

Abstract
The etiology of rheumatoid arthritis (RA) is poorly understood, and 30% of patients are unresponsive to established treatments targeting Tumor Necrosis Factor α (TNFα). Akt kinase is implicated in TNFα signaling, and may act as a barometer of patient responses to biologic therapies. Fluorescent peptide sensors and chemical cytometry were employed to directly measure Akt activity as well as proteolytic activity in individual fibroblast-like synoviocytes (FLS) from RA and normal subjects. The specificity of the peptide reporter was evaluated and shown to be a valid measure of Akt activity in single cells. The effect of TNFα treatment on Akt activity was highly heterogeneous between normal and RA subjects, which was not observable in bulk analyses. In 2 RA subjects, a bimodal distribution of Akt activity was observed, primarily due to a subpopulation (21.7%: RA Subject 5 and 23.8%: RA Subject 6) of cells in which >60% of the reporter was phosphorylated. These subjects also possessed statistically elevated proteolytic cleavage of the reporter relative to normal subjects, suggesting heterogeneity in Akt and protease activity that may play a role in the RA-affected joint. We expect that chemical cytometry studies pairing peptide reporters with capillary electrophoresis will provide valuable data regarding aberrant kinase activity from small samples of clinical interest.
INTRODUCTION
The advent of biologic therapies for treatment of rheumatoid arthritis (RA) signaled a shift towards precision medicine for treatment of rheumatologic disorders. Three very successful RA biologics include etanercept, adalimumab, and infliximab, all targeting TNFα, an inflammatory cytokine commonly upregulated in RA.1 Though considered revolutionary, these engineered proteins yield no clinically measurable benefit in 30% of patients; coined ‘primary nonresponders’.2 Due to the cost and potential co-morbidities associated with these drugs, the rheumatology community has worked for over a decade to identify biomarkers which may be predictive of patient response to anti-TNFα therapies. Clinical factors such as sex, smoking, and the presence of anti-cyclic citrullinated peptide antibodies as well as TNFα itself have not been predictive of patient response to anti-TNFα biologics; thus, the focus has shifted towards identification of molecular biomarkers or biochemical response profiles.3
One potential biomarker is the serine/threonine protein kinase Akt (also called protein kinase B), which is downstream of the TNFα receptor and is a component of TNF biochemical signaling. Both upregulated expression4 and constitutive activation5–7 of Akt in fibroblast-like synoviocytes (FLS) have been linked to chronic inflammation, increased efflux of matrix metalloproteinases (MMPs), and resistance to apoptosis, resulting in pseudo-tumoral proliferation of FLS and the irreversible cartilage damage characteristic of RA. These Akt abnormalities appear to exist primarily in FLS rather than macrophages or other cell types, suggesting that overactive Akt in FLS may be a component of disease progression.7 Thus, this signaling arm is being pursued as a therapeutic target for novel therapies and inhibitors with anti-inflammatory effects.8 With Akt mediating multiple downstream effects of TNFα signaling, the biochemical activity of this kinase in cells may also act as a barometer of the downstream effects of upregulated TNFα, with potential applications in patient classification as anti-TNFα therapy responsive or non-responsive.
A complicating factor in the analysis of these cells is the heterogeneity expected in RA FLS that possess this aggressive phenotype.9 Since only a fraction of the FLS population may have aberrant Akt activity, with even fewer present at early stages of the disease, it becomes imperative to analyze these cells with single-cell resolution. Traditional methodologies to determine the presence of Akt, such as immunohistochemistry, are valuable in determining the location of the kinase in tissue, but they are generally qualitative and do not provide compelling evidence that the labeled kinase’s biochemical activity is aberrant.10 Western blotting is another useful technique to garner gene expression information, but the detection of distinct subpopulations is not possible as the blot returns an ensemble average of the bulk population. Moreover, it is often difficult to obtain sufficient cells in biopsy samples for assay performance. Hughes and co-authors demonstrated a multiplexed solution capable of single-cell Western blots11, but the nature of their antibody-based kinase detection did not allow detection of constitutively or transiently active enzymes through quantification of substrate and products. These protein-only detection schemes are the primary limitations of other single-cell techniques, including Phospho-flow and mass cytometry.12,13 Liquid chromatography coupled tandem mass spectrometry (LC-MS/MS) has been reported to more accurately measure the stoichiometry of Akt phosphorylation in biological samples, but it requires immunoprecipitation, resulting in substantial samples losses.14 Furthermore, the low sensitivity necessitates large sample sizes. Meaningful measurement of Akt activity within RA FLS instead requires single-cell resolution, excellent limits of detection, quantification of substrate and products (as well as differentiation between any additional metabolites) and head-to-head comparison with normal FLS cells.
