATACTGGAACACTCAATCCCAATGCCTACTGCCTCGGA; shPRMT1-3, 5′TGCTGTTGACAGTGAGCGACCAAAGCAGCTGGTCACCAATTAGTGAAGCCACAGATGTAATTGGTGACCAGCTGCTTTGGGTGCCTACTGCCTCGGA.
Inhibitor
The TC-E5003 compound was procured from Tocris Bioscience (Minneapolis, USA). The compound was dissolved in DMSO for in vitro experiments and used at indicated concentrations and added to differentiating T cell cultures for the last 24 hours before harvesting.
Retroviral packaging and transduction
Retroviral expression and transduction of T cells were performed essentially as in the reference (25). Briefly, retroviral expression plasmids were transfected into Platinum-E packaging cells (Cell Biolabs, Inc., San Diego, CA) using Lipofectamine 2000 (Invitrogen Life Technologies, CA, USA). After 48 h, viral supernatants were collected, filtered through 0.4-μm syringe filters, and stored at -80°C until use. For transduction, CD4+ T cells were first activated with 0.5 μg/ml hamster anti-CD3 (145-2C11; BioLegend, CA, USA), 0.5μg/ml hamster anti-CD28 (37.51; BioLegend, CA, USA) in a 24-well plate precoated with goat anti-hamster Ab (0855397; MP Biomedicals) for 24 h, then spin-infected with viral supernatants (2500 rpm, 30°C for 2 h) in the presence of 8 μg/ml polybrene (TR-1003-G; EMD Millipore Corporation, CA, USA). After spin infection, appropriate cytokines were added to the culture media to induce Th17 differentiation.
T cell differentiation and flow cytometric analysis
Cell suspensions from thymus, spleen, draining lymph nodes were prepared as standard protocol. For in vitro T cell differentiation, naïve CD4+ T cells were purified from single-cell suspensions of spleens of 6- to 10-wk-old C57BL/6 mice by negative magnetic selection using a Mouse CD4+ T Cell Isolation Kit II (Miltenyi Biotec Bergisch-Gladbach, Germany) and were further sorted on a FACS AriaIII. Replicate suspensions of 1×106/ml cell in IMDM with 10% FBS, 100 U/ml penicillin G, and 100 μg/ml streptomycin were cultured in 24-well plates that had been precoated with 0.2 mg/ml goat anti-hamster Ab (MP Biomedicals) and activated with medium supplemented with soluble 0.5μg/ml hamster anti-CD3 (145-2C11; BioLegend, CA, USA), 0.5μg/ml hamster anti-CD28 (37.51; BioLegend, CA, USA). T cells were cultured for 3 days at 37°C with 5% CO2. For TH1 polarization, 10 ng/ml of recombinant IL-12 (210-12; PeproTech, NJ, USA) and 5 μg/ml anti-IL-4 (11B11; Biolegend, CA, USA) were used. For TH2 polarization, 20 ng/ml of recombinant IL-4 (550067, BD Biosciences, CA, USA) and 5 μg/ml anti-IFN-γ (XMG1.2; Biolegend, CA, USA) were used. For TH17 polarization, 2 ng/ml of recombinant TGF-β1 (130-095-067; Miltenyi Biotec Inc., CA, USA), and 25 ng/ml of recombinant IL-6 (130-096-682; Miltenyi Biotec Inc., CA, USA) with 5 μg/ml anti-IFN-γ (XMG1.2; Biolegend, CA, USA) and 5 μg/ml anti-IL-4 (130-095-067; Miltenyi Biotec Inc., CA, USA) were used. For Treg differentiation, 2 ng/ml of recombinant TGF-β (130-095-067; Miltenyi Biotec Inc., CA, USA) and 20 ng/ml of recombinant IL-2 (550069; BD Pharmingen) with 5 μg/ml anti-IFN-γ (XMG1.2; Biolegend, CA, USA) and 5 μg/ml anti-IL-4 (11B11; Biolegend, CA, USA) were used. After 72 h of culture, cells were collected for analysis by flow-cytometry. For intracellular cytokine analysis, cells were re-stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml, Sigma-Aldrich) and ionomycin (0.5 μg/ml, Sigma-Aldrich) in the presence of GolgiPlug (555029; BD biosciences) for IFN-γ and IL-4 or GolgiStop (554724; BD biosciences) for IL-17 for 5 hrs. For some experiments, we first surface stained the cells with antibodies to mouse anti-CD4 (1:20, GK1.5, BioLegend, CA, USA) and then fixed and permeabilized with Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences, CA, USA) followed by staining with antibodies to either mouse anti-IFN-γ (1:50, XMG1.2, BD Pharmingen), anti-IL-4 ((1:20, 11B11, BD Pharmingen) and anti-IL-17 (1:50, TC11-18H10.1, BioLegend). FOXP3 intracellular staining was performed using BD Pharmingen Transcription Factor Buffer Set (562574, BD Pharmingen) according to the manufacturer's instructions with anti-FOXP3 (1:20, FJK-16s, eBioscience). Samples were acquired using a BD FACSCanto II flow cytometer (BD Biosciences, CA, USA) and analyzed by FACSDIVA (BD Biosciences, CA, USA) or FlowJo software (Tree Star, OR, USA).
Cell proliferation and viability assay
To determine the proliferation of T cells in the absence and presence of PRMT1 inhibitors, naïve T cells were labeled with 1 μM carboxyfluorescein succinimidyl ester (CFSE Cell Division Tracker Kit, 423801, BioLegend, Inc) as per manufacturer instructions. Proliferation was measured on day 3of differentiation using flow cytometry. The viability of differentiated T cells in the absence and presence of PRMT1 inhibitors, was detected using CellTiter-Glo® Luminescent Cell Viability Assay (G7570, Promega Corporation) as per manufacturer's intructions.
