

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, endothelial cells, inflammation, macrophages, sphingolipids
Abstract
Objective—
Atherosclerosis is a chronic multifactorial and inflammatory disease of large and medium arteries and the leading cause of cardiovascular diseases worldwide. The aim of this study was to investigate whether and how the nSMase2 (type 2-neutral sphingomyelinase), a key enzyme of sphingolipid metabolism, may contribute to the development of atherosclerotic lesions.
Approach and Results—
The role of nSMase2 in atherosclerosis was investigated in Apoe−/−;Smpd3fro/fro mice, mutant for nSMase2, and in Apoe−/−;Smpd3+/+ mice intraperitoneally injected with GW4869, a pharmacological nSMase2 inhibitor. The defect or inhibition of nSMase2 resulted in a reduction of atherosclerotic lesions and a decrease in macrophage infiltration and lipid deposition, although cholesterolemia remained unchanged. nSMase2 inhibition decreased the inflammatory response of murine endothelial cells to oxLDL (oxidized low-density lipoprotein), as assessed by the significant reduction of MCP-1 (monocyte chemoattractant protein 1), ICAM-1 (intercellular adhesion molecule-1), and VCAM-1 (vascular cell adhesion molecule-1) mRNA expressions and macrophage recruitment. Likewise, in RAW264.7 or in macrophages isolated from Apoe−/−/Smpd3fro/fro or Apoe−/−/Smpd3+/+ mice stimulated by lipopolysaccharides, nSMase2 inhibition resulted in a decrease in the expression of inflammatory molecules. Mechanistically, the anti-inflammatory response resulting from nSMase2 inhibition involves Nrf2 (nuclear factor [erythroid-derived 2]-like 2 or NF-E2–related factor-2) activation in both endothelial cells and macrophages, as assessed by the lack of protective effect of GW4869 in endothelial cells silenced for Nrf2 by small interfering RNAs, and in lipopolysaccharide-stimulated macrophages issued from Nrf2-KO mice.
Conclusions—
The genetic deficiency or inhibition of nSMase2 strongly decreases the development of atherosclerotic lesions in Apoe−/− mice, by reducing inflammatory responses through a mechanism involving the Nrf2 pathway. Inhibitors of nSMase2 may, therefore, constitute a novel approach to slow down atherosclerosis progression.
Atherosclerosis is a multifactorial chronic disease characterized by lipid deposition, inflammatory response, and abnormal remodeling of the intima of large and medium-sized arteries.1–5 Among the various mechanisms involved in atherogenesis, LDLs (low-density lipoproteins) retention and oxidation generate a local inflammatory response including endothelial activation, monocyte recruitment, and cholesterol accumulation in macrophages.1,3–5 Activated endothelial cells express adhesion molecules, ICAM-1 (intercellular adhesion molecule-1), and VCAM-1 (vascular cell adhesion molecule-1), IL (interleukins), and chemokines such as MCP-1 (monocyte chemoattractant protein-1) that promote the recruitment of mononuclear cells, their migration in the intima and their differentiation into macrophages.1–4 In the intima, oxLDLs (oxidized LDLs) and other modified LDLs are taken up by macrophages which accumulate cholesterol, thereby forming foam cells and fatty streaks, a feature of early atherosclerotic lesions.1–4 Activated endothelial cells and leukocytes in the intima may oxidize LDLs and produce proinflammatory cytokines, such as IL-1β, IL-6, or TNF-α (tumor necrosis factor-α), that participate in the local inflammatory response.1 These inflammatory factors activate various cellular signaling pathways, among them the sphingolipid pathway that generates bioactive lipid messengers involved in the regulation of cell adhesion, migration, proliferation, survival, and death.6 Sphingolipid mediators such as ceramide and derivatives can be generated by de novo synthesis or by the degradation of sphingomyelin or other complex sphingolipids.6 In cultured vascular cells, oxLDLs and inflammatory cytokines trigger the activation of SMases (sphingomyelinases) and the generation of ceramide and derivatives that mediate biological responses.7–9 Studies based on genetically engineered cells, pharmacological inhibitors, and cell-permeant ceramides suggest that sphingolipid mediators may contribute to atherogenesis.7–9 A recent study on human carotid plaques showed that high sphingolipid levels are associated with plaque inflammation and instability.10
The de novo sphingomyelin synthesis is involved in the regulation of lipoprotein metabolism11 and in atherogenesis, as shown by reduced atherosclerosis in macrophagic sphingomyelin synthase 2-deficient LDLR−/− mice,12 and in Apoe−/− mice treated by myriocin, a serine palmitoyltransferase inhibitor that decreases the biosynthesis of sphingosine, ceramide, and sphingomyelin.13
The role of SMases has been reported in atherogenesis.7,8 The secretory aSMase (acid SMase), which is upregulated by inflammatory cytokines, could be implicated in lipoprotein retention, foam cell formation, and, conversely, its deficiency is associated with reduced atherogenesis in aSMase−/−/Apoe−/− and aSMase−/−/LDLR−/− mice.14,15 This could be in part because of the lowering of cholesterol or the reduction of inflammation. However, unexpectedly, AAV (adeno-associated viral)-mediated expression of secretory aSMase reduces atherosclerosis in Apoe−/− mice.16
A nSMase (neutral sphingomyelinase), more specifically the nSMase2 (type 2-neutral sphingomyelinase), is activated in vascular cells by stress agents such as oxLDLs,17 TNF-α,18,19 IL-1β,20 interferon γ,21 shear stress,22 and oxidative stress,23 and may contribute to endothelium activation,24,25 phagocyte chemotaxis26 and inflammation.27 This inflammatory response may play an atherogenic role but, reversely, the defect of nSMase2 induces an accumulation of cholesterol in fibroblasts, thus may also be proatherogenic.28 As the role of nSMase2 in atherosclerosis is still debated, this study was performed to clarify its involvement in atherogenesis. For this purpose, we used a genetic double mutant mouse model deficient in both nSMase2 activity and Apoe−/− expression. The genetic deficiency of nSMase2 (encoded by the Smpd3, sphingomyelin phosphodiesterase 3 gene) is observed in mice homozygous for the Fro (fragilitas ossium) mutation of the Smpd3 gene (Smpd3fro/fro)29 and in Smpd3-KO mice.30 The Fro mutation of the smpd3 gene induces the loss of the C-terminal domain containing the active site of nSMase2 so that Fro mice bearing the homozygous fro/fro mutation are deficient in nSMase2 activity. Alternatively, we used a pharmacological model of long-term inhibition of nSMase2 by GW4869 in Apoe−/− mice.31
In the 2 experimental models, nSMase2 deficiency or inhibition significantly reduced the size of atherosclerotic lesions and lipid deposition, via, in part, a Nrf2-dependent anti-inflammatory mechanism.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Antibodies and Reagents
A full list of all reagents and antibodies used are listed in the expanded Methods in the online-only Data Supplement.
Animals
Animal protocol was approved by the Committee on Research Animal Care of INSERM UMR1048 Center (protocol APAFIS number 3802-2016012614366248v5). Double mutant Apoe−/− and nSMase2-deficient mice were obtained as described in the expanded Methods in the online-only Data Supplement. Male C57BL/6 WT and C57BL/6 Nrf2−/− transgenic 6 to 12-week-old mice were used as described previously.32,33 Animal experiment was conducted in agreement with the recommendation on design and execution of animal atherosclerosis studies.34
Plaque Formation Analysis
The extent of atherosclerosis development and plaque inflammation were analyzed as described in the expanded Methods in the online-only Data Supplement.
LDL Isolation and Oxidation
LDL from human pooled sera were prepared by ultracentrifugation.17 The extent of LDL oxidation was monitored by measuring the thiobarbituric acid-reactive substance content.17
Genes Expression
Gene expression was analyzed as described in the expanded Methods in the online-only Data Supplement.
nSMase and Lipids Determinations
nSMase activity was determined as reported.17 Additionally, lipid analysis were performed on lipids samples obtained after Bligh and Dyer extraction.35 Expanded Methods in the online-only Data Supplement.
Western Blot Analysis
Protein expressions were quantified by Western blot analysis on cell extracts or after nuclear or cytosol extractions as described in the expanded Methods in the online-only Data Supplement.
Cytokine and Nitrite Quantification
IL-6, IL-10, MCP-1, and TNF-α were quantified in the culture media using a BD cytometric bead array (CBA 552364), and IL-1β was quantified using the Mouse IL-1β Elisa Ready-set-GO (ref eBioscience: 887013–22). Nitrite quantification was performed by the Griess method (Molecular probes D1692).
Monocyte Adhesion and Migration Assays
Migration was studied in Boyden chamber using membrane 8 μm pore size Costar transwells permeable support (Corning, Lowell, MA, ref: 353180) as reported.36 Monocyte adhesion assay was performed according to Srinivasan et al.37 Expanded Methods is reported in the online-only Data Supplement.
Statistical Analysis
All data passed a normality test followed by a normal Gaussian test repartition to verify the equal variance or not (Minitab software), that orientate the statistical analysis test. Statistical analysis were done using Graph Pad Prism 5 for Windows (Graph Pad Software). Parametric tests consisted in unpaired student t test (Dunnett correction was applied when >2 experimental conditions were analyzed) and nonparametric tests in Mann–Whitney (2 experimental conditions) or Kruskal–Wallis (combined with Dunn correction if >2 experimental conditions were evaluated). Results are expressed as mean±SEM when experiments were performed on cell extracts and median±interquartile intervals when experiments were performed on an animal. Results were considered significant at P<0.05.
Results
nSMase2 Deficiency or Pharmacological Inhibition Significantly Reduce the Size of Atherosclerotic Lesions in Apoe−/− Mice
The role of nSMase2 in atherogenesis was investigated using either double mutant mice, nSMase2-deficient (smpd3fro/fro), and apoE-KO (Apoe−/−) and long-term pharmacological inhibition of nSMase2 by GW4869 in Apoe−/−/Smpd3+/+ mice.