Chemical cytometry in the form of capillary electrophoresis with laser induced fluorescence detection (CE-LIF) routinely attains excellent (10-20 mol) limits of detection, as is required when analyzing intracellular metabolites of single mammalian cells which are typically ≤1 pL in volume. When coupled with peptide based probes, this separation based technology can directly assess kinase activity15–17, and is highly quantitative with the inclusion of internal standards. In this study we evaluated Akt and peptidase activity in single FLS cells from RA and normal subjects. Prior to single-cell analysis, the specificity of the peptide-based Akt reporter was characterized to ensure rigorous measurements of the intended pathway. The Akt activity was then quantified in single cells from normal and RA FLS cells by chemical cytometry, and compared to traditional bulk analyses, which revealed novel information about the heterogeneity of this pathway in RA FLS.
EXPERIMENTAL SECTION
Supporting information regarding cell culture, siRNA transfections, western blotting, flow cytometry, and details regarding statistical analysis are available online.
FLS Cell Culture
FLS from 6 individuals were collected at the time of synovectomy or total joint replacement. Due to the their de-identification and pre-existence in a repository, the tissues did not constitute human subjects research as defined under federal regulations, and were exempt from IRB approval (Study #14-1569). Three were biologic-naïve RA patients which fulfilled the American College of Rheumatology 1997 criteria for RA classification. Three were individuals who were undergoing surgical resection of tissue for reasons unrelated to rheumatologic disorders. Tissues were stored under liquid N2 until the time of FLS culture. Synovial tissue was minced and immobilized in a tissue culture plate and covered with Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1% PenStrep Solution (Gibco, Grand Island, NY) and were maintained in a humidified atmosphere of 37°C in 5% CO2. Media was changed regularly until the FLS expanded to fill the entire plate. These cell lines from primary FLS were utilized from the third to ninth passage.
Cell Loading and Single-Cell Analysis by Capillary Electrophoresis
FLS from normal and rheumatoid arthritis subjects were separately cultured in custom cell chambers as described previously.18 Chambers were placed on the stage of a custom-built single-cell CE-LIF system (fluorescence excitation at 473 nm, emission at 530 nm).19 Cells were microinjected with 100 μM peptide VI-B (6FAM-GRP-MeArg-AFTF-MeAla-Amide) using a Transjector 5246 microinjection system (Eppendorf AG, Hamburg, Germany) and bathed in a continuous flow of extracellular buffer (ECB; 10 mM HEPES, 135 mM NaCl, 5 mM KCl, 1 mM CaCl2, pH 7.4, 37°C) during incubation and analysis. After microinjection (5 min), individual cells were rapidly lysed with a 9 ns pulse from a Nd:YAG laser (New Wave Research, Bozeman, MT) and the cellular contents simultaneously electrokinetically injected (5 s at -125 V/cm applied to the outlet) into an overlying 30 μm inner diameter capillary (Polymicro Technologies, Phoenix, AZ). Electrophoresis was performed in 100 mM borate, 15 mM SDS, pH 11.6 with a field strength of -250 V/cm applied to the outlet. Electropherograms were integrated using customized software written in MATLAB (Natick, MA).17 Detailed information regarding peptide fragment nomenclature and identification is available in supplemental information.
Statistical Analysis
Mean phosphorylation and degradation are reported with standard deviations. To compare phosphorylation and degradation in single cells in these chemical cytometry experiments, bootstrapping, a nonparametric resampling method commonly utilized for the analysis of small clinical samples, was utilized to generate population distributions for statistical comparisons (details; Supplemental Section 1.6).
RESULTS AND DISCUSSION
Evaluating Peptide Reporter Specificity
In order to measure Akt activity in single FLS, a phosphorylatable-threonine-containing peptide, herein after referred to as VI-B (6FAM-GRP-MeArg-AFTF-MeAla-Amide), was utilized. The sequence was determined using an iterative design strategy originating from the consensus sequence of Akt.16 VI-B possessed favorable phosphorylation kinetics as well as demonstrated degradation resistance, and has been employed to measure Akt activity in single tissue-cultured cells and pancreatic adenocarcinoma patient-derived xenografts.21,22
Since many eukaryotic protein kinases possess similar catalytic domains,23 we sought to evaluate whether the majority of VI-B phosphorylation was due to Akt or other analogous, potentially interfering kinases. A basal level of phosphorylation was established by depriving FLS cells of serum for 12 h, loading VI-B, and analyzing cells by single-cell CE-LIF. Basal phosphorylation varied between cells (0-37%), but was similar between normal and RA subjects (p = 0.31) (Figure 1A,B). To determine whether VI-B phosphorylation was a result of Akt activity or due to other kinases within the intracellular environment, reporter phosphorylation in response to physiologic stimulation or pharmacologic inhibition of the Akt pathway was measured. Insulin stimulates cell growth and protein synthesis by activation of Akt.24 When FLS were treated with 100 ng/mL insulin, significantly increased mean VI-B phosphorylation was observed in both normal (n = 16; 38.2 ± 31%) and RA subjects (n = 11; 39.5 ± 37.7%). Conversely, when cells were treated with 10 μM LY294002, a reversible inhibitor of PI3K, the kinase directly upstream of Akt,25 mean VI-B phosphorylation was less than 5% of the basal level. These data supported that the majority of the reporter phosphorylation observed was the result of Akt activity (Figure 1A,B).
