

Abstract
Hydrogen sulfide (H2S) has emerged as a relevant signaling molecule in physiology, taking its seat as a bona fide gasotransmitter akin to nitric oxide (NO) and carbon monoxide (CO). After being merely regarded as a toxic poisonous molecule, it is now recognized that mammalian cells are equipped with sophisticated enzymatic systems for H2S production and breakdown. The signaling role of H2S is mainly related to its ability to modify different protein targets, particularly by promoting persulfidation of protein cysteine residues and by interacting with metal centers, mostly hemes. H2S has been shown to regulate a myriad of cellular processes with multiple physiological consequences. As such, dysfunctional H2S metabolism is increasingly implicated in different pathologies, from cardiovascular and neurodegenerative diseases to cancer. As a highly diffusible reactive species, the intra- and extracellular levels of H2S have to be kept under tight control and, accordingly, regulation of H2S metabolism occurs at different levels. Interestingly, even though H2S, NO, and CO have similar modes of action and parallel regulatory targets or precisely because of that, there is increasing evidence of a crosstalk between the three gasotransmitters. Herein are reviewed the biochemistry, metabolism, and signaling function of hydrogen sulfide, as well as its interplay with the other gasotransmitters, NO and CO.
1. Introduction
1.1. Brief Historical Account of Hydrogen Sulfide
Hydrogen sulfide (H2S) is known in popular culture as a toxic poisonous gas and a source of foul smell, such as that of rotten eggs, sewers, swamps, and volcanic sources. Ancient alchemic practices produced several preparations now known to release hydrogen sulfide, for example, the H2S- and ammonia-rich gases released upon distillation to obtain Liquor Hepatis (Balsam of the Soul), made from limestone and camel dung. In his treaty on workers' diseases “De Morbis Artificum,” the 17th–18th century Italian physician Bernardino Ramazzini first described painful eye inflammation often leading to secondary bacterial infection and eventually blindness in workers who cleaned privies and cesspits [1]. Ramazzini hypothesized that, when the workers stirred the excrements, an unknown volatile compound was released which, besides having the undesired health effects, was also responsible for turning black their copper and silver coins on the surface. The same toxic volatile substance, originated in the Paris sewers, was likely responsible for a series of incidents in the late 18th century, ranging from mild eye and mucous membrane inflammation to severe asphyxia. Almost at the same time, in 1777, the Swedish chemist Carl Wilhelm Scheele described the stinking substance (sulfur air) resulting from the reaction of pyrite (ferrous disulfide) with a mineral acid [2].
While long considered a poisonous substance, not only hazardous for workers but also burdensome for some industrial processes (e.g., “oil souring” in oil extraction), hydrogen sulfide was in the late 20th century discovered to be endogenously formed in humans, and in the early 21st century, it was recognized as an extremely relevant signaling molecule in physiology and general biology.
1.2. General Chemistry of Hydrogen Sulfide
Hydrogen sulfide is a colorless flammable gas. It is a weak acid, existing in aqueous solution in equilibrium with hydrosulfide (HS−) and sulfide (S2−), according to pKa1~7.0 (H2S/HS−) and pKa2~19 (HS−/S2−), at 25°C ([3] and references therein). At physiological pH values, the S2− concentration in solution is therefore negligible. According to this equilibrium, at the physiological pH 7.4, c.a. 70% of hydrogen sulfide is in its HS− form, the remainder occurring as H2S. In the alkaline mitochondrial matrix (pH 8.0), HS− reaches 92%, while the remaining 8% corresponds to H2S. Contrarily, under the acidic conditions in lysosomes (pH 4.7), >99% of hydrogen sulfide is in its H2S form and is slightly polar, allowing it to freely diffuse across and accumulate in aqueous or hydrophobic milieu such as biological membranes. Herein, unless otherwise stated, the terms “hydrogen sulfide” or “sulfide” and the abbreviation “H2S” henceforth collectively designate the pool of the H2S, HS−, and S2− species.
H2S is the sulfur species corresponding to the lowest sulfur oxidation state (−2). Sulfur, with its electronic configuration [Ne] 3s2 3p4, is a highly redox versatile element, with oxidation states ranging from −2 to +6 (as in sulfate), due to the six valence electrons. This versatility certainly accounts for its biological usefulness and is probably related to the major role of sulfur in the emergence and evolution of life on Earth (reviewed in [4]). H2S is a reducing species, displaying a reduction potential of −280 mV (pH 7.0, against the standard hydrogen electrode) for the two-electron HS−/S0 redox couple (−230 mV for H2S/S0) [5], close to the values for the glutathione disulfide/glutathione (GSSG/GSH) and the cystine/cysteine redox couples.
Presently, despite major advances, the physiological H2S concentrations are still a matter of debate, and the posited levels have been decreasing with the increasing sophistication and accuracy of the detection methods. Currently, free H2S is reported to be submicromolar, although it may exist in equilibrium with a pool of labile sulfur-containing molecules that can liberate H2S “on demand,” that is, under particular physiological conditions [6]. To add more complexity into the role of H2S in human (patho)physiology, it has become clear that a great part of the signaling effects attributed to H2S (see below) actually results from the occurrence of persulfides and polysulfides, among other sulfur-containing molecules, that have been collectively termed “reactive sulfur species” (RSS). More details on the formation of persulfides and polysulfides, and their physiological relevance intertwined with that of hydrogen sulfide, are given below.
2. Hydrogen Sulfide Metabolism in Human Physiology
The reactivity and potential toxicity of hydrogen sulfide demand its levels to be kept under strict control. Indeed, even temporary imbalances in local hydrogen sulfide concentrations can trigger a cascade of cellular events with pathological consequences. The production and breakdown of H2S is thus essentially ensured by specialized enzymes, tightly regulated and compartmentalized. Although H2S is freely permeable to membranes and thus highly diffusible inside the cell, compartmentalization of its synthesis or breakdown could be relevant to ensure local effects in cellular organelles. A clue for this has been for instance provided by showing that the mitochondria-targeted H2S donor AP39 ([10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5yl)phenoxy)decyl) triphenylphosphonium bromide]), consisting of the H2S-releasing moiety ADT-OH (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione)coupled to the mitochondria-targeting triphenylphosphonium (TPP+) moiety, protects mitochondria in oxidatively stressed endothelial cells more effectively than ADT-OH itself, not coupled to TPP+ and thus unable to specifically target mitochondria [7]. Despite decades of molecular and cellular studies on the enzymatic systems involved in H2S synthesis and breakdown, it appears at times that this field of biology is still in its infancy, with new reactive molecules and new targets of H2S and related species being consistently identified and new mechanistic details and regulatory subtleties being unraveled.
2.1. Human H2S-Synthesizing Enzymes
The biogenesis of hydrogen sulfide in human physiology occurs via two major routes: by endogenous specialized enzymes and as an end-product or intermediate of microbial metabolic pathways within the gut microbiota, particularly in sulfate-reducing bacteria. Interestingly, by comparing germ-free versus conventional mice, it was shown that the intestinal microbiota regulates H2S homeostasis not only in the gut but also systemically in various tissues and organs [8]. Namely, the presence of microbiota was found to be associated with higher levels of free H2S not only in some intestinal tracts (colon and cecum) but also in plasma. Moreover, as compared to germ-free mice, the conventional animals displayed higher levels of bound sulfane sulfur in plasma, fat, and lung, and higher CSE activity and reduced cysteine levels in most organs and tissues [8]. Another secondary source of H2S is proposed to be persulfides and polysulfides, either endogenously generated or derived from dietary intake (reviewed in, e.g., [9]).
The three human enzymes reported to endogenously generate hydrogen sulfide are cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE, also known as cystathionase), and 3-mercaptopyruvate sulfurtransferase (MST) (Figure 1) [10].
Figure 1.
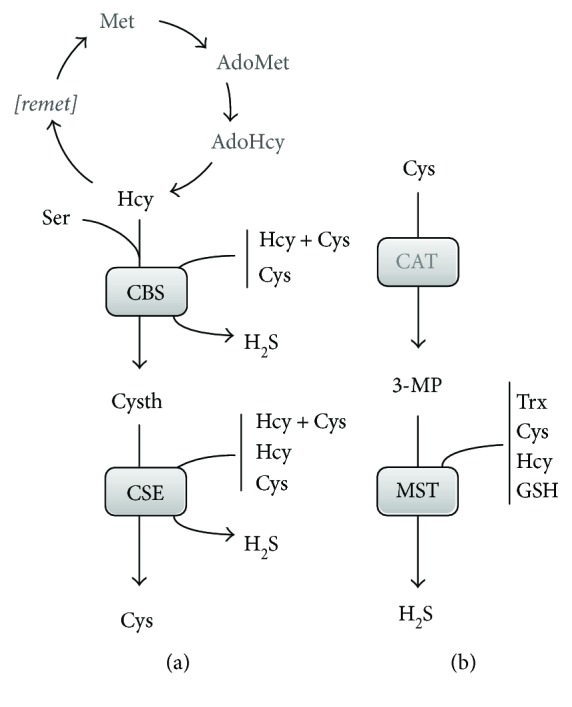
Hydrogen sulfide biosynthetic pathways in mammalian physiology. (a) Transsulfuration branch of the methionine cycle. l-Methionine (Met) is converted by l-methionine adenosyltransferase to s-adenosyl-l-methionine (AdoMet), which is used by methyltransferases in methylation reactions, generating s-adenosyl-l-homocysteine (AdoHcy). AdoHcy hydrolase then converts AdoHcy into l-homocysteine (Hcy), which can either be converted back to l-methionine through the remethylation cycle ([remet]), or enter the transsulfuration branch. Cystathionine β-synthase (CBS) converts Hcy and l-serine into l-cystathionine (Cysth), which is taken up by cystathionine γ-lyase (CSE) to generate l-cysteine (Cys). Hydrogen sulfide (H2S) is synthesized by CBS and CSE in several alternative reactions (see Figure 2). (b) H2S synthesis by 3-mercaptopyruvate sulfurtransferase (MST). Cys is converted by cysteine aminotransferase (CAT) into 3-mercaptopyruvate (3-MP), which is used by MST to synthesize H2S along with its cosubstrates thioredoxin (Trx), Cys, Hcy, and l-glutathione (GSH).
2.1.1. Expression in Cells and Tissues and Cellular Localization
The occurrence of all three H2S-synthesizing enzymes in cells, tissues, and organs and their cellular distribution remain evolving subjects, particularly taking into account that these may vary under different (patho)physiologic conditions. It is generally considered that CBS regulation occurs at the protein level, the enzyme being a target of multiple posttranslational modifications and having two regulatory domains that respond to different stimuli (reviewed, e.g., in [11]), unlike CSE, for which no posttranslational regulatory mechanisms are known [10]. Moreover, since CBS and CSE employ various combinations of the same substrates in their H2S-generating reactions (Figures 1 and 2), regulation of one enzyme will likely affect the other, due to increased or decreased substrate availability (e.g., in [11]).
Figure 2.

Catalytic versatility of the H2S-synthesizing enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). Chemical structure of the metabolites and reaction schemes involved in H2S synthesis by CBS and CSE. Top panel, “canonical” reactions catalyzed by CBS and CSE as part of the transsulfuration branch of the methionine cycle. Center-bottom panel, alternative H2S-generating reactions. Chemical structures labeled as α-ketobutyrate and pyruvate are represented as the α-ketobutyric and pyruvic acid, respectively. ∗Serine and homoserine have not been detected as intermediates in the CSE-catalyzed reactions 3 and 5, respectively, only their downstream products α-ketobutyric and pyruvic acids (resp.).
The tissue and organ distribution of the H2S-synthesizing enzymes has been studied for different organisms, employing immunohistochemical detection and analyzing transcriptional levels and H2S-generating activity. In man and rodents, CBS is mostly abundant in the liver, pancreas, kidney, brain, and nervous system [9, 12–14]. Recently, CBS expression in the carotid body, as well as in uterine and mesenteric and umbilical arteries, was reported [15]. As for CSE, it is mostly expressed in the liver, kidney, and smooth muscle (both vascular and nonvascular), its expression and H2S-generating activity being negligible in the brain, heart, and spleen [9, 13, 16, 17]. MST tissue distribution has been systematically studied for different organisms. The activity of bovine MST has been reported to be highest in the adrenal cortex, followed by the liver, heart, and kidney [18]. Abundant rat MST expression has been detected in the kidney (proximal tubular epithelium), liver (pericentral hepatocytes), aorta, and brain glial cells [19]. Recently, Tomita et al. detected abundant murine MST in the brain (neural and glial cells), liver, kidney, testes, and endocrine organs (particularly in pancreatic islets) and lower levels in bronchiolar cells, spleen, thymus, and small intestine [20]. Finally, Western blot analysis and enzymatic assays revealed the presence of active MST, but undetectable CBS and CSE, in red blood cells [21].
In terms of cellular localization, the classical view is that both CBS and CSE are cytosolic enzymes, whereas MST localizes to the mitochondria (e.g., [9, 10, 22]). However, several examples show that this classical view may be challenged. It has been reported that when blood homocysteine levels rise, microvascular endothelial cells and hepatocytes may secrete CBS and CSE, thus circulating as part of the plasma proteome and contributing to H2S generation [23]. Extracellularly synthesized H2S was shown to contribute to increased cell viability and decreased oxidative damage to DNA after serum starvation and hypoxia/reoxygenation. Furthermore, immunoprecipitation of circulating CBS and CSE from serum prior to its supplementation with homocysteine enhanced serum-induced stress towards endothelial cells. Altogether, these observations suggest that secreted CBS and CSE have a protective role in the endothelium by both producing H2S and clearing excess homocysteine. Another notable characteristic of CBS is its ability to be SUMOylated leading to protein accumulation in the nucleus [24]. Perhaps of greatest significance for their role as H2S-synthesizing enzymes is the observation that both CBS and CSE can translocate to the mitochondria under various (patho)physiological conditions. This becomes particularly relevant taking into account that H2S can not only stimulate mitochondrial bioenergetics (i) by supplying electron equivalents to the quinol pool via sulfide:quinone oxidoreductase (reviewed in [10, 25]), (ii) by activating the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase [26], and (iii) by persulfidation of ATP synthase [27, 28] but also block mitochondrial respiration by tight inhibition of cytochrome c oxidase (CcOX, reviewed in [29–31]). CBS has been observed to partially localize to liver mitochondria, where the protein transiently accumulates under hypoxia/ischemia, with the resulting increased H2S generation protecting mitochondria from oxidative stress [32]. Once cells return to normoxia, CBS levels are restored via protein degradation by mitochondrial Lon protease, which recognizes and targets specifically the oxidized form of the regulatory heme at the CBS N-terminal domain [32].
In the colorectal cancer cell line HCT 116, a significant fraction of CBS localizes to the mitochondria, being associated with the outer mitochondrial membrane. The resulting increased H2S levels are proposed to stimulate cancer cell energy metabolism, particularly oxidative phosphorylation and glycolysis [33]. In line with this observation, CBS has been shown to contribute in various cancer cells to a shift in the fate of glucose utilization from glycolysis to the pentose phosphate pathway [34] (detailed in Section 4.3.1). By also promoting tumor angiogenesis, the increased H2S availability may contribute to increased oxygen supply to tumor cells. In a mouse model of amyotrophic lateral sclerosis, CBS accumulation in mitochondria isolated from the spinal cord and the resulting increased H2S generation are proposed to be related with impaired cytochrome c oxidase-dependent respiration [35].
CSE has been reported to translocate to the mitochondria of vascular smooth muscle cells (SMCs) in response to stimuli that increased intracellular calcium levels. This translocation appears to be mediated by the mitochondrial membrane translocase of the outer membrane 20 (Tom20). Whereas SMCs display impaired ATP production in hypoxic conditions, the presence of CSE inside the mitochondria contributed to cysteine catabolism, H2S synthesis, and stimulated ATP synthesis under such conditions, suggesting that CSE translocation may sustain mitochondrial bioenergetics in response to certain stresses [28].
MST is generally regarded as a mitochondrial enzyme. However, Fräsdorf et al. have shown that there are actually two splice variants of the protein, MST-Iso1 and MST-Iso2, both of which localize to the cytosol, while only MST-Iso2 also localizes to the mitochondria in HEK293a and HeLa cells [36].
2.1.2. Associated Metabolic Pathways
CBS and CSE participate in the transsulfuration branch of the methionine methylation/remethylation cycle (Figure 1(a)), a key pathway which generates extremely relevant signaling molecules for mammalian physiology. In this cycle, methionine (Met) derived essentially from dietary intake is converted to s-adenosyl-l-methionine (AdoMet) by methionine adenosyltransferase ([37] and references therein). AdoMet is the methyl group donor for virtually all methylation reactions in mammalian physiology, through the action of methyltransferases. Concurrently with methylation, s-adenosyl-l-homocysteine (AdoHcy) is generated, which is used by AdoHcy hydrolase (SAHH) to produce homocysteine (Hcy) and adenosine. Homocysteine then either enters the transsulfuration branch or proceeds through the remethylation pathway to revert to methionine, depending on different metabolic fluxes and other physiological conditions. The metabolites AdoMet, AdoHcy, and Hcy participate in the regulation of numerous physiological processes. As detailed below, AdoMet is an allosteric regulator of CBS, increasing its enzymatic activity by 2–5-fold upon binding. This allosteric activation results in an increase in Vmax, without any notable effect on the substrate affinity (KM) [38, 39]. The “canonical” reactions attributed to CBS and CSE within the transsulfuration pathway result in conversion of homocysteine into cysteine as follows (Figure 2): CBS catalyzes the β-replacement of serine by homocysteine, yielding cystathionine, and CSE then proceeds by converting cystathionine to cysteine, α-ketobutyrate, and ammonia in an α,γ-elimination reaction. The role of CBS and CSE in hydrogen sulfide production results from an extensive repertoire of alternative reactions that these enzymes are able to catalyze (Figure 2) [39, 40], possibly related to the fact that both are pyridoxal 5′-phosphate- (PLP-) dependent enzymes. This may be perceived as catalytic promiscuity or versatility-based robustness [11]. A notable thorough in vitro kinetic characterization of CBS- and CSE-catalyzed H2S-generating reactions combined with kinetic simulations has been carried out by Singh et al., allowing to envisage which reactions are relevant in vivo under different (patho)physiological conditions [39]. Both CBS and CSE are able to synthesize H2S through cysteine β-elimination or β-replacement, respectively employing one or two cysteine molecules (Figure 2, reactions (3) and (4)), or through β- or γ-replacement using cysteine and homocysteine (Figure 2, reaction (7)). CSE alone is able to synthesize H2S via homocysteine α,γ-elimination or γ-replacement, respectively employing one or two homocysteine molecules (Figure 2, reactions (5) and (6)). Both CBS and CSE also catalyze cystine α,β-elimination to yield cysteine persulfide, which ultimately may generate H2S (Figure 2, reaction (8)). Besides H2S synthesis, some of these reactions originate thioether metabolites such as lanthionine and homolanthionine, which may have been suggested as biomarkers of hydrogen sulfide generation in homocystinuric patient samples [41]. Under substrate saturating conditions, the turnover number of recombinant human CBS for the H2S-generating cysteine and homocysteine β-/γ-replacement reaction (reaction (7) in Figure 2) is 3.7-fold higher than that of the cystathionine-generating canonical reaction [39]. Kinetic simulations show that this difference drops to 1.3-fold at physiologically relevant substrate concentrations, which still indicates that CBS-catalyzed H2S synthesis at the expense of homocysteine and cysteine accounts for ~56% of cystathionine production. Notably, reaction (7) is responsible for 96% of the net CBS-catalyzed H2S production, whereas the cysteine-alone-based reactions (reactions (3) and (4) in Figure 2) account for a mere 1.6–2.6% of H2S synthesis. Still, the H2S-producing capacity of CBS appears to be insensitive to the systemic homocysteine levels, possibly related to the fact that homocysteine can only bind to CBS after serine or cysteine. The same kinetic simulations show that, contrarily to CBS, CSE catalyzes the canonical reaction of cysteine synthesis via cystathionine α,γ-elimination (reaction (2) in Figure 2) with a much higher (20–30 fold) catalytic efficiency (kcat/KM) than determined for the H2S-synthesizing reactions (reactions (3–6) in Figure 2). At physiologically relevant substrate concentrations (10 μM homocysteine, 100 μM cysteine, and 5 μM cystathionine), the canonical reaction turnover is still 5-fold or 12-fold higher than those for H2S-generating cysteine or homocysteine cleavage reactions, respectively. Under those conditions, approximately 70% of CSE-catalyzed H2S production derives from cysteine and 29% from homocysteine. Also in contrast with CBS, the CSE-catalyzed H2S generation depends on homocysteine availability. Indeed, kinetic simulations assuming equimolar CBS (fully activated by AdoMet) and CSE at “normal” homocysteine concentrations (10 μM) reveal that CSE contributes to 32% of the synthesized H2S, whereas the CSE contribution increases to 45% and 74% under conditions of moderate and severe hyperhomocysteinemia, respectively. This indicates that CSE may have a significant role in homocysteine clearance at pathophysiological elevated homocysteine levels, particularly in tissues or organs where CBS is absent or poorly expressed.