The morphometric analysis on cryosections of aortic sinus stained by oil red O showed that the mean area of atherosclerotic lesions in nSMase2-deficient Apoe−/−/Smpd3fro/fro mice was reduced by 69% compared with Apoe−/−/Smpd3+/+ mice (Figure 1A). The en face analysis of aortas stained with oil red O showed that lipid accumulation in Apoe−/−/Smpd3fro/fro mice was decreased by 68% compared with Apoe−/−/Smpd3+/+ littermates (Figure 1B).
Figure 1.
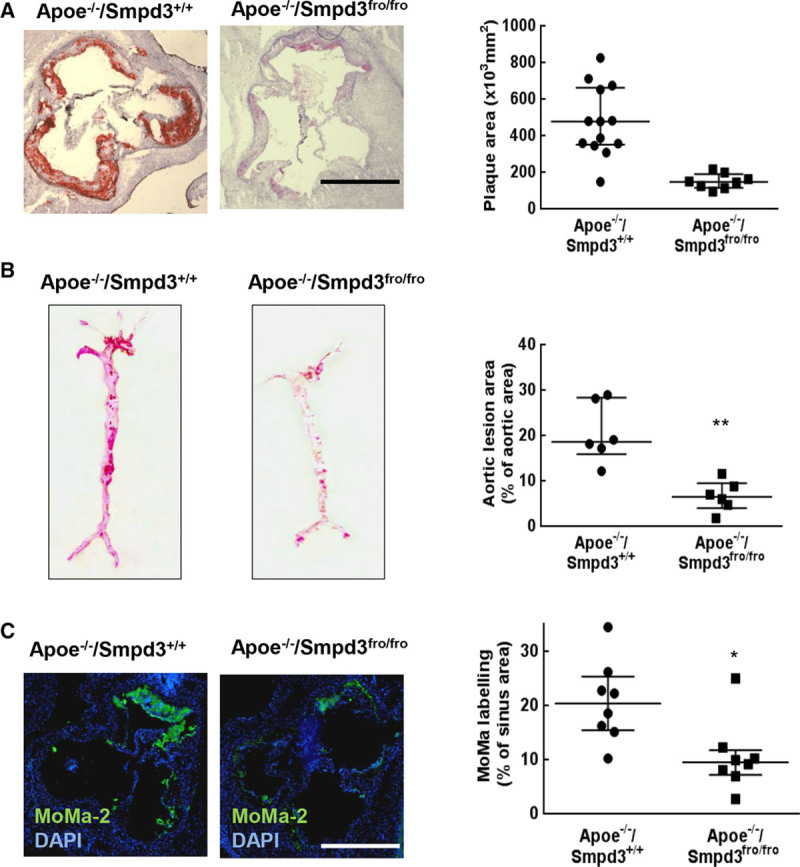
The genetic deficiency of nSMase2 (type 2-neutral sphingomyelinase) reduces the formation of atherosclerotic lesions in Apoe-deficient mice. A, Representative pictures of aortic sinus sections stained with Oil Red O and hematoxylin (left) and quantification of the atherosclerotic lesion area (right) of Apoe−/−/Smpd3fro/fro (n=8) and Apoe−/−/Smpd3+/+ (n=13) mice. In B, en face pictures of Oil Red O-stained aortas from Apoe−/−/Smpd3fro/fro (n=6) and Apoe−/−/Smpd3+/+ (n=6) mice, and evaluation of lipid-rich (red stained) atherosclerotic lesions (expressed as percent of total aortic area). In C, representative aortic root sections and quantification of MoMa-2 expressing cells in 28-wk-old mice genetically invalidated (Apoe−/−/Smpd3fro/fro, n=7) or not (Apoe−/−/Smpd3+/+, n=8), stained with the anti MoMa-2 antibody and counterstained with DAPI. The MoMa-2-stained areas were quantified using image J. A and C, White scale bar: 500 µm. The data were expressed as the median with interquartile range. All the statistical analysis of the figure were done using Mann–Whitney test. *P<0.05, **P<0.01, ***P<0.001.
To investigate whether a pharmacological approach allows similar protection, Apoe−/−/Smpd3+/+ mice were treated with GW4869, a specific inhibitor of nSMase,31,38 for 15 weeks. As shown in Figure IIA in the online-only Data Supplement, atherosclerotic lesion area in the aortic sinus of GW4869-treated mice was reduced by 49% compared with littermates treated only with the vehicle (DMSO).
Immunofluorescence using the MoMa-2 (anti-monocyte/macrophages2) antibody showed that the number of monocytes/macrophages in atherosclerotic lesions of Apoe−/−/Smpd3fro/fro mice was reduced by 49% compared with Apoe−/−/Smpd3+/+ littermates (Figure 1C) and was decreased by 68% in Apoe−/−/Smpd3+/+ mice treated with GW4869 (Figure IIB in the online-only Data Supplement).
These data indicate that nSMase2 deficiency or inhibition results in a decreased macrophage accumulation in the lesions. No major differences were observed concerning the collagen and SMC content, in GW4869-treated and untreated Apoe−/−/Smpd3+/+ mice, as well as Apoe−/−/Smpd3fro/fro animals (data not shown).
Altogether, these data show that the genetic deficiency of nSMase2 or its pharmacological inhibition by GW4869 significantly reduced the size of atherosclerotic areas and the accumulation of macrophages in the lesions of Apoe−/− mice.
To investigate the mechanism of this atheroprotective effect, we evaluated the effect of nSMase2 deficiency on several metabolic and inflammatory atherogenic factors in the Apoe−/− murine model.
nSMase2 Deficiency or Inhibition Alter Sphingolipid Circulating Levels but Does Not Affect Hypercholesterolemia in Apoe−/− Mice
The effect of nSMase2 deficiency on plasma cholesterol, ceramide, sphingomyelin, and phospholipids was evaluated in Apoe−/−/Smpd3fro/fro mice (compared with Apoe−/−/Smpd3+/+ littermates) and in Apoe−/−/Smpd3+/+ mice treated (or not) with GW4869 (Table; Figure III in the online-only Data Supplement).
Table.
Plasma Total Cholesterol, LDL, and HDL Levels in Mice Models
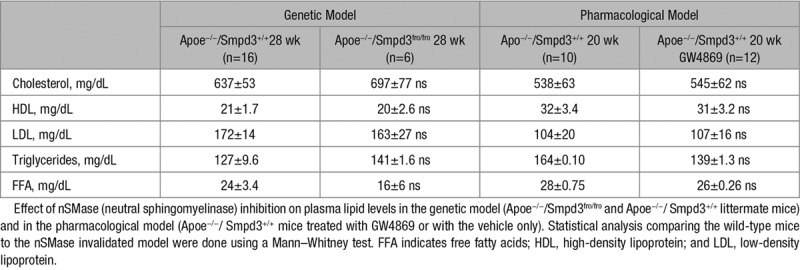
As expected, the deficiency or inhibition of nSMase2 activity resulted in a significant decrease in plasma ceramide levels, particularly in 24:1, 22:0, and 24:0 ceramide levels, as previously reported by Qin et al.28 Plasmatic SM and phospholipid levels were not affected by nSMase2 deficiency or inhibition (Figure III in the online-only Data Supplement).
Interestingly, nSMase2 deficiency or pharmacological inhibition by GW4869 induced no significant variation of plasma cholesterol, HDL (high-density lipoprotein), LDL, triglycerides nor free fatty acids (Table). These data indicate that the atheroprotective mechanism of nSMase2 mutation/inhibition does not result from plasma cholesterol changes.
Note that no weight variations were observed after GW4869 treatment (Figure IV in the online-only Data Supplement).
Inhibition of nSMase2 Reduces Endothelial Cell Activation and Monocyte Adhesion
In agreement with previous reports,39 oxLDLs induced an increase of ceramide in CRL2181 which resulted from nSMase activation, as it was inhibited by GW4869 (Figure V in the online-only Data Supplement).
As the adhesion of mononuclear cells to the activated endothelium plays a major role in early atherogenesis,1–4 and as nSMase2 was implicated in VCAM-1 expression,40 we investigated whether nSMase2 inhibition altered the expression of VCAM-1 in CRL2181 and the adhesion of murine macrophages. OxLDLs stimulated nSMase activity in CRL2181 and this is inhibited by nSMase2 siRNA treatment (Figure VIA in the online-only Data Supplement). OxLDLs also induced ICAM-1 and VCAM-1 mRNAs and the adhesion of fluorescent calcein-stained RAW264.7 to CRL2181 (Figure 2A through 2C). All these events were inhibited when endothelial cells were treated with the nSMase inhibitor GW4869 or were silenced for nSMase2 using a smpd3 si-RNA (Figure 2).
Figure 2.
