

Abstract
NK cells, which contribute to immune defense against certain viral infections and neoplasia, are emerging as modifiers of chronic immunologic diseases including transplant rejection and autoimmune diseases. Immunobiology and genetic studies have implicated NK cells as a modifier of Crohn’s disease, a condition often treated with thiopurine agents such as 6-mercaptopurine (6-MP). Here, we demonstrate that thiopurines mediate NK cell apoptosis via a caspase 3 and 9 inclusive pathway, and that this process is triggered by thiopurine-mediated inhibition of Rac1. We also show that CD patients in clinical remission maintained on 6-MP have decreased NK cell Rac1 activity, and decreased NK cell numbers in their intestinal biopsies. These observations suggest that thiopurine targeting of NK cells may be a previously unappreciated therapeutic action of these agents in IBD.
Keywords: Caspase, 6-Mercaptopurine, Inflammatory bowel disease, NK cells, Rac1, Thiopurine
1. Introduction
NK cells belong to the innate immune system and are classically understood for their role in host defense responses against certain viruses and cells with malignant potential [1–4]. Key to these roles is the NK cells’ receptor repertoire for detection of surface molecules specific to microbial agents or categorically induced by the molecular stress response shared by viral infection and neoplastic states [2,5,6]. Modern studies in NK cell biology have increasingly highlighted that NK cells influence the pathophysiologic processes underlying diverse chronic inflammatory conditions such as transplant rejection [7,8], rheumatoid arthritis [9], diabetes [10], and inflammatory bowel disease (IBD) including ulcerative colitis (UC) and Crohn’s disease (CD) [11–15]. The mechanisms by which NK cells affect these inflammatory processes are incompletely understood, but may include direct tissue damage via parenchymal cell cytolysis and production of cytokines inducing T cell or myeloid cell recruitment and activation [16–18].
Intestinal inflammation in CD results from dysregulated actions of adaptive and innate immune cells driven by genetic, environmental, and microbial processes. Although adaptive immune cells are considered the definitive mediators of mucosal inflammation, NK cells have received increasing attention as potential contributors to this immune pathophysiology. Genome-wide studies have associated characteristic NK receptors (killer immunoglobulin-like receptors, KIR) and their cognate HLA alleles with IBD susceptibility [19,20]. At the cellular level, populations of NK cells with cytolytic potential are enriched in colonic lamina propria of individuals with active IBD [11,15], and subsets of NK cells exert inflammatory effect by promoting CD4+ T cell proliferation and CD-relevant Th17 differentiation via production of pro-inflammatory cytokines [16]. Notably, this inflammatory action is dependent on KIR and HLA-dependent genetic programming of NK cells termed ‘licensing’, and results in distinct subsets of human subpopulations genetically distinct for inflammatory and anti-viral proficiency of their NK cell compartment [16,21–23]. These observations suggest that drugs which affect NK and other innate immune cells may be a significant and underappreciated component of their therapeutic action.
6-mercaptopurine (6-MP) and its precursor drug azathiopurine are immunosuppressive thiopurines frequently used in treatment of hematologic malignancies, chronic inflammatory diseases, and maintenance of graft function following solid organ transplants. 6-MP has been used for over a decade in CD and UC patients to maintain disease remission and continues to be a mainstay in therapeutic options for inflammatory bowel diseases [24,25]. Both 6-MP and azathiopurine remain in inactive prodrug form until they are converted by intracellular enzyme hypoxanthine-guanine phosphoribosyltransferase, which is involved in the purine recycling pathway and ubiquitously present in many cell types. Thereafter, the drugs undergo rapid enzymatic conversion to form 6-thioguanine (6-TG) nucleotide, a purine analog and the principal metabolite responsible for immunosuppressive and cytotoxic effects.
Incorporation of 6-TG into replicating DNA structures of immune cells, thereby inhibiting cell proliferation, was long believed to be the therapeutic mechanism of 6-MP. However, studies of the past decade uncovered high affinity and competitive antagonist activity of 6-TG for the small GTPase protein Rac1 [26,27]. Rac1 is involved critical cell functions such as migration, production of soluble signaling mediators, and survival. It alternates between an active and an inactive form in a cycle catalyzed by guanine nucleotide exchange factor (GEF), which catalyzes exchange of GDP and GTP in Rac1, thereby creating the active GTP bound form of Rac1. 6-MP and its metabolite 6-TG interfere with this exchange, leading to inhibition of Rac1 activity [26,28]. In CD4+ T cells, inhibition of Rac1 by 6-MP metabolites induces apoptosis in the presence of co-stimulatory CD28 signal [26]. In macrophages, 6-MP reduces expression of inducible nitric oxide synthase in a Rac1 dependent manner while in intestinal epithelial cells, 6-MP inhibition of Rac1 leads to decreased proliferation and diminished interleukin-8 production [27]. These observations indicate that Rac1 targeting by thiopurines and the consequent effect on cellular function may be an important component of their therapeutic mechanism.
When 6-MP was first adopted as maintenance therapy in IBD, patients on such treatment were reported to have decreased number of peripheral NK cells that correlated with diminished disease activity [11,15,29,30]. However, the mechanism underlying this clinical finding has not been investigated. In the present study, we demonstrate that 6-MP mediates apoptosis of NK cells via a caspase 3 and 9 inclusive pathway, and that this process is triggered by inhibition of Rac1. We also show that CD patients in clinical remission on 6-MP therapy have decreased NK cell Rac1 activity and decreased numbers of NK cells in their intestinal biopsies. These findings provide evidence for the mechanism of NK cell depletion by thiopurines, and for its effect on both peripheral and intestinal NK cell compartments during thiopurine treatment. In view of recent biologic and genetic evidence for roles of NK cells in IBD pathogenesis, these observations suggest that thiopurine targeting of NK cells may be a previously unappreciated therapeutic action of these agents in IBD.
2. Materials and methods
2.1. Clinical samples
Clinical samples were collected according to protocols approved by the institutional review committees in Cedars-Sinai Medical Center (CSMC) and in University of California, Los Angeles (UCLA). Peripheral blood samples from CD patients were collected from patients recruited at CSMC and from healthy donors recruited at UCLA. Intestinal and colon biopsy samples were collected from CD patients and healthy individuals during clinically indicated colonoscopy procedures at UCLA Medical Center. CD patients and healthy individuals not taking 6-MP did not have any exposure to 6-MP or azathiopurine at least 4 weeks prior to collection of blood or biopsy samples.