Recently, Majtan et al. reported on a thorough kinetic analysis of CBS-catalyzed reactions combined with simulations [38]. This study demonstrated the competitive advantage of serine over cysteine as a substrate for its condensation with homocysteine (to yield H2O or H2S, resp.), with a 2–5-fold higher catalytic efficiency of the canonical reaction compared to the H2S-generating reaction. However, despite this apparent advantage at saturating substrate concentrations, the H2S-producing reaction is able to compete with the canonical reaction under physiologically relevant conditions [38]. Indeed, one of the key findings is that a main determinant of CBS-catalyzed H2S production is the cysteine to serine ratio, adding another layer of complexity to be taken into account when considering the in vivo role of each H2S-producing enzyme.
Despite the remarkable value of these kinetic studies, H2S generation obviously also depends on the relative expression levels of each enzyme in each cell/tissue and on the systemic and local availability of substrates (cysteine, homocysteine, cystine, and serine), regulatory metabolites (e.g., AdoMet), and other effectors which negatively modulate enzymatic activity (e.g., NO and CO inhibit CBS; detailed below).
The role of 3-mercaptopyruvate sulfurtransferase (MST) is also tightly associated with cysteine metabolism. 3-Mercaptopyruvate (3-MP) is generated by cysteine (or aspartate) aminotransferase (CAT, Figure 3(a)) through cysteine deamination, using α-ketoglutarate as cosubstrate and yielding glutamate as coproduct [10]. An additional pathway whereby 3-MP is generated has been recently demonstrated to be present mostly in the cerebellum and the kidney, involving D-cysteine and a D-amino acid oxidase [42].
Figure 3.

Reaction mechanism of H2S production by 3-mercaptopyruvate sulfurtransferase (MST). Chemical structure of the metabolites and reaction schemes involved in H2S synthesis by MST. (a) Production of 3-mercaptopyruvate (3-MP) is catalyzed by cysteine aminotransferase (CAT), with α-ketoglutarate as cosubstrate. (b) MST reaction with its activating substrate 3-MP and acceptor substrates. Upon reacting with 3-MP, the side-chain sulfhydryl of Cys248 (CysSH) becomes persulfidated (CysSSH), which activates MST (MST∗) towards the reaction with the acceptor substrates. The reaction with low-molecular-weight (LMW) thiol-containing substrates (RSH) involves the sequential reaction with two substrate molecules. In the first step, MST∗ transfers the sulfane sulfur from the cysteine persulfide to the RSH, regenerating the enzyme and releasing a persulfide RSSH product. In the second step, another RSH reacts with RSSH to yield H2S and oxidized RSSR. The reaction of MST∗ with reduced thioredoxin (Trx) proceeds similarly to that with LMW RSH molecules. The MST cysteine persulfide is initially transferred to Trx, yielding CysSSH, which is then attacked by a nearby cysteine thiol, releasing H2S and yielding oxidized Trx. Chemical structures labeled as α-ketoglutarate, 3-mercaptopyruvate, and l-glutamate are represented as α-ketoglutaric, 3-mercaptopyruvic, and l-glutamic acids, respectively.
The interaction between MST and its 3-MP substrate gives rise to an enzyme-bound cysteine persulfide intermediate at Cys248 (MST∗ in Figure 3(b)), concomitantly with pyruvate release [43]. H2S can then be released by transfer of the active site cysteine persulfide to physiologic small thiols like cysteine, homocysteine, and glutathione or reducing molecules such as thioredoxin (Trx) or dihydrolipoic acid (DHLA) or by reaction with nonphysiological reductants like 2-mercaptoethanol and dithiothreitol [43]. The MST persulfide intermediate transfers its persulfide to an acceptor R-SH, forming an R-S-SH intermediate, which then reacts with another acceptor RSH molecule to yield a disulfide (RSSR) and H2S. From the tested physiological acceptor substrates, thioredoxin displays the highest catalytic efficiency, orders of magnitude higher than those for DHLA, cysteine, homocysteine, and glutathione [43]. This observation is consistent with the observation that some protozoan parasites encode MST variants with a fused thioredoxin domain.
The broad impact of H2S on mammalian physiology has been widely assessed in numerous in vivo studies, utilizing knockout mouse models of the three H2S-synthesizing enzymes CBS, CSE, and MST and of ethylmalonic encephalopathy protein 1 (ETHE1, also known as persulfide dioxygenase or sulfur dioxygenase), implicated in H2S breakdown. Making use of these models, particularly the ones for CSE and CBS, H2S has been shown to take part in a huge number of physiological and pathophysiological processes and thus to have a high impact on human health and disease. For a comprehensive overview of the wide spectrum of studies carried out on these models, the readers can refer to [9], a recent review specifically addressing this topic.
2.1.3. Structural and Functional Properties of H2S-Generating Enzymes
(1) Cystathionine β-Synthase (CBS). Human cystathionine β-synthase (CBS) is considered to assemble as a homotetramer of 551 amino acid-long (~61 kDa) monomers, each consisting of three domains (Figure 4(a)): an N-terminal domain (residues 1–70) that binds a noncatalytic heme cofactor, a central catalytic PLP-binding domain (residues 71–381), and a C-terminal regulatory domain (residues 412–551, also known as Bateman module, comprising two motifs, CBS1 and CBS2) responsible for enzyme activation upon AdoMet binding. Structural studies on the human enzyme have revealed that the catalytic domain has a highly conserved structural fold of the β-family of PLP-dependent enzymes, consisting of thirteen α-helices and two β-sheets composed of four and six strands [44, 45]. Several intrinsically flexible regions that were initially not visible in the structure of a truncated human CBS lacking the C-terminal domain became visible in the structure of a “full-length” CBS construct (with a 10-residue deletion at the C-terminus, (Figure 4(a)). These include the strands β4, β5, and β6 preceding the loops L145–148, L171–174, and L191–202, which are proposed to constitute the catalytic site entrance (together with loop L295–316) and are stacked between the catalytic core and the C-terminal domain, as well as helix α7 following the L191–202 loop. The active site PLP moiety is bound to the catalytic core through a Schiff base bond to the ε-amino group of lysine 119 (Figure 4(b)). A long 30-residue α-helical stretch (residues 382–411; helices α15 and α16) tethers the catalytic core to the C-terminal AdoMet-binding domain. The latter actually comprises two so-called CBS domains which share a common structural fold despite the poor sequence identity (~7% over 133 residues). The key finding in the structure of full-length CBS was the revelation that, in AdoMet-free CBS, the C-terminal domain from one monomer blocks the substrate entry site of the opposing monomer within one dimer (Figure 4(c)) [44]. This blockage is accomplished by stabilization of the above mentioned intrinsically flexible region composed by the β4, β5, and β6 strands preceding the loops L145–148, L171–174, and L191–202. Indeed, the interaction between the core and regulatory domains has been shown to involve (i) hydrophobic interactions with residues from the C-terminal domain CBS2 motif and (ii) H-bonds with residues from the C-terminal domain CBS1 motif [44, 46]. Notably, in the presence of AdoMet, the two C-terminal domains assemble together in a “disc”-like form, similarly to the constitutively activated Drosophila melanogaster AdoMet-free CBS [47], the substrate entrance opens, and access to the active site is enhanced (Figure 4(c)), thus increasing enzymatic activity [44, 46, 48]. Each Bateman module contains two putative AdoMet-binding cavities (S1 and S2) with different binding affinities [49]. One of these cavities (S2) is exposed and thus likely represents the primary AdoMet binding site, while the other one (S1) appears to be hindered by bulky hydrophobic residues from the catalytic core [46, 48, 49]. The AdoMet-induced association between adjoining Bateman modules disrupts the interactions between the catalytic and C-terminal regulatory domains and opens the catalytic core to the substrates.
Figure 4.
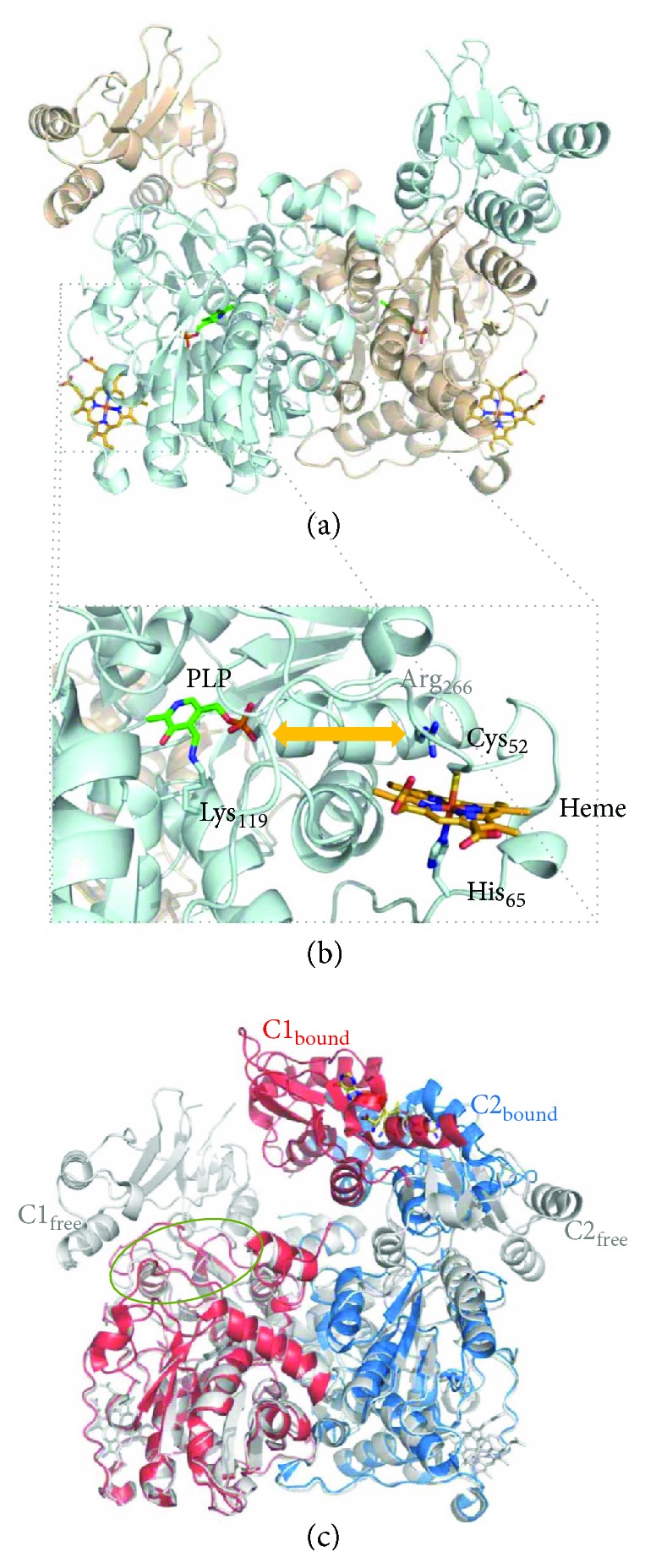
Crystallographic structure of human cystathionine β-synthase (CBS). (a) Cartoon representation of the “full-length” human CBS homodimer (PDB ID: 4COO; Δ516–525; 2.0 Å resolution). Green sticks, active site PLP moiety, where H2S production occurs. Orange sticks, regulatory heme b where CO or NO binds, resulting in enzyme inhibition. (b) Zoom-in into the catalytic (PLP) and regulatory (heme) sites. The PLP moiety is covalently attached to CBS through Lys119 (human CBS numbering), while the b-type heme is axially coordinated by Cys52 and His65. Orange arrow indicates the communication between the heme and PLP sites mediated by the α-helix comprising residues Thr257-Gly258-Gly259-Thr260-Ile261-Thr262-Gly263-Ile264-Ala265-Arg266. (c) Structural effect of AdoMet binding. While in AdoMet-free CBS, the C-terminal domain of each monomer (C1free and C2free, colored in light gray) blocks the substrate entrance into the active site (green oval circle) of the adjacent monomer, AdoMet binding to the C-terminal domains leads to association of the latter in a disk-like form (C1bound and C2bound; PDB ID: 4PCU; Δ516–525 Glu201Ser; 3.58 Å resolution), unblocking the active site and derepressing the enzymatic activity. Figure generated with PyMol 1.8.2.0 [238].
(2) Cystathionine γ-Lyase (CSE). Cystathionine γ-lyase (CSE) is a homotetrameric enzyme constituted by 405-amino-acid-long ~44 kDa monomers, each consisting of two structural domains (Figure 5(a)) [50]. The larger PLP-binding catalytic domain (comprising residues 9–263) assembles as an α/β/α fold consisting of a seven-stranded β-sheet (6 parallel and 1 antiparallel) flanked by eight α-helices. The smaller C-terminal domain (residues 264–401) is composed of a β-sheet (4 antiparallel strands) with three helices on one side. Similarly to CBS, the PLP in CSE is anchored through a Schiff base bond between the PLP carbonyl and the ε-amino group of a lysine residue (Lys212) (Figure 5(b)), although other forces are at play to stabilize PLP: π-stacking interactions between the Tyr114 phenyl moiety and the PLP pyridine ring, and H-bonds between the PLP phosphate moiety and residues Gly90, Leu91, Ser209, and Thr211 from the same subunit and Tyr60 and Arg62 from the adjacent subunit. Structural studies on CSE in the presence of its inhibitor propargylglycine (PPG) attempted to explain its mode of action [50]. While the lysine-PLP bond appears to remain unaffected, PPG is proposed to covalently bind to Tyr114, becoming a vinyl ether, while also forming H-bonds between its amino group and Glu339 and between its carboxyl moiety and Arg119 and Arg62 from the other monomer. This static position of the covalently bound PPG vinyl ether is such that it extends towards the internal PLP aldimine, thus blocking the cofactor reactivity [50].
Figure 5.
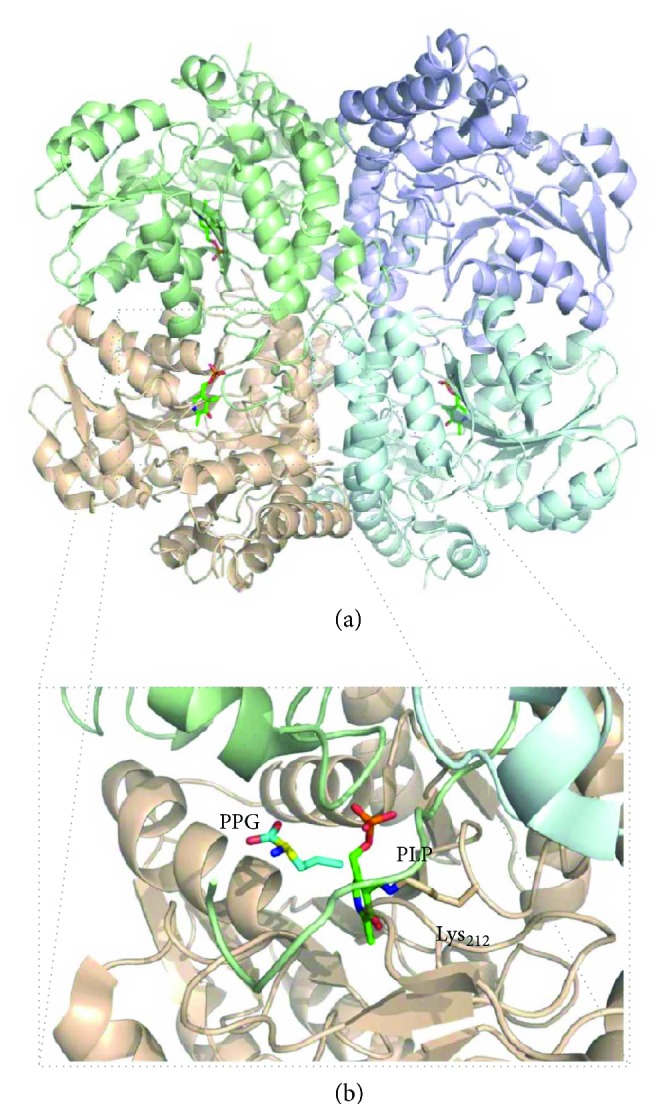
Crystallographic structure of human cystathionine γ-lyase (CSE). (a) Cartoon representation of human CSE homotetramer (PDB ID: 3COG; 2.0 Å resolution) cocrystallized with the inhibitor propargylglycine (PPG). Each chain is represented in a different colour. Green sticks, active site PLP moiety where H2S production occurs. (b) Zoom-in into the catalytic PLP site. The PLP moiety (green sticks) is covalently attached to CSE through Lys212 (human CSE numbering); PPG is represented in blue sticks. Figure generated with PyMol 1.8.2.0 [238].
(3) Mercaptopyruvate Sulfurtransferase (MST). Human mercaptopyruvate sulfurtransferase (MST) is composed of 297 amino acid residues and assembles as a ~33 kDa monomer consisting of two structurally related domains with a rhodanese-like fold (Figure 6(a)) [43]. The N-terminal (residues 1–138) and C-terminal (residues 165–285) domains are tethered by a 26-amino acid linker that strongly interacts with both domains. The assembly of these structurally related domains could have arisen from gene duplication. Structural studies of MST in the presence of its 3-MP substrate have shown that it binds to cysteine 248 in the active site, which is located in a cleft between the two domains (Figure 6(b)) [43]. The reaction originates a persulfidated CysSSH-activated intermediate. Despite the significant substrate-induced chemical modification of the active site, a comparison between the structures of MST with and without bound 3-MP reveals that no major structural differences occur [43]. In a recent report on a compound screening campaign targeting MST, the structures of MST in complex with hit compounds revealed a strong interaction between the persulfidated Cys248 and the inhibitors' pyrimidone-like aromatic rings [51], without major structural changes.
Figure 6.
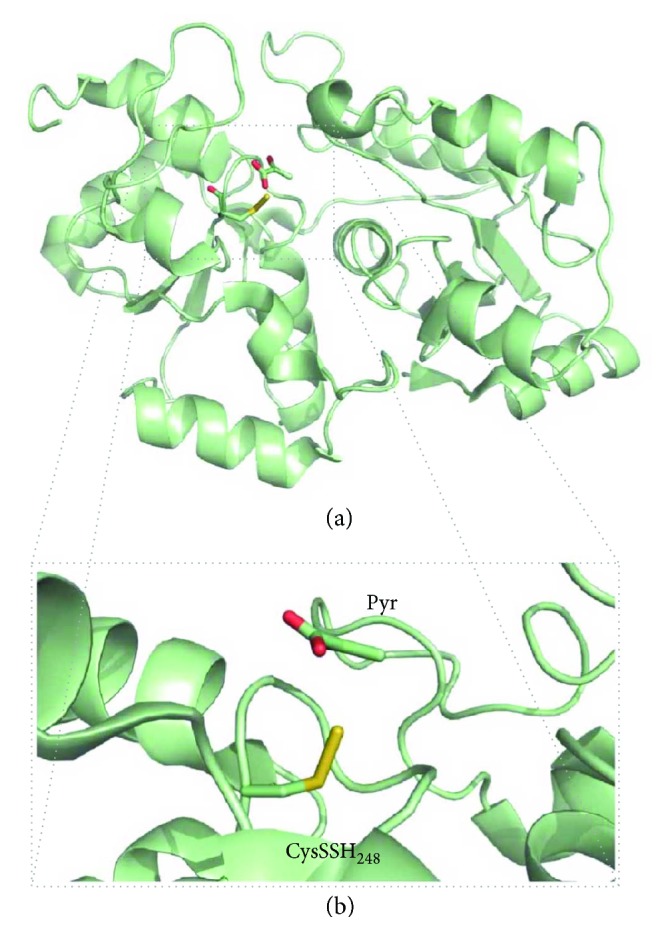
Crystallographic structure of human 3-mercaptopyruvate sulfurtransferase (MST). (a) Cartoon representation of human MST monomer (PDB ID: 4JGT; 2.16 Å resolution) cocrystallized with its substrate 3-mercaptopyruvate (3-MP). (b) Zoom-in into the catalytic persulfidated Cys248 (CysSSH248; human MST numbering) site, after reaction with 3-MP. Pyruvate, a by-product of the reaction of MST with 3-MP, is represented in green sticks. Figure generated with PyMol 1.8.2.0 [238].
2.2. H2S Catabolism
2.2.1. H2S: A “Self-Regulatory” Janus Molecule for Cellular Bioenergetics
As mentioned above, while playing a key signaling role only at relatively low, physiological concentrations, at higher concentrations H2S is potentially toxic, being able to impair cell respiration through inhibition of CcOX [52]. To prevent toxicity, H2S bioavailability must therefore be finely and differently regulated in different tissues and organs, depending on specific physiological demands. Control of H2S bioavailability is exerted not only at the level of its biosynthesis but also through enzymatic disposal of such a potentially toxic molecule. In mammals, the ability to detoxify H2S to thiosulfate and sulfate was demonstrated by metabolic labelling in early studies [53]. More recently, it was discovered that H2S breakdown is mainly afforded by a mitochondrial enzymatic system (reviewed in [10, 25]), currently designated as “sulfide-oxidizing unit” [54] or “sulfide-oxidizing pathway”. Following its initial identification in the lugworm Arenicola marina [55], this mitochondrial system has been investigated in more detail. According to current views, this pathway comprises four distinct, yet functionally associated enzymes that together cooperate to catalyze the breakdown of H2S to thiosulfate and sulfate, the main sulfide catabolites: a sulfide:quinone oxidoreductase (SQR), a persulfide dioxygenase (also known as ETHE1 or sulfur dioxygenase), a thiosulfate sulfurtransferase (rhodanese), and a sulfite oxidase (SOx) (Figure 7). It is to be noted that H2S oxidation to polysulfide can also be catalyzed by globins (see below) and other proteins, such as catalase and superoxide dismutase, utilizing O2 or H2O2 as electron acceptor [56–59].