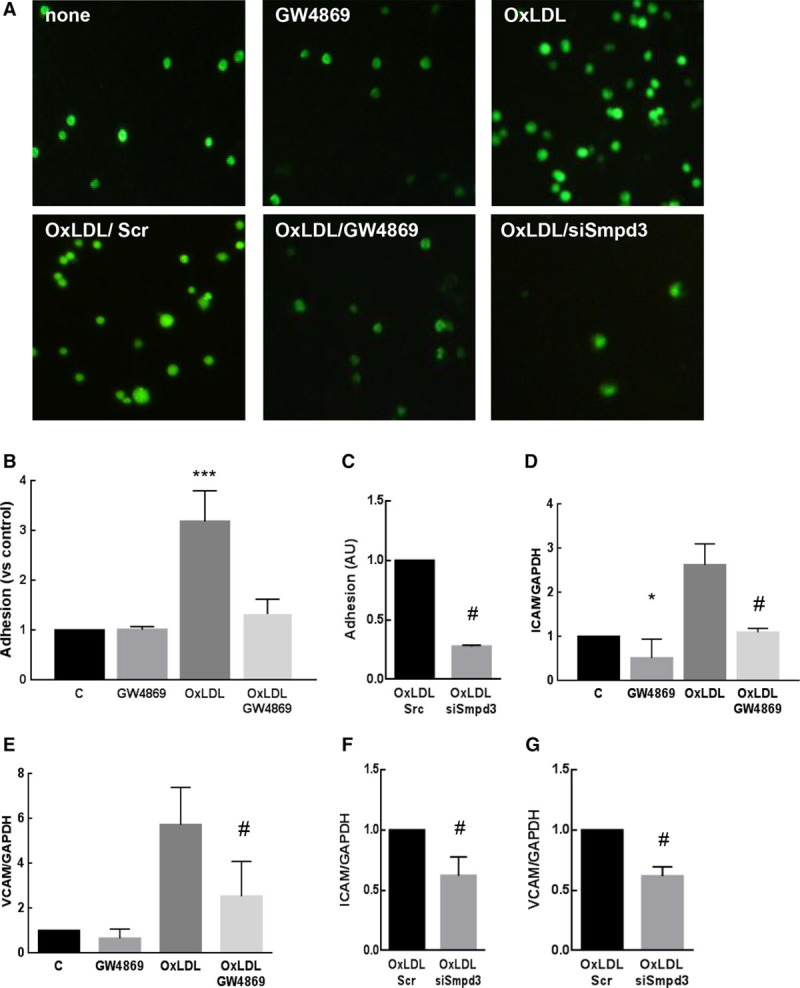
Effect of nSMase (neutral sphingomyelinase) inhibition on CRL2181 activation. A–C, Monocyte adhesion assay to activated endothelial cells. CRL2181 were grown on 6-multiwell culture plates and stimulated for 4 h by oxLDLs (oxidized low-density lipoproteins; 100 μg/mL) after 30 min preincubation with GW4869 (10 μmol\L, A, B). Alternatively, cells were treated with scramble (Scr) or nSMase2 siRNAs (siSmpd3, A, C). RAW264.7 macrophages stained by calcein were added to activated CRL2181 (50 000 cells/well) for 30 min. Adhesion was quantified by measuring the fluorescence of cell lysates (B, C). The results are normalized to the nonstimulated control (A) or oxLDL treated by Scr siRNA (C). D, G, Quantification of VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1) mRNAs by RT-qPCR in comparison with GAPDH. The data are expressed as mean±SEM of at least 3 independent experiments. Statistical analysis was performed using t test with Dunnett correction when >2 groups were compared (* means compared with untreated control and # means compared with oxLDL treated cells). *,#P<0.05, **,##P<0.01, ***P<0.001.
In Boyden Chamber assays, the migration of RAW264.7 monocytes was stimulated by the culture medium of CRL2181 treated by oxLDLs, and this was inhibited by GW4869 (Figure VI in the online-only Data Supplement). Moreover, GW4869 and smpd3 si-RNA inhibited the expression of MCP-1 stimulated by oxLDLs in CRL2181 (Figure VID in the online-only Data Supplement).
Altogether these results suggest that nSMase2 inhibition reduces endothelium activation, and the subsequent monocyte recruitment, adhesion, and migration.
Activation of Nrf2 in Response to nSMase2 Inhibition in Endothelial Cells
As the redox-sensitive Nrf2 transcription factor could be downregulated by ceramide41 which may also stimulate the expression of VCAM-1 and adhesion of monocytes to activated endothelium,42 we checked whether Nrf2 could play a role in the anti-inflammatory response resulting from nSMase2 inhibition.
The treatment of CRL2181 by GW4869 stimulated per se the nuclear translocation of Nrf2 as assessed by the decreased expression of Nrf2 and Keap1 (Kelch-like ECH-associated protein 1) in the cytosol (Figure VIIA through VIIC in the online-only Data Supplement) and the increase of nuclear Nrf2 expression (Figure 3A).
Figure 3.
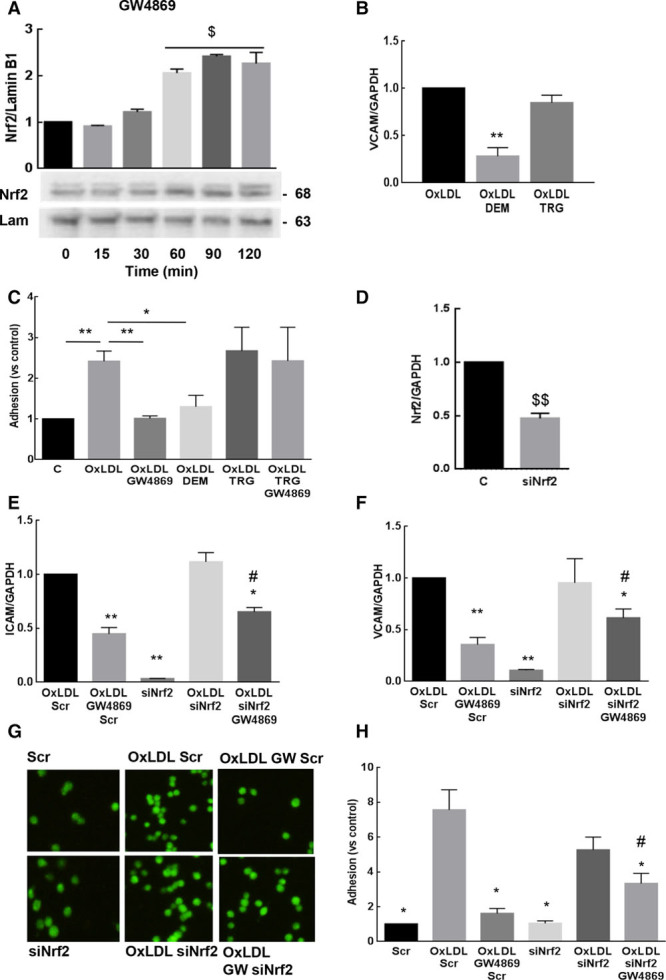
Implication of Nrf2 (nuclear factor [erythroid-derived 2]-like 2 or NF-E2–related factor-2) in the inhibitory effect of GW4869 on CRL2181 activation. A, Time-course of Nrf2 nuclear translocation evoked by GW4869 (10 µmol\L) in CRL2181. B, VCAM-1 (vascular cell adhesion molecule-1) mRNAs expression in CRL2181 stimulated by oxLDLs (oxidized low-density lipoproteins; 100 μg/mL) for 4 h±DEM (100 µmol\L) or trigonelline (TRG; 10 µmol\L), quantified by RT-qPCR in comparison with GAPDH. C, Monocyte adhesion to endothelial cells activated as described in B. D, Expression of Nrf2 mRNA in CRL2181 incubated for 48 h with Nrf2 siRNA. E, F, Effect of Nrf2 siRNA on the expression of ICAM-1 (intercellular adhesion molecule-1; E) and VCAM-1 (F) mRNAs induced by oxLDL. G, H, Effect of Nrf2 siRNAs on the adhesion of calcein-stained RAW 264.7 macrophages on CRL2181 activated for 4 h by oxLDL (100 μg/mL). The data (3 separate experiments) were expressed as mean±SEM. In A, statistical analysis was performed using ANOVA test (Dunnett correction) and (B–H) statistical analysis was performed using t test and Dunnett correction when >2 conditions were tested. *,$ or #P<0.05; **P<0.01. $: compared with untreated cells, *: compared with oxLDLs or oxLDLs+scr-treated cells, #: compared with oxLDL+GW4869-treated cells.
We then checked the effect of Nrf2 translocation on endothelium inflammation. We first used a pharmacological approach as reported by Kobayashi et al43: DEM (diethylmaleate), a potent activator of Nrf2, inhibited the expression of VCAM-1 and the adhesion of RAW264.7 cells to CRL2181 (Figure 3B and 3C), thus indicating that Nrf2 induction mimicked the protective effect of GW4869. In contrast, TRG (trigonelline), a pharmacological inhibitor of Nrf2 activation, prevented the inhibitory effect of GW4869 on monocyte adhesion to CRL2181 (Figure 3B and 3C). Note that DEM and trigonelline did not block nSMase activation induced by oxLDLs (Figure VIII in the online-only Data Supplement). Alternatively, Nrf2 silencing by siRNAs, under conditions leading to 50% decrease of Nrf2 expression in CRL2181, reversed by 50% the protective effect of GW4869 on ICAM-1 and VCAM-1 mRNA expression, and on the adhesion of RAW264.7 (Figure 3D through 3H).
Altogether these results suggest that nSMase inhibition prevents the endothelial inflammation through a Nrf2-dependent mechanism.
nSMase2 Inhibition Reduces the Inflammatory Response of Macrophagic Cells
In atherosclerotic lesions, macrophages release pro- and anti-inflammatory cytokines, depending on their polarization, M1 being proinflammatory, and M2 considered as repair and anti-inflammatory.44 Previous reports suggested that the inflammatory response evoked by lipopolysaccharides (LPS) could involve nSMase2.45–47 We investigated whether nSMase2 inhibition may directly affect the inflammatory phenotype of macrophages, that is, the production of cytokines, using either LPS-stimulated peritoneal macrophages isolated from Apoe−/−/Smpd3fro/fro and Apoe−/−/Smpd3+/+ littermates, or LPS-activated RAW264.7 cells treated or not by GW4869.