Quantitative Real-time PCR
Total RNA was isolated using RNeasy Isolation Kit according to the manufacturer's instructions (Qiagen). cDNA was synthesized with the Superscript III First-Strand Synthesis system (Invitrogen), and real-time PCR was performed on a Bio-Rad iCycler with SSOFast SYBR Green Supermix (Bio-Rad) and primer pairs specific for cDNA (5′ to 3′) were as follows: IL17A Forward: TTTAACTCCCTTGGCGCAAAA, IL17A Reverse: CTTTCCCTCCGCATTGACAC; IL17F Forward: TGCTACTGTTGATGTTGGGAC, IL17F Reverse: AATGCCCTGGTTTTGGTTGAA; CCR6 Forward: CCTGGGCAACATTATGGTGGT, CCR6 Reverse: CAGAACGGTAGGGTGAGGACA; CCL20 Forward: GCCTCTCGTACATACAGACGC, CCL20 Reverse: CCAGTTCTGCTTTGGATCAGC. Quantification of relative mRNA expression was determined by the comparative CT (critical threshold) method where the amount of target mRNA, normalized to endogenous β-actin expression, is determined by the formula 2-ΔCT and was represented as 2−ΔCt, where ΔCt = CtIL2 – CtActin. In some experiments, normalized Ct values were presented as an induction relative to expression in control samples.
RNA isolation and RNA sequencing
Total RNA was purified with the RNeasy Plus Mini Kit according to the manufacturer's protocol (Qiagen). RNA was quantified with a Qubit 2.0 fluorometer (Invitrogen). Barcoded stranded mRNA-seq cDNA libraries were prepared from 500 ng of total RNA using Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat, Illumina) and KAPA stranded RNA-Seq Kit for Illumina (KAPABIOYSTEMS) according to manufacturer's manual. Quantity was assessed using Invitrogen's Qubit HS Assay Kit and library size was determined using Agilent's 2100 Bioanalyzer HS DNA assay. The final library is between 200-500bp with peak at approximately 280bp. Barcoded RNA-Seq libraries were clustered and sequenced on the Illumina HiSeq2500 using HiSeq SR Cluster kit v4 and HiSeq SBS Kit V4 (51 cycle with 7 cycle index reading). The raw output data of the HiSeq was preprocessed according to the Illumina standard protocol. Quality control on the sequencing data was performed with the FastQC tool (available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), as well as the comprehensive Qorts suite65. Inspecting the produced reports, all samples were deemed of good quality for further processing. RNA seq reads were aligned to mouse genome mm10 using STAR aligner (26) (https://github.com/alexdobin/STAR/archive/STAR_2.4.2a.zip). Bam files generated after alignment were processed with featurecounts from subread package (http://subread.sourceforge.net/) to get the raw reads (27). Differential expression analysis was performed with edgeR package (https://bioconductor.org/packages/edgeR/) from R (28). Gene expression profiles were plotted as heatmap and scatterplot with ggplot2 (http://docs.ggplot2.org), a plotting system for R. Further pathway analysis was performed using Gene Set Enrichment Analysis available through the Broad Institute.
Cell culture, transient transfection, and reporter assays
HEK293T (CRL-3216; ATCC, VA, USA) cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, Cells (1 × 105 in each well of 96 well-plates) were transfected with the reporter plasmid (100 ng), pSV40-Renilla luciferase vector (50 ng), and expression vectors (0.5 μg) by using BioT (Bioland Scientific LLC, CA, USA). EL4 (TIB-39; ATCC, VA, USA) cells were cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The total amount of transfected DNA was kept constant by adjusting the amount of the empty vector. Cells were electroporated in Amaxa Nucleofector® II Device. Cells were collected after 24 h and lysed in 100 μl passive lysis buffer (Promega, 137 mM NaCl, 50 mM Tris-HCl, 0.5% NP-40); 50 μl aliquots of the clarified extracts were used to assay luciferase activity using Dual Luciferase assay kits/reagents from Promega Biotech (Promega, WI, USA) according to the manufacturer's protocols and measured on CLARIOstar (BMG LABTECH GmbH, Ortenberg, Germany). Luciferase activity was normalized to the levels of Renilla luciferase activities. The average is reported along with the standard deviation of the mean of triplicate transfections.
Cell lysis, western blotting and immunoprecipitation from whole cell lysates
Cell lysis, western blotting and immunoprecipitation were carried as described previously (29). The antibodies used are: anti-RORγt (Cat No: MAB6109, R&D Systems, Inc.), anti-Foxp3 (236A/E7, Cat No:20034, Abcam), anti-Actin (Cat No: A1978, Sigma-Aldrich, Inc.), anti-Stat3 (79D7, Cat No:4904P, Cell Signaling Technology), anti-Stat5 (D2O6Y, Cat No: 94205, Cell Signaling Technology), anti-PRMT1 (Cat No: ab190892, Abcam), anti-PRMT5 (Cat No:ab109451, Abcam), anti-H4R3(me)2a (Cat No: 39705, Active Motif), anti-H4 (Cat No: 61299, Abcam), anti-Foxp3 (236A/E7, Cat No:20034, Abcam), anti-Actin (Cat No: A1978, Sigma-Aldrich, Inc.), anti-Stat3 (79D7, Cat No:4904P, Cell Signaling Technology), anti-Stat5 (D2O6Y, Cat No: 94205, Cell Signaling Technology), anti-PRMT1 (Cat No: ab190892, Abcam), anti-PRMT5 (Cat No:ab109451, Abcam), anti-H4R3(me)2a (Cat No: 39705, Active Motif), anti-H4 (Cat No: 61299, Abcam),
Induction and assessment of EAE
For adoptive transfer EAE induction, age- and sex-matched C57BL/6 mice were immunized subcutaneously with 100μg MOG35–55 peptide (2HN-MEVGWYRSPFSRVVHLYRNGKCOOH) in complete Freund's adjuvant [Hooke Kit™ MOG35–55/CFA Emulsion PTX; cat. no. EK-2110; Hooke Laboratories]. Pertussis toxin (150 ng; Hooke Laboratories) in PBS was administered intravenously on days 0 (after 6 hrs) and day 1. At the peak of the disease, CD4+ T cells were collected from CNS, draining lymph nodes and spleen and stimulated with MOG35–55 for 48 hrs in presence IL-23, and treated with or without 2μM PRMT1 inhibitor (TC-E5003) for another 24 hours; 1×107 cells were transferred to Rag1−/− mice via i.p. injection. Mice were examined daily and scored for disease severity using a standard scale: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; 5, moribund or death. After the onset of EAE, food and water were provided on the cage floor. For visualization of CNS infiltration by cells of the immune system and demyelination, spinal cords of mice in which EAE was induced were collected on day 15. Mononuclear cells were prepared from the CNS (brain and spinal cord) of mice in which EAE was induced and were analyzed by flow cytometry.