Figure 7.
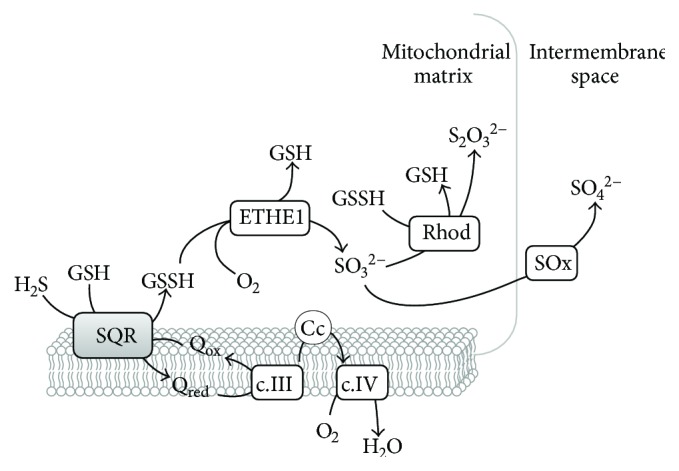
Mitochondrial sulfide-oxidizing pathway. Scheme depicting the enzymatic components and metabolites involved in sulfide oxidation in the mitochondria. H2S is initially oxidized by sulfide:quinone oxidoreductase (SQR), which transfers electron equivalents to quinones and generates glutathione persulfide (GSSH) as coproduct. Electrons are transferred to O2 via complex III (c.III), cytochrome c (Cc), and complex IV (c.IV), contributing to membrane energization and ATP synthesis. GSSH, with O2 as cosubstrate, is then converted by persulfide dioxygenase (ETHE1) to sulfite (SO32−) and GSH. Sulfite can be converted, with GSSH as cosubstrate, into thiosulfate (S2O32−) by rhodanese (Rhod), or oxidized into sulfate (SO42−) by sulfite oxidase (SOx).
From several perspectives, it is fascinating that H2S oxidation by mitochondria is coupled to energy production, namely, ATP synthesis, as initially demonstrated by investigating the invertebrate Solemya reidi [60]. Electrons derived from sulfide oxidation are indeed injected into the respiratory chain at the level of coenzyme Q, thus promoting O2 consumption and, in turn, energization of the inner mitochondrial membrane. On this basis, H2S has been recognized as the first inorganic respiratory substrate discovered in mammals [61]. This makes H2S a very peculiar molecule from a bioenergetic point of view [62], with a dual effect on mitochondrial respiration: stimulatory at low (nanomolar) concentrations when the mitochondrial sulfide oxidation pathway is fully operative or inhibitory at higher (micromolar) concentrations when CcOX activity is impaired and reduced coenzyme Q accumulates, leading to blockage of H2S detoxification. As remarked recently [63], the system is therefore finely regulated through positive feedback loops so that at lower concentrations sulfide oxidation prevents its own accumulation and consequent inhibition of respiration, whereas at higher concentrations sulfide impairs its own detoxification via inhibition of CcOX. That H2S can act as an effective substrate of the mitochondrial respiratory chain is in full agreement with the observation that, despite the low Ki value measured with isolated CcOX (Ki = 0.2 μM at pH 7.4 [52]), in isolated mitochondria or intact cells, inhibition of respiration only occurs if much higher concentrations of H2S are administered (micromolar to tens of micromolar; see, for instance, [64, 65]).
The dual effect of H2S on cell respiration makes investigation of the mitochondrial sulfide-oxidizing activity somewhat challenging from a methodological point of view. Sulfide stimulation of mitochondrial oxygen consumption and consequent energization are indeed best appreciated at low sulfide concentrations, unable to inhibit CcOX. For this reason, most of studies on mitochondrial sulfide oxidation and related bioenergetic effects have been carried out by continuous supply of sulfide at given infusion rates, rather than by sulfide addition as a single bolus (method reviewed in [66]). Using this approach, sulfide oxidation has been quantitatively investigated in several systems, including isolated mitochondria and permeabilized or intact cells from humans and other higher organisms [54, 61, 63, 67–72]. Notably, remarkable differences in terms of H2S oxidation ability have been reported between human cell lines, spanning from cells in which this catalytic activity is undetectable (as, e.g., in neural cells) to others remarkably active in H2S removal, like colonocytes [54]. The latter cells are indeed physiologically exposed to massive amounts of H2S produced by the intestinal microbiota and are therefore likely to have undergone adaptive mechanisms to prevent H2S toxicity. In this regard, it is interesting that colonocytes were found to ensure a prominent H2S-catabolyzing activity even through activity reversal of complex I in the respiratory chain [54].
2.2.2. Components of the Mitochondrial Sulfide-Oxidizing Pathway
The first enzyme operating in the mitochondrial sulfide oxidizing pathway is sulfide:quinone oxidoreductase (SQR, Figure 7). SQR is a member of the protein family of disulfide oxidoreductases, which has been identified in all life domains [73]. It is a membrane-associated protein located at the periplasmic side of the cytoplasmic membrane in prokaryotes and at the inner mitochondrial membrane in eukaryotes. Although no structure of a eukaryotic SQR has thus far been obtained, crystallographic structures of prokaryotic SQRs have been reported [74–76], enabling the construction of a structural model of human SQR (Figures 8(a) and 8(b)). Several structural and amino acid sequence features linked to sulfide oxidation and quinone reduction have allowed for the classification of the SQR family into six subclasses, with human SQR classified as a type II SQR [77]. The main features of type II SQRs, particularly as compared to type I SQRs, are essentially the lack of any extended loops, the poor conservation of quinone-interacting residues, and the substitution of the conserved cysteine covalently linking the FAD cofactor for a tyrosine (Tyr170 according to human SQR numbering), which probably establishes a covalent link with FAD in human SQR via an 8-α-O-tyrosyl bond [77]. SQRs can be monomeric or assemble into dimers or trimers of ~50 kDa subunits [78], each harboring a cysteine disulfide in its active site and a noncovalently bound flavin adenine dinucleotide (FAD) moiety implicated in electron transfer [77]. In mammals, SQR couples H2S oxidation to the reduction of coenzyme Q, with the concomitant transfer of a sulfur atom to an acceptor that has been proposed to be either sulfite- (SO32−-) yielding thiosulfate (S2O32−) [79–81] or reduced glutathione (GSH) yielding glutathione persulfide (GSSH) [82]. Although the protein displays a higher catalytic efficiency using as cosubstrate sulfite (kcat/KM = 2.5 × 106 M−1 · s−1) as compared to GSH (kcat/KM = 1.6 × 104 M−1 · s−1) [83], at physiological concentrations of both cosubstrates, GSH was suggested to be the preferential sulfur acceptor [82–84], at variance from [79–81]. In contrast to the mammalian protein, bacterial SQRs directly release the oxidized sulfur as a soluble polysulfide with up to ten sulfur atoms, instead of reacting with a sulfur acceptor [75, 85]. The reaction mechanism of SQR has been investigated with the isolated protein not only in detergent solution [80, 82, 84] but also after incorporation into nanodiscs [83], where slightly higher catalytic rates have been measured. The postulated mechanism involves (i) reaction of the active site cysteine disulfide with H2S associated with persulfidation of Cys379 (human SQR numbering) to form CysSSH and release of the Cys201 thiolate that in turn forms a charge transfer complex with FAD, (ii) transfer of the sulfane sulfur from the persulfidated Cys379 to the acceptor with the concomitant reduction of FAD to FADH2, and (iii) oxidation of FADH2 by coenzyme Q. According to kinetic data, the sulfane sulfur transfer step is the rate-limiting one in the reaction.
Figure 8.

Structural models of human sulfide quinone oxidoreductase (SQR) and rhodanese (Rhod). (a) Cartoon representation of structural model of human SQR (UniProt accession code: Q9Y6N5.1) generated with Swiss-Model based on the structure of Acidithiobacillus ferrooxidans SQR (H198A variant; PDB code: 3SZF; ~19% sequence identity; ~87% sequence coverage) [78]. Flavin adenine dinucleotide (FAD) cofactor in yellow sticks. (b) Zoom-in on the SQR active site comprising the FAD moiety, the active site cysteine residues Cys201 and Cys379, and the Tyr170 residue which may establish a covalent link with the FAD cofactor. (c) Cartoon representation of structural model of human rhodanese (UniProt accession code: Q16762.4) generated with Swiss-Model based on the structure of the bovine enzyme (PDB code: 1BOH; ~90% sequence identity; 100% sequence coverage). (d) Zoom-in on the Rhod Cys248 active site. Figure generated with PyMol 1.8.2.0 [238].
The next step in the sulfide oxidation pathway is catalyzed by ETHE1 (Figure 7). Mutations in the gene encoding this protein were found to result into an autosomal recessive disorder, known as ethylmalonic encephalopathy, characterized by a severe clinical picture and leading to death during early childhood [86, 87]. The crystallographic structure of human ETHE1 was recently solved (Figure 9) [88]. ETHE1 is a dimeric enzyme, each monomer comprising an αββα fold structurally related to dizinc classical metallo-β-lactamases, harboring a mononuclear nonheme iron octahedrally coordinated by two histidines, one aspartate, and three water molecules [88]. The enzyme is proposed to catalyze the oxygenation of the sulfane sulfur atom in the GSSH released from SQR, yielding sulfite [55]. It is important to note that one O2 molecule is consumed in the reaction.
Figure 9.
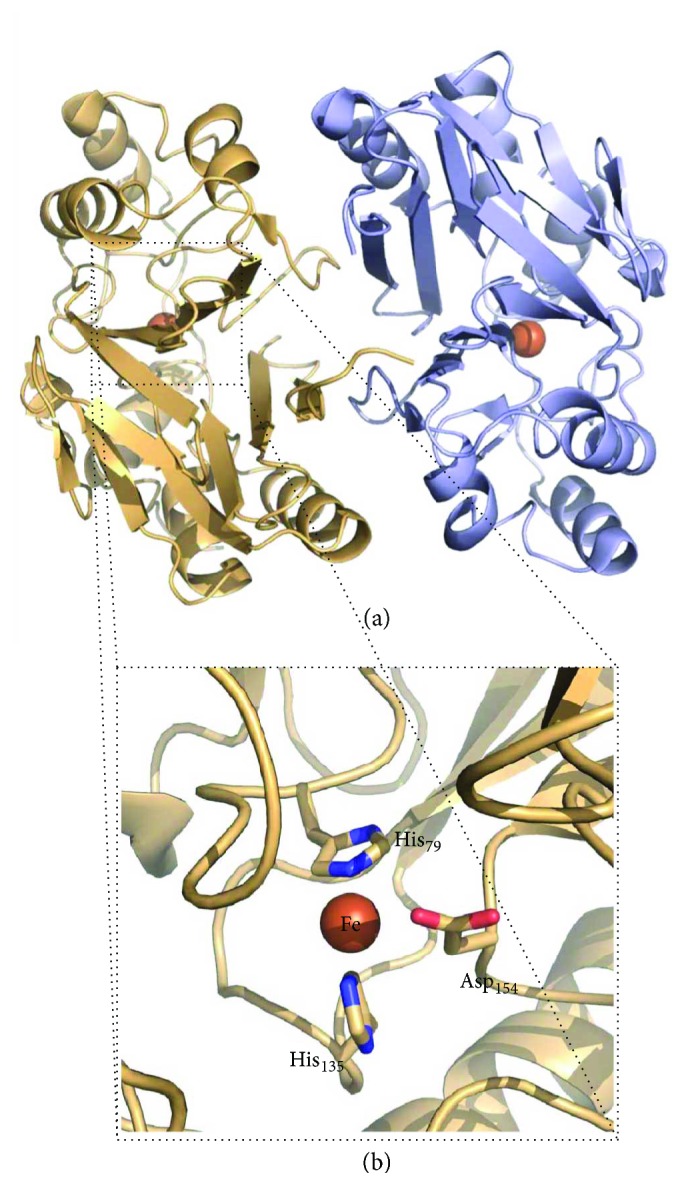
Crystallographic structure of human persulfide dioxygenase (ETHE1). (a) Cartoon representation of human ETHE1 homodimer (PDB ID: 4CHL; 2.61 Å resolution). (b) Zoom-in into the mononuclear nonheme iron catalytic site. Iron ligands (His79, His135, and Asp154; human ETHE1 numbering) are represented in sticks. Figure generated with PyMol 1.8.2.0 [238].
GSSH and sulfite are further converted into thiosulfate and GSH by rhodanese (Rhod, Figure 7). No structural studies of the human enzyme have been reported, yet a structural model can be obtained based on the ~90% identical bovine homologue (Figures 8(c) and 8(d)) [89]. This protein has a redox-active cysteine (Cys248) in the active site and is highly promiscuous in that it can effectively react with several different substrates. Yet, at physiologically relevant substrate concentrations, the most likely reaction catalyzed by rhodanese was suggested to be the transfer of a sulfur atom from GSSH to sulfite to form thiosulfate [82]. Sulfite oxidase (SOx, Figure 7) catalyzes the last step in the sulfide oxidation pathway. SOx is a multidomain cytochrome b5 with a molybdenum cofactor [90] and a heme mediating the intramolecular electron transfer from sulfite to cytochrome c. Concomitantly, an oxygen atom is supplied by an H2O molecule and sulfate (SO42−) forms as the end-product of the reaction [90].
Overall, the mitochondrial sulfide-oxidizing pathway couples the oxidation of H2S to thiosulfate and sulfate with the injection of electrons into coenzyme Q, leading to consumption of 0.79 O2 per H2S molecule oxidized, as reported by Goubern and coworkers [61], which is compatible with the predicted value of 0.75. Indeed, based on the postulated architecture of the mitochondrial sulfide oxidation pathway presented above, the oxidation of 2 H2S molecules (involving 2 electrons) is expected to require the consumption of 1.5 O2 molecules (0.5 by CcOX plus 1 by ETHE1). As O2 is required for sulfide removal, the efficacy of mitochondrial sulfide oxidation is expected to depend on O2 availability, clearly vanishing under anoxic conditions. Evidence for a decline of mitochondrial sulfide oxidation at lower O2 concentrations was preliminarily provided in [69] and, then, substantiated in a later study [63]. To be noted that at very low O2 concentrations (down to 0.73 ± 0.04 μM), given a fixed amount of sulfide, a faster onset of cell respiration inhibition was observed at decreasing O2 concentrations [69], which is consistent with a lower efficacy of sulfide removal but also with a higher control of the respiratory electron transfer chain by CcOX at low O2 concentration.
Our knowledge of H2S catabolism and, particularly, of its implication in human physiology and pathophysiology is still rather elusive. Current views of the mitochondrial sulfide oxidation pathway are yet rather patchy and represent a matter of debate. It is likely that the physiological function of this pathway goes well beyond the mere removal/detoxification of H2S. The impact of this pathway on cell bioenergetics under specific physiological or pathological conditions needs to be better established, as well as its ability to produce bioactive sulfur metabolites (e.g., thiosulfate) whose physiological role has only recently begun to emerge. Finally, it can be anticipated that the mitochondrial sulfide oxidation pathway plays a key role in mediating the intricate interplay between sulfide, O2, and the other gasotransmitters, but more studies are needed in this direction.
3. Signaling Mediated by Hydrogen Sulfide
The relevance of hydrogen sulfide in mammalian physiology results essentially from its role as a second messenger that transduces signals by interaction with target proteins. The initial classical view of H2S-mediated signaling initially merely considered the inhibitory properties of hydrogen sulfide towards respiratory CcOX (detailed below) leading to a state of suspended animation. Presently, the signal transducing function of hydrogen sulfide is known to occur via at least two major mechanisms, namely, through (i) interaction with protein metal centers, particularly heme moieties, and (ii) protein persulfidation (detailed below).
3.1. H2S and Heme Proteins
Besides being able to promote thiol persulfidation, H2S can react with protein hemes. As reviewed in Bianco et al. [91], the mechanism and fate of the reaction of H2S with a heme protein largely depend on several factors, such as the redox and ligation state of the heme iron, the environment in the heme pocket, the protonation state of bound sulfide, and the presence/absence of O2 or reducing agents in solution. Heme-Fe(III) can bind H2S as such or as HS− (Figure 10). Stability of one or the other heme adduct generated by reaction with hydrogen sulfide (heme-Fe(III)-H2S or a heme-Fe(III)-HS−) depends on the protein residues in the heme surroundings and, particularly, on the presence of either basic residues able to accept protons from bound H2S to yield the heme-Fe(III)-HS− species or, alternatively, nonpolar residues stabilizing the fully protonated state of the ligand. Owing to its reducing character, bound sulfide can reduce the heme iron, yielding a heme-Fe(II)-HS· radical adduct that can further react with excess HS−/HS· or O2/H2O leading, respectively, to formation of polysulfides or thiosulfate. In contrast to these reactions, H2S can also promote a covalent modification of the heme, yielding the so-called sulfheme derivative with a sulfur atom incorporated into one of the porphyrin pyrrole rings (Figure 10). The mechanism of sulfheme formation is still unclear. Sulfheme can be formed by reaction of H2S with either the ferrous oxygenated (heme-Fe(II)-O2 [92]) or the ferryl heme (heme-FeIV=O [93, 94]). However, also in the former case the reaction requires H2O2 and thus, likely, the formation of higher valent heme iron intermediates [95, 96]. As reviewed in [5, 91, 95–97], the reactions above have been described by several groups working on numerous hemeproteins, including, among others: globins from mammals, invertebrates, and bacteria; heme-based sensor proteins of diatomic molecules such as O2, NO, and CO; cytochrome c oxidase; catalase; and peroxidases (lactoperoxidase, myeloperoxidase and thyroid peroxidase).
Figure 10.
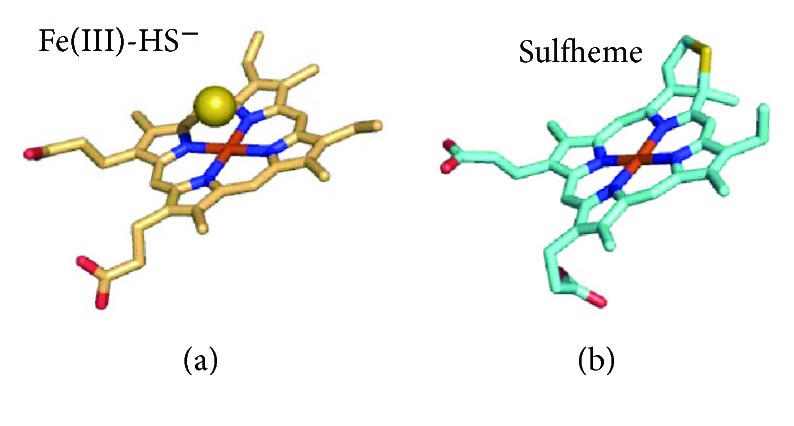
Structure of sulfide-reacted heme moieties in globins. (a) Structure of ferric heme moiety (light orange sticks) in human hemoglobin with bound HS− (yellow sphere); PDB entry: 5UCU [115]. (b) Structure of sulfheme moiety (light blue sticks) generated by reaction of horse myoglobin and sulfide, resulting in sulfmyoglobin; PDB entry 1YMC [239].
Because heme proteins can usually react not only with H2S but also with NO, CO, and O2, they can play a key role in mediating the crosstalk between these gaseous ligands, namely, controlling each other's biological function. A paradigm of this concept is CcOX. The enzyme, highly reactive with O2, is indeed a recognized target for all three gasotransmitters (NO, CO, and H2S). Each of these species can inhibit CcOX, though with markedly different kinetics and mechanisms [29]. While CO can only bind to the fully reduced heme a3-CuB active site, leading to competitive inhibition of the enzyme [98], CcOX inhibition by NO can proceed through two alternative reaction pathways (reviewed in [99–104]): an O2-competitive pathway, favoured at lower O2 tension and higher electron flux, leading to a nitrosyl adduct of CcOX with NO bound to ferrous heme a3, and an O2-uncompetitive one, prevailing at lower electron flux and higher O2 tension, whereby an inhibited adduct of enzyme forms with nitrite bound at ferric heme a3 [29, 105–107]. The mechanism of CcOX inhibition by H2S has not been investigated as thoroughly as for NO and CO. Differently from these two species, H2S does not bind ferrous heme a3. Based on the postulated mechanism, when the enzyme in turnover with O2 is exposed to H2S, oxidized or reduced CuB is the primary target site in CcOX. Afterwards, sulfide is thought to be transferred intramolecularly to the nearby ferric heme a3, resulting in enzyme inhibition [30]. H2S-inhibited CcOX thus exhibits one sulfide molecule bound to ferric heme a3 [108, 109] and, possibly, a second one bound to cuprous CuB, as revealed by electron paramagnetic resonance (EPR) spectroscopy [110]. Regardless of the exact molecular mechanism, CcOX inhibition by H2S was demonstrated to be relatively fast (initial rate constant of 2.2 × 104 M−1·s−1, as measured at pH 7.4 with the enzyme in turnover with O2 and cytochrome c [30]), effective (Ki = 0.2 μM at pH 7.4), independent of O2 concentration, and reversible [52]. Similarly to other heme proteins, CcOX inhibition by H2S was found to be greatly dependent on pH. Acidic conditions remarkably enhance the inhibitory action of sulfide, and consistently, as the pH shifts from 8.05 to 6.28, Ki decreases from 2.6 μM to 0.07 μM [111]. The enhanced efficacy of sulfide inhibition at lower pH suggests that sulfide binds to CcOX either as H2S (e.g., in fully protonated state) or as HS− with the concomitant protonation by a protein residue. This is consistent with the active site of the enzyme being located in a nonpolar environment, therefore facilitating the binding of electroneutral or proton-neutralized anionic species [112].