LPS treatment stimulated ceramide generation in RAW264.7 (Figure VC and VD in the online-only Data Supplement). The inflammatory response to LPS was characterized by the increase of IL-1β, IL-6, TNF-α, MCP-1 (Figure IXA through IXG in the online-only Data Supplement), by a high level of nitrite production and an increased iNOS expression leading to a decreased arginase/iNOS ratio (Figure XA through XC in the online-only Data Supplement), and by the phosphorylation of Stat1 and Stat3 transcription factors (Figure XD through XF in the online-only Data Supplement). All these responses were inhibited by GW4869. Similar results were observed in nSMase2-deficient macrophages isolated from Apoe−/−/Smpd3fro/fro mice (Figure 4) and in mouse peritoneal macrophages treated by LPS and by GW4869 (Figure XI in the online-only Data Supplement). In contrast, the expression of anti-inflammatory cytokines IL-4 and IL-10 was modestly increased when nSMase activity was inhibited (Figure 4; Figure IXH and IXI in the online-only Data Supplement). These data were confirmed by inhibition of the morphological changes associated with LPS-induced M1 polarization. Incubation of RAW264.7 cells (small and rounded shaped) with LPS (18 hours) induced an M1 morphological phenotype, characterized by irregular and rough form with accelerated cell spreading and forming large pseudopodia, and highly vacuolated cytoplasm. All these morphological changes were inhibited by GW4869 treatment (Figure IXJ in the online-only Data Supplement).
Figure 4.
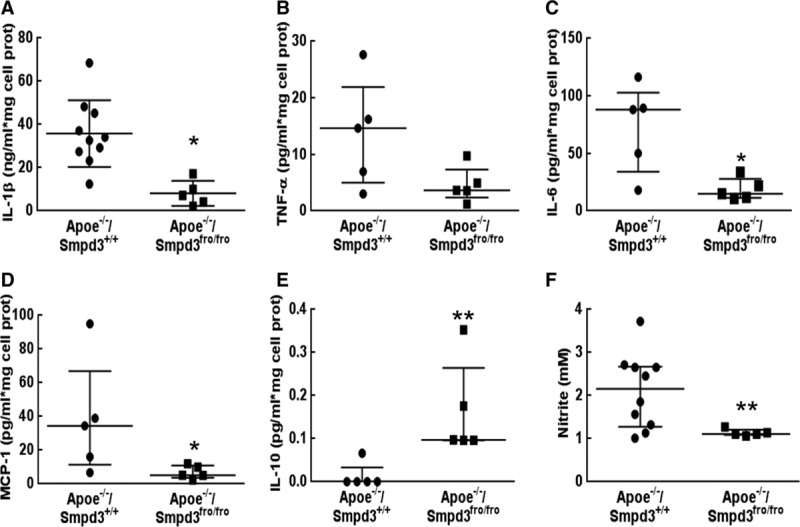
Cytokine production by mouse peritoneal macrophages isolated from (Apoe−/−/Smpd3+/+) or (Apoe−/−/Smpd3fro/fro) mice. Macrophages extracted from the different animals (figured by 1 symbol) were stimulated with lipopolysaccharides (LPS; 20 ng/mL) during 18 h. The cytokine levels were quantified in culture supernatants by Elisa (A) or BD Cytometric Bead Array (B–E) test. F, LPS-induced production of nitrites was quantified in the incubation medium by Griess assay. The data were expressed as median with interquartile range. For all the figures, statistical analysis was performed using Mann–Whitney test. *P<0.05, **P<0.01. IL indicates interleukin; and TNF, tumor necrosis factor.
We then checked whether inflammation was decreased in vivo in response to nSMase2 inhibition, either in nSMase2-deficient Apoe−/−/Smpd3fro/fro mice or after treatment by GW4869. Circulating IL-1β was significantly reduced (47% decrease; Figure XIIA in the online-only Data Supplement) in the plasma of Apoe−/−/Smpd3+/+ mice treated by GW4869 in comparison with vehicle-treated mice and in the plasma of Apoe−/−/Smpd3fro/fro mice (57% decrease; Figure XIIB in the online-only Data Supplement). Likewise, the expression of VCAM-1, IL-1β, IL-6, and TNF-α mRNAs was reduced in aortas of mice injected with GW4869 (Figure XIII in the online-only Data Supplement), together with a decreased ceramide content (around 30%; Figure XIV in the online-only Data Supplement). Finally, the number of MoMa-2/IL-1β positive cells was decreased in the aortic sinus of mice treated by GW4869 (Figure 5A and 5B) and in Apoe−/−/Smpd3fro/fro mice (Figure 5C and 5D). Altogether, these data indicated that nSMase2 inhibition decreases vascular inflammation by reducing the recruitment of monocytes to endothelium and macrophage M1 differentiation.
Figure 5.
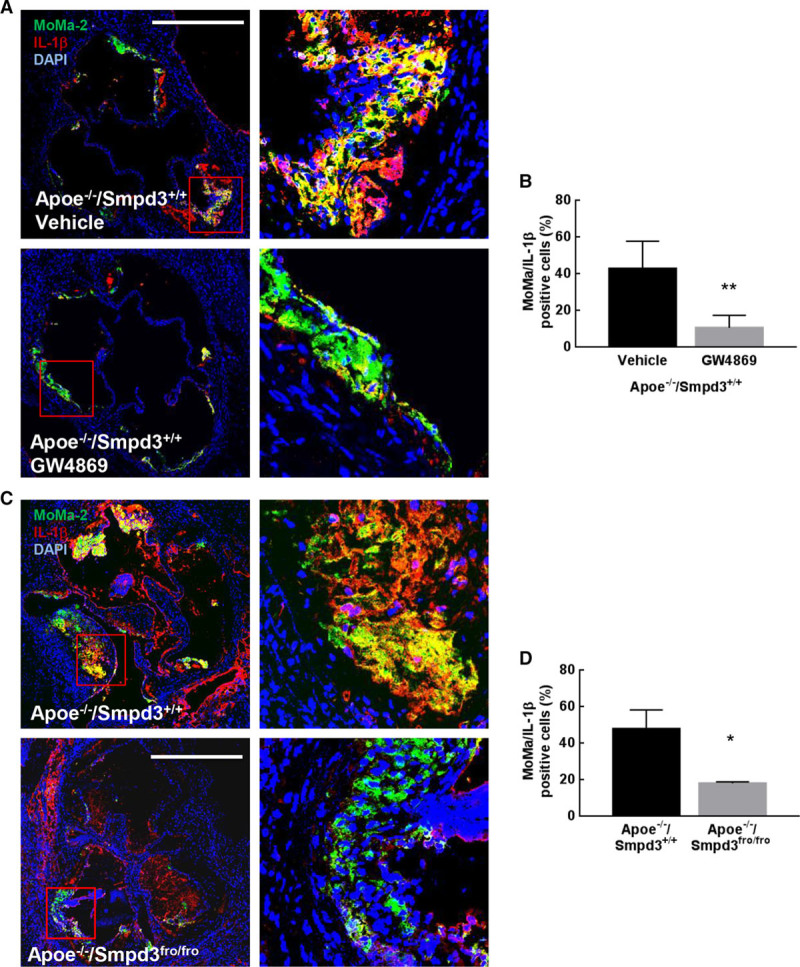
Effect of nSMase2 (type 2-neutral sphingomyelinase) inhibition on inflammation in vivo. A, Representative pictures of MoMa-2 and IL-1β immunostaining in atherosclerotic lesions from Apoe−/−/Smpd3+/+ and Apoe−/−/Smpd3fro/fro. B, Quantification of IL-1β mean fluorescent intensity (MFI) in MoMa-2+ area (scale bar, 500 µm). C, Representative pictures of MoMa-2 and IL-1β immunostaining in atherosclerotic lesions from Apoe−/−/Smpd3+/+ mice treated or not with GW4869. D, Quantification of IL-1β MFI in MoMa-2 positive area (scale bar, 500 µm). n=6 for control groups and n=5 for GW4869 treated group for the GW4869 study and n=5 for each group for the Apoe−/−/Smpd3+/+ vs Apoe−/−/Smpd3fro/fro study. Quantifications are expressed as median±interquartile range. Statistical analysis was performed using Mann–Whitney test. *P<0.05, **P<0.01.
Nrf2 Participates to the Reduced Inflammatory Response of Macrophages After nSMase2 Inhibition
As nSMase2 inhibition significantly reduced endothelial cell activation via a Nrf2-dependent mechanism, we hypothesized that Nrf2 could be also involved in the reduced inflammatory response of macrophages. In vitro experiments performed on RAW264.7 showed that cells treated by GW4869 alone exhibited a rapid nuclear translocation of Nrf2 (Figure 6A and 6B; Figure VIID through VIIF in the online-only Data Supplement). Confocal microscopy experiments showed that Nrf2 accumulated in the nucleus after 2 hours of GW4869 treatment (Figure 6A). In vivo, the total Nrf2 expression was modestly increased in the aorta of animals injected with GW4869 (Figure XVA in the online-only Data Supplement).
Figure 6.
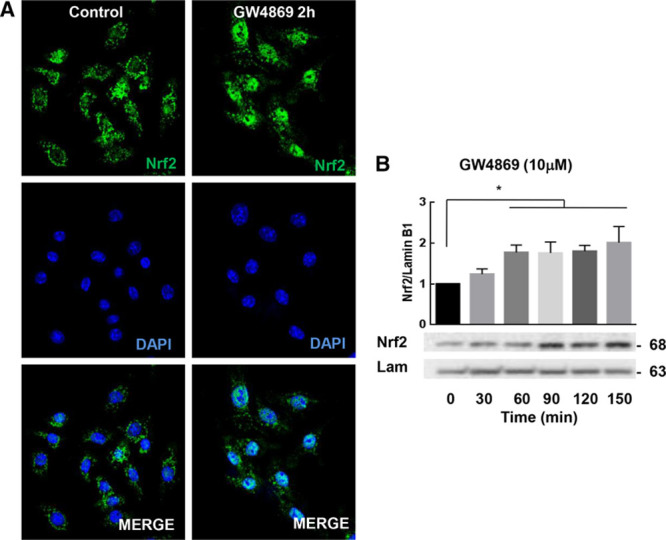
nSMase (neutral sphingomyelinase) inhibition induces Nrf2 (nuclear factor [erythroid-derived 2]-like 2 or NF-E2–related factor-2) nuclear translocation. A, Confocal microscopy of RAW264.7 stimulated or not with GW4869 (2 h). B, Cells were stimulated by GW4869 at various times and Nrf2 nuclear translocation was quantified by Western blot as described in the Materials and Methods section. The data are expressed as mean±SEM. Statistical analysis was performed using ANOVA test followed by Dunnett post hoc test. Representative of at least 3 independent experiments. *P<0.05.