Figure 1. Specificity Evaluation of Akt Reporter VI-B.
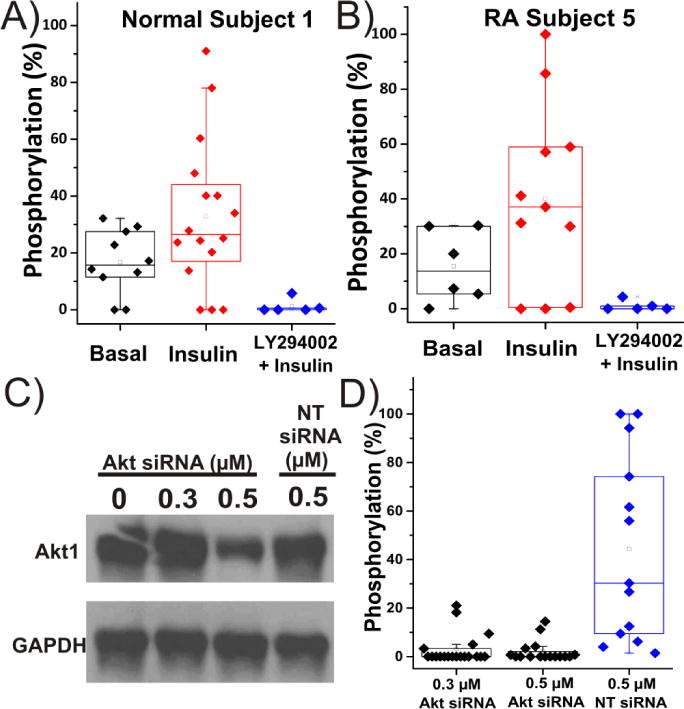
Basal measurements, physiologic stimulation, and pharmacologic inhibition of the Akt pathway in single FLS cells from a A) normal subject 1 or B) RA subject 5. Before microinjection with VI-B, cells were either serum starved for 12 h for basal measurements, stimulated with 100 ng/mL insulin, or were treated with 10 μM LY294002 prior to insulin treatment (100 ng/mL). Within the boxplots, the middle bar indicates the median, while the upper and lower boxes represent the 75% and 25% percentile of the data, respectively. Whiskers extend to the 5th and 95th percentiles. Any individual data points outside of the whiskers are outliers. C) Western blot results from siRNA knockdown of Akt in RA Subject 5. The housekeeping protein GAPDH was probed as a loading control. Cells underwent no transfection as a control or were transfected with various concentrations of Akt-targeting siRNA or non-targeting, scrambled siRNA (NT siRNA). D) Individual FLS cells analyzed for Akt activity 48 hrs after siRNA transfection with Akt-targeting siRNA or non-targeting, scrambled siRNA (NT siRNA).
In a second experiment, Akt protein was transiently knocked down in FLS by administering a specific siRNA for Akt. The presence of the kinase was monitored 48 h post siRNA-transfection by western blot (Figure 1C), which demonstrated a dose-dependent reduction in Akt expression. The transfection process did not disrupt Akt expression, as demonstrated by similar expression of Akt between un-transfected cells (0 nM Akt siRNA) and 500 nM of a non-targeting (NT) scrambled siRNA. Using chemical cytometry, VI-B phosphorylation was assessed in single cells 48 h following transfection with 300 and 500 nM Akt-targeting siRNA. Cells were serum starved and stimulated with insulin (100 ng/mL) prior to analysis as described above. Significant reduction in reporter phosphorylation was seen in comparison to FLS transfected with non-targeting scrambled siRNA (Figure 1D), again supporting that a dominant portion of reporter phosphorylation was due to Akt activity. These data from physiologic and pharmacological perturbation of the Akt pathway indicated that VI-B phosphorylation was a valid measure of Akt activity in FLS.
Akt Activity within Single FLS from RA and Normal Subjects
Individual FLS cells (analyzed: n=107) were stimulated with TNFα to determine whether this factor would activate Akt in FLS from normal and RA subjects (Figure 2). TNFα is known to be elevated in the joints of RA patients; therefore, addition of TNFα to FLS is expected to partially mimic the inflammatory environment of the RA-affected joint. FLS obtained from RA patients and normal subjects were plated on glass cell chambers. Cells were serum-starved overnight, then stimulated with 100 ng/mL TNFα for 30 min and loaded with VI-B. After a 5-min incubation, individual cells were assayed for phosphorylation of VI-B. When cells originating from normal subjects 1-3 (N1-N3) were stimulated with TNFα, minimal phosphorylation of the reporter was detected (Figure 2D). N1-N3 demonstrated 0.8±2.5% (n=14), 1.6±6.1% (n=16), and 0.8±2.6% (n=15) phosphorylation, respectively (Figure 3b). When individual FLS were analyzed from RA Subject 4 under identical conditions s (Figure 3a), a similar level of phosphorylation was detected (0.5±2.1% [n=18]), suggesting that in this particular subject, TNFα contributed minimally to Akt activation. In contrast, when single FLS were analyzed from RA subjects 5 and 6, the mean phosphorylation rose to 25.6±36.4% (n=23) and 27.1±37.8% (n=21), respectively. This was primarily due to a subpopulation (21.7% of cells: RA Subject 5 and 23.8% of cells: RA Subject 6) which phosphorylated >60% of the reporter within the 5 min incubation period.