H2S can also act as a reducing substrate for CcOX [30, 111, 113], when the enzyme is in the so-called pulsed state [30], but the reaction products are yet to be characterized. By reacting with H2S at relatively low concentrations, heme a3 in the active site of the enzyme is promptly reduced and, by further reacting with O2, leads to a ferryl species with optical features indistinguishable from those of the “P” catalytic intermediate [30]. Afterwards, the “F” ferryl intermediate spontaneously forms on a longer timescale, but it is unclear if the reaction results from autoreduction or electron donation from residual sulfide [30]. CcOX is therefore in principle able to catalyze the oxidative breakdown of H2S, but the reaction is probably too slow to be physiologically relevant, particularly when compared with the high H2S-metabolizing activity of SQR.
Another group of proteins whose reactivity with H2S has been investigated in detail is the globin family. These studies focused primarily, but not exclusively, on human hemoglobin (Hb) and myoglobin (Mb). The reaction of sulfide with Hb has long been known to lead to irreversible covalent conversion of the protein heme to sulfheme (reviewed in [96]); as described above, the modification consists in the incorporation of a sulfur atom into the heme pyrrole B. The resulting protein derivative, named “sulfhemoglobin,” exhibits a characteristic green color and lower O2 affinity as compared to the native protein. Accumulation of sulfhemoglobin is therefore associated to toxicity, leading to a rare pathological condition known as “sulfhemoglobinemia.” A similar reactivity has been demonstrated also for Mb, leading to formation of “sulfmyoglobin” (Figure 10). Whereas sulfhemoglobinemia likely represents a form of sulfide toxicity, methemoglobinemia, that is, the accumulation of ferric hemoglobin (metHb) in the blood, has been suggested to protect against sulfide toxicity in mice [114]. In these in vivo studies, 2–4 mol of H2S was inferred to be inactivated per mol of metHb [114]. This superstoichiometric value suggests that metHb is able not only to bind H2S but also to promote its catalytic breakdown. Consistently, under aerobic conditions metHb was recently demonstrated to bind H2S, oxidizing it to a mixture of thiosulfate and polysulfides [21]. The reaction, though proceeding at relatively low rates (3 min−1 in the presence of 0.5 mg·mL−1 metHb, at pH 7.4 and 25°C [21]), could be of physiological relevance, as Hb-rich red blood cells lack mitochondria and therefore cannot dispose H2S via the mitochondrial sulfide-oxidizing pathway. Sulfide is a relatively low-affinity ligand for metHb: at pH 7.4 and 37°C, it binds with a kon of 3.2 × 103 M−1·s−1 and dissociates from the oxidized heme with a koff of 0.053 s−1, yielding a KD of 17 μM (whose value may be overestimated, given that the H2S species represents only a fraction of sulfide at pH 7.4). The rate constant for H2S binding to metHb was found to increase with decreasing pH, and on this basis, H2S rather than the HS− species, more abundant at pH 7.4, initially binds to the heme. After binding to the heme iron, H2S is proposed to deprotonate to HS−. The postulated Fe(III)-HS− species has been recently detected by X-ray crystallographic analysis of the sulfide-reacted metHb (Figure 10) [115].
Possible mechanisms for polysulfide and thiosulfate formation by metHb have been proposed in [21]. Interestingly, the newly formed polysulfide remains bound to the protein upon heme iron reduction by methionine sulfoxide reductase (MSR). In contrast, it reacts with physiologically relevant, low-molecular-weight (LMW) thiol compounds, such as reduced glutathione (GSH) or cysteine (CysSH), yielding the corresponding persulfides, GSSH and CysSSH, that are eventually released into the solution. In light of the high intracellular concentration of GSH, Hb can act as a source of GSSH that in turn could serve as a persulfide donor in protein persulfidation (detailed below). Therefore, based on its intricate chemistry with sulfide, depending on conditions, Hb can mediate sulfide toxicity via formation of sulfhemoglobin or be protective by promoting sulfide disposal, a particularly relevant function in red blood cells lacking mitochondria. Furthermore, Hb can be viewed as a source of physiologically relevant sulfide oxidation products, namely, thiosulfate and glutathione persulfide (GSSH), whose impact on human physiology is emerging. A somewhat different scenario occurs in atherosclerotic lesions, particularly when they are infiltrated by red blood cells. Indeed, in those lesions, oxidized forms of Hb with the heme in the ferric and ferryl state accumulate and mediate toxicity by promoting radical reactions leading to formation of cross-linked Hb species and lipid modification. Under these circumstances, H2S was recently reported to exert protective effects, acting as a reductant of ferryl Hb [116].
Another heme protein whose reactivity with sulfide was investigated in depth is human leukocyte myeloperoxidase (MPO). Based on recent studies [117, 118], upon reaction with sulfide, the enzyme does not lead to formation of the sulfheme derivative, at variance with other heme proteins. Of interest, MPO was reported to catalyze the oxidation of sulfide to sulfane sulfur species both in the presence and in the absence of H2O2 [117, 118]. The reaction involves as a central intermediate compound III, a resonance form between the ferrous/dioxygen (Fe(II)-O2) and ferric/superoxide (Fe(III)-O2·−) complex, it is facilitated by ascorbate or SOD and might be of physiological relevance as the formed sulfane sulfur species were proven to oxidize protein cysteine residues to their corresponding per/polysulfide derivatives [117].
3.2. Persulfidation and Persulfide (Bio)chemistry
As mentioned above, many of the signaling functions attributed to H2S have been shown to be tightly associated with formation of persulfides through the modification of specific cysteine side chains from target proteins, often involving free reactive LMW persulfides (RSSH), particularly cysteine persulfide (CysSSH) and glutathione persulfide (GSSH). Polysulfides are also considered as potentially relevant from a physiological viewpoint, as reviewed elsewhere [119]. However, because they are contaminants of most inorganic H2S donors employed in research and therefore possibly responsible for some of the effects attributed to H2S, their (bio)chemistry is still far from being fully understood and will not be detailed herein. The relevance of persulfidation is attested by the fact that deficiency in persulfidated proteins has been associated with different pathologies, including cardiovascular and neurodegenerative (Parkinson's) diseases [120, 121]. Besides its signaling function, persulfidation may also constitute a mechanism to prevent thiol oxidation or electrophilic modification and irreversible damage [122]. The ability of LMW persulfides to readily donate one electron and form a stable perthiyl radical (RSS·), unreactive towards oxygen or NO, led to the proposal of a possible role in redox processes [123]. Recently, the oxidation of cysteine persulfides to perthiosulfenic acid (CysSSOH) observed in different proteins as a consequence of NADPH oxidase activation led to the proposal of this modification as an additional redox-based signaling event [124]. The chemical and biochemical properties, biosynthesis and natural occurrence, analytical detection methods, and cellular longevity of persulfides and polysulfides have all been covered in excellent articles and reviews (e.g., [3, 6, 120, 121, 125–129]). As in other fields focused on reactive molecules, there seem to be controversies as to which species and reactions are more plausible and relevant from a chemical-to-physiological viewpoint. Presently, there is however a consensus that H2S cannot react directly with protein cysteine thiols. Protein persulfidation, in order to occur, has special requirements both at a “local” (the environment surrounding the target cysteine) and a “global” (redox status, free glutathione/cysteine/H2S availability) level. Currently, although several possibilities for the formation of persulfidated proteins through posttranslational modification can be envisaged, only four appear to be more plausible (Figure 11(a)). Two of these possibilities involve previous oxidation of the protein target cysteine thiol to a sulfenic moiety (CysSOH), for example, by reaction with hydrogen peroxide, or to a disulfide (CysSSR) by reaction with an oxidized sulfur-containing LMW molecule, for example, oxidized glutathione (GSSG). Both of these oxidized cysteine residues can then be targeted directly by H2S, resulting in a persulfidated protein. Reduced cysteine residues (e.g., protein thiols) can be modified to their persulfide derivatives through reaction with a free LMW persulfide (RSSH), such as GSSH or (homo)cysteine persulfide (derived from the H2S biosynthetic or catabolic pathway [125, 126]), or via reaction with HS·, generated upon reaction of H2S with metal centers, particularly oxidized protein heme iron. The protein cysteine thiol reaction with HS· should yield a transient CysSSH·− that, upon reaction with oxygen, yields the protein-bound CysSSH and superoxide anion, similar to that reported for HSSH·− [3, 130]. In addition to posttranslational persulfidation, it has been quite recently posited that in mammals a major source of persulfidated (and polysulfidated) proteins in vivo are actually cysteinyl-tRNA synthetases (CARSs, particularly the mitochondrial CARS2), through cotranslational incorporation of cysteine persulfide or polysulfide at cysteine sites into the nascent polypeptide [131] (Figure 11(b)). From a cellular signaling perspective, the advantage of protein persulfidation by H2S and/or persulfides is that the resulting species can be reverted to the thiol moiety in different ways, such as direct reaction with reduced glutathione (GSH) or through the thioredoxin system [126], thus affording a reliable switching modification.
Figure 11.

Reactions leading to protein persulfidation. Scheme depicting the most plausible in vivo reactions leading to cysteine persulfidation in target proteins, through (a) posttranslational modification of cysteine residues or (b) cotranslational incorporation of CysSSH via CysSSH-bound tRNA derived from cysteinyl-tRNA synthetases (CARSs). (a) Proteins with oxidized cysteine residues (CysSSR and CysSOH) can react directly with H2S (derived from its synthesizing enzymes CBS, CSE, and MST) to yield a protein cysteine persulfide (CysSSH). Proteins with reduced cysteine residues (CysSH) can react with free LMW persulfides (RSSH, such as glutathione persulfide and cysteine persulfide, products of H2S biosynthetic—CBS, CSE, and MST—or catabolic—SQR—enzymes) or with the HS· radical formed by reaction of H2S with metal centers (Mn+). The resulting protein-bound cysteine perthiyl radical (CysSSH·−) can then react with oxygen to yield CysSSH and superoxide anion. (b) Generation of protein cysteine persulfides through cotranslational incorporation of CysSSH. Mammalian cysteinyl-tRNA synthetases (CARSs, dark blue box, which also synthesize free CysSSH from cysteine) catalyze the synthesis of tRNA-bound cysteine persulfides (light blue line), which incorporate CysSSH into the nascent polypeptide (dark blue line) at cysteine sites upon translation (ribosome depicted in green and mRNA in grey).
As for the LMW persulfides, which are key molecules in protein persulfidation, they are increasingly coming into the limelight due to their intrinsic reactivity (e.g., [125]). Until recently, the two major sources of these persulfides, particularly cysteine persulfide (CysSSH) and glutathione persulfide (GSSH), were considered the enzymes involved in H2S metabolism. CysSSH is readily generated by the H2S-synthesizing CBS and CSE, using cystine (CysSSCys) as substrate [125]. Notably, in the course of this reaction, longer polythiolated products with catenated sulfur atoms, such as CysSSSH (synthesized by both CBS and CSE) and CysSSSSH (synthesized by CSE), have been detected. The enzymatic generation of these species further results in appreciable amounts of oxidized polysulfide species like CysSSSCys, CysSSSSCys, and CysSSSSSCys. The newly formed CysSSH and longer cysteine persulfides can then react with reduced glutathione to yield GSSH and GS(S)nH. It should however be noted that, at physiologically relevant substrate concentrations, H2S rather than CysSSH is expected to be the major product of the transsulfuration pathway enzymes [132], also taking into account that in the cytoplasm, cysteine and reduced glutathione are much more abundant than their oxidation products cystine and GSSG [132]. Recently, MST was also recognized as a source of CysSSH and GSSH, as well as of longer polysulfides, related with its ability to generate other RSS, such as hydrogen persulfide (H2S2) and hydrogen trisulfide (H2S3) [133, 134]. MST-derived persulfides and polysulfides have been associated with higher cellular levels of protein-bound sulfane sulfur [134, 135]. The MST-catalyzed CysSS(n)H and GSS(n)H production was demonstrated with recombinant MST (wild-type and site-directed variants), MST-expressing COS cells lysates, and mouse brains (cell suspensions and whole brains) [133], showing that in the brain LMW persulfides and protein persulfidation are essentially generated via MST and not CBS or CSE [133]. A principal role in CysSSH (and CysSS(n)H) synthesis has been recently proposed for mammalian CARSs, particularly the mitochondrial isoform CARS2 [131]. The same report suggests that mitochondria are the key cellular compartments where CysSSH is formed before being released into the cytosol to exert its effects. It is also posited that whereas CARSs may be the major source of CysSSH under physiological conditions, CBS and CSE may still act as major players in CysSSH synthesis in pathophysiological conditions associated with oxidative and electrophilic stress with concomitantly increased cystine concentrations [125, 131, 132].
Besides the LMW persulfides derived from the H2S-synthesizing enzymes CBS, CSE, and MST [125, 132, 133], GSSH is a metabolic intermediate of the mitochondrial sulfide-oxidizing pathway [126] (Figure 7). Indeed, GSH has been suggested as the preferential sulfur acceptor in the H2S oxidation reaction catalyzed by SQR, yielding GSSH [82]. This persulfide is, in turn, the preferential substrate (with sulfite as cosubstrate) for rhodanese to generate thiosulfate as the final oxidation product of sulfide oxidation, together with sulfate [82].
Regardless of how LMW persulfides are generated, their reactivity prompts them to be major signal-transducing species linked to H2S (patho)physiology. In a nutshell, persulfides are stronger nucleophiles than their thiol counterparts are (yet displaying also a weak electrophilic character). Despite this assumption, only recently it was clearly demonstrated by Cuevasanta et al. [136] that a human serum albumin persulfide derivative displays 20-fold increased reactivity with respect to its thiol counterpart. Persulfides have lower pKa values than the analogous thiols, thus existing mostly in their anionic nucleophilic RSS− form [3, 6]. Conversely, their weak electrophilicity is exhibited solely in their protonated RSSH form. Interestingly, both the outer sulfhydryl and inner sulfenyl sulfurs have electrophilic character, the reaction occurring on either one depending on factors such as steric hindrance or acidity of the leaving group [3]. The ambiguous nucleophilic/electrophilic nature of LMW persulfides, while at the basis of their physiological relevance, accounts for their instability. These species indeed quickly decay or react with other species, particularly oxidants, hampering their characterization both in vitro and in vivo. This notwithstanding, the molecular details and functional consequences of persulfidation at specific cysteine residues have been demonstrated for a number of targets and shown to regulate several physiological processes (reviewed in, e.g., [127]), such as glycolysis [26], ER stress [137], apoptosis [138], vasorelaxation, and vasodilation [139] (detailed in Table 1). Regarding the latter, H2S was the third gasotransmitter, following NO and CO, to be regarded as an endothelium-derived relaxing factor (EDRF) and as an endothelium-derived hyperpolarizing factor (EDHF), thus regulating vasodilation (detailed below). Despite the relatively few examples available in the literature, protein persulfidation is likely prevalent, yet technically challenging to be detected due to the intrinsic reactivity of persulfides. Using an adaptation of the biotin switch assay modified to detect persulfidated rather than nitrosated proteins, Mustafa et al. reported that 25–50% of hepatic proteins are persulfidated [26]. The extent of protein persulfidation in vivo has been further demonstrated by increasingly elaborate and quantitative methodologies [125, 140–143]. It is expected that the number of identified new targets of protein persulfidation will continuously increase, pairing this posttranslational modification with other highly relevant cellular switches.
Table 1.
Protein targets for cysteine persulfidation and molecular and cellular consequences.
| Protein | Target cysteine | Physiological process | Consequences of persulfidation | Refs. |
|---|---|---|---|---|
| Protein Tyr phosphatase 1B (PTP1B) | Cys215 | ER stress response | PTP1B is inhibited, enhancing PERK(a)-mediated phosphorylation of eIF2α(b), suppressing protein translation. | [137] |
| p65 subunit of nuclear factor кB (NF-кB) | Cys38 | NF-κB signaling; apoptosis | Stimulates binding of p65 to ribosomal protein S3, resulting in increased translation of prosurvival genes. | [138] |
| Inwardly rectifying potassium channel subunit Kir6.1 | Cys43 | Vasorelaxation and vasodilation | Enhances channel activity through decreased affinity for ATP and increased binding of PtdIns(4,5)P2(c). | [139] |
aProtein kinase RNA-like ER kinase; beukaryotic translational initiation factor 2α; cphosphatidylinositol-4,5-bisphosphate.
4. Interplay between Gasotransmitters
4.1. Brief Historical Account of Gasotransmitters
The term “gasotransmitters” [144] has entered the biochemistry and physiology jargon to designate essentially the three small molecules gaseous in nature with demonstrated roles in signal transduction: nitric oxide (NO; although the formally correct abbreviation should be NO·, herein NO will be used for simplicity), carbon monoxide (CO), and hydrogen sulfide (H2S). Arguably, molecular oxygen (O2), although not synthesized endogenously in mammalian cells, could be considered a gasotransmitter due to its gaseous nature, reactivity, and physiological relevance. Besides, O2 is a substrate of the mammalian enzymes that synthesize NO and CO, so O2 levels dictate the bioavailability of the latter. Gasotransmitter-mediated signaling is an intricate and integrated process involving not only NO, CO, H2S, and O2 but also the collective pool of reactive species (reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species (RSS) resulting from their cross-reactions, establishing a so-called reactive species interactome (RSI) [145]. Irrespectively of the identity of the gasotransmitter molecule or derived species, its signaling mode of action can be narrowed down essentially to two possibilities: interaction with metal centers or modification of cysteine residues in proteins, both resulting either in the activation or in the inactivation of the target protein. The three gasotransmitters NO, CO, and H2S share a common history in that they were initially merely regarded as toxic and poisonous molecules, until the discovery that virtually all life forms, from bacteria to man, are endowed with dedicated synthesizing and detoxifying enzymes and produce or scavenge NO, CO, and H2S to accomplish specific functions. Indeed, in the late 1980s and early 1990s, major advances concerning the role of NO in the regulation of cardiovascular function led to NO being considered Molecule of the Year in 1992 and to the Nobel Prize in Physiology or Medicine being awarded in 1998 to Robert Furchgott, Ferid Murad, and Louis Ignarro Jr “for their discoveries concerning nitric oxide as a signaling molecule in the cardiovascular system” [146]. One of the key findings was the identification of the endothelium-derived relaxing factor (EDRF) as NO [147, 148]. This observation, which revealed beneficial rather than deleterious effects of an otherwise considered toxic molecule, triggered a subfield of biology dedicated to signaling by gaseous second messengers. Indeed, besides other regulatory roles attributed to NO, similar findings were demonstrated for CO, which was also shown to have a role in vasorelaxation [149]. The same function came to be assigned to H2S [17], which also led it to be labeled as an EDRF [127]. The multiple functions of gasotransmitters in human physiology are well attested by the “explosion” of interest that arose since the discovery of NO as a signaling molecule in the mid-1980s, CO in the mid-1990s, and H2S in the 2000s. This is well illustrated by carrying out a Pubmed search querying “gasotransmitters,” “nitric oxide,” “carbon monoxide,” and “hydrogen sulfide” (Figure 12). Altogether, the three gasotransmitters regulate, often through similar molecular processes, vasodilation, energy metabolism, redox homeostasis, apoptosis, cell cycle, reproduction, neuronal function, and so on, and dysregulation of the metabolism and/or levels of NO, CO, or H2S is associated with several pathological conditions, from cardiovascular and neurodegenerative diseases to diabetes and cancer, among others.
Figure 12.
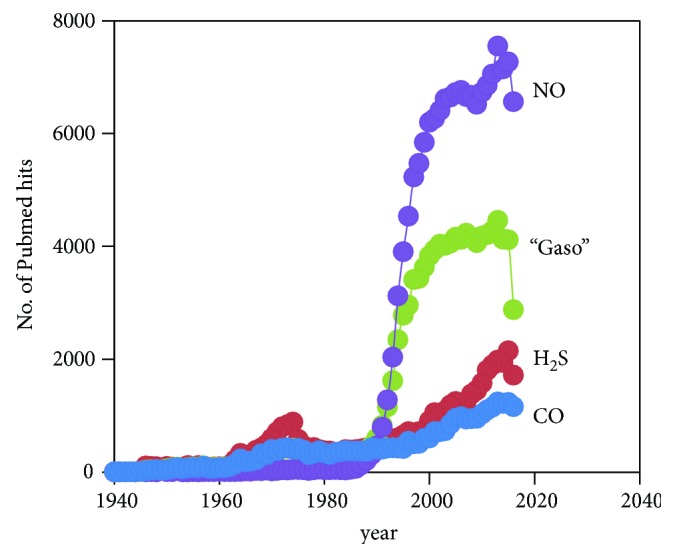
Literature on gasotransmitters, H2S, NO, and CO. Number of articles retrieved from Pubmed queries with the following terms: “hydrogen sulfide” (H2S), “gasotransmitters” (“gaso”), “nitric oxide” (NO), and “carbon monoxide” (CO).