The anti-inflammatory mechanism, because of Nrf2 activation was then investigated. The nuclear translocation of Nrf2 triggers the expression of cytoprotective genes and their protein products, among them HO-1 (heme oxygenase 1) which exerts strong antioxidant and anti-inflammatory functions.48 To confirm the activation of Nrf2 in our model, we checked whether nSMase2 inhibition stimulated the expression of HO-1 in vitro and in vivo. As shown in Figure 7A, GW4869 significantly increased the expression of HO-1 in RAW264.7, in a time-dependent manner. Moreover, the decrease in HO-1 expression resulting from long-term LPS treatment (24 hours) was reversed by GW4869 (Figure 7B). In vivo, the expression of HO-1 was highly increased in aortic sinus of Apoe−/−/Smpd3fro/fro mice (Figure XVB in the online-only Data Supplement). All these data suggest that the Nrf2/HO-1 axis is activated and may be involved in the anti-inflammatory response after pharmacological or genetic nSMase2 inhibition. A recent report from Kobayashi et al43 indicated that Nrf2 could directly act as an anti-inflammatory factor leading to the early inhibition of IL-1β and IL-6 expression. To test this hypothesis, cells were incubated from 0 to 6 hours with LPS and GW4869: at this incubation time, LPS induced an early expression and secretion of IL-1β which was in part reversed by GW4869 (Figure 7C and 7D). These events (protective effect of GW4869 on IL-1β expression and secretion) are probably not due to the increased expression of HO1, as they occurred much earlier during the stimulation of macrophages by LPS. Altogether these data indicated that Nrf2 exerts an anti-inflammatory effect only partly dependent on HO-1 expression.
Figure 7.
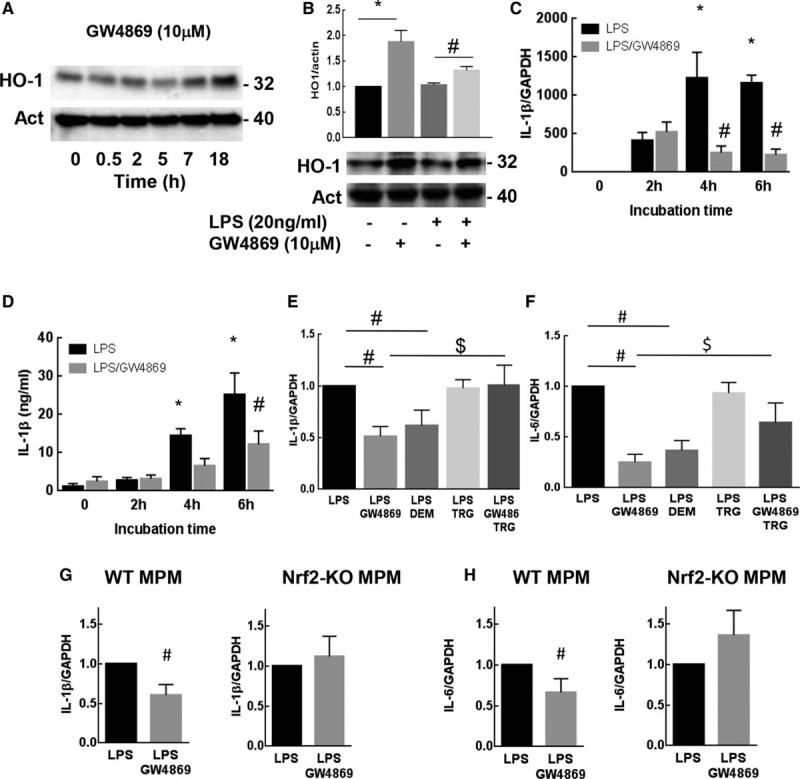
Nrf2 (nuclear factor [erythroid-derived 2]-like 2 or NF-E2–related factor-2) activation protects against inflammation. A, Time-course of HO-1 (heme oxygenase 1) expression in RAW264.7 stimulated by GW4869 (10 µmol\L) and analyzed by Western blot. B, Expression of HO-1 in RAW264.7 stimulated by lipopolysaccharides (LPS; 18 h, 20 ng/mL) after preincubation with GW4869 (10 µmol/L). IL (interleukin)-1b mRNAs expression (C) and protein secretion (D) were analyzed from RAW264.7 stimulated by LPS (20 ng/mL)±GW4869 (0 to 6 h). IL-1β (E) and IL-6 (F) mRNA expression in RAW264.7±LPS (18 h, 20 ng/mL), ±GW4869 (10 µmol\L), DEM (100 µmol\L), or trigonelline (TRG; 10 µmol\L). G, H, IL-1β (G) or IL-6 mRNA expression was analyzed from mouse peritoneal macrophages extracted from wild-type or Nrf2-KO mice stimulated by LPS (20 ng/mL)±GW4869 (16 h). The data are expressed as mean±SEM. Statistical analysis was performed using ANOVA test (B) or t test (C–H) followed by a Dunnett post hoc test. Representative of at least 3 independent experiments., *P<0.05 vs 0, #LPS or $LPS+GW4869 experimental conditions.
To confirm the role of Nrf2 in the decreased inflammation resulting from nSMase inhibition, 2 experimental approaches were tested: First, LPS-induced inflammation in macrophages was monitored by measuring IL-1β and IL-6 mRNA expression, using the pharmacological inhibitor/activator strategy. We observed that DEM, the Nrf2 activator, mimicked the protective effect of GW4869 on IL-1β and IL-6 mRNA expression in agreement with previous reports,43 whereas the Nrf2 inhibitor trigonelline, reduced its protective effect (Figure 7E and 7F). Second, the effect of GW4869 was evaluated on peritoneal macrophages from Nrf2-KO mice stimulated by LPS. As shown in Figure 7G and 7H, GW4869 was unable to prevent the expression of IL-1β and IL-6 mRNAs evoked by LPS in Nrf2-KO macrophages. Altogether these results demonstrate that Nrf2 is involved in the anti-inflammatory response evoked by nSMase2 inhibition in macrophages.
Discussion
Clinical evidence and studies on animal models show that inflammation is present at each step of atherosclerosis development.1,3–5 The mechanisms leading to the inflammatory response in the vascular wall are only partly identified. This study reports that inflammation and atherosclerosis lesions are strongly decreased in response to nSMase2 inhibition, elicited either pharmacologically when using the nSMase inhibitor GW4869,31 or in genetically nSMase2-deficient Apoe−/−/Smpd3fro/fro mice. GW4869 is the most widely used nSMase2 inhibitor. GW4869 has been used in many studies to demonstrate the implication of nSMase2 in response to TNF-α,49 HLA,50 oxLDLs,17 in the secretion of hyaluronic acid,28 or miRNAs.51 Recently, Airola et al52 confirmed that GW4869 acts as a phosphatidylserine-competitive inhibitor of nSMase2, with an IC50 around 1 μmol\L.
An important point is that the pharmacological or genetic inhibition of nSMase2 was not associated with plasma lipoprotein changes, SM accumulation nor cholesterol level modification in our mice models, in agreement with Stoffel group who reported that nSMase2 invalidation did not generate any accumulation of SM, cholesterol, phosphatidylethanolamine, and phosphatidylcholine.30 These data suggest that the antiatherogenic effect resulting from nSMase2 inhibition is attributable to a direct effect on the arterial wall.
The lower inflammation patterns observed in the vascular wall of Apoe−/−/Smpd3fro/fro mice and confirmed by the treatment of Apoe−/−/Smpd3+/+ mice with GW4869, emphasize the role of nSMase2 and ceramide in inflammation and atherosclerosis development. nSMase2 hydrolyses an SM pool located in the inner leaflet of the plasma membrane, which generates ceramide40 in all cell types, including vascular cells and macrophages.7 Ceramide is thought to play a proinflammatory role within the plaque, as supported by studies showing its colocalization with CD68, MCP-1, or IL-6 in human atherosclerotic lesions.10 In RAW264.7, ceramide may induce IFN-γ and iNOS expression, and Stat1 phosphorylation via the activation of PP1 (protein phosphatase 1) and PP2A (protein phosphatase 2A)53 which are necessary for IFN-γ-synergized with LPS to induce iNOS/NO biosynthesis.54 Likewise, our data indicate that iNOS mRNA expression and Stat1 phosphorylation elicited by LPS are decreased by GW4869. Several proatherogenic/inflammatory agents, including TNF-α, oxLDLs, IL-1β, or endothelin-1, may activate nSMase2 and ceramide production.17–21,23–27 For instance, in hepatocytes, IL-1β activates nSMase2 via a signaling pathway implicating IL1-R1 (IL1-receptor1), JNK (c-Jun N-terminal kinase), IRAK-1 (interleukin-1 receptor-activated protein kinase), and PP2A.55 In neuronal cells, IL-1β activates nSMase2 which leads to IL-6 secretion via src.56,57 In HL-60, INFγ, and TNF-α activate nSMase, which stimulates their differentiation into monocytes.21 Conversely, little is known concerning the implication of nSMase2 in the expression of cytokines and inflammatory factors. Here, we show that nSMase2 inhibition decreased the expression of inflammatory cytokines evoked by LPS in macrophages, and the activation of endothelial cell induced by oxLDLs, together with a decrease of ceramide level in the blood, in macrophages, endothelial cells, and in vascular tissues of animal models. These data suggest that nSMase2 and ceramide may directly modulate inflammation, possibly via an autoamplification loop in which they stimulate the expression of inflammatory cytokines, which in turn activate the sphingolipid pathway to maintain inflammation. Conversely, the inhibition of nSMase2 may inhibit inflammation at 2 levels, via a short-time mechanism implicating Nrf2, and long-term action, by decreasing the production of proinflammatory ceramide.