Figure 2. Akt Activity Measurements in Single FLS.
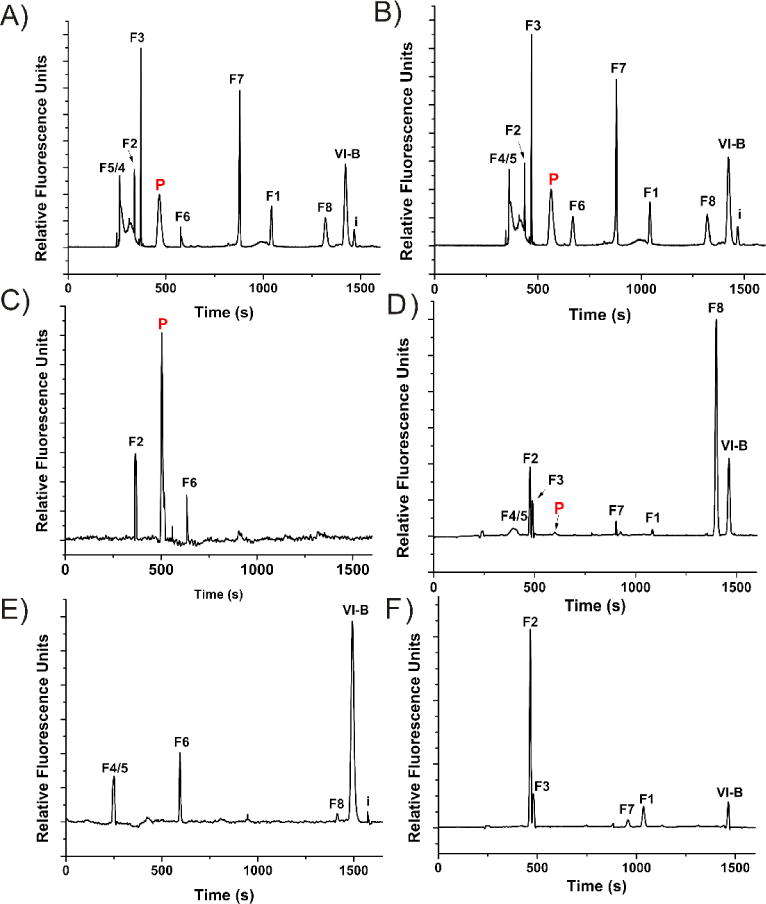
CE-LIF analysis from individual cells originating from RA subject 5 (A,C,E) and normal subject 1 (B,D,F). Migration time standards of parent peptide (VI-B), phosphorylated peptide (P) and all possible fluorescent fragments (F1-8) were loaded into the capillary after loading the contents of single cells not loaded with VI-B (A,B). Individual cells were stimulated with 100 ng/mL TNFα for 30 min (C,D) or treated with 10 μM LY294002 for 25 min prior to TNFα stimulation (E,F) and subsequently analyzed with CE-LIF. Each trace represents CE-LIF analysis of a single cell.
Figure 3. Akt Activity in FLS from Normal and RA Subjects after Stimulation with TNFα.
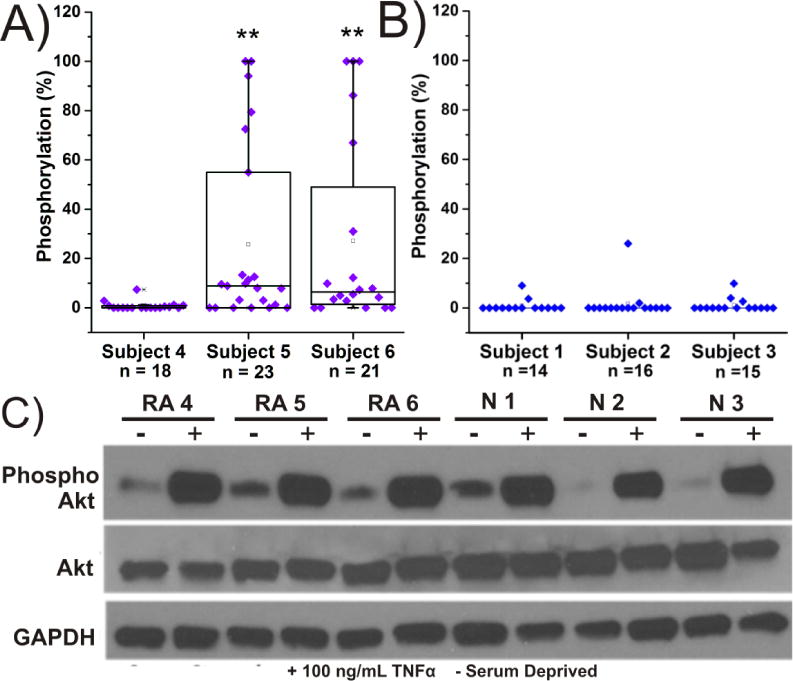
Each data point represents a single cell analyzed for Akt activity from A) RA subjects or B) normal subjects. Boxplots are organized as described in Figure 1. All cells were serum deprived overnight and subsequently stimulated with 100 ng/mL TNFα for 30 min before analysis with chemical cytometry. * = p < 0.05, ** = p <0.01.C) Western blotting was performed in lysates prepared from cells with and without TNFα stimulation (100 ng/mL).