2.2. Isolation of primary NK cells and cell culture methods
Human peripheral blood mononuclear cells (PBMCs) from healthy volunteers (20–60 years of age), patients with CD (25–60 years of age), and patients with CD taking 6-MP (25–58 years of age) were isolated using SepMate™ and Lymphoprep™ (Stemcell Technologies, Vancouver, BC, Canada) density gradient medium according to the manufacturer’s protocol. NK cells were purified from PBMCs by negative selection technique using EasySep™ (Stemcell Technologies) human NK cell enrichment kit according to the manufacturer’s protocol. The purity of isolated NK and T cells were confirmed by flow cytometry to be above 90%.
Cultures of NK cells were performed in complete medium composed of RPMI-1640, 10% heat inactivated FBS, 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin, 2 mM L-glutamine, 10 mM HEPES buffer (Cellgro, Manassas, VA), 5 × 10−5 M 2-ME (Sigma-Aldrich, St. Louis, MO), and 1 ng mL−1 (13 IU) recombinant human IL-2 (R&D Systems, Minneapolis, MN) [31]. NK cells were cultured at 5–10 × 104 cells mL−1 in 96-well plates for 24–72 h. For cell survival and Rac-1 assays, cells were cultured with 5–25 μM of 6-MP (Sigma Aldrich), 5–10 μM of 6-TG (Sigma Aldrich), or 100 μM of Rac1 inhibitor ITX-3 (Sigma Aldrich) to mimic physiologic levels of 6-MP and 6-TG as previously described [26].
2.3. Caspase-3/-7 and -9 activity and blocking assays
Following incubation of NK cells with 6-MP for 72 h, 100 μl of Caspase-Glo 3/7 or Caspase-Glo 9 (Promega Corporation, Madison, WI) detection reagents were added to each well. The cells were incubated in room temperature for 40 min and luminescence was measured using a GloMax 96 microplate luminometer (Promega Corp). 10 μM of caspase-3 inhibitor Z-DEVD-FMK (R&D Systems) and 10 μM of caspase-9 inhibitor Z-LEHD-FMK (R&D Systems) were added to the cell culture with 6-MP and incubated for 72 h. Thereafter, caspase-3 and caspase-9 activities were measured using Caspase-Glo assay reagents, and apoptosis was determined by FACS analysis.
2.4. FACS analysis of cell apoptosis
Following 48 to 7 hour cultures with 6-MP or 6-TG, apoptotic cells were detected by staining with Annexin V and 7-amino-actinomycin D (7-AAD) using the Annexin V PE Apoptosis Detection kit (BD Pharmingen). Cells were washed in PBS and resuspended in Annexin V binding buffer (BD Pharmingen) at concentration of 106 cells mL−1. 5 μl of PE-conjugated Annexin V and 5 μl of PI were added per 105 cells. Labeled cells were analyzed with LSR II (BD Biosciences) using FACSDiva software (BD Biosciences) at UCLA Flow Cytometry core, and data analysis was performed using FlowJo software (Tree Star, Ashland, OR).
2.5. Preparation of cell lysates
Following indicated culture periods, cells were washed with ice cold PBS and resuspended in 50 μl of ice cold cell lysis buffer containing 50 mM Tris pH 7.5, 10 mM MgCl2, 0.3 M NaCl, and 2% IGEPAL (Cytoskeleton Inc., Denver, CO). A protease inhibitor cocktail (Cytoskeleton Inc.) containing Pepstatin A, Leupeptin, Benzamidine, and Na-p-tosyl-L-arginine methyl ester (TAME) was added to the cell lysis buffer to yield concentrations of 1 μM, 1.5 μM, 1 mM, and 0.4 mM, respectively. The amount of lysis buffer was adjusted prior to the activation assays to yield protein concentration of 0.5 to 1.0 mg mL−1. The protein concentration in cell lysates was determined with Precision Red™ Advanced Protein Assay Reagent (Cytoskeleton Inc.) and Bio-Rad micro-plate reader model 550 (Bio-Rad, Hercules, CA). The cells were lysed by intermittent vortexing in lysis buffer for 8 min while being kept in below 4 °C environment. The lysates were clarified by centrifugation at 10,000 ×g, at 4 °C for 1 min. The cell lysates supernatants were snap-frozen and stored at −70 °C until use in activation assays.
2.6. Rac-1, RhoA, and Cdc42 activation assays
Levels of active GTP-bound Rac1, RhoA, and Cdc42 in NK cells lysates were determined using colorimetric assays. NK cells were cultured for indicated periods of time in the presence or absence of 6-MP or ITX-3. 50 μl of cell lysates were placed in each well of the 96 well plate provided in the Rac1, RhoA, and Cdc42 G-LISA activation assay kit (Cytoskele-ton Inc.). The respective assays were performed in accordance with the manufacturer’s protocol. The signals from the plates were read by measuring absorbance at 490 nm using Bio-Rad microplate spectrophotometer model 550 (Bio-Rad, CA).
2.7. Western blot analysis
Equal amount of cell extract (10 μg) were added to 10 μl of 2× Laemmli buffer (Boston BioProducts, Ashton, MA). After boiling, extracts were loaded onto 4–10% SDS-PAGE gels and electrophoretically separated. Proteins were transferred to nitrocellulose membranes and blocked with 10% low fat milk in TBS containing 0.1% Tween 20. The proteins were probed with rabbit anti-Rac1 primary antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA) or β-actin (US Biologicals Life Sciences USA). Protein bands were visualized using Goat Anti Rabbit Horseradish peroxidase-conjugated Secondary antibody (Southern Biotechnology Associates, Birmingham, AL) followed by ECL chemiluminescence detection (Millipore, Massachusetts, USA). Band intensities were quantified using ImageJ software.
2.8. Immunohistochemical analysis of biopsy specimens
Formalin-fixed, paraffin embedded sections were de-paraffinized in xylene and rehydrated in graded alcohol. Antigen retrieval was performed by steaming the sections in 0.01 mol/L citric acid buffer (pH 6.0) for 25 min in vegetable steamer. Sections were first blocked with 5% normal donkey serum in DPBS, and then incubated with primary antibody against CD3 (1:50 dilution A0542, Dako, Denmark) overnight at 4 °C. Sections were then incubated with Alex549 Anti-rabbit secondary antibody (1:1000 dilution A10040, Thermo Fisher) for 1 h at room temperature. The slides were incubated with primary antibody against CD56 (1:50 dilution CM164, Biocare Medical, Concord, CA) overnight at 4 °C and then incubated with Alex 488 anti-mouse secondary antibody (1:1000 dilution, Thermo Fisher) for 1 h at room temperature. Slides were scanned by Leica Fluorescent Whole Slide Imaging system. Image was analyzed using Aperio Imagescope software (Leica, Buffalo Grove, IL).