Statistical analysis
Prism software v6.01 was used for data analysis (GraphPad, San Diego, CA). Unpaired, two-tailed Student's t test was performed to ascertain the significance of the differences between the means of the experimental groups. For some analysis, one way analysis of variance (ANOVA) was performed. Nonparametric statistical analysis was applied to data sets of EAE clinical scores and enumeration of CNS infiltrating cells and determined by Mann–Whitney U test. In the animal studies, five mice were required for each group based on the calculation to achieve a 2.5-fold change (effect size) in a two-tailed t-test with 95% power and a significance level of 5%. (Calculated by comparison of Means: 2-Sample, 2-Sided Equality; http://powerandsamplesize.com/Calculators/Compare-2-Means/2-Sample-Equality). We considered values of P<0.05 to be statistically significant. P value <0.001 were considered extremely significant (***), P value ranging between 0.001 to 0.01 were very significant (**), P value 0.01 to 0.05 as significant (*) and P value >0.05 were not significant (ns) (Student's unpaired t test or Ordinary One-way ANOVA with Tukey's multiple comparison).
Results
PRMT1 interacts with RORγt and regulate Th17 differentiation
During our initial analysis of RORγt interacting proteins (25, 30), PRMT1, but not other members of PRMT family, was identified with high confidence by mass spectrometry (Fig. 1A). Indeed, immunoprecipitation of RORγt brought down PRMT1 as a binding partner in Th17 cells (Fig. 1B). To determine whether PRMT1 regulates Th17 differentiation, we used GFP-retrovirus to introduce PRMT1 to the naïve CD4+ T cells which were then differentiated under Th17 priming conditions. Indeed, expression of wild type (WT) PRMT1 increased percentage of IL-17+ cells, whereas an enzymatically inactive PRMT1 (PRMT1-E153Q) due to a point mutation (31) decreased percentage of IL-17+ cells (Fig. 2C, D, E, supplementary Fig. 1A). As a control, percentage of IL-17+ cells was not changed in GFP- population that were not transduced with retrovirus. Three different shRNAs, PRMT1-shRNA1 to PRMT1-shRNA3, were then used to knockdown PRMT1 (Fig. 1F). Knockdown by PRMT1-shRNA2 significantly reduced IL17+ cells (Fig. 1G, H, supplementary Fig. 1B). In contrast, PRMT1-shRNA1 and PRMT1-shRNA3 could not effectively knockdown PRMT1 or GFP- cells that were not transduced by shRNA had no obvious effects on the generation of IL-17+ cells. These results thus suggest the critical role of PRMT1 in the regulation of Th17 differentiation.
Figure 1. PRMT1 interacts with RORγt and regulate Th17 differentiation.

A) PRMT1 identified by mass spectrometric analysis of RORγt-associated proteins represented in tabular form. B) Immunoprecipitation analysis of RORγt-PRMT1 interaction in Th17 cells. C) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells transduced with retrovirus expression GFP only (empty virus, EV) or together with PRMT1 or enzymatically inactive PRMT1-E153Q and differentiated under Th17 priming conditions for three days. D) Quantification of the results shown in C. E) Immunoblot analysis of PRMT1 expression in GFP+ cells shown in C. F) Immunoblot analysis of PRMT1 expression in CD4+ T cells transduced with retrovirus expressing indicated shRNA for knockdown PRMT1. G) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells transduced with retrovirus expressing indicated shRNA and differentiated under Th17 priming conditions for three days. H) Quantification of the results shown in G. *P<0.05, (D and H, One-way ANOVA, with Tukey's post-analysis multiple comparison), NS: non-significant. Error bars represent s.e.m. B, E and F are the representatives of three independent experiments.
Figure 2. Pharmacological PRMT1 inhibitor TC-E 5003 impairs Th17 differentiation.

A) Flow cytometric analysis of percentage of cells positive for indicated cytokines or Foxp3 among naïve CD4+ T cells differentiated under Th1, Th2, Th17 or Treg priming conditions in the presence of indicated concentrations of PRMT1 inhibitor. B) Quantification of the results shown in A. C) Percentage of IL-17+ cells among naïve CD4+ cells differentiated under Th17 priming conditions in the presence of indicated concentrations of PRMT5 inhibitor, as determined by flow cytometric analysis. D) Flow cytometric analysis of percentage of CSFE positive cells in indicated gate area for proliferating cells among naïve CD4+ cells differentiated under Th17 priming conditions in the presence or absence of indicated concentrations of PRMT1 inhibitor. E) Quantification of the results shown in D. F) Cell viability among naïve CD4+ T cells differentiated under Th17 priming conditions in the presence and absence of indicated concentrations of PRMT1 inhibitor, as determined by flow cytometric analysis of Cell Titer-Glo Luminescent Cell Viability Assay. G) Flow cytometric analysis of the percentage of IL-2+ cells among naïve CD4+ T cells differentiated under Th17 priming conditions in the presence or absence of indicated concentrations of PRMT1 inhibitor. H) Quantification of the results shown in G. I) Heatmap visualization of the up-regulated (yellow) and down-regulated (blue) genes in CD4+ cells differentiated under Th17 priming conditions in the presence (inhibitor) or absence (control, CTL) of 2 μM PRMT1 inhibitor, as determined by RNA-seq analysis. J) Comparison of gene expression profiles between naïve CD4+ T cells differentiation under Th17 priming conditions in the presence and absence of 2 μM PRMT inhibitor. Black dots represent selected genes as indicated. Average counts of transcript per million mapped reads (CPM) values of two replicates as shown in I. Right panel is the heatmap of Th17 signature genes indicated in the left panel. K) Expression levels of different members of PRMT1 in CD4+ T cells differentiation under Th17 priming conditions in the presence and absence of 2 μM PRMT inhibitor. Color indicates relative levels of gene calculated based on average counts of transcript per million mapped reads (CPM) values of two replicates as shown in I. L) Gene set enrichment analysis (GSEA) analysis of RNA-seq results indicates the negative correlation of glycolysis and hypoxia pathways with PMRT1 inhibitor-treated samples. *P<0.05, (B, E, F, H, One-way ANOVA, with Tukey's post-analysis multiple comparison, C, two-tailed unpaired t-test), NS: non-significant. Error bars represent s.e.m.