Active Akt Determined by Western Blot Analysis in Lysates from FLS
Western blot analysis was performed on FLS from all subjects to compare the amount of active Akt detected between single cells and ensemble population level measurements (Figure 3C). Cells were unstimulated or stimulated with TNFα as described above prior to lysis. The amount of endogenous Akt was determined with an anti-Akt antibody competent to bind to Akt1, Akt2, and Akt3. Phospho-Akt was estimated utilizing a monoclonal antibody against phosphorylated serine at location 473. In all cases, TNFα treatment resulted in increased amounts of p473, but there were no differences detectable between normal and RA subjects.
Tumor Necrosis Factor Receptor 1 (TNFR1) Expression in FLS
TNFα signaling is initiated by binding to receptors expressed on nucleated cells. Of two distinct receptors, Akt is thought to be regulated by the 55-kDa type 1 receptor (TNFR1).8 To evaluate whether differences in TNFR1 expression between RA and normal subjects might be due to receptor expression, we probed FLS with antibodies specific for the receptor and analyzed the FLS by flow cytometry (Figure 4). Positive, negative, and isotype controls were also performed (Supplementary Figure 1). Cells were concomitantly stained with SYTOX green to detect non-viable cells. All subjects possessed TNFR1 positive populations with few TNFR1-negative cells. Statistical analysis (Kruskal Wallis one-way ANOVA) performed on live cells determined to be TNFR1-positive, revealed that normal subjects 2 and 3 possessed elevated expression of TNFR1 (p = 0.009 and 0.012, respectively), relative to RA cells. This is in agreement with reports of decreased membrane TNFR1 expression on RA FLS, which may occur due to receptor internalization or increased rates of cleavage of membrane TNFR1 to form soluble extracellular 17 kDa TNFα, not detected by flow cytometry.26–28 The mechanism of Akt activation in cells with decreased TNFR1 expression remains debated, but it is possible that the increased TNFα ligand generated by cleavage of the receptor,29 or that shuttling of TNFα by the soluble receptor7 plays a role.
Figure 4. Tumor Necrosis Factor Receptor 1 (TNFR1) Expression in FLS.
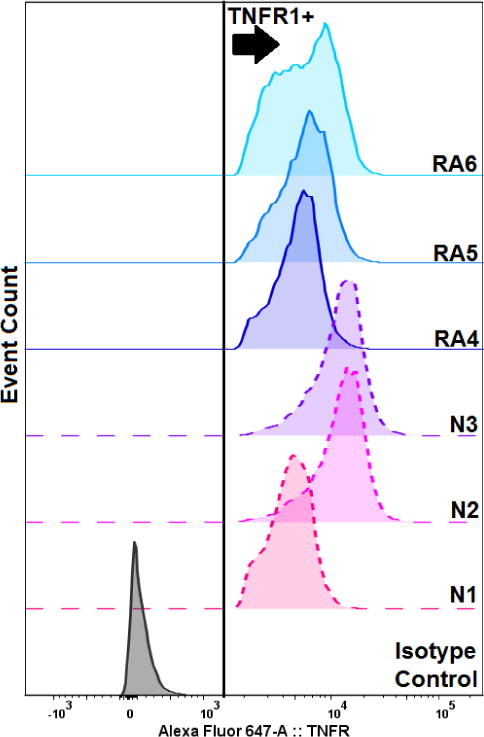
Normal subjects 1-3 (N1-N3, dashed lines) and RA subjects 4-6 (RA4-6, solid lines) FLS were co-stained with a monoclonal antibody to detect TNFR1 and the dead-cell indicator SYTOX Green (10 μM), then were analyzed by flow cytometry. Cells considered to be TNFR1 positive, viable cells are depicted to the right of the arrow.