2.9. Statistical analysis
Statistical analysis was performed using Prism version 7.0 software (GraphPad, San Diego, CA) and Microsoft Excel (Microsoft, Redmond, WA). Results were analyzed by one-way analysis of variance (ANOVA) and Student’s t-test. p < 0.01(*) and p < 0.005(**) were regarded as statistically significant and highly significant, respectively.
3. Results
3.1. 6-MP and its metabolite 6-TG induce apoptosis of peripheral blood NK cells
Because 6-MP and its main metabolite 6-TG have cytotoxic and functional inhibitory effects on CD4+ T cells, we first queried whether 6-MP and 6-TG induces apoptosis in NK cells. We isolated peripheral blood NK cells from healthy individuals and cultured them with recombinant IL-2 (rIL-2) at 1 ng mL−1 (13 IU) in the presence or absence of 5 μM 6-MP from 24 to 72 h Fig. 1A and B show representative flow cytometry data, and data from an experimental series analyzed for cell death (positive for Annexin V and 7-AAD). At 24 h of culture, no difference in Annexin V and 7-AAD positive staining cells were observed between 6-MP treated and untreated cells. However, at 48 and 72 h we observed greater induction of Annexin V and 7-AAD positive cells in the group treated with 6-MP, with the most significant difference observed at 72 h of culture. Similar to 6-MP, NK cells treated with 5 μM 6-TG had greater number of Annexin V and 7-AAD positive staining cells at 72 h compared with untreated cells. There was no significant difference in Annexin V and 7-AAD positive cells between 6-TG and 6-MP treated cells, indicating that equal concentrations of 6-TG and 6-MP had comparable effects on NK cells (Fig. 1C). To determine whether there is a dose-dependent effect of 6-MP on NK cell apoptosis and death, we treated NK cells with 5, 10, and 25 μM of 6-MP for 72 h. Increasing concentrations of 6-MP resulted in higher levels of NK cells positive for Annexin V and 7-AAD (Fig. 2A, B)
Fig. 1.
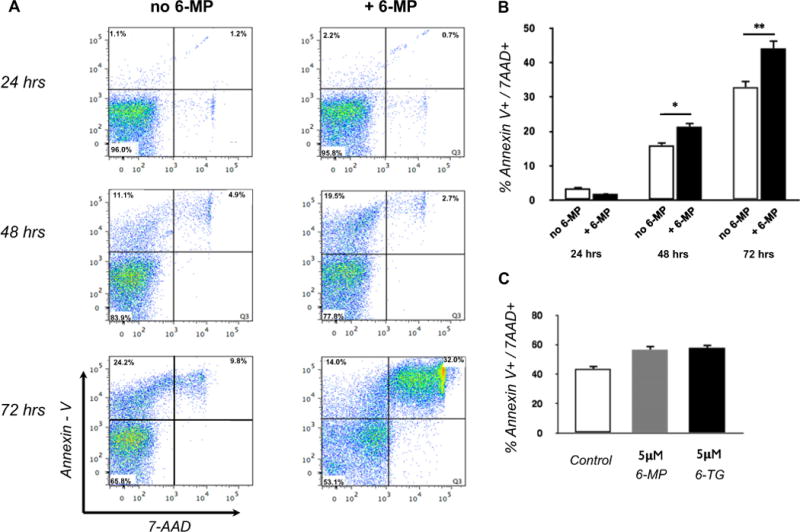
6-MP and its metabolite 6-TG induce NK cell apoptosis. (A) Peripheral blood NK cells isolated from healthy individuals were cultured with and without 6-MP and 1 ng mL−1 of rIL-2. Representative flow cytometry at 24, 48, and 72 h of culture. (B) Bar plot showing data from 3 experiments tabulated for % Annexin V and 7-AAD positive cells at 24, 48, and 72 h. (Student’s t-test, two-tailed. *p < 0.01, **p < 0.005) (C) Bar plot of Annexin V and 7-AAD positive NK cells at 72 h culture with 5 μmol/L 6-MP or 6-TG. Greater percentage of cells cultured with 6-MP and 6-TG were Annexin V and 7-AAD positive. Plot is representative of separate experiments from 3 healthy individuals. One way ANOVA [F(2,6) = 25.97, p = 0.001].
Fig. 2.
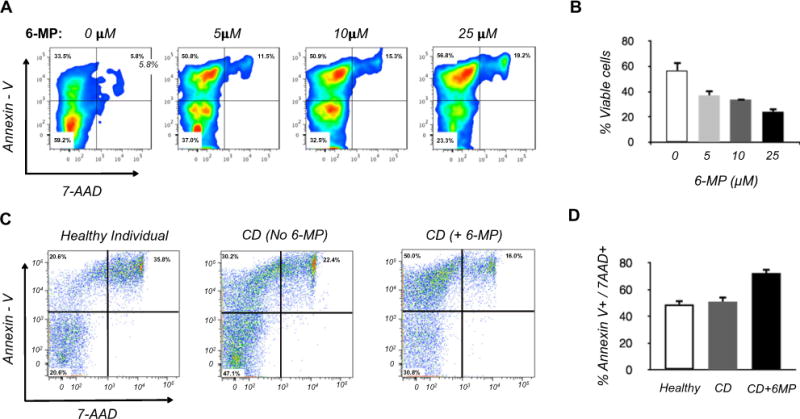
Effect of 6-MP on NK cells in vitro. (A, B) Dose response of blood NK cells from healthy individuals cultured for 72 h with 6-MP. (A) Representative flow cytometry; (B) Tabulation of % viable cells (Annexin V and 7-AAD double-negative cells) from separate experiments using 3 unrelated healthy individuals. One way ANOVA [F(3,11) = 33.43, p < 0.0001]. (C, D) Viability of blood NK cells isolated from healthy individuals, CD patients not undergoing 6-MP therapy (no 6-MP), or undergoing 6-MP therapy (+6-MP). Isolated cells from all groups were cultured for 72 h without 6-MP, and tested for viability by flow cytometry. (C) Representative flow cytometry. (D) Tabulation of % Annexin V and 7-AAD positive cells from separate experiments using three unrelated subjects in each group. ANOVA [F(2,6) = 22.62, p = 0.0016].