Pharmacological PRMT1 inhibitor selectively impairs Th17 differentiation and promotes the generation of Tregs in vitro
We next tested the specific PRMT1 pharmacological inhibitor, TC-E5003 (24) on the differentiation pattern of T cells. PRMT1 inhibitor had no effects on Th1 and Th2 differentiation, however, it impaired Th17 differentiation in a dose dependent manner (Fig. 2A, B). Interestingly, the inhibitor treatment boosted the differentiation of Foxp3+ Treg cells at higher concentrations (3 and 6 uM), which reflected the often observed reciprocal relationship between Th17 and Treg differentiation (12). Notably, the specific PMRT5 inhibitor (32, 33) had no effects on Th17 differentiation (Fig. 2C), indicating the specificity of PRMT1. The concentrations of PRMT1 inhibitor used in above experiments did not affect T cell proliferation (Fig. 2D, E, supplementary Fig. 2A), cell viability (Fig. 2F, supplementary Fig. 2B) and IL-2 production (Fig. 2G, H, supplementary Fig. 2C), indicating the specificity of PRMT1 inhibitor in repressing Th17 differentiation program only. Since we only observed the expression of IL-17A so far, to further evaluate the global effects of PRMT1 inhibitor on Th17 differentiation, we compared transcriptome of Th17 differentiation by RNA-seq in the presence and absence of PRMT1 inhibitor. We found very similar expression patterns between two biological repeats of control (CTL, without inhibitor) or inhibitor-treated samples (inhibitor) (Fig. 2I), indicating the reproducibility of our assays. Many RORγt-regulated Th17 signature genes including Il17a, Il17f, Il22 and Il1r1 were conspicuously down-regulated in inhibitor-treated samples along with moderate down-regulation of Il23r and Ccr6 (Fig. 2J), suggesting that PRMT1 inhibitor affected Th17 differentiation program. However, the RORγt levels and expression of PRMT1 (Fig. 2K) were changed nominally by inhibitor treatment, indicating that the PRMT1 inhibitor does not repress the expression of RORγt target genes by interfering with RORγt expression, but likely affect the RORγt activity via inhibiting the co-factor activity of PRMT1.
Recent studies have highlighted the importance of metabolism in the regulation of CD4+ T cell differentiation (34). In particular, Th17 cells are characterized by a preferential use of glycolysis and expression of hypoxia-inducible genes (35). Interestingly, unbiased Gene set enrichment analysis (GSEA) of our RNA-seq data revealed a global decrease in genes associated with glycolysis and hypoxia in PRMT1 inhibitor-treated cells compared with untreated cells (Fig. 2L), hence reaffirming the role of PMT1 inhibitor in suppressing the overall differentiation pattern of Th17 cells. Taken together, PRMT1 inhibitor treatment prevents the generation of IL-17+ CD4 T cells by inhibiting the transcriptional programs critical for Th17 differentiation.
PRMT1 inhibitor prevents Th17-induced EAE
PRMT1 inhibitor impaired the generation of IL-17+ cells (Fig. 2A). To investigate how PRMT1 activity contributes to the differentiation of CD4+ T cells into Th17 cells capable of functioning as pathogenic effectors in vivo, we used Th17-mediated passive EAE model that requires adoptive transfer of activated T cells (36). For this purpose, T cells obtained from mice challenged with MOG35-55 peptide were expanded in vitro by IL-23 and MOG35-55 in presence or absence of PRMT1 inhibitor, and then adoptively transferred to Rag1-/- mice to induce EAE. Indeed, PRMT1 inhibitor-treated CD4+ T cells induced much less severe EAE compared to that without inhibitor treatment (Fig. 3A). Histological examination of spinal cords revealed lesser inflammation (Fig. 3B, H&E staining), tissue damage due to demyelination (Fig. 3B, LFB staining) and infiltration of CD4+ cells (Fig. 3B, right panels) in Rag-/- mice reconstituted with PRMT1 inhibitor-treated CD4+ T cells. Impaired inflammation was also indicated by significantly reduced central nervous system (CNS) infiltrating lymphocytes including CD45+CD4+ T cells (Fig. 3C, top panels), CD45+Ly6G+ monocytes (Fig. 3C, middle panels) and CD45+F4/80+ macrophages (Fig. 3C, bottom panels) in experimental mice group receiving inhibitor treated T cells. At the peak of the disease, Rag1-/- mice reconstituted with CD4+ cells treated with or without PRMT1 showed equal percentage of CD4+IFNγ+ cells (Fig. 3D, middle panels), however, inhibitor-treated CD4+ T cells showed greatly reduced IL-17+CD4+ T cells (Fig. 3D top panels) and increased CD4+Foxp3+ cells at the recovery phase (Fig. 3D, bottom panels), which is consistent with the reciprocal effects of PRMT1 inhibitor on the generation of IL-17+ and Foxp3+ cells (Fig. 2A). We also observed reduced GM-CSF producing T cells in mice receiving PRMT1 inhibitor treated T cells (Fig. 3E), another pathogenic factor for EAE (14, 15). Reduced expression of Th17 signature genes including Il17a, Il17f, Il23r, Il1r and Ccr6 were also detected in CNS infiltrated lymphocytes obtained from Rag1-/- mice reconstituted with PRMT1 inhibitor-treated CD4+ T cells (Fig. 3F). Actually, inhibition of IL-17 signature genes was observed prior to adoptive transfer of PRMT1-treated cells (supplementary Fig. 1C). These results thus indicate that PRMT1 activity promotes the pathogenic effector function of Th17 cells.