A surprising element in gasotransmitter history was the finding that mammals, among many other organisms, encode specialized enzymes that synthesize these previously considered noxious gaseous molecules. NO is enzymatically produced by NO synthases, of which three isoforms have been described and historically classified according to their location or regulation, yet presently considered to be ubiquitous and constitutively expressed: inducible NO synthase (iNOS), endothelial NO synthase (eNOS), and neuronal NO synthase (nNOS) (reviewed, e.g., in [150]). Regardless of the isoform and its localization or regulation, NO synthases catalyze the conversion of arginine and O2 into NO and citrulline, using electron equivalents derived from NADPH. Besides being synthesized on demand by NO synthases, NO is endogenously generated (i) under hypoxic conditions through nitrite reduction by several heme proteins (including hemoglobin, as reviewed in [151]), (ii) in the digestive tract as an intermediate of bacterial nitrogen metabolism (particularly in the mouth and the gut), and (iii) chemically from nitrite under the acidic environment in the stomach (reviewed, e.g., in [152]). As for CO, it is generated by either isoform of heme oxygenase, HO-1 and HO-2, which catalyze the conversion of heme, with O2 as cosubstrate, into biliverdin and CO, employing NADPH-derived electron equivalents (reviewed, e.g., in [153]). H2S synthesis is catalyzed by CBS, CSE and MST, as described above.
As mentioned before, it has become increasingly clear that no gasotransmitter should be regarded as an isolated entity with an independent cellular function and lifespan. Rather, gasotransmitters interact with each other not only by direct cross-reactions that generate other reactive species but also by having cooperative or opposite molecular and cellular consequences towards their targets. Moreover, each gasotransmitter in many ways is able to modulate the synthesis of the others, which represents an additional intricate level of regulation. An example of this modulatory action is described below, concerning the regulation of CBS-catalyzed H2S production by NO and CO.
4.2. Chemical Bases of the Interplay between H2S and NO
The biological effects of H2S and NO are intimately interconnected. The interplay between these two gaseous molecules takes place through numerous mechanisms acting at different levels (reviewed in [154]). Many studies aimed at gaining insight into the chemical foundation of the interaction between H2S and NO. An extensive analysis has been carried out of the reactivity of sulfide with several NO metabolites, including nitrite [155], peroxynitrite [156, 157], and nitrosothiols [158–163], and with NO releasers such as nitroprusside [163–167]. Despite these efforts, the picture is currently far from being completely clear. As discussed in [5, 168–171], some remarkable discrepancies have indeed emerged, mainly regarding the nature of the species generated in these reactions and their suitability as signaling molecules under physiological conditions.
The first S/N hybrid species suggested to form by reaction of nitrosothiols (e.g., s-nitrosoglutathione (GSNO)) with sulfide was thionitrous acid/thionitrite (HSNO/ONS−) [161]. In line with previous studies by Whiteman et al. [172, 173], Filipovic et al. [161] reported the formation of HSNO by reaction of H2S with either GSNO or acidified nitrite, as well as by investigation of the reaction of NO with HS· generated by pulse radiolysis. In addition to HSNO, the reaction of GSNO with sulfide was found to yield also a yellow product assumed to be a mixture of polysulfides. Of interest regarding its potential physiological implications, HSNO was proposed to serve not only as a source of both NO and nitroxyl anion (NO−) but also as a donor of nitrosonium ion (NO+) to thiols, thereby promoting transnitrosation reactions even across biological membranes [161]. On these bases, HSNO was suggested to be a key species in the interplay between H2S and NO, sitting at the crossroad between the H2S and NO signaling pathways.
This view was challenged in subsequent studies by Cortese-Krott et al. [145, 155, 159, 160, 169, 174], who failed to detect HSNO accumulation during the reaction of GSNO with H2S and claimed HSNO to be too short-lived to accumulate and be identified under physiological conditions, in agreement with Munro and Williams [175] and Williams [176]. Compared to other physiologically relevant nitrosthiols, HSNO was indeed suggested to be much more unstable as it undergoes a facile isomerization by hydrogen shift, leading to formation of four distinct isomers. The yellow product generated by reaction of GSNO with H2S initially described in [161] was assigned by Cortese-Krott et al. [159] to nitropersulfide (SSNO−), a compound discovered and chemically characterized in the '80s [177]. The same research groups reported later [160] that, under physiologically relevant conditions, the reaction between sulfide and NO generates not only SSNO− but also polysulfides and SULFI/NO [ON(NO)SO3−], an additional bioactive compound consisting of an adduct of two molecules of NO with sulfite. Based on these and further investigations [174], Cortese-Krott et al. reported that SSNO− is stable in the presence of thiols and cyanides and is able to release NO upon decomposition, targeting the Keap1/Nrf2 redox system, among other possible ones. Overall, this led to the proposal that SSNO−, rather than HSNO, plays a key role in the interplay between NO and H2S under physiological conditions [159]. In line with this proposal, SSNO− was reported to be very stable under anaerobic conditions, to decay slowly (half life of about 40 minutes) in aqueous buffer under aerobic conditions, and to directly react with deoxygenated metHb with a rate constant of 11 M−1·s−1, yielding ferrous nitrosyl Hb (HbNO) presumably via NO release [178]. In contrast, by characterizing pure crystalline SSNO− prepared as a bis(triphenylphosphine)iminium (PNP+) salt according to [179], Filipovic et al. reported that SSNO− is a highly unstable species, leading to rapid decomposition with formation of HNO/NO− in the presence of water, light, or acid and producing HSNO by reaction with H2S, cyanide, and GSH [180, 181].
From the observations reported above, it is clear that additional investigations are needed to reconcile the discrepancies present in the literature and gain further insight into the chemical foundation of the interplay between NO and H2S.
4.3. Modulation of H2S Metabolism by NO and CO
Besides the chemical reactions that define new players in gasotransmitter-mediated signaling, there is an intricate web of cross-regulatory mechanisms whereby each gasotransmitter regulates the homeostasis of the others. While part of that cross-regulation may occur at the transcriptional and translational level, herein we will mainly cover the molecular mechanisms that result from direct interactions between the gasotransmitters and their target proteins, focusing on the modulation by NO and CO of H2S biosynthesis and breakdown.
4.3.1. Modulation of H2S-Generating Enzymes by NO, CO, and Derived Species
The most thoroughly studied H2S-generating enzyme in terms of regulatory mechanisms has certainly been CBS. In what regards its modulation by the other gasotransmitters NO and CO, CBS regulation is centered at its noncatalytic heme bound to the N-terminal domain. This interplay between gasotransmitters results essentially from the fact that H2S production by CBS is reversibly inhibited by NO and CO. The direct effect of this inhibitory mechanism would be that local increases in NO or CO result in decreased H2S synthesis, which has been demonstrated in different cellular models and tissues [182–185]. However, recent evidence suggests that CBS inhibition may in fact result in overall increased H2S production, due to a metabolic switch in the transsulfuration pathway (Figure 1(a)) [11, 186]. In this perspective, inhibiting CBS deviates the transsulfuration branch from cysteine to H2S production via CSE, which has a higher H2S-synthesizing activity, particularly using the accumulated homocysteine also resulting from CBS inhibition [11, 186]. Moreover, the pathophysiological significance of CBS inhibition by gasotransmitters may extend beyond “merely” affecting H2S homeostasis. CBS acts as a metabolic hinge determining the ratio between the remethylation and transsulfuration steps of the methionine cycle (Figure 1(a)). CO inhibition of CBS in various cancer cells has been shown to affect the methylation state of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), suppressing 6-phosphofructo-2-kinase, resulting in a shift of glucose from glycolysis to the pentose phosphate pathway [34, 187].
The regulatory heme-binding domain may be considered a hallmark of CBS evolution, since it is absent from CBS from prokaryotes or unicellular eukaryotes. The low-spin hexacoordinate b-type heme is axially bound to the His65 imidazole and Cys52 thiolate moieties (human CBS numbering; Figure 4(b)). Besides its regulatory role, the CBS heme has been proposed to have a structural stabilizing effect and contribute to improve CBS folding [188]. After initial submission of this review, a quite interesting observation has been reported, which will once again stir the field of CBS heme regulation. Kumar et al. observed that the N-terminal residues 1–40, so far absent from all CBS crystallographic structures, constitute an intrinsically disordered region that is able to bind heme with Cys and His as axial ligands, in a cysteine-proline-based motif [189]. This observation, deserving further exploration, constitutes an additional level of complexity regarding all the literature on the CBS heme regulation (see below).
The heme regulatory role is essentially related to its redox state and the nature of its endogenous and exogenous ligands. The initial designation of the CBS heme as a redox sensor stemmed from its sensitivity to reducing conditions. Indeed, heme reduction by excess sodium dithionite at 37°C slowly (>20 minutes) inactivates the enzyme, which has been assigned to a ligand switch process in which one of the endogenous ligands of heme iron is replaced by another as yet unknown ligand (Figure 13) [190]. The resulting species has been termed “C424” due to its characteristic UV-visible absorption spectrum, in which the Soret band obtained for reduced CBS with its λmax at 449 nm is shifted to a lower-intensity band centered at 424 nm. Irrespectively of the bound endogenous ligands, once reduced, the heme is able to bind the gasotransmitters CO and NO, which results in reversible enzyme inhibition (Figure 13) [191–195]. Although the low reduction potential (−350 mV, [196]) measured for the CBS heme raised some doubts about the occurrence of reduced CBS heme in vivo, the human enzyme methionine synthase reductase (MSR) has been shown to be able to catalyze CBS reduction in vitro at the expense of NADPH oxidation. Moreover, MSR is able to generate both the ferrous-CO and the ferrous-NO species of human CBS [191, 197]. Despite the significant distance between the heme and PLP cofactors (~11 Å from edge to edge and ~20 Å from the Fe ion to the pyridoxine ring), communication between them is ensured by a network of molecular interactions occurring at helix 8 (depicted by the yellow arrow in Figure 4(b)) [198, 199]. While at the PLP end of helix 8 the residues Thr257 and Thr260 establish hydrogen bonds with the PLP phosphate group, at the heme extremity of this helix Arg266 is in electrostatic contact with the heme ligand Cys52 thiolate moiety. The relevance of these residues in the allosteric communication between the active (PLP) and regulatory (heme) sites has been further demonstrated in clinically relevant variants [199, 200].
Figure 13.

Mechanisms of heme reduction and ligand binding to human CBS.
The kinetic characterization of the interplay between the three gasotransmitters centered at CBS has been made possible by taking advantage of the distinct characteristic UV-visible absorption spectra displayed by the heme cofactor in different oxidation and ligation states. Most studies on the CBS redox state and ligand reactivity have thus employed static and time-resolved (stopped-flow) absorption spectroscopy [192–195, 199–204].
The reaction of reduced CBS with CO has been thoroughly characterized in vitro and shown to result in a hexacoordinate species in which the endogenous Cys52 ligand has been replaced by CO, with no evidence for stable intermediates. Indeed, the reaction is limited by the slow dissociation of Cys52 (0.003–0.017 s−1 [192, 195, 197, 204]), as confirmed by laser flash photolysis and time-resolved Raman spectroscopy [192]. Equilibrium titrations revealed that CO binds to ferrous CBS according to two dissociation constants (Kd1 = 0.7–1.5 μM and Kd2 = 45–68 μM; C50 ≈ 30 μM), which has been assigned to anticooperativity between paired monomers [192] or differences in the heme microenvironment [194]. The inhibitory effect of CO binding to the ferrous heme in human CBS is characterized by Ki values of 5.6–9.5 μM [194, 204].
Recently, our groups have reported that a clinically relevant variant with a mutation in the N-terminal domain, p.P49L, displays a markedly higher affinity for CO with respect to WT, while showing an essentially WT-like behavior in other functional and structural terms [203]. While this mutation may induce increased flexibility of the loop containing both heme endogenous ligands, the overall higher proneness to become inhibited by CO may represent a new pathogenic mechanism in classical homocystinuria. Current efforts to develop alternative therapies for classical homocystinuria have essentially focused on chemical and pharmacological chaperones to recover misfolded protein variants originating from missense mutations [205–211] and recently on the development of a PEGylated CBS for enzyme replacement therapy [212–214].
NO binding to reduced CBS is remarkably faster and displays much higher affinity than CO binding. However, initial reports on the reaction between ferrous CBS and NO indicated otherwise. Very low affinity for NO was reported (Kd = 281 μM), employing a high excess of sodium dithionite to keep CBS reduced throughout the NO titrations and using a slow NO releaser (diethylamine NONOate) as NO source [193]. Under these conditions, free NO, either unreacted or dissociated from the ferrous-NO CBS adduct, reacts with the excess dithionite and leads to an overestimation of the dissociation constants, as later shown even with minute amounts of excess dithionite [195]. Recently, our groups have shown that NO binding to ferrous CBS occurs with an affinity compatible with a physiological role of NO in controlling CBS function [195]. An apparent Kd of 0.23 μM was determined at the lowest dithionite concentration used in that study, which implies that the actual affinity is even higher than measured. Contemporarily, a Kd = 30 μM was determined and still was considered an upper limit for this dissociation constant, as it was measured using NADPH and human MSR as reduction system, therefore involving multiple equilibria (NADPH ↔ MSR, MSR ↔ CBS, and CBS ↔ NO) [191]. As compared to CO, NO binding to reduced CBS not only occurs with markedly higher affinity but is also orders of magnitude faster, as investigated by stopped-flow absorption spectroscopy. Differently from CO, NO binding results in a pentacoordinate ferrous-NO adduct, where both endogenous ligands have been displaced and a single NO molecule is bound. No reaction intermediates have been detected upon mixing NO with reduced CBS, at least within the time resolution of the stopped-flow equipment. Moreover, also differently from CO, NO binding proceeds with rate constants linearly dependent on NO concentration (up to 800 μM), that is, unlimited by dissociation of the endogenous ligands, yielding a bimolecular rate constant of ~8 × 103 M−1·s−1 (at 25°C). In line with the higher affinity and faster binding of NO with respect to CO, NO dissociation from ferrous CBS is slower than CO dissociation. In mechanistic terms, since NO binding is not rate-limited by the displacement of Cys52 (which instead limits CO binding), it has been proposed that NO may initially displace the His65 heme ligand (Figure 13). In turn, the absence of evidence for a hexacoordinate intermediate suggests that Cys52 is immediately displaced upon NO binding. Finally, although it has been established that the final NO adduct is a pentacoordinate species, it remains to be demonstrated whether NO ends up on the His65 or Cys52 side of the heme, since both residues are supposed to dissociate from the heme iron. It should be noted that if NO ends up on the Cys52 side, the binding mechanism must involve the formation of a transient bis-NO intermediate (Figure 13). Irrespectively of the mechanistic details, the kinetic and spectroscopic investigations of CBS reduction and CO and NO binding have determined parameters compatible with an in vivo role of this regulatory mechanism.
The reaction of reduced CBS with O2 was also investigated by stopped-flow absorption spectroscopy and shown to be reasonably fast, occurring with a bimolecular rate constant of ~1 × 105 M−1·s−1 (25°C) [202], that is, approximately 10-fold faster than the reaction with NO. Although this reaction reverts CBS back to its active ferric state, it also generates superoxide anion as a reaction product, which may have damaging effects. This oxidation process has indeed been reported to involve a partial heme loss. Superoxide anion promptly reacts with NO (at rates close to the diffusion limit) to yield the powerful oxidant peroxynitrite. Interestingly, it has been shown that CBS heme not only is inactivated by peroxynitrite but also may be a source of it. The reaction of oxidized CBS with peroxynitrite has been shown to result in enzyme inactivation with an IC50 of 150 μM, occurring according to a bimolecular rate constant of 2.4–5 × 104 M−1·s−1 and leading to nitration of Trp208, Trp43, and Tyr223, as well as damaging the heme moiety [215]. Intriguingly, a recent report demonstrated that the reaction between ferrous-NO CBS and O2, previously shown to revert the heme to its ferric state, yields peroxynitrite [201] (Figure 13). Moreover, the reaction of reduced CBS with nitrite may itself constitute a source of NO [191, 201], which further adds to the complex heme-mediated regulation of H2S synthesis by CBS (Figure 13).
4.3.2. Effect of AdoMet on CO/NO Binding and Inhibition of H2S Production by CBS
CBS continues to surprise researchers in the field since regulation of the protein activity not only is intricate but also at times appears odd. It has long been established that AdoMet is an activator of CBS, increasing its enzymatic activity by 2- to 5-fold. Moreover, AdoMet binding also has a stabilizing effect, particularly under “normal” (nonpathological) conditions [216]. Recently, the structural details of this regulatory mechanism have been elucidated. While the C-terminal regulatory domain from one monomer blocks the substrate entry site in the catalytic core of an adjacent monomer, AdoMet binding to the C-terminal domain (also called Bateman module) causes a domain shift and leads to the association of two C-terminal domains in a disk-like form, unblocking the substrate entrance and thus “activating” (or derepressing) the enzyme [44, 46]. The relevance of this regulatory mechanism is well attested by the fact that some homocystinuric patients express CBS variants with mutations in the C-terminal domain, that are naturally activated (more active than WT) and have limited or absent AdoMet response as the molecular basis of disease [217, 218]. While working on the kinetic characterization of NO and CO binding to human CBS, we noticed that AdoMet has the intriguing effect of eliciting faster and tighter binding of both CO and NO, thus making the enzyme more prone to inhibition by either gasotransmitter [204]. This observation is directly related to the binding of exogenous ligands, since AdoMet had been shown to have no effect on the heme redox properties [196]. Under the same reaction conditions, AdoMet makes CO inhibition >10 times more potent than in its absence (Ki(CO, +AdoMet) = 0.7 μM versus Ki(CO, −AdoMet) = 9.5 μM) [204]. The kinetic details of this functional effect have been investigated, and it was shown that AdoMet increases CO affinity by ~5-fold and association kinetics by ~10-fold, whereas the effect on NO binding is less drastic (~2-fold higher affinity with respect to AdoMet-free CBS). The latter observation also enforces the previous mechanistic proposal that CO and NO “attack” different sides of the heme. Recently, this observation has been extended to a clinically relevant variant, p.P49L, which displays the same AdoMet response in terms of becoming more sensitive to CO, while being slightly impaired in the AdoMet activation of H2S production, compared to WT [203]. Irrespectively of the mechanistic details, the AdoMet-induced enhanced CO and NO binding appears as an intricate regulatory mechanism which adds to the complexity of CBS regulation.
4.3.3. Modulation of Sulfide Oxidation by NO
Whereas the effect of CO and NO on H2S biosynthesis has been investigated in detail, much scarcer is the information on the effect of these two gaseous molecules on H2S catabolism. Because CO and NO, similarly to cyanide, can both potently inhibit CcOX through different mechanisms (see above), relatively low concentrations of these gasotransmitters are expected to affect mitochondrial sulfide oxidation. Following cell exposure to CO or NO, a transient inhibition of CcOX is indeed expected to occur and lead to electron accumulation in the mitochondrial respiratory chain, eventually impairing quinol oxidation. As a result, SQR-mediated oxidation of sulfide is expected to be impaired as long as CcOX inhibition persists. Accordingly, addition of the NO releaser MAMA NONOate (≥20 μM) or cyanide (260 μM) to colonocytes was recently reported to inhibit cellular respiration and its stimulation by 20 μM NaHS [219]. Furthermore, it is worth noting that long-term intraperitoneal or intravenous administration of nitroglycerine in rats was shown to induce decreased rhodanese activity and diminished levels of sulfane sulfur in liver extracts [220]. The observed inhibition of rhodanese was proposed to arise from s-nitrosation of the catalytic cysteine in the protein active site. A cysteine in the isolated rhodanese from bovine liver was indeed shown to be susceptible to s-nitrosation by the NO donors s-nitroso-N-acetylpenicillamine (SNAP) and GSNO, leading to enzyme inhibition [220].
The preliminary information reported above unveils an additional level at which the interplay between the gasotransmitters can be established. A full description of these molecular mechanisms awaits further investigations.
4.3.4. Modulation of NO and CO Synthesis by Hydrogen Sulfide
In light of the crosstalk between gasotransmitters, hydrogen sulfide has been shown to have a role in the control of NO and CO bioavailability under specific (patho)physiological conditions.
As mentioned above, CO is generated by either isoform of heme oxygenase, HO-1 and HO-2 [153]. Several lines of evidence point to a role of hydrogen sulfide in increasing the expression of heme oxygenase, particularly HO-1, and consequently CO levels (mostly evaluated from the abundance of carboxylated hemoglobin), thereby exerting either protective or deleterious effects (e.g., [221–227]). The underlying regulatory mechanism is proposed to involve the Keap1/Nrf2 system. In particular, persulfidation of Keap1 Cys151, by reaction either of H2S with oxidized Cys151 or of reduced Cys151 with H2S-derived HS· (Figure 11(a)), causes the protein dissociation from Nrf2 that, in turn, translocates into the nucleus, ultimately resulting in increased HO-1 transcription and protein levels (Figure 14) [225]. Another possibility whereby CO production may be modulated resides in the fact that a thiol/disulfide redox switch comprising three Cys-Pro signatures in HO-2 regulates heme binding [228, 229]. The sensitivity of this thiol/disulfide switch to the cellular redox state and possibly to the intracellular sulfide levels may afford another level of H2S-mediated control of CO levels. Interestingly, the beneficial health effects of certain garlic-derived organosulfur compounds, particularly diallyl disulfide and trisulfide, have been proposed to be related with the modulation of CO production by H2S via the Keap1/Nrf2 pathway [230–232].