Since in vitro exposure to 6-MP diminishes survival of NK cells, we next investigated whether NK cells from CD patients taking 6-MP were more susceptible to apoptosis compared to cells from CD patient not taking 6-MP and healthy individuals. We collected peripheral blood NK cells from 3 healthy individuals, 3 CD patients taking 6-MP, and 3 CD patients who were on non-6-MP therapies. All CD patients were in clinical remission at the time of collection. Cells from all groups were cultured for 72 h without 6-MP, and tested for viability by flow cytometry. The numbers of Annexin V and 7-AAD positive NK cells were similar in healthy individuals and CD patients untreated with 6-MP. However, CD patients treated with 6-MP had significantly elevated Annexin V and 7-AAD positive NK cells compared to either of the non-treated groups (Fig. 2C, D). This finding indicates that 6-MP exerts a pro-apoptotic effect on NK cells in vivo.
3.2. 6-MP induces a pathway of apoptosis dependent mainly on caspase-9
We next determined the mechanism of apoptosis triggered by 6-MP in NK cells. As caspases are well-studied executioners of apoptosis and have been implicated in mediating 6-MP induced apoptosis in other cell types, we assessed caspase activities in primary blood NK cells following treatment with 6-MP. NK cells from healthy individuals were cultured in low dose rIL-2 in the presence or absence of 6-MP, and caspase-9 and -3/-7 activities were determined after 72 h. Cells exposed to 6-MP had marked elevation of both caspase-9 and caspase-3/-7 suggesting initiation of the mitochondrial pathway of apoptosis (Fig. 3A) [32]. To relate caspase activity to NK cell death, inhibitors of caspase-9 and caspase-3 were added to the cultures with 6-MP. Caspase-9 inhibitor led to decreased Annexin V and 7-AAD positive cells (Fig. 3B, C). Caspase-9 inhibitor in particular reduced 6-MP mediated NK cell death and increased cell survival to levels comparable to that of NK cells cultured in the absence of 6-MP. In contrast, addition of caspase 3 inhibitor with 6-MP did not significantly decrease the percentage of Annexin V and 7-AAD positive cells compared to 6-MP alone (Fig. 3B). This indicates that caspase-9 may play a more dominant role in 6-MP induced apoptosis of NK cells.
Fig. 3.
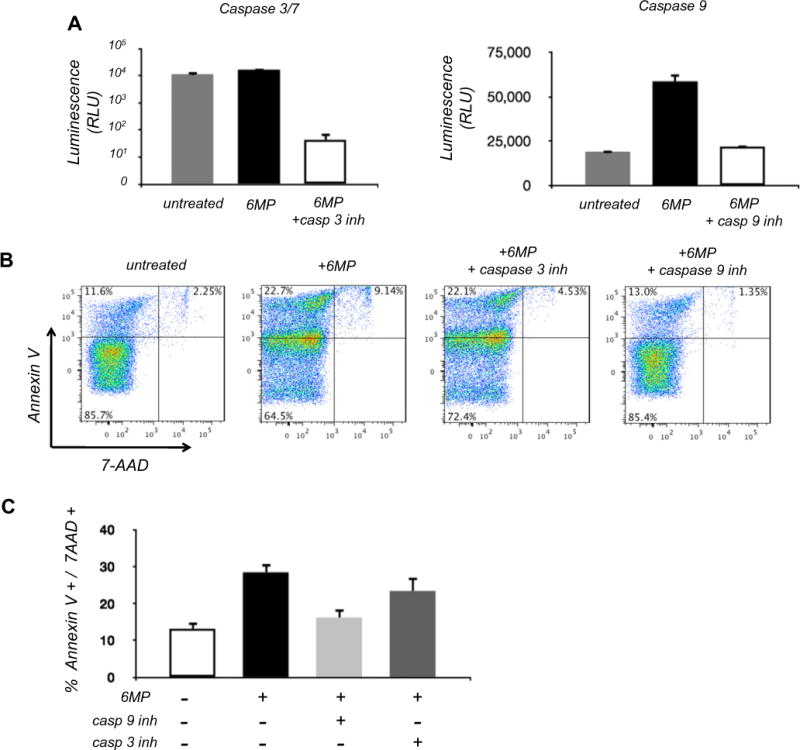
Effect of 6-MP on caspase induction in NK cells. NK cells were cultured with or without 5 μmol 6-MP for 72 h; some cultures were also treated simultaneously with 10 μM caspase-9 or caspase-3/7 inhibitor. (A) Caspase activity levels measured using the Caspase-Glo assay. Exposure to 6-MP increases caspase-3/7 activity ANOVA [F(2,9) = 103.7, p < 0.0001] and caspase-9 activity ANOVA [F(2,7) = 406.4, p < 0.0001]. Mean and SEM of caspase 3/7 activity in untreated NK cells is 11,360 ± 1331 and that of 6MP treated cells is 16,467 ± 522. (B) Representative flow cytometry for Annexin V and 7-AAD NK cells. (C) Tabulation of Annexin V+ and 7-AAD+ NK cells from three independent experiments one way ANOVA [F(3,11) = 29.7, p < 0.0001].
3.3. 6-MP inhibits activity of Rac1 but not RhoA or Cdc42
Rac1 is a known target of the 6-MP metabolite 6-TG in T cells, macrophages, and gut epithelial cells [26–28]. 6-TG physically binds Rac1, inhibiting GDP to GTP exchange and preventing its ability to activate downstream signaling targets. To determine whether 6-MP inhibits Rac1 activity in NK cells, we quantified levels of active GTP-bound Rac1 in NK cells culture with and without 6-MP. We assessed Rac1 activity at 24 h, as this earlier time point precedes the onset of cell death. As a positive control, we also assessed ITX3, a nontoxic inhibitor of the guanine nucleotide exchange factor which catalyzes formation of active GTP bound Rac1 from the inactive GDP bound form [33,34]. Rac1 activity was strongly suppressed in NK cells cultured with 6-MP, equivalent to the reduction in Rac1 activity with ITX3 (Fig. 4A) [33,34]. Expression of total Rac1 was not significantly suppressed by 6-MP as assessed by Western blot (Fig. 4B). In T cells, 6-TG is able to bind other small GTPases such as RhoA and Cdc42 [28,35]. We therefore assessed whether 6-MP modulates RhoA and Cdc42 in NK cells. As shown in Fig. 4C, neither RhoA nor Cdc42 activities were altered by 6-MP (Fig. 4C).