Figure 3. PRMT1 inhibitor prevents T cell-induced EAE.

A) Mean EAE clinical score of Rag1-/- mice adoptively transferred with PBS only, or CD4+ T expanded in the presence of MOG35-55 peptides and IL-23 (control, CTL) or together with PRMT1 inhibitor (inhibitor). B) H&E, LFB and CD4 staining of spinal cord transverse sections of CTL or inhibitor-treated mice as described in A at the peak of EAE. C) Percentage of different infiltrated lymphocytes in harvested CNS of indicated EAE-induced mice at the peak of disease, as determined by flow cytometric analysis of indicated surface markers. Right panels are the quantification results of the left panels. D) Percentage of IL-17A+ and IFNγ+ among infiltrated CD4+ T cells at the peak of disease and Foxp3+ cells at the recovery phase of the disease in harvested CNS of indicated EAE-induced mice, as determined by flow cytometric analysis of indicated molecules. Right panels are the quantification results of the left panels. E) Percentage of IL-17A+/GM-CSF3+ cells among infiltrated CD4+ T cells in harvested CNS of indicated EAE-induced mice at the peak of disease, as determined by flow cytometric analysis of indicated molecules. Right panel is the quantification results of the left panel. F) Expression of indicated Th17 signature genes among infiltrated lymphocytes of indicated EAE-induced mice at the peak of disease, as determined by qPCR. n=5 mice per group in each experiment. Error bars represent s.d. NS: non-significant. P< 0.05 and **P<0.01 (A, C, D, E, F, nonparametric Mann–Whitney U-test).
PRMT1 inhibitor regulates reciprocal recruitment of STAT3 and STAT5 to IL-17 locus
PRMT1 is a methyltransferase that epigenetically marks histone 4 with H4R3me2a modification to remodel chromatin, which then allows the access of transcription factors for gene activation (7, 18, 19). We hence performed ChIP assays to determine whether PRMT1 and the resultant histone modification affects the recruitment of transcription factors known to regulate IL-17 gene. As expected, PRMT1-induced H4R3me2a modification at the IL-17 promoter (Fig. 4A) was greatly decreased by the pharmacological inhibition of PRMT1. Notably, the loss of H4R3me2a mark, did not affect the recruitment of IRF4, BATF, p300, and RORγt in the inhibitor-treated Th17 cells. Strikingly, the recruitment of STAT3 was reduced whereas the recruitment of STAT5 was reciprocally increased (Fig. 4A), although the activation of STAT3 and STAT5 as indicated by their phosphorylation status was not affected by PRMT1 inhibitor treatment (Fig. 4B). These results were further corroborated by immunoblotting analysis after chromatin precipitation with anti-RORγt antibody that brought down H4R3me2a modified histones and STAT3 in control Th17 cells, whereas noticeably lesser H4R3me2a modified histones and STAT3, and more STAT5 were precipitated in the presence of PRMT1 inhibitor (Fig. 4C). Next, we performed global ChIP-seq analysis to monitor STAT3 and STAT5 binding at IL-17 locus. Interestingly, blocking the PRMT1 activity by pharmacological inhibitor significantly diminished the binding of STAT3, whereas reciprocally increased the binding of STAT5 to the IL-17 locus (Fig. 4D). Thus, these results indicate the regulation of reciprocal binding pattern of STAT3 and STAT5 at the IL-17 locus by PRMT1.
Figure 4. PRMT1 inhibitor regulates the reciprocal recruitment of STAT3 and STAT5 to the IL-17 locus.
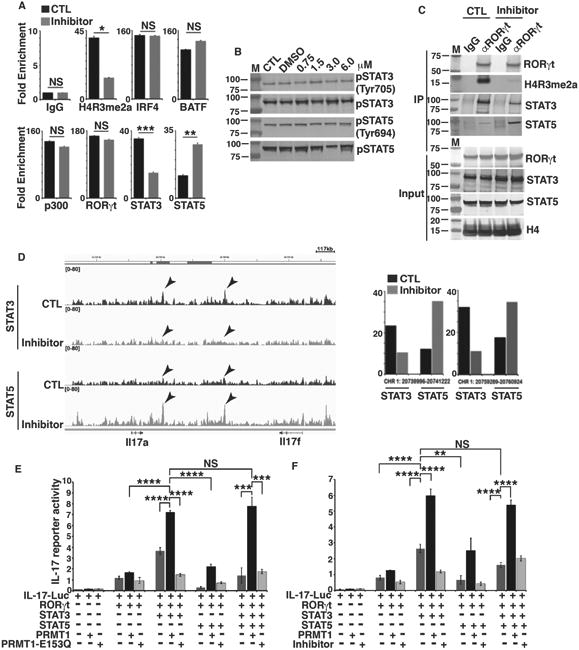
A) ChIP analysis of the enrichment of H4R3me2a, or indicated transcription factors in IL-17 promoter region in GFP+ fractions of CD4+ T cells differentiated under Th17 priming conditions in the presence (inhibitor) and absence (control, CTL) of PRMT1 inhibitor. B) Immunoblot analysis of phosphorylated or whole STAT3 and STAT5 in naïve CD4+ T cells differentiated under Th17 priming conditions in the presence and absence of indicated concentrations of PRMT1 inhibitor. C) Immunoblot analysis of indicated proteins from immune-complexes eluted from chromatin precipitates with control anti-IgG or RORγt antibody from naïve CD4+ T cells differentiated under Th17 priming conditions in the presence and absence of PRMT1 inhibitor. D) STAT3 and STAT5 DNA-binding peaks at IL-17 gene locus in CD4+ T cells differentiated under Th17 priming conditions in the presence and absence of PRMT1 inhibitor, as determined by ChIP-seq assay. Arrows indicate the locations of two most prominent peaks. Right panel is the quantitative value of arrow indicated peaks on left. E) Relative luciferase activity of IL-17 reporter in HEK 293T cells transfected with indicated expression plasmids. The luciferase activity is normalized to Renilla luciferase activity (mean ± sd). F) Relative luciferase activity of IL-17 reporter in HEK 293T cells transfected with indicated expression plasmids and/or presence of PRMT1 inhibitor. The luciferase activity is normalized to Renilla luciferase activity (mean ± sd). *P<0.05, (A, two-tailed unpaired t-test; E, F, One-way ANOVA with Tukey's post-analysis multiple comparison), NS: non-significant by unpaired t-test. B is the representatives of three independent experiments.