Our data indicate that nSMase2 inhibition leads to Nrf2 activation, which suggests that nSMase2 negatively regulates Nrf2 signaling. The anti-inflammatory function of Nrf2 has been largely reported, in macrophages43,58 or in endothelial cells in which the Nrf2/HO-1 axis could inhibit NF-κB activation and the expression of inflammatory genes (VCAM-1, ICAM-1, and MCP-1).59 Under unstressed conditions, Nrf2 is maintained inactive in the cytosol by its repressor, Keap1. Once activated in response to injury and inflammation stimuli, Nrf2 translocates into the nucleus, binds to the antioxidant response element-controlled genes, thereby generating antioxidant and cytoprotective signaling pathways. Our data show that GW4869 stimulates a sustained nuclear translocation and activation of Nrf2 in both endothelial cells and macrophages. OxLDLs and LPS are known Nrf2 inducers, so one hypothesis is that Nrf2 activation by these agents, could be increased or more sustained when ceramide generation is lower or inhibited. This is in agreement with Park et al,41 who reported that the nuclear translocation and activation of Nrf2, and the expression of its target genes such as glutathione S-transferases, are inhibited by ceramide. A balance between ceramide and Nrf2 is also observed during reperfusion, after coronary artery occlusion and myocardial ischemia, as shown by Reforgiato’s60 group who reported that injury (associated to an increase of inflammation) was reduced by myriocin treatment. These authors established a link between the ceramide decrease and the expression of the Nrf2 target HO-1.60 Interestingly, our different approaches such as mouse peritoneal macrophages extracted from Nrf2-KO mice, RAW264.7 treated by the Nrf2 inhibitor trigonelline, or the silencing of Nrf2 by siRNAs in endothelial cells, reversed the anti-inflammatory effect of GW4869, including monocyte adhesion on endothelium, or macrophage inflammation. Moreover, we show that HO-1 expression is increased, suggesting that it may contribute to the anti-inflammatory and antiatherogenic effect observed in response to nSMase inhibition. This is in agreement with reports showing that HO-1 protects against atherosclerosis development, for instance in LDLR−/− mice deficient for HO-1 in bone marrow cells, which develop more atherosclerotic lesions with increased signs of inflammation. Likewise, HO-1–deficient peritoneal macrophages exhibit an increased oxidative burst and inflammatory cytokine production (MCP-1 or IL-6) in response to oxLDLs.61 However, in our model, it seems that IL-1β and IL-6 production evoked by LPS were early inhibited by GW4869, before any induction of HO-1 expression (Figure 7). This suggests that Nrf2 activated in response to GW4869, could also directly block the expression of IL-1β and IL-6, as recently reported by Kobayashi et al.43
The mechanisms by which ceramide negatively regulates, and conversely, nSMase2 inhibition activates Nrf2, are not yet elucidated. One hypothesis is that ceramide and nSMase2 inhibit the PI3K/Akt pathway, as suggested by Park et al,41 possibly via an activation of the phosphatase PP2A by ceramide.62,63 In contrast, the lack of ceramide could inhibit PP2A, this resulting in a persistent Akt phosphorylation,28,64 that could promote Nrf2 stabilization and nuclear translocation.65,66 Among the other hypothesis, Nrf2 activation may also result from modifications of the intracellular redox status or of the mitochondrial function in response to nSMase2 inhibition. Further experiments will be necessary to clarify the mechanisms leading to Nrf2 regulation and the place of nSMase2 in this process.
The fact that Nrf2 is implicated in the anti-inflammatory response because of nSMase2 inhibition raises the question of its role in atherosclerosis, which is still debated and controversial. Indeed, if it is generally admitted that the Nrf2 pathway is rather antiatherogenic, via its antioxidant and cytoprotective properties,67 several reports indicate that Nrf2 may be proatherogenic, by promoting the expression of CD36,68 or by increasing the plasma and hepatic cholesterol content,69 or as a positive regulator of the NLRP3 (nucleotide-binding oligomerization domain-like receptors PYD) inflammasome system.67 As reviewed by Jakobs et al70 and Mimura and Itoh,58 the role of Nrf2 could depend on the genetic animal model, the environment, or the stage of atherosclerosis development. In the study reported by Harada et al71 in ApoE−/−/Nrf2−/− double KO mice, the lack of Nrf2 was protective against atherosclerosis development, in spite of an increased inflammatory phenotype observed in the early weeks. A proinflammatory role for Nrf2 was also described by Freigang et al,72 who reported its implication in the activation of the inflammasome NLRP3 by cholesterol crystals, a hallmark of chronic inflammation in advanced atherosclerotic lesions.73 In contrast, in LDLR−/− mice, Nrf2 deficiency aggravates the development of atherosclerosis in both early and late stages.74 Likewise, Ruotsalainen et al75 reported that bone marrow transplantation of Nrf2−/− to LDLR−/− mice aggravates foam cell formation and inflammation. These discrepancies could be explained by genetic differences between the 2 animal models, as Apoe−/− mice may have a more severe atherogenic phenotype than LDLR−/− mice, which is accelerated and aggravated by high fat and cholesterol-enriched diet,76 prone to activate the NLRP3 inflammasome pathway.72 In these conditions, (Apoe−/− mice fed with high-fat diet), one hypothesis is that the anti-inflammatory and antioxidant properties of the Nrf2/HO-1 pathway could be overwhelmed by the lipid charge, whereas Nrf2 may exert proatherogenic functions such as CD36 overexpression or NLRP3 activation.72 Our data support a role for Nrf2 in the anti-inflammatory response resulting from the pharmacological or genetic inhibition of nSMase2 in Apoe−/− mice, which is also associated with a reduction of atherosclerotic lesions, implicating or not Nrf2. Indeed, it is likely that nSMase inhibition may generate other antiatherogenic responses, such as a decreased LDL infiltration in the intima, or an inhibition of the exosomal pathway involved in the secretion of proatherogenic miRNAs.77 More studies will be necessary for understanding the proinflammatory function of nSMase2 in the vessels and its links with atherosclerosis.
In summary, the present study demonstrated that nSMase2 and ceramide play a pivotal role in atherosclerosis in regards to the inflammatory process associated with the development of the lesion. Therefore, targeting nSMase2 may be a new therapeutic strategy to prevent the inflammatory processes involved in atherogenesis.
Acknowledgments
We wish to thank Corinne Bernis, Christophe Santiago, and Plateforme Imagerie, Plateforme Génomique for excellent technical assistance. Moreover, we wish to thank for sphingolipidomic analysis the MetaToul-Lipidomic Core Facility (I2MC, Inserm 1048, Toulouse, France), MetaboHUB-ANR-11-INBS-0010.
Sources of Funding
We acknowledge INSERM, Université Toulouse-3, Agence de Biomédecine, Fondation de France and IDEX-Palma 2016 for logistic and financial support.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- aSMase
- acid SMase
- HDL
- high-density lipoprotein
- HO-1
- heme oxygenase 1
- ICAM
- intercellular adhesion molecule
- IL
- interleukin
- Nrf2
- nuclear factor (erythroid-derived 2)-like 2 or NF-E2–related factor-2
- nSMase
- neutral SMase
- oxLDL
- oxidized low-density lipoprotein
- PP2A
- protein phosphatase 2A
- TNF-α
- tumor necrosis factor-α
- VCAM
- vascular cell adhesion molecule
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311208/-/DC1.
Highlights
nSMase2 (type 2-neutral sphingomyelinase) inhibition reduces atherosclerosis progression in Apoe−/− mice.
GW4869, a pharmacological inhibitor of nSMase, reduces atherogenesis in Apoe−/− mice.
nSMase2 inhibition reduces the inflammatory responses evoked by atherogenic agents in endothelial cells and macrophages.
The redox-regulated transcription factor Nrf2 (nuclear factor [erythroid-derived 2]-like 2 or NF-E2–related factor-2) is activated consequently to nSMase2 inhibition.
Sphingolipids are involved in inflammation and atherosclerosis.
Sphingolipids are potential therapeutical targets to prevent or delay atherogenesis.