Fig. 4.
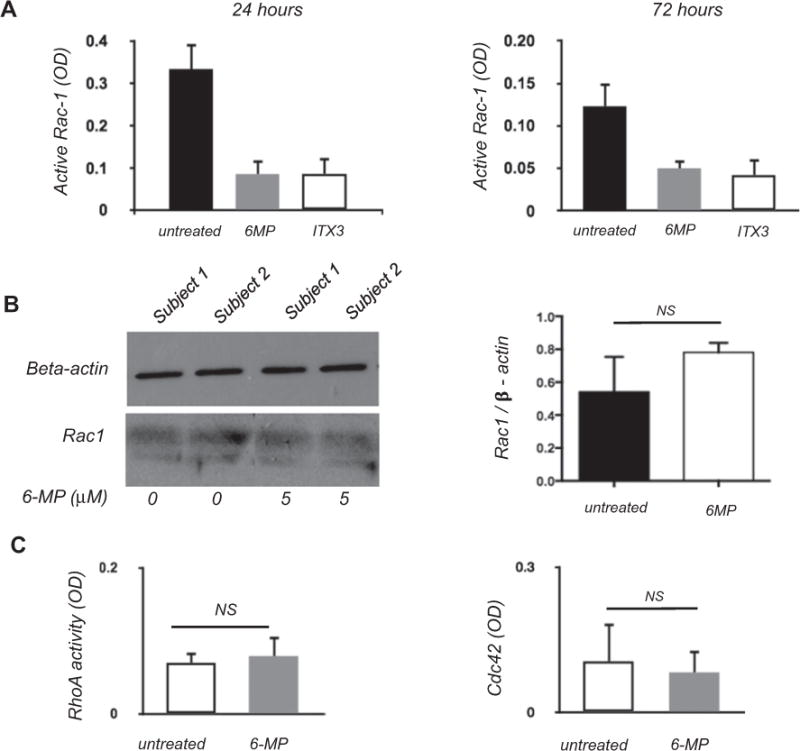
Exposure to 6MP decreases activity of Rac1 but not RhoA or Cdc42. (A) Rac 1 activity measured by immunoassay kit in surviving NK cells following culture with 5 μM 6-MP or 100 μM ITX3 (a known inhibitor of guanine nucleotide exchange factor), at 24 h ANOVA [F(2,6) = 27.9, p = 0.0009] and at 72 h ANOVA [F(2,9) = 19.6, p = 0.0005]. OD = Optical density (B) Western blot showing loading control (beta-actin) and Rac1 in NK cells isolated from 2 healthy individuals following 24 h of culture. Densitometry analysis does not show differences in ratio of Rac 1 to beta-actin between cells exposed and unexposed to 6MP (Student’s t-test, two tailed. p = 0.32).(C) RhoA and Cdc42 activity measured using an immunoassay kit following 24 h of culture with 6-MP.
3.4. Rac1 activity is inhibited in remission CD patients on 6-MP therapy
Since 6-MP inhibited Rac1 activity in NK cells from healthy individuals, we tested whether Rac1 activity is diminished in NK cells from CD patients exposed to 6-MP in vivo. We isolated NK cells from 3 healthy individuals, 4 CD patients in clinical remission on 6-MP, and 3 CD patients in clinical remission on non-6-MP therapies. NK cells from CD patients on 6-MP therapy had significantly lower Rac1 activity compared to healthy individuals or to CD patients on non-6-MP therapies (Fig. 5). Taken together, these findings demonstrate that clinically-attained concentrations of 6-MP inhibits Rac1 in NK cells, leading to cell apoptosis and death.
Fig. 5.
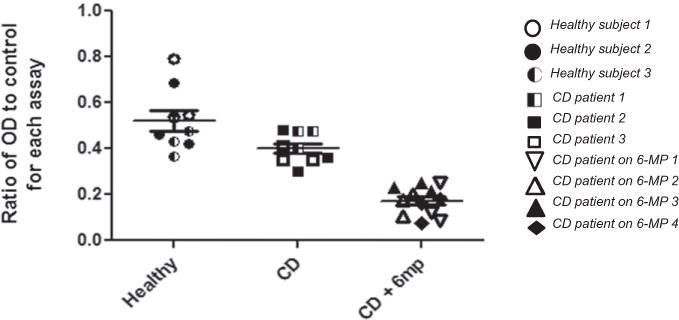
Rac1 activity is significantly decreased in CD patients undergoing 6-MP treatment. Blood NK cell Rac1 activity was quantitated in 3 healthy individuals, 4 CD patients taking 6-MP, and 3 CD patients not taking 6-MP. All CD patients were in clinical remission. The OD ratio was calculated by dividing the OD value from the sample by the control OD value (to normalize for inter-assay variability). Groups were compared by one way ANOVA [F(2,27) = 40.3, p < 0.0001].
3.5. Crohn’s disease patients on 6-MP treatment have decreased colon lamina propria NK cells
NK cells in intestinal mucosa are implicated in disease development and perpetuation of inflammation in human IBD as well as animal models of colitis [36–39]. Mucosal CD56+CD3− NK cells are effective producers of IFN-gamma and TNF-alpha and play a role in activating T cells [16,40]. Therefore, we sought to determine whether 6-MP therapy impacts NK cells resident in the mucosal compartment. We compared the numbers of colonic lamina propria CD56+CD3− NK cells in 4 CD patients taking 6-MP, 4 CD patients maintained on non-6-MP therapy, and 3 healthy individuals. All CD patients were noted to have quiescent disease activity in the colonic mucosa. Examples of immunofluorescence images with antibodies to CD3 and CD56 are shown in Fig. 6A–C; the abundance of NK cells (CD56+ CD3−) were quantified in each specimen, and quantitated from all biospecimens of the sample set (Fig. 6D). The tabulations demonstrate that CD patients taking 6-MP had significantly decreased numbers of LP NK cells compared to CD patient maintained on other types of therapies as well as healthy individuals. Notably, we also observed cells positive for both CD3 and CD56 (yellow), which likely represent lamina propria-resident NK T cells. Levels of these presumptive NK T cells were equivalent in normal and CD patients. However, for CD patients maintained on 6-MP, levels of these cells were significantly reduced (Fig. 6E).