Degradation of VI-B in Single FLS from Normal and RA Subjects
It is well known that within the RA-affected joint, FLS secrete potent MMPs and cathepsins which act extracellularly to degrade collagen and bone.9 Intracellular protease and peptidase activity constitutes a mostly unexplored area of FLS research, but correlations have been shown between advanced (>10 yr duration) RA and highly active intracellular proteolysis pathways involving cysteine and aspartate proteinases.30 As shown in Figure 2A,F, we generated standards of all potential fluorescent fragments of VI-B, which are well-resolved from phosphorylated VI-B by CE-LIF. Thus, we have the capability to identify the fluorescent proteolysis products of VI-B via chemical cytometry as previously described16, and therefore characterize the proteolysis rate as well as the primary sites of VI-B cleavage within FLS from RA as well as normal subjects. Normal subjects 1-3 and RA Subject 4 possessed rates of VI-B degradation that were not statistically different (p =0.11) (Figure 5A,B): 0.14±0.76, 0.35±0.78, 0.75±0.46, and 0.71±.69 zmol pg-1 s-1 for Subjects 1-4, respectively. However, RA Subjects 5 (2.99±0.52 zmol pg-1 s-1) and 6 (3.27±0.60 zmol pg-1 s-1) demonstrated significantly more rapid degradation of the reporter (p = .013), providing evidence that at least some RA subjects have may have greater proteolytic capability, which may correlate with intracellular Akt activity.
Figure 5. Proteolysis of VI-B in Single FLS cells from Normal and RA Subjects.
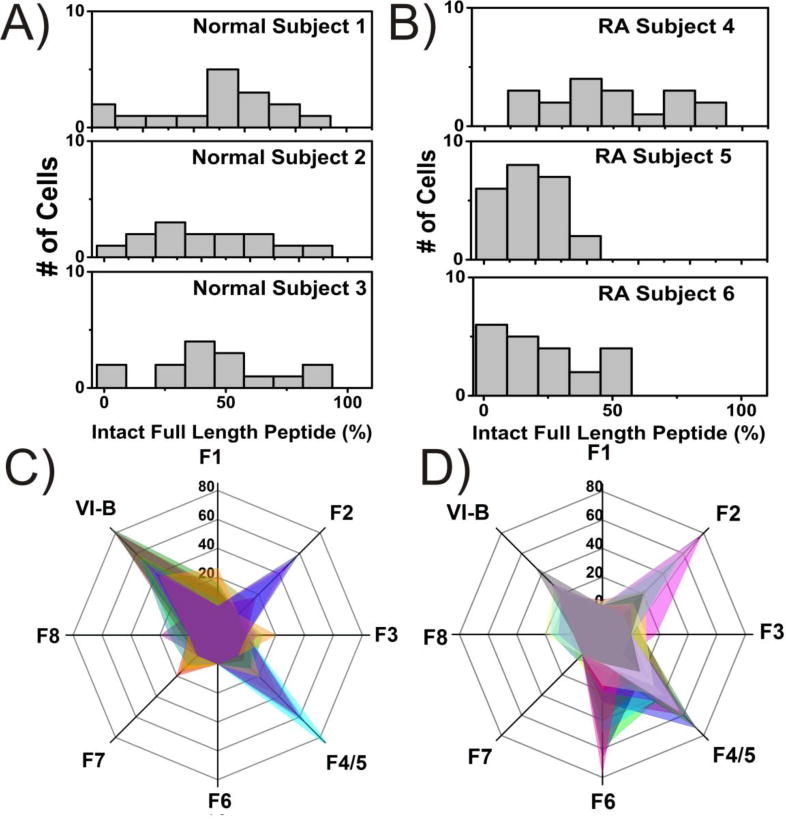
Histograms illustrating the percentage of intact, un-degraded reporter (VI-B) from each TNFα-stimulated cell analyzed by chemical cytometry from A) normal and B) RA subjects. Fragmentation profiles for C) normal subject 1 (n=14 cells each depicted as a different color) and D) RA subject 5 (n=23). VI-B represents the full length peptide, while F1-F8 indicates the 1 to 8 amino-acid fluorescent fragments, respectively.
Interestingly, fragment identities between RA and normal cells also differed (Figure 5C,D). Fragment F1 corresponds to the shortest possible fluorescent fragment (6FAM-G), which extends C-terminally to the longest fragment F8 (6FAM-GRP-MeArg-AFTF). RA Subjects 5 and 6 were the only group to cleave between phenylalanine and threonine (F6), with an average frequency of 19.4±5.8% (RA5) and 22.5±9.8% (RA6). Both RA and normal subjects often possessed the fragments arising due to cleavage C-terminal of arginine (F2) and N-methylarginine or alanine (F4/5) (RA: 52.2±22% Normal: 46.6±14%). RA Subjects rarely produced fragments due to cleavage after proline (F3), penultimate phenylalanine (F7), or glycine (F1), which were frequently observed in small quantities from the FLS of normal subjects (Figure 2D, F).