References
- 1.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462–2478. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- 3.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 5.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 6.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 7.Augé N, Nègre-Salvayre A, Salvayre R, Levade T. Sphingomyelin metabolites in vascular cell signaling and atherogenesis. Prog Lipid Res. 2000;39:207–229. doi: 10.1016/s0163-7827(00)00007-2. [DOI] [PubMed] [Google Scholar]
- 8.Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–183. doi: 10.1093/cvr/cvp030. doi: 10.1093/cvr/cvp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Becker KA, Zhang Y. Ceramide in redox signaling and cardiovascular diseases. Cell Physiol Biochem. 2010;26:41–48. doi: 10.1159/000315104. doi: 10.1159/000315104. [DOI] [PubMed] [Google Scholar]
- 10.Edsfeldt A, Dunér P, Ståhlman M, Mollet IG, Asciutto G, Grufman H, Nitulescu M, Persson AF, Fisher RM, Melander O, Orho-Melander M, Borén J, Nilsson J, Gonçalves I. Sphingolipids contribute to human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol. 2016;36:1132–1140. doi: 10.1161/ATVBAHA.116.305675. doi: 10.1161/ATVBAHA.116.305675. [DOI] [PubMed] [Google Scholar]
- 11.Worgall TS. Sphingolipid synthetic pathways are major regulators of lipid homeostasis. Adv Exp Med Biol. 2011;721:139–148. doi: 10.1007/978-1-4614-0650-1_9. doi: 10.1007/978-1-4614-0650-1_9. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Huan C, Chakraborty M, Zhang H, Lu D, Kuo MS, Cao G, Jiang XC. Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ Res. 2009;105:295–303. doi: 10.1161/CIRCRESAHA.109.194613. doi: 10.1161/CIRCRESAHA.109.194613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS, Robertson AW, Kindt EK, Homan R, Karathanasis SK, Rekhter MD. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2004;110:3465–3471. doi: 10.1161/01.CIR.0000148370.60535.22. doi: 10.1161/01.CIR.0000148370.60535.22. [DOI] [PubMed] [Google Scholar]
- 14.Wong ML, Xie B, Beatini N, Phu P, Marathe S, Johns A, Gold PW, Hirsch E, Williams KJ, Licinio J, Tabas I. Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. Proc Natl Acad Sci USA. 2000;97:8681–8686. doi: 10.1073/pnas.150098097. doi: 10.1073/pnas.150098097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devlin CM, Leventhal AR, Kuriakose G, Schuchman EH, Williams KJ, Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28:1723–1730. doi: 10.1161/ATVBAHA.108.173344. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leger AJ, Mosquea LM, Li L, Chuang W, Pacheco J, Taylor K, Luo Z, Piepenhagen P, Ziegler R, Moreland R, Urabe A, Jiang C, Cheng SH, Yew NS. Adeno-associated virus-mediated expression of acid sphingomyelinase decreases atherosclerotic lesion formation in apolipoprotein E(-/-) mice. J Gene Med. 2011;13:324–332. doi: 10.1002/jgm.1575. doi: 10.1002/jgm.1575. [DOI] [PubMed] [Google Scholar]
- 17.Augé N, Maupas-Schwalm F, Elbaz M, Thiers JC, Waysbort A, Itohara S, Krell HW, Salvayre R, Nègre-Salvayre A. Role for matrix metalloproteinase-2 in oxidized low-density lipoprotein-induced activation of the sphingomyelin/ceramide pathway and smooth muscle cell proliferation. Circulation. 2004;110:571–578. doi: 10.1161/01.CIR.0000136995.83451.1D. doi: 10.1161/01.CIR.0000136995.83451.1D. [DOI] [PubMed] [Google Scholar]
- 18.Tellier E, Nègre-Salvayre A, Bocquet B, Itohara S, Hannun YA, Salvayre R, Augé N. Role for furin in tumor necrosis factor alpha-induced activation of the matrix metalloproteinase/sphingolipid mitogenic pathway. Mol Cell Biol. 2007;27:2997–3007. doi: 10.1128/MCB.01485-06. doi: 10.1128/MCB.01485-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devillard R, Galvani S, Thiers JC, Guenet JL, Hannun Y, Bielawski J, Nègre-Salvayre A, Salvayre R, Augé N. Stress-induced sphingolipid signaling: role of type-2 neutral sphingomyelinase in murine cell apoptosis and proliferation. PLoS One. 2010;5:e9826. doi: 10.1371/journal.pone.0009826. doi: 10.1371/journal.pone.0009826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikolova-Karakashian M, Morgan ET, Alexander C, Liotta DC, Merrill AH., Jr Bimodal regulation of ceramidase by interleukin-1beta. Implications for the regulation of cytochrome p450 2C11. J Biol Chem. 1997;272:18718–18724. doi: 10.1074/jbc.272.30.18718. [DOI] [PubMed] [Google Scholar]
- 21.Kim MY, Linardic C, Obeid L, Hannun Y. Identification of sphingomyelin turnover as an effector mechanism for the action of tumor necrosis factor alpha and gamma-interferon. Specific role in cell differentiation. J Biol Chem. 1991;266:484–489. [PubMed] [Google Scholar]
- 22.Czarny M, Schnitzer JE. Neutral sphingomyelinase inhibitor scyphostatin prevents and ceramide mimics mechanotransduction in vascular endothelium. Am J Physiol Heart Circ Physiol. 2004;287:H1344–H1352. doi: 10.1152/ajpheart.00222.2004. doi: 10.1152/ajpheart.00222.2004. [DOI] [PubMed] [Google Scholar]
- 23.Cinq-Frais C, Coatrieux C, Grazide MH, Hannun YA, Nègre-Salvayre A, Salvayre R, Augé N. A signaling cascade mediated by ceramide, src and PDGFRβ coordinates the activation of the redox-sensitive neutral sphingomyelinase-2 and sphingosine kinase-1. Biochim Biophys Acta. 2013;1831:1344–1356. doi: 10.1016/j.bbalip.2013.04.014. doi: 10.1016/j.bbalip.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 24.Clarke CJ, Truong TG, Hannun YA. Role for neutral sphingomyelinase-2 in tumor necrosis factor alpha-stimulated expression of vascular cell adhesion molecule-1 (VCAM) and intercellular adhesion molecule-1 (ICAM) in lung epithelial cells: p38 MAPK is an upstream regulator of nSMase2. J Biol Chem. 2007;282:1384–1396. doi: 10.1074/jbc.M609216200. doi: 10.1074/jbc.M609216200. [DOI] [PubMed] [Google Scholar]
- 25.Ohanian J, Forman SP, Katzenberg G, Ohanian V. Endothelin-1 stimulates small artery VCAM-1 expression through p38MAPK-dependent neutral sphingomyelinase. J Vasc Res. 2012;49:353–362. doi: 10.1159/000336649. doi: 10.1159/000336649. [DOI] [PubMed] [Google Scholar]
- 26.Sitrin RG, Sassanella TM, Petty HR. An obligate role for membrane-associated neutral sphingomyelinase activity in orienting chemotactic migration of human neutrophils. Am J Respir Cell Mol Biol. 2011;44:205–212. doi: 10.1165/rcmb.2010-0019OC. doi: 10.1165/rcmb.2010-0019OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikolova-Karakashian M, Karakashian A, Rutkute K. Role of neutral sphingomyelinases in aging and inflammation. Subcell Biochem. 2008;49:469–486. doi: 10.1007/978-1-4020-8831-5_18. doi: 10.1007/978-1-4020-8831-5_18. [DOI] [PubMed] [Google Scholar]
- 28.Qin J, Berdyshev E, Poirer C, Schwartz NB, Dawson G. Neutral sphingomyelinase 2 deficiency increases hyaluronan synthesis by up-regulation of hyaluronan synthase 2 through decreased ceramide production and activation of akt. J Biol Chem. 2012;287:13620–13632. doi: 10.1074/jbc.M111.304857. doi: 10.1074/jbc.M111.304857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, Salvayre R, Negre-Salvayre A, Goldberg M, Guénet JL, Poirier C. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet. 2005;37:803–805. doi: 10.1038/ng1603. doi: 10.1038/ng1603. [DOI] [PubMed] [Google Scholar]
- 30.Stoffel W, Jenke B, Blöck B, Zumbansen M, Koebke J. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc Natl Acad Sci USA. 2005;102:4554–4559. doi: 10.1073/pnas.0406380102. doi: 10.1073/pnas.0406380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luberto C, Hassler DF, Signorelli P, Okamoto Y, Sawai H, Boros E, Hazen-Martin DJ, Obeid LM, Hannun YA, Smith GK. Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J Biol Chem. 2002;277:41128–41139. doi: 10.1074/jbc.M206747200. doi: 10.1074/jbc.M206747200. [DOI] [PubMed] [Google Scholar]
- 32.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 33.Olagnier D, Lavergne RA, Meunier E, Lefèvre L, Dardenne C, Aubouy A, Benoit-Vical F, Ryffel B, Coste A, Berry A, Pipy B. Nrf2, a PPARγ alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog. 2011;7:e1002254. doi: 10.1371/journal.ppat.1002254. doi: 10.1371/journal.ppat.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, III, Rosenfeld ME, Virmani R American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/ATV.0000000000000062. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 35.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 36.Trayssac M, Galvani S, Augé N, Sabbadini R, Calise D, Mucher E, Sallusto F, Thomsen M, Salvayre R, Nègre-Salvayre A. Role of sphingosine-1-phosphate in transplant vasculopathy evoked by anti-HLA antibody. Am J Transplant. 2015;15:2050–2061. doi: 10.1111/ajt.13264. doi: 10.1111/ajt.13264. [DOI] [PubMed] [Google Scholar]
- 37.Srinivasan S, Hatley ME, Reilly KB, Danziger EC, Hedrick CC. Modulation of PPARalpha expression and inflammatory interleukin-6 production by chronic glucose increases monocyte/endothelial adhesion. Arterioscler Thromb Vasc Biol. 2004;24:851–857. doi: 10.1161/01.ATV.zhq0504.2260. doi: 10.1161/01.ATV.zhq0504.2260. [DOI] [PubMed] [Google Scholar]
- 38.Tabatadze N, Savonenko A, Song H, Bandaru VV, Chu M, Haughey NJ. Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J Neurosci Res. 2010;88:2940–2951. doi: 10.1002/jnr.22438. doi: 10.1002/jnr.22438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Augé N, Andrieu N, Nègre-Salvayre A, Thiers JC, Levade T, Salvayre R. The sphingomyelin-ceramide signaling pathway is involved in oxidized low density lipoprotein-induced cell proliferation. J Biol Chem. 1996;271:19251–19255. doi: 10.1074/jbc.271.32.19251. [DOI] [PubMed] [Google Scholar]