Fig. 6.
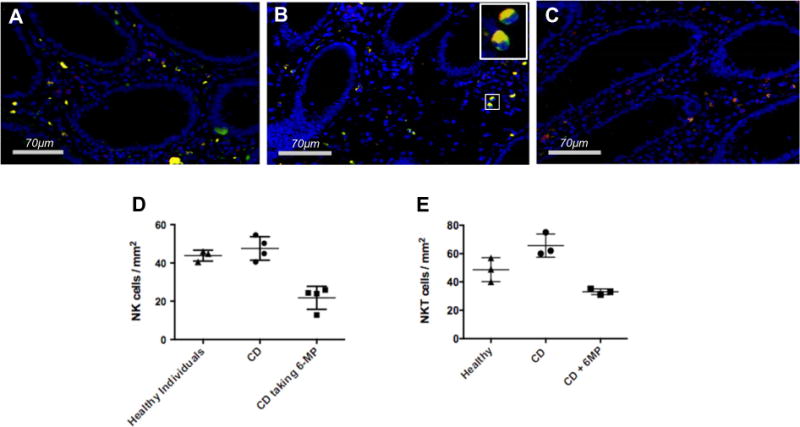
Colon biopsies from CD patients in remission on 6-MP therapy have decreased numbers of CD3−CD56+ NK cells compared to patients not undergoing 6-MP therapy. (A) Multicolor immunofluorescence was used to image T cells (CD3+CD56−, red), NK cells (CD3−CD56+, green), and NKT cells (CD3+CD56+, yellow) in the colonic lamina propria (LP) at 630× magnification. (A) healthy individual. (B) CD patient not on 6-MP but in remission. Enlarged image of the inset better delineates CD3−CD56+ cells. (C) CD patient on 6-MP. (D, E) Biopsies from 3 healthy individuals, 4 CD patients maintained on non-6-MP therapy, and 4 CD patients taking 6-MP were analyzed using Aperio Imagescope software for NK cells and NKT cells in colonic LP (cells per mm2). Each dot represents LP NK cell count from one patient. (D) NK cells, ANOVA analysis [F(2,8) = 25.6, p = 0.0003]; (E) NKT cells, ANOVA analysis [F(2,6) = 16.8, p = 0.003].
4. Discussion
6-MP is an immunosuppressive medication used frequently in the treatment of Crohn’s disease and ulcerative colitis. NK cells are innate immune cells increasingly found to be involved in chronic inflammatory diseases including gut mucosal inflammation in IBD patients, via cytotoxic targeting of at-risk cell types and production of cytokines relevant to TH1 and TH17 pathogenesis [14–16]. NK cell depletion may thus be a relevant target for therapy, and 6-MP has long been known to modulate the behavior and survival of peripheral and mucosal NK cells [11,25,30]. However, the intra-cellular mechanisms triggered by 6-MP or its metabolites within NK cells have not been clearly delineated.
In the present study, we present several lines of evidence which for the first time identify the mechanism for 6-MP metabolite induction of NK cell apoptosis. Our findings show that 6-MP metabolite modulates Rac1 activity without affecting expression of Rac1 protein and results in caspase 3 and 9 inclusive pathway of apoptosis. In vitro exposure of NK cells to 6-MP and 6-TG, the primary active metabolite of 6-MP, resulted in significantly greater cell death and apoptosis at 72 h compared to NK cells unexposed to 6-MP. Presence of low concentration of IL-2, which is required for maturation and development of NK cells, and survival of NK cells in vitro [31,41–43], did not alter apoptosis response to 6-MP. The action of 6-MP was selective for Rac1, as biochemical inhibition was observed for Rac1 but not other closely related small G protein family members. This selectivity is concordant with prior studies demonstrating the distinctive selectivity of 6-MP and its metabolites for this small G protein isoform [26,28].
Rac1 inhibition leading to apoptosis has been described in activated T cells by interfering with Rac1-mediated protection from caspase 6/9-mediated apoptosis [26,28]. As in these previous studies, we found that 6-MP does not alter expression of Rac1, but inhibits the activity of the signaling protein. Additionally, results from our studies also showed delayed onset of apoptosis in NK cells, as significant cell death and apoptosis was noted at 72 h in culture with 6-MP. This is in accordance with pharmacokinetic studies that support delayed clinical effect of 6-MP and azathiopurine [44]. In T cells, apoptosis was noted 4–5 days following culture which is somewhat delayed compared to NK cells at the concentration of 6-MP physiologically found in serum of patients compliant on the medication. This suggests a greater sensitivity of NK cells to 6-MP and its metabolite compared to T cells, although the reason for this accelerated response is uncertain.
The biology of NK cells in circulating, lymphoid, and mucosal compartments is distinct [2,45–47], so it is important to understand whether 6-MP targets these compartments of NK cells. Ex vivo, we found that an increasing 6-MP dose induced elevated NK cells apoptosis and death, within a range achieved by clinical administration of 6-MP. Accordingly, it is notable that within this range, clinical studies show that higher levels of drug detected in CD patients was more often associated with clinical remission [25]. In vivo, patients under therapeutic dosing of 6-MP had reduced levels of circulating NK cells, and those still present had undergone Rac1 inhibition in situ. Similarly, in the colonic lamina propria compartment, the levels of NK cells were selectively depleted in patients under 6-MP versus non-thiopurine maintenance therapy. We note that all patients studied were in clinical remission, so these differences are associated with therapy type, rather than disease activity. However, we acknowledge that the number of patients studied was relatively small, and hence it was not feasible to test for additional clinical or immunologic correlates that may also contribute to the differences in observed NK cell levels.