Figure 14.

H2S-mediated modulation of nitric oxide and carbon monoxide biosynthesis. Hydrogen sulfide produced by CBS, CSE, and MST, and the related per- and polysulfide species (RS(n)H), can modulate NO and CO bioavailability through different mechanisms. Left, representation of sulfide-mediated enhanced eNOS activity: H2S reduces VEGFR2 Cys1024-Cys1045 disulfide, resulting in activation of PI3K; the accumulated PIP3 increases Akt-mediated phosphorylation of eNOS at Ser1077 and/or Ser1079, enhancing its NO-synthesizing activity. Center, direct persulfidation of eNOS Cys443 sustains the protein catalytic activity by competing with Cys443s-nitrosation, which causes dissociation of the active dimeric into the inactive monomeric form. Right, modulation of CO biosynthesis through increased expression of HO-1, mediated by the Keap1/Nrf2 pathway. Persulfidation of Keap1 Cys151, either by reaction of H2S with oxidized Cys151 or of reduced Cys151 with H2S-derived HS·, causes its dissociation from Nrf2, which can then translocate to the nucleus, where it enhances heme oxygenase HO-1 expression, thus resulting in higher CO levels that may become inhibitory for CBS.
The control of NO biosynthesis by hydrogen sulfide has been investigated in more detail and appears to occur at the transcriptional, translational, and posttranslational levels, involving other signaling pathways, besides a direct inhibition of all NOS isoforms by H2S. The abundance of data in the literature highlights the fact that the effect of H2S on the expression and activity of each NOS isoform can vary—sometimes yielding conflicting results—with the model organ, tissue, or cell type investigated and with the nature, concentration, and potency of the employed sulfide donor. Moreover, whereas in some studies “only” the levels of NO (or nitrite/nitrate) are measured as a function of sulfide exposure, other studies further investigate the NO-mediated cellular and physiological effects of sulfide exposure.
Regulation of eNOS by H2S has been thoroughly investigated. In different cell types and under different conditions and stimuli, eNOS activation in response to sulfide exposure seems to be mediated by the PIP3/Akt (phosphatidylinositol 3,4,5-trisphosphate/serine/threonine-specific protein kinase) pathway (Figure 14). The molecular mechanism is proposed to involve the reduction by H2S of the vascular endothelial growth factor receptor 2 (VEGFR2) Cys1024-Cys1045 disulfide that in the oxidized state represses its receptor activity. This H2S-induced disulfide reduction thus prolongs the VEGFR2 activity and consequently the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) activity, yielding an increase in PIP3 levels (which may also be influenced by persulfidation of Cys124 in the phosphatase and tensin homolog (PTEN)). The resulting increased Akt activation leads to enhanced eNOS phosphorylation at Ser1177 and/or Ser1179, thereby increasing the NO-synthesizing activity. Another level of sulfide-mediated eNOS regulation concerns the direct persulfidation at Cys443 (Figure 14). Whereas dimerization appears to be a prerequisite for optimal eNOS activity, s-nitrosation of Cys443 induces dimer dissociation into the virtually inactive monomeric form. Sulfide-mediated persulfidation of this residue actively competes with s-nitrosation, thereby maintaining eNOS in the active homodimeric form. Direct in vitro inhibition of recombinant eNOS by H2S is characterized by an extremely high (nonphysiological) IC50 of 170 μM [233]. Regarding the sulfide-mediated modulation of eNOS expression, various reports have either shown a down- or upregulation of eNOS mRNA and protein levels in response to H2S exposure. These somehow contradictory results obtained for different model systems (animals, organs, and cells) rule out the establishment of a clear trend, induction versus repression, for sulfide modulation of eNOS expression.
Besides its direct in vitro inhibition at nonphysiological H2S concentrations (IC50 = 210 μM) [233], sulfide-mediated modulation of iNOS is reported to occur mainly through regulation of its expression. Similarly to eNOS, initial contradictory results indicated either an increase or a decrease in iNOS-mediated NO production in response to sulfide exposure [234]. This controversy was addressed by comparatively investigating the effects of different H2S donors [235], showing that the rate of H2S release by the donors is a key determinant for the downstream effects. Subsequent investigations on various models commonly assigned an inhibitory effect of sulfide on iNOS mRNA expression and consequently iNOS-mediated NO production, thereby contributing to the establishment of H2S as an anti-inflammatory molecule (reviewed, e.g., in [154]).
Currently, the information on nNOS modulation by H2S is scarce. Besides its inhibition at high nonphysiological sulfide concentrations (IC50 = 130 μM) [233, 236], nNOS expression seems to be either insensitive to sulfide exposure or decreased upon overexpression of the H2S-synthesizing CBS in hypertensive (but not normotensive) rats [237].
Altogether, the multiple molecular mechanisms whereby each of the three gasotransmitters may control the bioavailability of the others highlight the intricate signaling mechanisms that govern mammalian (patho)physiology and offer valuable possibilities for pharmacological targeting of each pathway.
5. Concluding Remarks
Hydrogen sulfide evolved from being merely perceived as a toxic and poisonous molecule to a prominent signaling molecule in mammalian physiology, joining nitric oxide and carbon monoxide as the triad of gasotransmitters. The last couple of decades witnessed an accumulation of data showing that (i) H2S is endogenously synthesized and catabolized by specialized enzymes and kept under a tight control through multiple intricate regulatory mechanisms, (ii) the signaling functions attributed to H2S are mainly linked to the interaction with and modification of target proteins, via reaction with metal centers (mostly in heme proteins) and through cysteine persulfidation that alters protein structure and function, (iii) dysregulation of H2S homeostasis is at the basis of several pathologies, including cardiovascular and neurodegenerative diseases and cancer, and (iv) possible mechanisms underlying a functional crosstalk between the three gasotransmitters have been unveiled. The clear link between H2S metabolism and homeostasis with human health has triggered a strong interest in the discovery and development of pharmacological (synthetic and naturally derived) compounds, either to release H2S or to modulate the H2S metabolism enzymes. Despite the accumulated knowledge on hydrogen sulfide biochemistry over the years, all a contribution as this one can amount to be, particularly concerning a field in its infancy, is just a still picture of a fast moving body. Indeed, during the writing of this manuscript, several pieces of information even challenging previous observations arose in the literature, further contributing to the complexity of this quickly progressing field.
Acknowledgments
The iNOVA4Health Research Unit (LISBOA-01-0145-FEDER-007344), which is cofunded by Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior, through national funds, and by FEDER under the PT2020 Partnership Agreement, is acknowledged by João B. Vicente. The grants PNR-CNR Aging Program 2012–2014 and PRIN 20158EB2CM 003 by Ministero dell'Istruzione, dell'Università della Ricerca of Italy are acknowledged by Alessandro Giuffrè. The authors are grateful to present and past collaborators and to Catarina Tomé for critically reading the manuscript.
Abbreviations
- 3-MP:
3-Mercaptopyruvate
- AdoHcy:
S-Adenosyl-L-homocysteine
- AdoMet:
S-Adenosyl-L-methionine
- ADT-OH:
5-(4-Hydroxyphenyl)-3H-1,2-dithiole-3-thione
- Akt:
Serine/threonine-specific protein kinase
- AOAA:
Aminooxyacetic acid
- AP39:
[10-Oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5yl)phenoxy)decyl) triphenylphosphonium bromide]
- ATP:
Adenosine triphosphate
- CARSs:
Cysteinyl-tRNA synthetases
- CAT:
Cysteine (or aspartate) aminotransferase
- CBS:
Cystathionine β-synthase
- CcOX:
Mitochondrial cytochrome c oxidase (complex IV)
- CO:
Carbon monoxide
- CSE:
Cystathionine γ-lyase
- CysSOH:
Cysteine residue with modified thiol to sulfenic acid
- CysSSH:
Cysteine persulfide
- CysSS(n)H:
Cysteine polysulfide
- CysSSOH:
Cysteine perthiosulfenic
- CysSSR:
Cysteine residues with modified thiol to disulfide
- DHLA:
Dihydrolipoic acid
- EDHF:
Endothelium-derived hyperpolarizing factor
- EDRF:
Endothelium-derived relaxing factor
- eIF2α:
Eukaryotic translational initiation factor 2α
- eNOS:
Endothelial nitric oxide synthase
- EPR:
Electron paramagnetic resonance spectroscopy
- ETHE1:
Persulfide dioxygenase, or ethylmalonic encephalopathy protein 1
- FAD:
Flavin adenine dinucleotide
- GAPDH:
Glyceraldehyde 3-phosphate dehydrogenase
- GSNO:
S-Nitrosoglutathione
- GSSH:
Glutathione persulfide
- GSS(n)H:
Glutathione polysulfide
- Hb:
Hemoglobin
- H2S:
Hydrogen sulfide
- H2S2:
Hydrogen persulfide
- H2S3:
Hydrogen trisulfide
- HO-1:
Heme oxygenase 1
- HO-2:
Heme oxygenase 2
- HS−:
Hydrosulfide
- HSNO/ONS−:
Thionitrous acid/thionitrite
- iNOS:
Inducible nitric oxide synthase
- Keap1:
Kelch-like ECH-associated protein 1
- Kir6.1:
Inwardly rectifying potassium channel subunit Kir6.1
- LMW:
Low-molecular-weight
- metHb:
Ferric hemoglobin
- Mb:
Myoglobin
- MSR:
Methionine synthase reductase
- MST:
3-Mercaptopyruvate sulfurtransferase
- NADPH:
Nicotinamide adenine dinucleotide phosphate (reduced form)
- NF-κB:
Nuclear factor κB
- nNOS:
Neuronal nitric oxide synthase
- NO:
Nitric oxide (NO·)
- NO−:
Nitroxyl anion
- NO+:
Nitrosonium ion
- Nrf2:
Nuclear factor erythroid 2-related factor 2
- PERK:
Protein kinase RNA-like ER kinase
- PI3K:
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- PIP3:
Phosphatidylinositol 3,4,5-trisphosphate
- PLP:
Pyridoxal 5′-phosphate
- PNP+:
Bis(triphenylphosphine)iminium
- PPG:
Propargylglycine
- PtdIns(4,5)P2:
Phosphatidylinositol-4,5-bisphosphate
- PTEN:
Phosphatase and tensin homolog
- PTP1B:
Protein Tyr phosphatase 1B
- Rhod:
Rhodanese
- RNS:
Reactive nitrogen species
- ROS:
Reactive oxygen species
- RSS:
Reactive sulfur species
- RSS·:
Perthiyl radical
- S2−:
Sulfide
- SAHH:
S-Adenosyl-L-homocysteine hydrolase
- SMCs:
Vascular smooth muscle cells
- SOx:
Sulfite oxidase
- SQR:
Sulfide:quinone oxidoreductase
- SSNO−:
Nitropersulfide
- Tom20:
Translocase of the outer membrane 20
- TPP+:
Triphenylphosphonium
- Trx:
Thioredoxin
- VEGFR2:
Vascular endothelial growth factor receptor 2.
Contributor Information
Alessandro Giuffrè, Email: alessandro.giuffre@uniroma1.it.
João B. Vicente, Email: jvicente@itqb.unl.pt.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Ramazzini B. De Morbis Artificum Diatriba. Padova: Baptistam Conzattum; 1713. [Google Scholar]
- 2.Scheele C. W. Chemische Abhandlung von der Luft und dem Feuer. Upsala, Sweden: Magnus Swederus; 1777. [Google Scholar]
- 3.Cuevasanta E., Möller M. N., Alvarez B. Biological chemistry of hydrogen sulfide and persulfides. Archives of Biochemistry and Biophysics. 2017;617:9–25. doi: 10.1016/j.abb.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Olson K. R., Straub K. D. The role of hydrogen sulfide in evolution and the evolution of hydrogen sulfide in metabolism and signaling. Physiology. 2016;31(1):60–72. doi: 10.1152/physiol.00024.2015. [DOI] [PubMed] [Google Scholar]
- 5.Li Q., Lancaster J. R., Jr Chemical foundations of hydrogen sulfide biology. Nitric Oxide. 2013;35:21–34. doi: 10.1016/j.niox.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono K., Akaike T., Sawa T., et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility. Free Radical Biology and Medicine. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szczesny B., Módis K., Yanagi K., et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–130. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X., Carlström M., Borniquel S., Jädert C., Kevil C. G., Lundberg J. O. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radical Biology and Medicine. 2013;60:195–200. doi: 10.1016/j.freeradbiomed.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose P., Moore P. K., Zhu Y. Z. H2S biosynthesis and catabolism: new insights from molecular studies. Cellular and Molecular Life Sciences. 2017;74(8):1391–1412. doi: 10.1007/s00018-016-2406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kabil O., Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxidants & Redox Signaling. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Current Opinion in Chemical Biology. 2017;37:115–121. doi: 10.1016/j.cbpa.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao L., Vlček Č., Pačes V., Kraus J. P. Identification and tissue distribution of human cystathionine β-synthase mRNA isoforms. Archives of Biochemistry and Biophysics. 1998;350(1):95–103. doi: 10.1006/abbi.1997.0486. [DOI] [PubMed] [Google Scholar]
- 13.Kabil O., Vitvitsky V., Xie P., Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxidants & Redox Signaling. 2011;15(2):363–372. doi: 10.1089/ars.2010.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mudd S. H., Finkelstein J. D., Irreverre F., Laster L. Transsulfuration in mammals. Microassays and tissue distributions of three enzymes of the pathway. Journal of Biological Chemistry. 1965;240(11):4382–4392. [PubMed] [Google Scholar]
- 15.Saha S., Chakraborty P. K., Xiong X., et al. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. The FASEB Journal. 2016;30(1):441–456. doi: 10.1096/fj.15-278648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogasawara Y., Isoda S., Tanabe S. Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biological & Pharmaceutical Bulletin. 1994;17(12):1535–1542. doi: 10.1248/bpb.17.1535. [DOI] [PubMed] [Google Scholar]
- 17.Yang G., Wu L., Jiang B., et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taniguchi T., Kimura T. Role of 3-mercaptopyruvate sulfurtransferase in the formation of the iron-sulfur chromophore of adrenal ferredoxin. Biochimica et Biophysica Acta (BBA) - Enzymology. 1974;364(2):284–295. doi: 10.1016/0005-2744(74)90014-X. [DOI] [PubMed] [Google Scholar]
- 19.Nagahara N., Ito T., Kitamura H., Nishino T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochemistry and Cell Biology. 1998;110(3):243–250. doi: 10.1007/s004180050286. [DOI] [PubMed] [Google Scholar]
- 20.Tomita M., Nagahara N., Ito T. Expression of 3-mercaptopyruvate sulfurtransferase in the mouse. Molecules. 2016;21(12, article 1707) doi: 10.3390/molecules21121707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitvitsky V., Yadav P. K., Kurthen A., Banerjee R. Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. Journal of Biological Chemistry. 2015;290(13):8310–8320. doi: 10.1074/jbc.M115.639831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabil O., Banerjee R. Redox biochemistry of hydrogen sulfide. Journal of Biological Chemistry. 2010;285(29):21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bearden S. E., Beard R. S., Jr, Pfau J. C. Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. American Journal of Physiology-Heart and Circulatory Physiology. 2010;299(5):H1568–H1576. doi: 10.1152/ajpheart.00555.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabil O., Zhou Y., Banerjee R. Human cystathionine β-synthase is a target for sumoylation. Biochemistry. 2006;45(45):13528–13536. doi: 10.1021/bi0615644. [DOI] [PubMed] [Google Scholar]
- 25.Bouillaud F., Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxidants & Redox Signaling. 2011;15(2):379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 26.Mustafa A. K., Gadalla M. M., Sen N., et al. H2S signals through protein S-sulfhydration. Science Signaling. 2009;2(96):p. ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Módis K., Ju Y. J., Ahmad A., et al. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacological Research. 2016;113(Part A):116–124. doi: 10.1016/j.phrs.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu M., Zhang W., Wu L., Yang G., Li H., Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):2943–8. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper C. E., Brown G. C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. Journal of Bioenergetics and Biomembranes. 2008;40(5):533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 30.Nicholls P., Marshall D. C., Cooper C. E., Wilson M. T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochemical Society Transactions. 2013;41(5):1312–1316. doi: 10.1042/BST20130070. [DOI] [PubMed] [Google Scholar]
- 31.Vicente J. B., Malagrinò F., Arese M., Forte E., Sarti P., Giuffrè A. Bioenergetic relevance of hydrogen sulfide and the interplay between gasotransmitters at human cystathionine β-synthase. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2016;1857(8):1127–1138. doi: 10.1016/j.bbabio.2016.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Teng H., Wu B., Zhao K., Yang G., Wu L., Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(31):12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo C., Coletta C., Chao C., et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(30):12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto T., Takano N., Ishiwata K., et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nature Communications. 2014;5, article 3480 doi: 10.1038/ncomms4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davoli A., Greco V., Spalloni A., et al. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Annals of Neurology. 2015;77(4):697–709. doi: 10.1002/ana.24372. [DOI] [PubMed] [Google Scholar]
- 36.Fräsdorf B., Radon C., Leimkühler S. Characterization and interaction studies of two isoforms of the dual localized 3-mercaptopyruvate sulfurtransferase TUM1 from humans. Journal of Biological Chemistry. 2014;289(50):34543–34556. doi: 10.1074/jbc.M114.605733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finkelstein J. D. Pathways and regulation of homocysteine metabolism in mammals. Seminars in Thrombosis and Hemostasis. 2000;26(3):219–226. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 38.Majtan T., Krijt J., Sokolová J., et al. Biogenesis of hydrogen sulfide and thioethers by cystathionine beta-synthase. Antioxidants & Redox Signaling. 2018;28(4):311–323. doi: 10.1089/ars.2017.7009. [DOI] [PubMed] [Google Scholar]
- 39.Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. Journal of Biological Chemistry. 2009;284(33):22457–22466. doi: 10.1074/jbc.m109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. Journal of Biological Chemistry. 2009;284(17):11601–11612. doi: 10.1074/jbc.m808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kožich V., Krijt J., Sokolová J., et al. Thioethers as markers of hydrogen sulfide production in homocystinurias. Biochimie. 2016;126:14–20. doi: 10.1016/j.biochi.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Shibuya N., Koike S., Tanaka M., et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nature Communications. 2013;4, article 1366 doi: 10.1038/ncomms2371. [DOI] [PubMed] [Google Scholar]
- 43.Yadav P. K., Yamada K., Chiku T., Koutmos M., Banerjee R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. Journal of Biological Chemistry. 2013;288(27):20002–20013. doi: 10.1074/jbc.M113.466177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ereño-Orbea J., Majtan T., Oyenarte I., Kraus J. P., Martinez-Cruz L. A. Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(40):E3790–E3799. doi: 10.1073/pnas.1313683110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meier M., Janosik M., Kery V., Kraus J. P., Burkhard P. Structure of human cystathionine β‐synthase: a unique pyridoxal 5′‐phosphate‐dependent heme protein. The EMBO Journal. 2001;20(15):3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ereño-Orbea J., Majtan T., Oyenarte I., Kraus J. P., Martínez-Cruz L. A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(37):E3845–E3852. doi: 10.1073/pnas.1414545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koutmos M., Kabil O., Smith J. L., Banerjee R. Structural basis for substrate activation and regulation by cystathionine beta-synthase (CBS) domains in cystathionine β-synthase. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(49):20958–20963. doi: 10.1073/pnas.1011448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCorvie T. J., Kopec J., Hyung S.-J., et al. Inter-domain communication of human cystathionine β-synthase: structural basis of S-adenosyl-L-methionine activation. Journal of Biological Chemistry. 2014;289(52):36018–36030. doi: 10.1074/jbc.M114.610782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pey A. L., Majtan T., Sanchez-Ruiz J. M., Kraus J. P. Human cystathionine β-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): complex regulation of CBS activity and stability by SAM. Biochemical Journal. 2013;449(1):109–121. doi: 10.1042/BJ20120731. [DOI] [PubMed] [Google Scholar]
- 50.Sun Q., Collins R., Huang S., et al. Structural basis for the inhibition mechanism of human cystathionine γ-lyase, an enzyme responsible for the production of H2S. Journal of Biological Chemistry. 2009;284(5):3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 51.Hanaoka K., Sasakura K., Suwanai Y., et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteinepersulfide. Scientific Reports. 2017;7, article 40227 doi: 10.1038/srep40227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen L. C. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1977;460(2):299–307. doi: 10.1016/0005-2728(77)90216-X. [DOI] [PubMed] [Google Scholar]
- 53.Curtis C. G., Bartholomew T. C., Rose F. A., Dodgson K. S. Detoxication of sodium 35S-sulphide in the rat. Biochemical Pharmacology. 1972;21(17):2313–2321. doi: 10.1016/0006-2952(72)90382-6. [DOI] [PubMed] [Google Scholar]