- 40.Shamseddine AA, Airola MV, Hannun YA. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul. 2015;57:24–41. doi: 10.1016/j.jbior.2014.10.002. doi: 10.1016/j.jbior.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park IN, Cho IJ, Kim SG. Ceramide, an apoptotic rheostat, inhibits CCAAT/enhancer binding protein-beta and NF-E2-related factor-2 activation: the role in glutathione S-transferase A2 gene repression. Drug Metab Dispos. 2004;32:893–897. [PubMed] [Google Scholar]
- 42.Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol. 2015;225:R83–R99. doi: 10.1530/JOE-14-0662. doi: 10.1530/JOE-14-0662. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi: 10.1038/ncomms11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Paoli F, Staels B, Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J. 2014;78:1775–1781. doi: 10.1253/circj.cj-14-0621. [DOI] [PubMed] [Google Scholar]
- 45.Amtmann E, Baader W, Zöller M. Neutral sphingomyelinase inhibitor C11AG prevents lipopolysaccharide-induced macrophage activation. Drugs Exp Clin Res. 2003;29:5–13. [PubMed] [Google Scholar]
- 46.Lu Z, Li Y, Jin J, Zhang X, Hannun YA, Huang Y. GPR40/FFA1 and neutral sphingomyelinase are involved in palmitate-boosted inflammatory response of microvascular endothelial cells to LPS. Atherosclerosis. 2015;240:163–173. doi: 10.1016/j.atherosclerosis.2015.03.013. doi: 10.1016/j.atherosclerosis.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Won JS, Im YB, Khan M, Singh AK, Singh I. The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem. 2004;88:583–593. doi: 10.1046/j.1471-4159.2003.02165.x. [DOI] [PubMed] [Google Scholar]
- 48.Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. 2016;167:7–34. doi: 10.1016/j.trsl.2015.06.011. doi: 10.1016/j.trsl.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clarke CJ, Cloessner EA, Roddy PL, Hannun YA. Neutral sphingomyelinase 2 (nSMase2) is the primary neutral sphingomyelinase isoform activated by tumour necrosis factor-α in MCF-7 cells. Biochem J. 2011;435:381–390. doi: 10.1042/BJ20101752. doi: 10.1042/BJ20101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galvani S, Trayssac M, Augé N, Thiers JC, Calise D, Krell HW, Sallusto F, Kamar N, Rostaing L, Thomsen M, Nègre-Salvayre A, Salvayre R. A key role for matrix metalloproteinases and neutral sphingomyelinase-2 in transplant vasculopathy triggered by anti-HLA antibody. Circulation. 2011;124:2725–2734. doi: 10.1161/CIRCULATIONAHA.111.021790. doi: 10.1161/CIRCULATIONAHA.111.021790. [DOI] [PubMed] [Google Scholar]
- 51.Dinkins MB, Enasko J, Hernandez C, Wang G, Kong J, Helwa I, Liu Y, Terry AV, Jr, Bieberich E. Neutral sphingomyelinase-2 deficiency ameliorates alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci. 2016;36:8653–8667. doi: 10.1523/JNEUROSCI.1429-16.2016. doi: 10.1523/JNEUROSCI.1429-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Airola MV, Shanbhogue P, Shamseddine AA, Guja KE, Senkal CE, Maini R, Bartke N, Wu BX, Obeid LM, Garcia-Diaz M, Hannun YA. Structure of human nSMase2 reveals an interdomain allosteric activation mechanism for ceramide generation. Proc Natl Acad Sci USA. 2017;114:E5549–E5558. doi: 10.1073/pnas.1705134114. doi: 10.1073/pnas.1705134114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai CC, Kai JI, Huang WC, Wang CY, Wang Y, Chen CL, Fang YT, Lin YS, Anderson R, Chen SH, Tsao CW, Lin CF. Glycogen synthase kinase-3beta facilitates IFN-gamma-induced STAT1 activation by regulating src homology-2 domain-containing phosphatase 2. J Immunol. 2009;183:856–864. doi: 10.4049/jimmunol.0804033. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 54.Dong Z, Yang X, Xie K, Juang SH, Llansa N, Fidler IJ. Activation of inducible nitric oxide synthase gene in murine macrophages requires protein phosphatases 1 and 2A activities. J Leukoc Biol. 1995;58:725–732. doi: 10.1002/jlb.58.6.725. [DOI] [PubMed] [Google Scholar]
- 55.Karakashian AA, Giltiay NV, Smith GM, Nikolova-Karakashian MN. Expression of neutral sphingomyelinase-2 (NSMase-2) in primary rat hepatocytes modulates IL-beta-induced JNK activation. FASEB J. 2004;18:968–970. doi: 10.1096/fj.03-0875fje. doi: 10.1096/fj.03-0875fje. [DOI] [PubMed] [Google Scholar]
- 56.Tsakiri N, Kimber I, Rothwell NJ, Pinteaux E. Interleukin-1-induced interleukin-6 synthesis is mediated by the neutral sphingomyelinase/Src kinase pathway in neurones. Br J Pharmacol. 2008;153:775–783. doi: 10.1038/sj.bjp.0707610. doi: 10.1038/sj.bjp.0707610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barth BM, Gustafson SJ, Kuhn TB. Neutral sphingomyelinase activation precedes NADPH oxidase-dependent damage in neurons exposed to the proinflammatory cytokine tumor necrosis factor-α. J Neurosci Res. 2012;90:229–242. doi: 10.1002/jnr.22748. doi: 10.1002/jnr.22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mimura J, Itoh K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic Biol Med. 2015;88(pt B):221–232. doi: 10.1016/j.freeradbiomed.2015.06.019. doi: 10.1016/j.freeradbiomed.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 59.Huang CS, Lin AH, Yang TC, Liu KL, Chen HW, Lii CK. Shikonin inhibits oxidized LDL-induced monocyte adhesion by suppressing NFκB activation via up-regulation of PI3K/Akt/Nrf2-dependent antioxidation in EA.hy926 endothelial cells. Biochem Pharmacol. 2015;93:352–361. doi: 10.1016/j.bcp.2014.12.005. doi: 10.1016/j.bcp.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 60.Reforgiato MR, Milano G, Fabriàs G, Casas J, Gasco P, Paroni R, Samaja M, Ghidoni R, Caretti A, Signorelli P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res Cardiol. 2016;111:12. doi: 10.1007/s00395-016-0533-x. doi: 10.1007/s00395-016-0533-x. [DOI] [PubMed] [Google Scholar]
- 61.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, Deshane J, Bolisetty S, Shaposhnik Z, Shih DM, Agarwal A, Lusis AJ, Araujo JA. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–1711. doi: 10.1161/CIRCRESAHA.107.151720. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 62.Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120(pt 16):2935–2943. doi: 10.1242/jcs.03473. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- 63.Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signaling. Front Oncol. 2014;4:388. doi: 10.3389/fonc.2014.00388. doi: 10.3389/fonc.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garoby-Salom S, Rouahi M, Mucher E, Auge N, Salvayre R, Negre-Salvayre A. Hyaluronan synthase-2 upregulation protects smpd3-deficient fibroblasts against cell death induced by nutrient deprivation, but not against apoptosis evoked by oxidized LDL. Redox Biol. 2015;4:118–126. doi: 10.1016/j.redox.2014.12.004. doi: 10.1016/j.redox.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 66.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 67.Salvayre R, Negre-Salvayre A, Camaré C. Oxidative theory of atherosclerosis and antioxidants. Biochimie. 2016;125:281–296. doi: 10.1016/j.biochi.2015.12.014. doi: 10.1016/j.biochi.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 68.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 2008;3:e3791. doi: 10.1371/journal.pone.0003791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, Wang X, Castellani LW, Reue K, Lusis AJ, Araujo JA. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler Thromb Vasc Biol. 2011;31:58–66. doi: 10.1161/ATVBAHA.110.210906. doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jakobs P, Serbulea V, Leitinger N, Eckers A, Haendeler J. Nuclear factor (erythroid-derived 2)-like 2 and thioredoxin-1 in atherosclerosis and ischemia/reperfusion injury in the heart. Antioxid Redox Signal. 2017;26:630–644. doi: 10.1089/ars.2016.6795. doi: 10.1089/ars.2016.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harada N, Ito K, Hosoya T, Mimura J, Maruyama A, Noguchi N, Yagami K, Morito N, Takahashi S, Maher JM, Yamamoto M, Itoh K. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic Biol Med. 2012;53:2256–2262. doi: 10.1016/j.freeradbiomed.2012.10.001. doi: 10.1016/j.freeradbiomed.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 72.Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, Hersberger M, Yamamoto M, Bachmann MF, Kopf M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. 2011;41:2040–2051. doi: 10.1002/eji.201041316. doi: 10.1002/eji.201041316. [DOI] [PubMed] [Google Scholar]
- 73.Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15:313. doi: 10.1007/s11926-012-0313-z. doi: 10.1007/s11926-012-0313-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collins AR, Gupte AA, Ji R, Ramirez MR, Minze LJ, Liu JZ, Arredondo M, Ren Y, Deng T, Wang J, Lyon CJ, Hsueh WA. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler Thromb Vasc Biol. 2012;32:2839–2846. doi: 10.1161/ATVBAHA.112.300345. doi: 10.1161/ATVBAHA.112.300345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Mäkinen PI, Heinonen SE, Laitinen HM, Heikkilä J, Vatanen T, Hörkkö S, Yamamoto M, Ylä-Herttuala S, Jauhiainen M, Levonen AL. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. 2013;98:107–115. doi: 10.1093/cvr/cvt008. doi: 10.1093/cvr/cvt008. [DOI] [PubMed] [Google Scholar]
- 76.Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]