An interesting observation in the colonic LP of healthy individuals and CD patient unexposed to 6-MP was the presence of cells that co-express CD3 and CD56, T and NK cell receptors respectively, suggesting these cells represent NKT cells. CD patients maintained in remission on 6-MP therapy had reduced numbers of colonic LP CD3+ CD56+ cells compared to individuals unexposed to 6-MP. Mucosal NKT cells are known to contribute to intestinal immune responses in health and disease although their precise role in inflammatory bowel disease is an ongoing area of research [48]. Stimulation of CD1d-restricted Type I NKT appeared to improve disease scores in murine model of DSS colitis [49]. Conversely, Type II NKT cells were found to contribute to mucosal inflammation in human and mouse studies by production of cytokines skewed either towards Th1 or Th2 responses [50,51]. To our knowledge, this study is the first to report reduction in colonic LP CD3+CD56+ cells in patients taking therapeutic 6-MP doses, suggesting these cells may also be targets of 6-MP metabolites. Our results validate previous body of data that indicate NKT cells are important mediators of mucosal inflammation, and introduce a potentially new venue of investigation into the effect of 6-MP in NKT cell biology.
5. Conclusion
This study establishes the mechanism of NK cell depletion by 6-MP, and its impact on systemic and colonic mucosal NK cell populations. There is increasing evidence for roles of NK cells in the disease phenotype of inflammatory bowel disease and other chronic immunologic diseases. The present findings suggest the merit of future studies to test whether thiopurine agents may selectively benefit patient subsets with genetic (KIR/HLA) or functional NK cell features associated with elevated NK cell activity.
Acknowledgments
Supported by National Institutes of Health grants P01DK46763 (DPBM, JB), UL1TR000124 (DPBM, JB, SY), T32DK007180 (SY), the Crohn’s and Colitis Foundation of America 323814 (JB), the Helmsley Charitable Trust (DPBM), the Cedars-Sinai F. Widjaja Inflammatory Bowel and Immunobiology Institute Research Fund (DPBM), and Richard and Barbara Braun (JB). SY and DPBM selected and coordinated collection of clinical samples; SY designed and performed the ex vivo assays and tissue assays. JB and SY conceived the study and directed the experimental design; SY and JB wrote the manuscript. All authors reviewed and discussed the manuscript. We are grateful to a reviewer of this paper who called our attention to the observation regarding NKT cells.
Abbreviations
- 6-MP
6-mercaptopurine
- 6-TG
6-thioguanine
- 7-AAD
7-amino-actinomycin
- CD
Crohn’s disease
- IBD
inflammatory bowel disease
- KIR
killer immunoglobulin-like receptor
- UC
ulcerative colitis
References
- 1.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod’homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, Wang EC, Griffin CA, Davison AJ. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol. 2008;41:206–212. doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orr MT, Murphy WJ, Lanier LL. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol. 2010;11:321–327. doi: 10.1038/ni.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warren HS, Smyth MJ. NK cells and apoptosis, Immunol. Cell Biol. 1999;77:64–75. doi: 10.1046/j.1440-1711.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- 6.Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands, Immunol. Cell Biol. 2014;92:221–229. doi: 10.1038/icb.2013.98. [DOI] [PubMed] [Google Scholar]
- 7.Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by killing donor antigen-presenting cells. J Exp Med. 2006;203:1851–1858. doi: 10.1084/jem.20060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benichou G, Yamada Y, Aoyama A, Madsen JC. Natural killer cells in rejection and tolerance of solid organ allografts. Curr Opin Organ Transplant. 2011;16:47–53. doi: 10.1097/MOT.0b013e32834254cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 2002;46:1763–1772. doi: 10.1002/art.10410. [DOI] [PubMed] [Google Scholar]
- 10.van der Slik AR, Koeleman BP, Verduijn W, Bruining GJ, Roep BO, Giphart MJ. KIR in type 1 diabetes: disparate distribution of activating and inhibitory natural killer cell receptors in patients versus HLA-matched control subjects. Diabetes. 2003;52:2639–2642. doi: 10.2337/diabetes.52.10.2639. [DOI] [PubMed] [Google Scholar]
- 11.Steel AW, Mela CM, Lindsay JO, Gazzard BG, Goodier MR. Increased proportion of CD16(+) NK cells in the colonic lamina propria of inflammatory bowel disease patients, but not after azathioprine treatment, Aliment. Pharmacol Ther. 2011;33:115–126. doi: 10.1111/j.1365-2036.2010.04499.x. [DOI] [PubMed] [Google Scholar]
- 12.Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14:221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 13.Shanahan F, Leman B, Deem R, Niederlehner A, Brogan M, Targan S. Enhanced peripheral blood T-cell cytotoxicity in inflammatory bowel disease. J Clin Immunol. 1989;9:55–64. doi: 10.1007/BF00917128. [DOI] [PubMed] [Google Scholar]
- 14.Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, Matuzaki Y, Suzuki S, Sugita A, Koganei K, Hisamatsu T, Kanai T, Hibi T. Imbalance of NKp44(+)NKp46(−) and NKp44(−)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology. 2010;139:882–892. e883. doi: 10.1053/j.gastro.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 15.Ng SC, Plamondon S, Al-Hassi HO, English N, Gellatly N, Kamm MA, Knight SC, Stagg AJ. A novel population of human CD56+ human leucocyte antigen D-related (HLA-DR+) colonic lamina propria cells is associated with inflammation in ulcerative colitis. Clin Exp Immunol. 2009;158:205–218. doi: 10.1111/j.1365-2249.2009.04012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin L, Ma C, Wei B, Aziz N, Rajalingam R, Yusung S, Erlich HA, Trachtenberg EA, Targan SR, McGovern DP, Heath JR, Braun J. Human NK cells licensed by killer Ig receptor genes have an altered cytokine program that modifies CD4+ T cell function. J Immunol. 2014;193:940–949. doi: 10.4049/jimmunol.1400093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol. 2002;2:957–964. doi: 10.1038/nri956. [DOI] [PubMed] [Google Scholar]