CONCLUSIONS
While prior reports have indicated that Akt activity can be elevated in RA FLS cells in response to TNFα,31 this is an initial demonstration of the heterogeneity of this response, both within and between RA subjects. Such differential upregulation of Akt between patients may indicate differences in the responsiveness of the Akt pathway in these cells, for example, in the ability to transmit the downstream signal after TNFα binding to modify cell behavior, i.e. apoptosis resistance and/or secretion of MMPs.32 With respect to the heterogeneity of Akt activity within RA Subjects 5 and 6, such cell-to-cell variation in Akt activity is not unprecedented in the literature; it is relatively common in cancers33, Proteus syndrome34, and other diseases35,36 that hijack this pathway to promote survival of cells under stressful conditions. Our results demonstrate the merit of chemical cytometry to measuring synoviocyte heterogeneity with single cell resolution. An important avenue of future work will be on improvements in assay throughput and assay of greater numbers of patients. Importantly, we demonstrated that traditional bulk analyses by western blotting was unable to resolve these differences in Akt activation, solidifying the concept that single cell analysis is necessary to observe the heterogeneity in Akt signaling upon TNFα stimulation.
Though the sample size was limited, it was also determined that RA subjects 5 and 6 possessed more rapid degradation of the reporter and that fragmentation occurred in locations not detected in normal subjects. It could be possible that these findings are due to the differential activity and/or different types of proteases or peptidases found in FLS of RA and normal subjects. For example, the protease cathepsins B and L are known for their role in extracellular degradation of collagen, but are also overexpressed intracellularly as a result of increased TNFα stimulation.37,38 Specificity studies indicate their canonical preference for aromatic residues including phenylalanine.39 The increased cleavage at hydrophobic residues (F6) observed in RA subjects 5 and 6 could potentially be due to contributions from such cathepsins B and L, although analysis of additional cells from these and additional patients will be necessary to draw such conclusions.
FLS cells within the RA afflicted joint are unique in their dependence on kinases such as Akt, which allow for unfettered growth, but also proteases, which catalyze the destruction of cartilage, therefore the simultaneous measurement of both these parameters may be clinically valuable. Tracking the Akt upregulation within single cells from many RA patients following treatment to inhibit TNFα should help inform our understanding as to whether the activity of this kinase might be a predictive marker of patient response to biologic therapies. Importantly, cells possessing elevated Akt and protease activity could be identified in small clinical (0.1-1 million cells) samples, such as a fine needle biopsy40 or synovial fluid collection41; samples that current methodologies struggle to analyze. Broadening this study to a large cohort of patients with a high throughput system may provide clinically relevant results to inform the variable responses to targeted therapies.
Supplementary Material
Acknowledgments
This work was supported by NIH (CA177993 to NLA) and the Rheumatology Research Foundation to TKT. Research reported in this publication was conducted in part at the UNC Flow Cytometry Core Facility and was supported by the North Carolina Biotech Center Institutional Support Grant 2012-IDG-1006.
References
- 1.Neovius M, Arkema EV, Olsson H, Eriksson JK, Kristensen LE, Simard JF, Askling J, Bäcklund E, Cöster L, Forsblad-d’Elia H, Feltelius N, Jacobsson L, Klareskog L, Lindblad S, Rantapää-Dahlqvist S, Saxne T, van Vollenhoven R. Ann Rheum Dis. 2015;74:354–360. doi: 10.1136/annrheumdis-2013-204128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubbert-Roth A, Finckh A. Arthritis Res Ther. 2009;11:1–12. doi: 10.1186/ar2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potter C, Hyrich KL, Tracey A, Lunt M, Plant D, Symmons DPM, Thomson W, Worthington J, Emery P, Morgan AW, Wilson AG, Isaacs J, Barton A. Ann Rheum Dis. 2009;68:69–74. [Google Scholar]
- 4.García S, Liz M, Gómez-Reino JJ, Conde C. Arthritis Res Ther. 2010;12:R33. doi: 10.1186/ar2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García S, Mera A, Gómez-Reino JJ, Conde C. Rheumatology (Oxford) 2009;48:483–489. doi: 10.1093/rheumatology/ken502. [DOI] [PubMed] [Google Scholar]
- 6.Miyashita T, Kawakami A, Tamai M, Izumi Y, Mingguo H, Tanaka F, Abiru S, Nakashima K, Iwanaga N, Aratake K, Kamachi M, Arima K, Ida H, Migita K, Origuchi T, Tagashira S, Nishikaku F, Eguchi K. Biochem Biophys Res Commun. 2003;312:397–404. doi: 10.1016/j.bbrc.2003.10.141. [DOI] [PubMed] [Google Scholar]
- 7.Zhang HG, Wang Y, Xie JF, Liang X, Liu D, Yang P, Hsu HC, Ray RB, Mountz JD. Arthritis Rheum. 2001;44:1555–1567. doi: 10.1002/1529-0131(200107)44:7<1555::AID-ART279>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 8.Ozes ON, Akca H, Gustin JA, Mayo LD, Pincheira R, Korgaonkar CK, Donner DB. In: Cell Signaling in Vascular Inflammation. DPhil JBM, editor. Humana Press; 2005. pp. 13–22. [Google Scholar]