STAT3 is activated by IL-6 and is required for Th17 differentiation, whereas STAT5 is activated by IL-2 that inhibits Th17 differentiation (37, 38). We thus determined the effects of PRMT1 on a IL-17 promoter-luciferase reporter in the presence and absence of STAT3 and/or STAT5 (Fig. 4E and 4F). RORγt increased IL-17 reporter activity which was further stimulated by STAT3 but inhibited by STAT5, consistent with the positive role of STAT3 and negative role of STAT5 in the regulation of IL-17 expression (Fig. 4E). The optimal IL-17 reporter activity was observed in the presence of RORγt, STAT3 and PRMT1 but not inactive PRMT1-E153Q (Fig. 4E) or PMRT1 inhibitor (Fig. 4F). Actually, PRMT1-E153Q or PRMT1 inhibitor impaired RORγt and STAT3-mediated activation of IL-17 reporter. More importantly, PRMT1, but not PRMT1-E153Q, stimulated IL-17 reporter even in the presence of STAT5, suggesting that PRMT1 activity is critical for overcoming STAT5-mediated inhibition of IL-17 reporter. Taken together, these results indicate that, PRMT1 deposits active H4R3me2a mark on IL-17 locus, which enhances the recruitment of stimulatory STAT3 whereas disengages the repressive STAT5, resulting in the activation of IL-17 gene.
PRMT1 overcomes IL-2-mediated inhibition of Th17 differentiation by controlling reciprocal STAT3 and STAT5 recruitments
To further test the function of PRMT1 in the reciprocal regulation of STAT3 and STAT5 activity during Th17 differentiation, we took advantage of that IL-2 inhibits Th17 differentiation by stimulating STAT5 (38). Consistent with published results (38), IL-2 inhibited generation of IL-17+ cells in a concentration dependent manner (Fig. 5A). We next determined whether activation of STAT3 by IL-6 can overcome IL-2-inhibited generation of IL-17+ cells. IL-6 failed to increase IL-17+ cells in presence of IL-2 (Fig. 5B). However, ectopic expression of PRMT1 (Fig. 5C), but not inactive PRMT1-E153Q (Fig. 5D) or PRMT1 inhibitor treatment (Fig. 5E), overcame IL-2-mediated inhibition and increased IL-17+ cells. These results thus suggest that PRMT1 activity is essential for IL-6 to overcome IL-2-mediated inhibition of Th17 differentiation. We next monitored four different sites at IL-17 locus that previously showed reciprocal interactions with STAT3 and STAT5 (37, 38). Indeed, relatively high H4R3me2a signals correlated with high STAT3 and low STAT5 binding signals during normal Th17 differentiation (Fig. 5F). Whereas, addition of high concentration of IL-2, that activates STAT5 and inhibits Th17 differentiation, reversed the trend, i.e. reduced H4R3me2a signals and consequently decreased STAT3 while increased STAT5 binding signals (Fig. 5G). However, ectopic expression of WT PRMT1 (Fig. 5H), but not inactive PRMT1-E153Q (Fig. 5I) or PRMT1 inhibitor treatment (Fig. 5J), overcame IL-2-mediated inhibition, accompanied by increased H4R3me2a signals, augmented STAT3 whereas diminished STAT5 binding to the IL-17 locus. It was also reaffirmed by immunoblot analysis after chromatin-immunoprecipitation with anti-RORγt antibody from the different experimental sets as above (Fig. 5F-J), where prominently more STAT3 and H4R3me2a were recovered from cell overexpressing PRMT1, while lesser H4R3me2a, STAT3 and reciprocally higher STAT5 were detected from cells with higher IL-2 and overexpression of PRMT1-E153Q or with treatment by PRMT1 inhibitor (Fig. 5K). As controls for IL-2-activated STAT5 and IL-6-activated STAT3, we detected phosphorylated STAT5 (Tyr694) (Fig. 5L, first panel) and phosphorylated STAT3 (Tyr705) (Fig. 5L, panels 2-5). Our results thus suggest that PRMT1 is required to overcome IL-2-mediated inhibition of Th17 differentiation by depositing H4R3me2a to induce stimulatory STAT3 recruitment by disengaging inhibitory STAT5.
Figure 5. PRMT1 overcomes IL-2-mediated inhibition of Th17 differentiation by controlling reciprocal STAT3 and STAT5 recruitments.

A) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells differentiated under Th17 priming conditions in the presence of increasing concentrations of exogenous IL-2 (0, 10 or 100 IU/ml). Right panel is the quantification results of the left panels. B) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells differentiated under Th17 priming conditions in the presence of IL-2 (100 IU/ml of exogenous rhIL-2), rested for 24hrs and re-stimulated with increasing concentrations of IL-6 (0, 2.5, 25 ng/ml). Right panel is the quantification results of the left panels. C and D) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells transduced with retrovirus expressing GFP and WT PRMT1 (C) or inactive PRMT1-E153Q (D), and differentiated under Th17 priming conditions in the presence of IL-2 (100 IU/ml), rested for 24 hrs and re-stimulated with increasing concentrations of IL-6 (0, 2.5, 25 ng/ml). Right panels are the quantification results of the left panels. E) Flow cytometric analysis of the percentage of IL-17+ cells among naïve CD4+ T cells differentiated under Th17 priming conditions in the presence of IL-2 (100 IU/ml), increasing concentrations of IL-6 (0, 2.5, 25 ng/ml) and PRMT1 inhibitor (3μM). Right panel is the quantification results of the left panel. F-J) ChIP analysis of the enrichment of H4R3me2a, STAT3 and STAT5 at four different sites at IL-17 gene locus in CD4+ T cells differentiated under Th17 priming conditions (Th17) (F), Th17 conditions + IL-2 (100 IU/ml) (G), Th17 conditions + IL-2 (100 IU/ml) + IL-6 (25ng/ml) + retroviral expression of WT PRMT1 (H), or inactive PRMT1-E153Q (I), or Th17 conditions + IL-2 (100 IU/ml) + IL-6 (25ng/ml) + PRMT1 inhibitor (3μM) (J). IgG is a negative control. K) Immunoblot analysis of indicated proteins from immune-complexes eluted from chromatin precipitates with anti-RORγt antibody from CD4+ T cells treated as described in F-J. L) Immunoblot analysis of phosphorylated or whole STAT3 and STAT5 in naïve CD4+ T cells differentiated under indicated conditions as described in F-J. K and L are the representative results of three independent experiments. *P<0.05, (A-E, One-way ANOVA, with Tukey's post-analysis multiple comparison), NS: non-significant.
Discussion
Our results demonstrate sequential events responsible for PRMT1-mediated activation of IL-17 genes during Th17 differentiation: 1) PRMT1 is recruited to IL-17 promoter via association with RORγt complex; 2) PRMT1 deposits active H4R3me2a mark on IL-17 locus, which is required for the subsequent events of; 3) recruiting stimulatory STAT3 to IL-17 promoter, and either concurrently or sequentially; 4) releasing inhibitory STAT5 from IL-17 promoter, which eventually leads to transcriptional activation of IL-17 gene, a critical event for initiating Th17 differentiation process.
Protein arginine methylation has been indicated in the regulation of T cell function (39, 40). However, the function of PRMTs in T cells largely remains unknown, needless to say the isoform specific function of PRMTs. We demonstrate here for the first time that PRMT1, being associated with RORγt transcriptional complex, regulates Th17 differentiation. Furthermore, PRMT1 is required to deposit H4R3me2a activation mark at IL-17 locus. Next, we also questioned and correlated the contextual purpose of such modification in Th17 program. Th17 differentiation is regulated by multiple transcription factors including RORγt, STAT3, IRF4 and BATF that have been shown to cooperatively bind to IL-17 locus to activate its expression during Th17 differentiation (7, 41). Interestingly, many of the above mentioned transcription factors except RORγt are also involved in the development of other lineages of T helper cells. For instance, STAT3 also regulates the differentiation of Treg and T follicular helpers (Tfh) (42, 43) whereas RORγt is selectively required for generation of IL-17+ cells (6). It is not clear whether the binding of RORγt guides the recruitment of other transcription factors. Our results indicate that PRMT1-mediated histone modification does not affect the recruitment of RORγt, IRF4, p300 and BATF to the IL-17A locus, however, is required for enhanced binding of STAT3 whereas releasing STAT5 at IL-17 promoter. The fact that enzymatically inactive PRMT1 or PRMT1 inhibitor prevented the recruitment of STAT3 and the release of STAT5 strongly suggest that PRMT1-mediated deposition of H4R3me2a instructs the recruitment of STAT3 and disengagement of STAT5 at the IL-17 locus. Our study thus demonstrated a mechanism that controls the sequential recruitment of RORγt and STAT3 to displace STAT5 at the IL-17 locus.
The pathogenesis of autoimmune diseases is extremely complex and remains largely unknown. Uncontrolled Th17 responses are responsible for various pathological conditions including autoimmunity such as multiple sclerosis, psoriasis and arthritis (1-3, 7, 44-47), as well as tumor growth (48-50) and even autism (51). Th17 cells produce effector cytokines IL-17A, IL-17F, IL-22 and GM-CSF to mediate pathological inflammation; targeting Th17 cells is thus a potentially valuable treatment for these diseases (13). For example, ustekinumab, a human monoclonal antibody that inhibits Th17 responses by blocking the IL-23 receptor, has been approved by the FDA for treatment of severe plaque psoriasis (52). Given the essential function of PRMT1 in Th17 cells, further studies are needed to determine whether dysregulated PRMT1 contributes to these conditions. Since PRMT1 has been linked to several types of cancers including breast, prostate and leukemia (20, 22), PRMT1 inhibitors are developed for treatment of these cancers (23, 24). Taking advantage of the available selective PRMT1 inhibitors, we demonstrated that PRMT1 inhibitor impairs the development of IL-17+ cells, and inhibitor-treated CD4+ T cells cannot induce EAE. Our results thus indicate that PRMT1 inhibitor can be used to alleviate Th17-mediated autoimmune diseases. Interestingly, PRMT1 inhibitor not only prevents Th17 differentiation but also promotes the differentiation of inhibitory Tregs in in vitro differentiation as well as in vivo during the induction of EAE. The mechanisms responsible for PRMT1 inhibitor-stimulated generation of Treg still remain to be determined. Thus, our study demonstrates the great therapeutic potential of PRMT1 inhibitor in the clinical treatment of autoimmune conditions by both inhibiting Th17 and stimulating Treg differentiation.
Supplementary Material
Acknowledgments
We appreciate the help by city of hope supported cores including animal, genomic, flow cytometer, pathology and mass spectrometric cores. We thank Dr. Chen Dong (Tsinghuan University) for sharing the IL-17 reporter.