- 18.Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, Yokoyama WM, Eltzschig HK, Clambey ET, Tissue-Resident NK. Cells mediate ischemic kidney injury and are not depleted by anti-asialo-GM1 antibody. J Immunol. 2015;195:4973–4985. doi: 10.4049/jimmunol.1500651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson TJ, Jobim M, Jobim LF, Portela P, Salim PH, Rosito MA, Damin DC, Flores C, Peres A, Machado MB, Chies JA, Schwartsmann G, Roesler R. Study of killer immunoglobulin-like receptor genes and human leukocyte antigens class I ligands in a Caucasian Brazilian population with Crohn’s disease and ulcerative colitis. Hum Immunol. 2010;71:293–297. doi: 10.1016/j.humimm.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Hollenbach JA, Ladner MB, Saeteurn K, Taylor KD, Mei L, Haritunians T, McGovern DP, Erlich HA, Rotter JI, Trachtenberg EA. Susceptibility to Crohn’s disease is mediated by KIR2DL2/KIR2DL3 heterozygosity and the HLA-C ligand. Immunogenetics. 2009;61:663–671. doi: 10.1007/s00251-009-0396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH, Yokoyama WM. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 22.Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011;32:364–372. doi: 10.1016/j.it.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms, Adv. Immunol. 2009;101:27–79. doi: 10.1016/S0065-2776(08)01002-X. [DOI] [PubMed] [Google Scholar]
- 24.Present DH, Korelitz BI, Wisch N, Glass JL, Sachar DB, Pasternack BS. Treatment of Crohn’s disease with 6-mercaptopurine. A long-term, randomized, double-blind study. N Engl J Med. 1980;302:981–987. doi: 10.1056/NEJM198005013021801. [DOI] [PubMed] [Google Scholar]
- 25.Dubinsky MC. Azathioprine, 6-mercaptopurine in inflammatory bowel disease: pharmacology, efficacy, and safety. Clin Gastroenterol Hepatol. 2004;2:731–743. doi: 10.1016/s1542-3565(04)00344-1. [DOI] [PubMed] [Google Scholar]
- 26.Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, Lehr HA, Wirtz S, Becker C, Atreya R, Mudter J, Hildner K, Bartsch B, Holtmann M, Blumberg R, Walczak H, Iven H, Galle PR, Ahmadian MR, Neurath MF. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marinkovic G, Hamers AA, de Vries CJ, de Waard V. 6-Mercaptopurine reduces macrophage activation and gut epithelium proliferation through inhibition of GTPase Rac1, Inflamm. Bowel Dis. 2014;20:1487–1495. doi: 10.1097/MIB.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 28.Poppe D, Tiede I, Fritz G, Becker C, Bartsch B, Wirtz S, Strand D, Tanaka S, Galle PR, Bustelo XR, Neurath MF. Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J Immunol. 2006;176:640–651. doi: 10.4049/jimmunol.176.1.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi FD, Van Kaer L. Reciprocal regulation between natural killer cells and autoreactive T cells. Nat Rev Immunol. 2006;6:751–760. doi: 10.1038/nri1935. [DOI] [PubMed] [Google Scholar]
- 30.Shih WW, Ellison GW, Myers LW, Durkos-Smith D, Fahey JL. Locus of selective depression of human natural killer cells by azathioprine. Clin Immunol Immunopathol. 1982;23:672–681. doi: 10.1016/0090-1229(82)90330-0. [DOI] [PubMed] [Google Scholar]
- 31.Felices M, Lenvik TR, Ankarlo DE, Foley B, Curtsinger J, Luo X, Blazar BR, Anderson SK, Miller JS. Functional NK cell repertoires are maintained through IL-2Ralpha and Fas ligand. J Immunol. 2014;192:3889–3897. doi: 10.4049/jimmunol.1302601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 33.van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FP, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell. 2012;23:2831–2844. doi: 10.1091/mbc.E11-11-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouquier N, Vignal E, Charrasse S, Weill M, Schmidt S, Leonetti JP, Blangy A, Fort P. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem Biol. 2009;16:657–666. doi: 10.1016/j.chembiol.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 36.Yadav PK, Chen C, Liu Z. Potential role of NK cells in the pathogenesis of inflammatory bowel disease, 2011. J Biomed Biotechnol. 2011:348530. doi: 10.1155/2011/348530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fort MM, Leach MW, Rennick DM. A role for NK cells as regulators of CD4+ T cells in a transfer model of colitis. J Immunol. 1998;161:3256–3261. [PubMed] [Google Scholar]
- 38.Eken A, Singh AK, Oukka M. Interleukin 23 in Crohn’s disease, Inflamm. Bowel Dis. 2014;20:587–595. doi: 10.1097/01.MIB.0000442014.52661.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fink LN, Zeuthen LH, Christensen HR, Morandi B, Frokiaer H, Ferlazzo G. Distinct gut-derived lactic acid bacteria elicit divergent dendritic cell-mediated NK cell responses. Int Immunol. 2007;19:1319–1327. doi: 10.1093/intimm/dxm103. [DOI] [PubMed] [Google Scholar]
- 40.Leon F, Roldan E, Sanchez L, Camarero C, Bootello A, Roy G. Human small-intestinal epithelium contains functional natural killer lymphocytes. Gastroenterology. 2003;125:345–356. doi: 10.1016/s0016-5085(03)00886-2. [DOI] [PubMed] [Google Scholar]
- 41.Meazza R, Azzarone B, Orengo AM, Ferrini S. Role of common-gamma chain cytokines in NK cell development and function: perspectives for immunotherapy. J Biomed Biotechnol. 2011;2011:861920. doi: 10.1155/2011/861920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL-2-dependent adaptive control of NK cell homeostasis. J Exp Med. 2013:1179–1187. doi: 10.1084/jem.20122571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, Waldmann TA, Tagaya Y. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A. 2000;97:11445–11450. doi: 10.1073/pnas.200363097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 45.Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim TJ, Upadhyay V, Kumar V, Lee KM, Fu YX. Innate lymphoid cells facilitate NK cell development through a lymphotoxin-mediated stromal microenvironment. J Exp Med. 2014;211:1421–1431. doi: 10.1084/jem.20131501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, Walzer T, Vivier E. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dowds CM, Blumberg RS, Zeissig S. Control of intestinal homeostasis through crosstalk between natural killer T cells and the intestinal microbiota. Clin Immunol. 2015;159:128–133. doi: 10.1016/j.clim.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saubermann LJ, Beck P, DeJong YP, Pitman RS, Ryan MS, Kim HS, Exley M, Snapper S, Balk SP, Hagen SJ, Kanauchi O, Motoki K, Sakai T, Terhorst C, Koezuka Y, Podolsky DK, Blumberg RS. Activation of natural killer T cells by alpha-galactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119–128. doi: 10.1053/gast.2000.9114. [DOI] [PubMed] [Google Scholar]
- 50.Liao CM, Zimmer MI, Shanmuganad S, Yu HT, Cardell SL, Wang CR. Dysregulation of CD1d-restricted type ii natural killer T cells leads to spontaneous development of colitis in mice. Gastroenterology. 2012;142:326–334. doi: 10.1053/j.gastro.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P, Strober W. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]