- 9.Bartok B, Firestein GS. Immunol Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos-Vara JA. Vet Pathology. 2005;4:405–426. doi: 10.1354/vp.42-4-405. [DOI] [PubMed] [Google Scholar]
- 11.Hughes AJ, Spelke DP, Xu Z, Kang C, Schaffer DV, Herr AE. Nat Methods. 2014;11:749–755. doi: 10.1038/nmeth.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lujan E, Zunder ER, Ng YH, Goronzy IN, Nolan GP, Wernig M. Nature. 2015;521:352–356. doi: 10.1038/nature14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davey HM, Kell DB. Microbiol Rev. 1996;60:641–696. doi: 10.1128/mr.60.4.641-696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atrih A, Turnock D, Sellar G, Thompson A, Feuerstein G, Ferguson MAJ, Huang JT-S. J Proteome Res. 2010;9:743–751. doi: 10.1021/pr900572h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickinson AJ, Armistead PM, Allbritton NL. Anal Chem. 2013;85:4797–4804. doi: 10.1021/ac4005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Proctor A, Wang Q, Lawrence DS, Allbritton NL. Anal Chem. 2012;84:7195–7202. doi: 10.1021/ac301489d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips RM, Dailey LA, Bair E, Samet JM, Allbritton NL. Anal Chem. 2014;86:1291–1297. doi: 10.1021/ac403705c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner AH, Lebhar MS, Proctor A, Wang Q, Lawrence DS, Allbritton NL. ACS Chem Biol. 2015;11:355–362. doi: 10.1021/acschembio.5b00667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mainz ER, Dobes NC, Allbritton NL. Anal Chem. 2015;87:7987–7995. doi: 10.1021/acs.analchem.5b01929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efron B. Bootstrap Methods: Another Look at the Jackknife. In: Kotz S, Johnson NL, editors. Breakthroughs in Statistics. Springer; New York: 1992. pp. 569–593. [Google Scholar]
- 21.Proctor A. Ph D Dissertation. The University of North Carolina; Chapel Hill, NC: May, 2012. Development of Peptidase-Resistant Peptide Substrates For Measurement of Protein Kinase B and Bcr-Abl Kinase Activity. [Google Scholar]
- 22.Proctor A, Herrera-Loeza G, Wang Q, Lawrence DS, Yeh J, Allbritton NL. Anal Chem. 2014;86:4573–4580. doi: 10.1021/ac500616q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ubersax JA, Ferrell JE., Jr Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 24.Haar EV, Lee S, Bandhakavi S, Griffin TJ, Kim D. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 25.Gharbi S, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, Waterfield MD. Biochemical Journal. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong CK, Chen DP, Tam LS, Li EK, Yin YB, Lam CW. Arthritis Res Ther. 2010;12:1–15. doi: 10.1186/ar3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arend WP, Dayer J. Arthritis Rheumatol. 1995;38:151–160. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- 28.Levine SJ. J Biol Chem. 2008;283:14177–14181. doi: 10.1074/jbc.R700052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumeyer JS. Tumor Necrosis Factor. In: Offerman S, Rosenthal W, editors. Encyclopedia of Molecular Pharmacology. Springer Science & Business Media; New York, USA: 2008. pp. 1248–1251. [Google Scholar]
- 30.Tak PP, Bresnihan B. Arthritis Rheum. 2000;43:2619–2633. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 31.Reddy SAG, Huang JH, Liao WS. J Immunol. 2000;164:1355–1363. doi: 10.4049/jimmunol.164.3.1355. [DOI] [PubMed] [Google Scholar]
- 32.Mountz JD, Zhang H, Wang Y, Xie JF, Liang X, Hsu H, Curiel DT. Arthritis Res. 2001;3:P8. [Google Scholar]
- 33.Lei Q, Jiao J, Xin L, Chang C, Wang S, Gao J, Gleave ME, Witte ON, Liu X, Wu H. Cancer Cell. 2006;9:367–378. doi: 10.1016/j.ccr.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 34.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindhurst MJ, Parker VER, Payne F, Sapp JC, Rudge S, Harris J, Witkowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, Cunningham ML, Darling TN, Geer J, Gucev Z, Sutton VR, Tziotzios C, Dixon AK, Helliwell T, O’Rahilly S, Savage DB, Wakelam MJO, Barroso I, Biesecker LG, Semple RK. Nat Genet. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hevner RF. Semin Perinatol. 2015;39:36–43. doi: 10.1053/j.semperi.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou W, Li Z, Gordon RE, Chan K, Klein MJ, Levy R, Keysser M, Keyszer G, Brömme D. Am J Pathol. 2001;159:2167–2177. doi: 10.1016/S0002-9440(10)63068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemaire R, Huet G, Zerimech F, Grard G, Fontaine C, Duquesnoy B, Flipo RM. Br J Rheumatol. 1997;36:735–743. doi: 10.1093/rheumatology/36.7.735. [DOI] [PubMed] [Google Scholar]
- 39.Biniossek ML, Nägler DK, Becker-Pauly C, Schilling O. J Proteome Res. 2011;10:5363–5373. doi: 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- 40.Rajer M, Kmet M. Radiol Oncol. 2004;39:269–272. [Google Scholar]
- 41.McNally EG. Skeletal Radiol. 2008;37:99–113. doi: 10.1007/s00256-007-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.