- 54.Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010;1797(8):1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Hildebrandt T. M., Grieshaber M. K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS Journal. 2008;275(13):3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 56.Olson K. R., Gao Y., Arif F., et al. Metabolism of hydrogen sulfide (H2S) and production of reactive sulfur species (RSS) by superoxide dismutase. Redox Biology. 2018;15:74–85. doi: 10.1016/j.redox.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olson K. R., Gao Y., DeLeon E. R., et al. Catalase as a sulfide-sulfur oxido-reductase: an ancient (and modern?) regulator of reactive sulfur species (RSS) Redox Biology. 2017;12:325–339. doi: 10.1016/j.redox.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Searcy D. G. HS−:O2 oxidoreductase activity of Cu,Zn superoxide dismutase. Archives of Biochemistry and Biophysics. 1996;334(1):50–58. doi: 10.1006/abbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 59.Searcy D. G., Whitehead J. P., Maroney M. J. Interaction of Cu,Zn superoxide dismutase with hydrogen sulfide. Archives of Biochemistry and Biophysics. 1995;318(2):251–263. doi: 10.1006/abbi.1995.1228. [DOI] [PubMed] [Google Scholar]
- 60.Powell M. A., Somero G. N. Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science. 1986;233(4763):563–566. doi: 10.1126/science.233.4763.563. [DOI] [PubMed] [Google Scholar]
- 61.Goubern M., Andriamihaja M., Nubel T., Blachier F., Bouillaud F. Sulfide, the first inorganic substrate for human cells. The FASEB Journal. 2007;21(8):1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 62.Szabo C., Ransy C., Módis K., et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. British Journal of Pharmacology. 2014;171(8):2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abou-Hamdan A., Ransy C., Roger T., Guedouari-Bounihi H., Galardon E., Bouillaud F. Positive feedback during sulfide oxidation fine-tunes cellular affinity for oxygen. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2016;1857(9):1464–1472. doi: 10.1016/j.bbabio.2016.04.282. [DOI] [PubMed] [Google Scholar]
- 64.Leschelle X., Goubern M., Andriamihaja M., et al. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochimica et Biophysica Acta (BBA) - General Subjects. 2005;1725(2):201–212. doi: 10.1016/j.bbagen.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 65.Yong R., Searcy D. G. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology. 2001;129(1):129–137. doi: 10.1016/S1096-4959(01)00309-8. [DOI] [PubMed] [Google Scholar]
- 66.Abou-Hamdan A., Guedouari-Bounihi H., Lenoir V., Andriamihaja M., Blachier F., Bouillaud F. Oxidation of H2S in mammalian cells and mitochondria. Methods in Enzymology. 2015;554:201–228. doi: 10.1016/bs.mie.2014.11.042. [DOI] [PubMed] [Google Scholar]
- 67.Groeger M., Matallo J., McCook O., et al. Temperature and cell-type dependency of sulfide effects on mitochondrial respiration. Shock. 2012;38(4):367–374. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- 68.Helmy N., Prip-Buus C., Vons C., et al. Oxidation of hydrogen sulfide by human liver mitochondria. Nitric Oxide. 2014;41:105–112. doi: 10.1016/j.niox.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 69.Matallo J., Vogt J., McCook O., et al. Sulfide-inhibition of mitochondrial respiration at very low oxygen concentrations. Nitric Oxide. 2014;41:79–84. doi: 10.1016/j.niox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mimoun S., Andriamihaja M., Chaumontet C., et al. Detoxification of H2S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxidants & Redox Signaling. 2012;17(1):1–10. doi: 10.1089/ars.2011.4186. [DOI] [PubMed] [Google Scholar]
- 71.Módis K., Coletta C., Erdélyi K., Papapetropoulos A., Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. The FASEB Journal. 2013;27(2):601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 72.Searcy D. G. Metabolic integration during the evolutionary origin of mitochondria. Cell Research. 2003;13(4):229–238. doi: 10.1038/sj.cr.7290168. [DOI] [PubMed] [Google Scholar]
- 73.Theissen U., Hoffmeister M., Grieshaber M., Martin W. Single eubacterial origin of eukaryotic sulfide:quinone oxidoreductase, a mitochondrial enzyme conserved from the early evolution of eukaryotes during anoxic and sulfidic times. Molecular Biology and Evolution. 2003;20(9):1564–1574. doi: 10.1093/molbev/msg174. [DOI] [PubMed] [Google Scholar]
- 74.Brito J. A., Sousa F. L., Stelter M., et al. Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry. 2009;48(24):5613–5622. doi: 10.1021/bi9003827. [DOI] [PubMed] [Google Scholar]
- 75.Cherney M. M., Zhang Y., Solomonson M., Weiner J. H., James M. N. G. Crystal structure of sulfide:quinone oxidoreductase from Acidithiobacillus ferrooxidans: insights into sulfidotrophic respiration and detoxification. Journal of Molecular Biology. 2010;398(2):292–305. doi: 10.1016/j.jmb.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 76.Marcia M., Ermler U., Peng G., Michel H. The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(24):9625–9630. doi: 10.1073/pnas.0904165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marcia M., Ermler U., Peng G., Michel H. A new structure-based classification of sulfide:quinone oxidoreductases. Proteins: Structure, Function, and Bioinformatics. 2010;78(5):1073–1083. doi: 10.1002/prot.22665. [DOI] [PubMed] [Google Scholar]
- 78.Cherney M. M., Zhang Y., James M. N. G., Weiner J. H. Structure–activity characterization of sulfide:quinone oxidoreductase variants. Journal of Structural Biology. 2012;178(3):319–328. doi: 10.1016/j.jsb.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 79.Augustyn K. D. C., Jackson M. R., Jorns M. S. Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry. 2017;56(7):986–996. doi: 10.1021/acs.biochem.6b01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackson M. R., Melideo S. L., Jorns M. S. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry. 2012;51(34):6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- 81.Jackson M. R., Melideo S. L., Jorns M. S. Role of human sulfide: quinone oxidoreductase in H2S metabolism. Methods in Enzymology. 2015;554:255–270. doi: 10.1016/bs.mie.2014.11.037. [DOI] [PubMed] [Google Scholar]
- 82.Libiad M., Yadav P. K., Vitvitsky V., Martinov M., Banerjee R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. Journal of Biological Chemistry. 2014;289(45):30901–30910. doi: 10.1074/jbc.M114.602664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Landry A. P., Ballou D. P., Banerjee R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. Journal of Biological Chemistry. 2017;292(28):11641–11649. doi: 10.1074/jbc.M117.788547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mishanina T. V., Yadav P. K., Ballou D. P., Banerjee R. Transient kinetic analysis of hydrogen sulfide oxidation catalyzed by human sulfide quinone oxidoreductase. Journal of Biological Chemistry. 2015;290(41):25072–25080. doi: 10.1074/jbc.M115.682369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griesbeck C., Schütz M., Schödl T., et al. Mechanism of sulfide-quinone reductase investigated using site-directed mutagenesis and sulfur analysis. Biochemistry. 2002;41(39):11552–11565. doi: 10.1021/bi026032b. [DOI] [PubMed] [Google Scholar]
- 86.Tiranti V., D’Adamo P., Briem E., et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. The American Journal of Human Genetics. 2004;74(2):239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tiranti V., Viscomi C., Hildebrandt T., et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nature Medicine. 2009;15(2):200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 88.Pettinati I., Brem J., McDonough M. A., Schofield C. J. Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Human Molecular Genetics. 2015;24(9):2458–2469. doi: 10.1093/hmg/ddv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trevino R. J., Gliubich F., Berni R., et al. NH2-terminal sequence truncation decreases the stability of bovine rhodanese, minimally perturbs its crystal structure, and enhances interaction with GroEL under native conditions. Journal of Biological Chemistry. 1999;274(20):13938–13947. doi: 10.1074/jbc.274.20.13938. [DOI] [PubMed] [Google Scholar]
- 90.Johnson-Winters K., Tollin G., Enemark J. H. Elucidating the catalytic mechanism of sulfite oxidizing enzymes using structural, spectroscopic, and kinetic analyses. Biochemistry. 2010;49(34):7242–7254. doi: 10.1021/bi1008485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bianco C. L., Toscano J. P., Fukuto J. M. Chapter 2 – an integrated view of the chemical biology of NO, CO, H2S, and O2. In: Ignarro L. J., Freeman B., editors. Nitric Oxide (Third Edition) London, UK: Academic Press; 2017. pp. 9–21. [DOI] [Google Scholar]
- 92.Michel H. O. A study of sulfhemoglobin. Journal of Biological Chemistry. 1938;126(1):323–348. [Google Scholar]
- 93.Berzofsky J. A., Peisach J., Blumberg W. E. Sulfheme proteins. I. Optical and magnetic properties of sulfmyoglobin and its derivatives. Journal of Biological Chemistry. 1971;246(10):3367–3377. [PubMed] [Google Scholar]
- 94.Nicholls P. The formation and properties of sulphmyoglobin and sulphcatalase. Biochemical Journal. 1961;81(2):374–383. doi: 10.1042/bj0810374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagy P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods in Enzymology. 2015;554:3–29. doi: 10.1016/bs.mie.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 96.Ríos-González B. B., Román-Morales E. M., Pietri R., López-Garriga J. Hydrogen sulfide activation in hemeproteins: the sulfheme scenario. Journal of Inorganic Biochemistry. 2014;133:78–86. doi: 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pietri R., Román-Morales E., López-Garriga J. Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxidants & Redox Signaling. 2011;15(2):393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chance B., Erecinska M., Wagner M. Mitochondrial responses to carbon monoxide toxicity. Annals of the New York Academy of Sciences. 1970;174:193–204. doi: 10.1111/j.1749-6632.1970.tb49786.x. [DOI] [PubMed] [Google Scholar]
- 99.Brunori M., Giuffrè A., Forte E., Mastronicola D., Barone M. C., Sarti P. Control of cytochrome c oxidase activity by nitric oxide. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2004;1655(1–3):365–371. doi: 10.1016/j.bbabio.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 100.Cooper C. E. Nitric oxide and cytochrome oxidase: substrate, inhibitor or effector? Trends in Biochemical Sciences. 2002;27(1):33–39. doi: 10.1016/S0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- 101.Cooper C. E., Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: molecular mechanism and tissue physiology. American Journal of Physiology-Cell Physiology. 2007;292(6):C1993–C2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 102.Sarti P., Arese M., Bacchi A., et al. Nitric oxide and mitochondrial complex IV. IUBMB Life. 2003;55(10–11):605–611. doi: 10.1080/15216540310001628726. [DOI] [PubMed] [Google Scholar]
- 103.Sarti P., Forte E., Mastronicola D., Giuffrè A., Arese M. Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2012;1817(4):610–619. doi: 10.1016/j.bbabio.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Sarti P., Giuffrè A., Barone M. C., Forte E., Mastronicola D., Brunori M. Nitric oxide and cytochrome oxidase: reaction mechanisms from the enzyme to the cell. Free Radical Biology and Medicine. 2003;34(5):509–520. doi: 10.1016/S0891-5849(02)01326-6. [DOI] [PubMed] [Google Scholar]
- 105.Mason M. G., Nicholls P., Wilson M. T., Cooper C. E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):708–713. doi: 10.1073/pnas.0506562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mastronicola D., Genova M. L., Arese M., et al. Control of respiration by nitric oxide in Keilin-Hartree particles, mitochondria and SH-SY5Y neuroblastoma cells. Cellular and Molecular Life Sciences (CMLS) 2003;60(8):1752–1759. doi: 10.1007/s00018-003-3127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarti P., Giuffrè A., Forte E., Mastronicola D., Barone M. C., Brunori M. Nitric oxide and cytochrome c oxidase: mechanisms of inhibition and NO degradation. Biochemical and Biophysical Research Communications. 2000;274(1):183–187. doi: 10.1006/bbrc.2000.3117. [DOI] [PubMed] [Google Scholar]
- 108.Nicholls P. The effect of sulphide on cytochromeaa3 isosteric and allosteric shifts of the reduced α-peak. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1975;396(1):24–35. doi: 10.1016/0005-2728(75)90186-3. [DOI] [PubMed] [Google Scholar]
- 109.Nicholls P., Petersen L. C., Miller M., Hansen F. B. Ligand-induced spectral changes in cytochrome c oxidase and their possible significance. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1976;449(2):188–196. doi: 10.1016/0005-2728(76)90132-8. [DOI] [PubMed] [Google Scholar]
- 110.Hill B. C., Woon T. C., Nicholls P., Peterson J., Greenwood C., Thomson A. J. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochemical Journal. 1984;224(2):591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nicholls P., Kim J. K. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Canadian Journal of Biochemistry. 1982;60(6):613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- 112.Rich P. R., Meunier B., Mitchell R., John Moody A. Coupling of charge and proton movement in cytochrome c oxidase. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1996;1275(1–2):91–95. doi: 10.1016/0005-2728(96)00055-2. [DOI] [Google Scholar]
- 113.Nicholls P., Kim J. K. Oxidation of sulphide by cytochrome aa3. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1981;637(2):312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- 114.Smith R. P., Gosselin R. E. On the mechanism of sulfide inactivation by methemoglobin. Toxicology and Applied Pharmacology. 1966;8(1):159–172. doi: 10.1016/0041-008X(66)90112-8. [DOI] [PubMed] [Google Scholar]
- 115.Vitvitsky V., Yadav P. K., An S., Seravalli J., Cho U. S., Banerjee R. Structural and mechanistic insights into hemoglobin-catalyzed hydrogen sulfide oxidation and the fate of polysulfide products. Journal of Biological Chemistry. 2017;292(13):5584–5592. doi: 10.1074/jbc.M117.774943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Potor L., Nagy P., Méhes G., et al. Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxidative Medicine and Cellular Longevity. 2018;2018:16. doi: 10.1155/2018/3812568.3812568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garai D., Ríos-González B. B., Furtmüller P. G., et al. Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radical Biology and Medicine. 2017;113:551–563. doi: 10.1016/j.freeradbiomed.2017.10.384. [DOI] [PubMed] [Google Scholar]
- 118.Pálinkás Z., Furtmüller P. G., Nagy A., et al. Interactions of hydrogen sulfide with myeloperoxidase. British Journal of Pharmacology. 2015;172(6):1516–1532. doi: 10.1111/bph.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kimura H. Hydrogen polysulfide (H2Sn) signaling along with hydrogen sulfide (H2S) and nitric oxide (NO) Journal of Neural Transmission. 2016;123(11):1235–1245. doi: 10.1007/s00702-016-1600-z. [DOI] [PubMed] [Google Scholar]
- 120.Paul B. D., Snyder S. H. H2S: a novel gasotransmitter that signals by sulfhydration. Trends in Biochemical Sciences. 2015;40(11):687–700. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paul B. D., Snyder S. H. Protein sulfhydration. Methods in Enzymology. 2015;555:79–90. doi: 10.1016/bs.mie.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 122.Millikin R., Bianco C. L., White C., et al. The chemical biology of protein hydropersulfides: studies of a possible protective function of biological hydropersulfide generation. Free Radical Biology and Medicine. 2016;97:136–147. doi: 10.1016/j.freeradbiomed.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bianco C. L., Chavez T. A., Sosa V., et al. The chemical biology of the persulfide (RSSH)/perthiyl (RSS·) redox couple and possible role in biological redox signaling. Free Radical Biology and Medicine. 2016;101:20–31. doi: 10.1016/j.freeradbiomed.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heppner D. E., Hristova M., Ida T., et al. Cysteine perthiosulfenic acid (Cys-SSOH): a novel intermediate in thiol-based redox signaling? Redox Biology. 2018;14:379–385. doi: 10.1016/j.redox.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ida T., Sawa T., Ihara H., et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(21):7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mishanina T. V., Libiad M., Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nature Chemical Biology. 2015;11(7):457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Paul B. D., Snyder S. H. H2S signalling through protein sulfhydration and beyond. Nature Reviews Molecular Cell Biology. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 128.Poole L. B., Schöneich C. Introduction: what we do and do not know regarding redox processes of thiols in signaling pathways. Free Radical Biology and Medicine. 2015;80:145–147. doi: 10.1016/j.freeradbiomed.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Filipovic M. R., Zivanovic J., Alvarez B., Banerjee R. Chemical biology of H2S signaling through persulfidation. Chemical Reviews. 2018;118(3):1253–1337. doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Das T. N., Huie R. E., Neta P., Padmaja S. Reduction potential of the sulfhydryl radical: pulse radiolysis and laser flash photolysis studies of the formation and reactions of ·SH and HSSH·− in aqueous solutions. The Journal of Physical Chemistry A. 1999;103(27):5221–5226. doi: 10.1021/jp9907544. [DOI] [Google Scholar]
- 131.Akaike T., Ida T., Wei F. Y., et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nature Communications. 2017;8(1):p. 1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yadav P. K., Martinov M., Vitvitsky V., et al. Biosynthesis and reactivity of cysteine persulfides in signaling. Journal of the American Chemical Society. 2016;138(1):289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kimura Y., Koike S., Shibuya N., Lefer D., Ogasawara Y., Kimura H. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Scientific Reports. 2017;7(1, article 10459) doi: 10.1038/s41598-017-11004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kimura Y., Toyofuku Y., Koike S., et al. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Scientific Reports. 2015;5(1, article 14774) doi: 10.1038/srep14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shibuya N., Tanaka M., Yoshida M., et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxidants & Redox Signaling. 2009;11(4):703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 136.Cuevasanta E., Lange M., Bonanata J., et al. Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. Journal of Biological Chemistry. 2015;290(45):26866–26880. doi: 10.1074/jbc.M115.672816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krishnan N., Fu C., Pappin D. J., Tonks N. K. H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Science Signaling. 2011;4(203):p. ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sen N., Paul B. D., Gadalla M. M., et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Molecular Cell. 2012;45(1):13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mustafa A. K., Sikka G., Gazi S. K., et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circulation Research. 2011;109(11):1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Doka E., Pader I., Biro A., et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Science Advances. 2016;2(1, article e1500968) doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gao X.-H., Krokowski D., Guan B.-J., et al. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife. 2015;4, article e10067 doi: 10.7554/elife.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Longen S., Richter F., Köhler Y., Wittig I., Beck K. F., Pfeilschifter J. Quantitative persulfide site identification (qPerS-SID) reveals protein targets of H2S releasing donors in mammalian cells. Scientific Reports. 2016;6(1, article 29808) doi: 10.1038/srep29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang D., Macinkovic I., Devarie-Baez N. O., et al. Detection of protein S-sulfhydration by a tag-switch technique. Angewandte Chemie International Edition. 2014;53(2):575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? The FASEB Journal. 2002;16(13):1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 145.Cortese-Krott M. M., Koning A., Kuhnle G. G. C., et al. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxidants & Redox Signaling. 2017;27(10):684–712. doi: 10.1089/ars.2017.7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nobel Media AB; 2014. The Nobel prize in physiology or medicine 1998. http://www.nobelprize.org/nobel_prizes/medicine/laureates/1998/ [Google Scholar]
- 147.Ignarro L. J., Byrns R. E., Buga G. M., Wood K. S. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circulation Research. 1987;61(6):866–879. doi: 10.1161/01.RES.61.6.866. [DOI] [PubMed] [Google Scholar]
- 148.Palmer R. M. J., Ferrige A. G., Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 149.Zakhary R., Gaine S. P., Dinerman J. L., Ruat M., Flavahan N. A., Snyder S. H. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(2):795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bogdan C. Nitric oxide and the immune response. Nature Immunology. 2001;2(10):907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 151.Kuhn V., Diederich L., Keller T. C. S., IV, et al. Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxidants & Redox Signaling. 2017;26(13):718–742. doi: 10.1089/ars.2016.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lundberg J. O., Weitzber E. Nitric oxide formation from inorganic nitrate. In: Ignarro L. J., Freeman B. A., editors. Nitric Oxide, Biology and Pathobiology. Third. London: Academic Press; 2017. [DOI] [Google Scholar]
- 153.Wu L., Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacological Reviews. 2005;57(4):585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 154.Kevil C. G., Cortese-Krott M. M., Nagy P., Papapetropoulos A., Feelisch M., Szabo C. Cooperative interactions between NO and H2S: chemistry, biology, physiology, pathophysiology. In: Ignarro L. J., Freeman B. A., editors. Nitric Oxide, Biology and Pathobiology. Third. London: Academic Press; 2017. [DOI] [Google Scholar]