This work was supported by grants from NIH R01-AI053147, NIH R01-AI109644 and institutional pilot funding. In addition, research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572, which includes work performed in the animal, genomic, flow cytometer and mass spectrometric cores supported by this grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Wang Z, Friedrich C, Hagemann SC, Korte WH, Goharani N, Cording S, Eberl G, Sparwasser T, Lochner M. Regulatory T cells promote a protective Th17-associated immune response to intestinal bacterial infection with C. rodentium. Mucosal Immunol. 2014;7:1290–1301. doi: 10.1038/mi.2014.17. [DOI] [PubMed] [Google Scholar]
- 2.Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, Weaver CT. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol. 2015;16:286–295. doi: 10.1038/ni.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O'Connor W, Jr, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elloso MM, Gomez-Angelats M, Fourie AM. Targeting the Th17 pathway in psoriasis. J Leukoc Biol. 2012;92:1187–1197. doi: 10.1189/jlb.0212101. [DOI] [PubMed] [Google Scholar]
- 5.Johnson-Huang LM, Lowes MA, Krueger JG. Putting together the psoriasis puzzle: an update on developing targeted therapies. Dis Model Mech. 2012;5:423–433. doi: 10.1242/dmm.009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivanov II, Mc Kenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 7.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, Littman DR. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CS, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay JP, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, Casanova JL. Immunodeficiencies. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349:606–613. doi: 10.1126/science.aaa4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ratajewski M, Walczak-Drzewiecka A, Salkowska A, Dastych J. Upstream stimulating factors regulate the expression of RORgammaT in human lymphocytes. J Immunol. 2012;189:3034–3042. doi: 10.4049/jimmunol.1200519. [DOI] [PubMed] [Google Scholar]
- 12.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 13.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 15.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. 2014;35:493–500. doi: 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR, Allis CD. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 21.Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20:4859–4869. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–1215. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- 23.Dillon MB, Bachovchin DA, Brown SJ, Finn MG, Rosen H, Cravatt BF, Mowen KA. Novel inhibitors for PRMT1 discovered by high-throughput screening using activity-based fluorescence polarization. ACS Chem Biol. 2012;7:1198–1204. doi: 10.1021/cb300024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bissinger EM, Heinke R, Spannhoff A, Eberlin A, Metzger E, Cura V, Hassenboehler P, Cavarelli J, Schule R, Bedford MT, Sippl W, Jung M. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg Med Chem. 2011;19:3717–3731. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 25.He Z, Wang F, Ma J, Sen S, Zhang J, Gwack Y, Zhou Y, Sun Z. Ubiquitination of RORgammat at Lysine 446 Limits Th17 Differentiation by Controlling Coactivator Recruitment. J Immunol. 2016;197:1148–1158. doi: 10.4049/jimmunol.1600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sen S, Roy K, Mukherjee S, Mukhopadhyay R, Roy S. Restoration of IFNgammaR subunit assembly, IFNgamma signaling and parasite clearance in Leishmania donovani infected macrophages: role of membrane cholesterol. PLoS Pathog. 2011;7:e1002229. doi: 10.1371/journal.ppat.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Z, Ma J, Wang R, Zhang J, Huang Z, Wang F, Sen S, Rothenberg EV, Sun Z. A two-amino-acid substitution in the transcription factor RORgammat disrupts its function in TH17 differentiation but not in thymocyte development. Nat Immunol. 2017;18:1128–1138. doi: 10.1038/ni.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duncan KW, Rioux N, Boriack-Sjodin PA, Munchhof MJ, Reiter LA, Majer CR, Jin L, Johnston LD, Chan-Penebre E, Kuplast KG, Porter Scott M, Pollock RM, Waters NJ, Smith JJ, Moyer MP, Copeland RA, Chesworth R. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med Chem Lett. 2016;7:162–166. doi: 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, West KA, Lingaraj T, Stickland K, Ribich SA, Raimondi A, Scott MP, Waters NJ, Pollock RM, Smith JJ, Barbash O, Pappalardi M, Ho TF, Nurse K, Oza KP, Gallagher KT, Kruger R, Moyer MP, Copeland RA, Chesworth R, Duncan KW. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11:432–437. doi: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 34.Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev. 2013;252:52–77. doi: 10.1111/imr.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1952–1960. doi: 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- 37.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea J. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Beta-catenin accumulation and mutation of exon 3 of the beta-catenin gene in hepatocellular carcinoma. Jpn J Cancer Res. 1999;90 doi: 10.1111/j.1349-7006.1999.tb00712.x. inside front cover. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanchet F, Cardona A, Letimier FA, Hershfield MS, Acuto O. CD28 costimulatory signal induces protein arginine methylation in T cells. J Exp Med. 2005;202:371–377. doi: 10.1084/jem.20050176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. 2014;40:367–377. doi: 10.1016/j.immuni.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huber M, Steinwald V, Guralnik A, Brustle A, Kleemann P, Rosenplanter C, Decker T, Lohoff M. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol. 2008;20:223–234. doi: 10.1093/intimm/dxm139. [DOI] [PubMed] [Google Scholar]
- 44.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zelante T, De Luca A, D'Angelo C, Moretti S, Romani L. IL-17/Th17 in anti-fungal immunity: what's new? Eur J Immunol. 2009;39:645–648. doi: 10.1002/eji.200839102. [DOI] [PubMed] [Google Scholar]
- 46.van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng SC, Joosten I, van den Berg WB, Williams DL, van der Meer JW, Joosten LA, Netea MG. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe. 2009;5:329–340. doi: 10.1016/j.chom.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 49.Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, Hishinuma T, Goto J, Lotze MT, Kolls JK, Sasaki H. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. 2005;175:6177–6189. doi: 10.4049/jimmunol.175.9.6177. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–939. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benson JM, Peritt D, Scallon BJ, Heavner GA, Shealy DJ, Giles-Komar JM, Mascelli MA. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs. 2011;3:535–545. doi: 10.4161/mabs.3.6.17815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.