- 155.Cortese-Krott M. M., Fernandez B. O., Kelm M., Butler A. R., Feelisch M. On the chemical biology of the nitrite/sulfide interaction. Nitric Oxide. 2015;46:14–24. doi: 10.1016/j.niox.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 156.Carballal S., Trujillo M., Cuevasanta E., et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radical Biology and Medicine. 2011;50(1):196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 157.Cuevasanta E., Zeida A., Carballal S., et al. Insights into the mechanism of the reaction between hydrogen sulfide and peroxynitrite. Free Radical Biology and Medicine. 2015;80:93–100. doi: 10.1016/j.freeradbiomed.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 158.Berenyiova A., Grman M., Mijuskovic A., et al. The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants. Nitric Oxide. 2015;46:123–130. doi: 10.1016/j.niox.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 159.Cortese-Krott M. M., Fernandez B. O., Santos J. L. T., et al. Nitrosopersulfide (SSNO−) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biology. 2014;2:234–244. doi: 10.1016/j.redox.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cortese-Krott M. M., Kuhnle G. G. C., Dyson A., et al. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(34):E4651–E4660. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Filipovic M. R., Miljkovic J. L., Nauser T., et al. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. Journal of the American Chemical Society. 2012;134(29):12016–12027. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Teng X., Scott Isbell T., Crawford J. H., et al. Novel method for measuring S-nitrosothiols using hydrogen sulfide. Methods in Enzymology. 2008;441:161–172. doi: 10.1016/S0076-6879(08)01209-3. [DOI] [PubMed] [Google Scholar]
- 163.Ondrias K., Stasko A., Cacanyiova S., et al. H2S and HS− donor NaHS releases nitric oxide from nitrosothiols, metal nitrosyl complex, brain homogenate and murine L1210 leukaemia cells. Pflügers Archiv - European Journal of Physiology. 2008;457(2):271–279. doi: 10.1007/s00424-008-0519-0. [DOI] [PubMed] [Google Scholar]
- 164.Filipovic M. R., Eberhardt M., Prokopovic V., et al. Beyond H2S and NO interplay: hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. Journal of Medicinal Chemistry. 2013;56(4):1499–1508. doi: 10.1021/jm3012036. [DOI] [PubMed] [Google Scholar]
- 165.Gao Y., Toubaei A., Kong X., Wu G. Solving the 170-year-old mystery about red-violet and blue transient intermediates in the Gmelin reaction. Chemistry - A European Journal. 2015;21(48):17172–17177. doi: 10.1002/chem.201503353. [DOI] [PubMed] [Google Scholar]
- 166.Olabe J. A., Amorebieta V. T. P38 mechanism of the reactions of the nitroprusside ion, Fe (CN)5NO2−, with sulfides. Nitric Oxide. 2013;31(Supplement 2):p. S52. doi: 10.1016/j.niox.2013.06.100. [DOI] [Google Scholar]
- 167.Quiroga S. L., Almaraz A. E., Amorebieta V. T., Perissinotti L. L., Olabe J. A. Addition and redox reactivity of hydrogen sulfides (H2S/HS−) with nitroprusside: new chemistry of nitrososulfide ligands. Chemistry - A European Journal. 2011;17(15):4145–4156. doi: 10.1002/chem.201002322. [DOI] [PubMed] [Google Scholar]
- 168.Bruce King S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radical Biology and Medicine. 2013;55:1–7. doi: 10.1016/j.freeradbiomed.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cortese-Krott M. M., Butler A. R., Woollins J. D., Feelisch M. Inorganic sulfur–nitrogen compounds: from gunpowder chemistry to the forefront of biological signaling. Dalton Transactions. 2016;45(14):5908–5919. doi: 10.1039/C5DT05034K. [DOI] [PubMed] [Google Scholar]
- 170.Fukuto J. M., Carrington S. J., Tantillo D. J., et al. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chemical Research in Toxicology. 2012;25(4):769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kolluru G. K., Yuan S., Shen X., Kevil C. G. H2S regulation of nitric oxide metabolism. Methods in Enzymology. 2015;554:271–297. doi: 10.1016/bs.mie.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ali M. Y., Ping C. Y., Mok Y.-Y. P., et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? British Journal of Pharmacology. 2006;149(6):625–634. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Whiteman M., Li L., Kostetski I., et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochemical and Biophysical Research Communications. 2006;343(1):303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 174.Cortese-Krott M. M., Pullmann D., Feelisch M. Nitrosopersulfide (SSNO−) targets the Keap-1/Nrf2 redox system. Pharmacological Research. 2016;113(Part A):490–499. doi: 10.1016/j.phrs.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 175.Munro A. P., Williams D. L. H. Reactivity of sulfur nucleophiles towards S-nitrosothiols. Journal of the Chemical Society, Perkin Transactions 2. 2000;(9):1794–1797. doi: 10.1039/b004415f. [DOI] [Google Scholar]
- 176.Williams D. L. H. Nitrosation Reactions and the Chemistry of Nitric Oxide. Amsterdam: Elsevier; 2004. [Google Scholar]
- 177.Seel F., Wagner M. Reaction of sulfides with nitrogen monoxide in aqueous-solution. Zeitschrift für Anorganische und Allgemeine Chemie. 1988;558(3):189–192. [Google Scholar]
- 178.Bolden C., King S. B., Kim-Shapiro D. B. Reactions between nitrosopersulfide and heme proteins. Free Radical Biology and Medicine. 2016;99:418–425. doi: 10.1016/j.freeradbiomed.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Seel F., Kuhn R., Simon G., Wagner M. PNP-Perthionitrit und PNP-Monothionitrit / PNP-Perthionitrite and PNP-M onothionitrite. Zeitschrift für Naturforschung B. 1985;40(12):1607–1617. doi: 10.1515/znb-1985-1203. [DOI] [Google Scholar]
- 180.Wedmann R., Ivanovic-Burmazovic I., Filipovic M. R. Nitrosopersulfide (SSNO−) decomposes in the presence of sulfide, cyanide or glutathione to give HSNO/SNO−: consequences for the assumed role in cell signalling. Interface Focus. 2017;7(2, article 20160139) doi: 10.1098/rsfs.2016.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wedmann R., Zahl A., Shubina T. E., et al. Does perthionitrite (SSNO−) account for sustained bioactivity of NO? A (bio)chemical characterization. Inorganic Chemistry. 2015;54(19):9367–9380. doi: 10.1021/acs.inorgchem.5b00831. [DOI] [PubMed] [Google Scholar]
- 182.Morikawa T., Kajimura M., Nakamura T., et al. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(4):1293–1298. doi: 10.1073/pnas.1119658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Shintani T., Iwabuchi T., Soga T., et al. Cystathionine β-synthase as a carbon monoxide–sensitive regulator of bile excretion. Hepatology. 2009;49(1):141–150. doi: 10.1002/hep.22604. [DOI] [PubMed] [Google Scholar]
- 184.Wang C. N., Duan G. L., Liu Y. J., et al. Overproduction of nitric oxide by endothelial cells and macrophages contributes to mitochondrial oxidative stress in adrenocortical cells and adrenal insufficiency during endotoxemia. Free Radical Biology and Medicine. 2015;83:31–40. doi: 10.1016/j.freeradbiomed.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 185.Kajimura M., Fukuda R., Bateman R. M., Yamamoto T., Suematsu M. Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxidants & Redox Signaling. 2010;13(2):157–192. doi: 10.1089/ars.2009.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kabil O., Yadav V., Banerjee R. Heme-dependent metabolite switching regulates H2S synthesis in response to endoplasmic reticulum (ER) stress. Journal of Biological Chemistry. 2016;291(32):16418–16423. doi: 10.1074/jbc.C116.742213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Suematsu M., Nakamura T., Tokumoto Y., Yamamoto T., Kajimura M., Kabe Y. CO-CBS-H2S axis: from vascular mediator to cancer regulator. Microcirculation. 2016;23(3):183–190. doi: 10.1111/micc.12253. [DOI] [PubMed] [Google Scholar]
- 188.Oliveriusová J., Kery Vladimír, Maclean K. N., Kraus J. P. Deletion mutagenesis of human cystathionine β-synthase. Impact on activity, oligomeric status, and S-adenosylmethionine regulation. Journal of Biological Chemistry. 2002;277(50):48386–48394. doi: 10.1074/jbc.M207087200. [DOI] [PubMed] [Google Scholar]
- 189.Kumar A., Wißbrock A., Goradia N., et al. Heme interaction of the intrinsically disordered N-terminal peptide segment of human cystathionine-β-synthase. Scientific Reports. 8(1):p. 2474. doi: 10.1038/s41598-018-20841-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cherney M. M., Pazicni S., Frank N., Marvin K. A., Kraus J. P., Burstyn J. N. Ferrous human cystathionine β-synthase loses activity during enzyme assay due to a ligand switch process. Biochemistry. 2007;46(45):13199–13210. doi: 10.1021/bi701159y. [DOI] [PubMed] [Google Scholar]
- 191.Gherasim C., Yadav P. K., Kabil O., Niu W.-N., Banerjee R. Nitrite reductase activity and inhibition of H2S biogenesis by human cystathionine ß-synthase. PLoS One. 2014;9(1, article e85544) doi: 10.1371/journal.pone.0085544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Puranik M., Weeks C. L., Lahaye D., et al. Dynamics of carbon monoxide binding to cystathionine β-synthase. Journal of Biological Chemistry. 2006;281(19):13433–13438. doi: 10.1074/jbc.M600246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Taoka S., Banerjee R. Characterization of NO binding to human cystathionine β-synthase: possible implications of the effects of CO and NO binding to the human enzyme. Journal of Inorganic Biochemistry. 2001;87(4):245–251. doi: 10.1016/S0162-0134(01)00335-X. [DOI] [PubMed] [Google Scholar]
- 194.Taoka S., West M., Banerjee R. Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine β-synthase reveals nonequivalent active sites. Biochemistry. 1999;38(22):p. 7406. doi: 10.1021/bi995077i. [DOI] [PubMed] [Google Scholar]
- 195.Vicente J. B., Colaço H. G., Mendes M. I. S., Sarti P., Leandro P., Giuffrè A. NO• binds human cystathionine β-synthase quickly and tightly. Journal of Biological Chemistry. 2014;289(12):8579–8587. doi: 10.1074/jbc.M113.507533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Singh S., Madzelan P., Stasser J., et al. Modulation of the heme electronic structure and cystathionine beta-synthase activity by second coordination sphere ligands: the role of heme ligand switching in redox regulation. Journal of Inorganic Biochemistry. 2009;103(5):689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Carballal S., Cuevasanta E., Marmisolle I., et al. Kinetics of reversible reductive carbonylation of heme in human cystathionine β-synthase. Biochemistry. 2013;52(26):4553–4562. doi: 10.1021/bi4004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Weeks C. L., Singh S., Madzelan P., Banerjee R., Spiro T. G. Heme regulation of human cystathionine β-synthase activity: insights from fluorescence and Raman spectroscopy. Journal of the American Chemical Society. 2009;131(35):12809–12816. doi: 10.1021/ja904468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yadav P. K., Xie P., Banerjee R. Allosteric communication between the pyridoxal 5′-phosphate (PLP) and heme sites in the H2S generator human cystathionine β-synthase. Journal of Biological Chemistry. 2012;287(45):37611–37620. doi: 10.1074/jbc.M112.414706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Smith A. T., Su Y., Stevens D. J., Majtan T., Kraus J. P., Burstyn J. N. Effect of the disease-causing R266K mutation on the heme and PLP environments of human cystathionine β-synthase. Biochemistry. 2012;51(32):6360–6370. doi: 10.1021/bi300421z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Carballal S., Cuevasanta E., Yadav P. K., et al. Kinetics of nitrite reduction and peroxynitrite formation by ferrous heme in human cystathionine β-synthase. Journal of Biological Chemistry. 2016;291(15):8004–8013. doi: 10.1074/jbc.M116.718734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Carballal S., Madzelan P., Zinola C. F., et al. Dioxygen reactivity and heme redox potential of truncated human cystathionine β-synthase. Biochemistry. 2008;47(10):3194–3201. doi: 10.1021/bi700912k. [DOI] [PubMed] [Google Scholar]
- 203.Vicente J. B., Colaço H. G., Malagrinò F., et al. A clinically relevant variant of the human hydrogen sulfide-synthesizing enzyme cystathionine β-synthase: increased CO reactivity as a novel molecular mechanism of pathogenicity? Oxidative Medicine and Cellular Longevity. 2017;2017:13. doi: 10.1155/2017/8940321.8940321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vicente J. B., Colaco H. G., Sarti P., Leandro P., Giuffrè A. S-Adenosyl-L-methionine modulates CO and NO• binding to the human H2S-generating enzyme cystathionine β-synthase. Journal of Biological Chemistry. 2016;291(2):572–581. doi: 10.1074/jbc.M115.681221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kopecká J., Krijt J., Raková K., Kožich V. Restoring assembly and activity of cystathionine β-synthase mutants by ligands and chemical chaperones. Journal of Inherited Metabolic Disease. 2011;34(1):39–48. doi: 10.1007/s10545-010-9087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Majtan T., Liu L., Carpenter J. F., Kraus J. P. Rescue of cystathionine β-synthase (CBS) mutants with chemical chaperones: purification and characterization of eight CBS mutant enzymes. Journal of Biological Chemistry. 2010;285(21):15866–15873. doi: 10.1074/jbc.M110.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Majtan T., Pey A. L., Ereño-Orbea J., Martínez-Cruz L. A., Kraus J. P. Targeting cystathionine beta-synthase misfolding in homocystinuria by small ligands: state of the art and future directions. Current Drug Targets. 2016;17(13):1455–1470. doi: 10.2174/1389450117666160302094910. [DOI] [PubMed] [Google Scholar]
- 208.Majtan T., Pey A. L., Kraus J. P. Kinetic stability of cystathionine beta-synthase can be modulated by structural analogs of S-adenosylmethionine: potential approach to pharmacological chaperone therapy for homocystinuria. Biochimie. 2016;126:6–13. doi: 10.1016/j.biochi.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 209.Melenovská P., Kopecká J., Krijt J., et al. Chaperone therapy for homocystinuria: the rescue of CBS mutations by heme arginate. Journal of Inherited Metabolic Disease. 2015;38(2):287–294. doi: 10.1007/s10545-014-9781-9. [DOI] [PubMed] [Google Scholar]
- 210.Mendes M. I. S., Smith D. E. C., Vicente J. B., et al. Small aminothiol compounds improve the function of Arg to Cys variant proteins: effect on the human cystathionine β-synthase p.R336C. Human Molecular Genetics. 2015;24(25):7339–7348. doi: 10.1093/hmg/ddv431. [DOI] [PubMed] [Google Scholar]
- 211.Singh L. R., Chen X., Kožich V., Kruger W. D. Chemical chaperone rescue of mutant human cystathionine β-synthase. Molecular Genetics and Metabolism. 2007;91(4):335–342. doi: 10.1016/j.ymgme.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bublil E. M., Majtan T., Park I., et al. Enzyme replacement with PEGylated cystathionine β-synthase ameliorates homocystinuria in murine model. Journal of Clinical Investigation. 2016;126(6):2372–2384. doi: 10.1172/JCI85396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Majtan T., Hůlková H., Park I., et al. Enzyme replacement prevents neonatal death, liver damage, and osteoporosis in murine homocystinuria. The FASEB Journal. 2017;31(12):5495–5506. doi: 10.1096/fj.201700565R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Majtan T., Park I., Carrillo R. S., Bublil E. M., Kraus J. P. Engineering and characterization of an enzyme replacement therapy for classical homocystinuria. Biomacromolecules. 2017;18(6):1747–1761. doi: 10.1021/acs.biomac.7b00154. [DOI] [PubMed] [Google Scholar]
- 215.Celano L., Gil M., Carballal S., et al. Inactivation of cystathionine β-synthase with peroxynitrite. Archives of Biochemistry and Biophysics. 2009;491(1–2):96–105. doi: 10.1016/j.abb.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Prudova A., Bauman Z., Braun A., Vitvitsky V., Lu S. C., Banerjee R. S-Adenosylmethionine stabilizes cystathionine β-synthase and modulates redox capacity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(17):6489–6494. doi: 10.1073/pnas.0509531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mendes M. I. S., Colaço H. G., Smith D. E. C., et al. Reduced response of cystathionine beta-synthase (CBS) to S-adenosylmethionine (SAM): identification and functional analysis of CBS gene mutations in homocystinuria patients. Journal of Inherited Metabolic Disease. 2014;37(2):245–254. doi: 10.1007/s10545-013-9647-6. [DOI] [PubMed] [Google Scholar]
- 218.Mendes M. I. S., Santos A. S., Smith D. E. C., et al. Insights into the regulatory domain of cystathionine beta-synthase: characterization of six variant proteins. Human Mutation. 2014;35(10):1195–1202. doi: 10.1002/humu.22616. [DOI] [PubMed] [Google Scholar]
- 219.Beaumont M., Andriamihaja M., Lan A., et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: the adaptive response. Free Radical Biology and Medicine. 2016;93:155–164. doi: 10.1016/j.freeradbiomed.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 220.Kwiecień I., Sokołowska M., Luchter-Wasylewska E., Włodek L. Inhibition of the catalytic activity of rhodanese by S-nitrosylation using nitric oxide donors. The International Journal of Biochemistry & Cell Biology. 2003;35(12):1645–1657. doi: 10.1016/S1357-2725(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 221.D’Araio E., Shaw N., Millward A., Demaine A., Whiteman M., Hodgkinson A. Hydrogen sulfide induces heme oxygenase-1 in human kidney cells. Acta Diabetologica. 2014;51(1):155–157. doi: 10.1007/s00592-013-0501-y. [DOI] [PubMed] [Google Scholar]
- 222.Jia H., Ye J., You J., Shi X., Kang W., Wang T. Role of the cystathionine β-synthase/H2S system in liver cancer cells and the inhibitory effect of quinolone-indolone conjugate QIC2 on the system. Oncology Reports. 2017;37(5):3001–3009. doi: 10.3892/or.2017.5513. [DOI] [PubMed] [Google Scholar]
- 223.Kupai K., Almási N., Kósa M., et al. H2S confers colonoprotection against TNBS-induced colitis by HO-1 upregulation in rats. Inflammopharmacology. 2018;26(2):479–489. doi: 10.1007/s10787-017-0382-8. [DOI] [PubMed] [Google Scholar]
- 224.Shao M., Zhuo C., Jiang R., et al. Protective effect of hydrogen sulphide against myocardial hypertrophy in mice. Oncotarget. 2017;8(14):22344–22352. doi: 10.18632/oncotarget.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Xie L., Gu Y., Wen M., et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65(10):3171–3184. doi: 10.2337/db16-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhang C. Y., Li X. H., Zhang T., Fu J., Cui X. D. Hydrogen sulfide upregulates heme oxygenase‑1 expression in rats with volume overload‑induced heart failure. Biomedical Reports. 2013;1(3):454–458. doi: 10.3892/br.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang S., Wu T., Xia T., Rong X., Chu M., Wu R. Regulation of the heme oxygenase-1/carbon monoxide system by hydrogen sulfide in murine coxsackievirus B3-induced myocarditis. Cellular and Molecular Biology (Noisy-le-Grand, France) 2015;61(2):69–73. [PubMed] [Google Scholar]
- 228.Yi L., Jenkins P. M., Leichert L. I., Jakob U., Martens J. R., Ragsdale S. W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. Journal of Biological Chemistry. 2009;284(31):20556–20561. doi: 10.1074/jbc.M109.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yi L., Ragsdale S. W. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. Journal of Biological Chemistry. 2007;282(29):21056–21067. doi: 10.1074/jbc.M700664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Saidu N. E. B., Asali I. A., Czepukojc B., Seitz B., Jacob C., Montenarh M. Comparison between the effects of diallyl tetrasulfide on human retina pigment epithelial cells (ARPE-19) and HCT116 cells. Biochimica et Biophysica Acta (BBA) - General Subjects. 2013;1830(11):5267–5276. doi: 10.1016/j.bbagen.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 231.Shin I. S., Hong J., Jeon C. M., et al. Diallyl-disulfide, an organosulfur compound of garlic, attenuates airway inflammation via activation of the Nrf-2/HO-1 pathway and NF-kappaB suppression. Food and Chemical Toxicology. 2013;62:506–513. doi: 10.1016/j.fct.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 232.Zeng T., Zhang C. L., Song F. Y., et al. The activation of HO-1/Nrf-2 contributes to the protective effects of diallyl disulfide (DADS) against ethanol-induced oxidative stress. Biochimica et Biophysica Acta (BBA) - General Subjects. 2013;1830(10):4848–4859. doi: 10.1016/j.bbagen.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 233.Kubo S., Kurokawa Y., Doe I., Masuko T., Sekiguchi F., Kawabata A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology. 2007;241(1–2):92–97. doi: 10.1016/j.tox.2007.08.087. [DOI] [PubMed] [Google Scholar]
- 234.Oh G. S., Pae H. O., Lee B. S., et al. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radical Biology and Medicine. 2006;41(1):106–119. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 235.Whiteman M., Li L., Rose P., Tan C. H., Parkinson D. B., Moore P. K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxidants & Redox Signaling. 2010;12(10):1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kubo S., Doe I., Kurokawa Y., Nishikawa H., Kawabata A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension. Toxicology. 2007;232(1–2):138–146. doi: 10.1016/j.tox.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 237.Duan X.-C., Liu S.-Y., Guo R., et al. Cystathionine-β-synthase gene transfer into rostral ventrolateral medulla exacerbates hypertension via nitric oxide in spontaneously hypertensive rats. American Journal of Hypertension. 2015;28(9):1106–1113. doi: 10.1093/ajh/hpu299. [DOI] [PubMed] [Google Scholar]
- 238.The PyMOL Molecular Graphics System, Version 1.8 Schrödinger. LLC; [Google Scholar]
- 239.Evans S. V., Sishta B. P., Mauk A. G., Brayer G. D. Three-dimensional structure of cyanomet-sulfmyoglobin C. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4723–4726. doi: 10.1073/pnas.91.11.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]