

Abstract
Summary
Transcription factors regulate gene expression by binding to specific short DNA sequences of 5–20 bp to regulate the rate of transcription of genetic information from DNA to messenger RNA. We present PWMScan, a fast web-based tool to scan server-resident genomes for matches to a user-supplied PWM or transcription factor binding site model from a public database.
Availability and implementation
The web server and source code are available at http://ccg.vital-it.ch/pwmscan and https://sourceforge.net/projects/pwmscan, respectively.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Knowing where transcription factors (TFs) bind to the genome is the key to understanding gene regulation. The binding specificity of a TF is commonly represented by a numerical matrix, either as a position weight matrix (PWM), a position frequency matrix (PFM) or a letter probability matrix (LPM). The three representations are information-wise equivalent and inter-convertible. A PWM contains weights for each base at each motif position. By summing up weights at corresponding positions, a binding score can be computed for any base sequence of the same length as the PWM. Large collections of TF specificity matrices are nowadays available from public libraries such as JASPAR (Khan et al., 2017) or HOCOMOCO (Kulakovskiy et al., 2017).
PWMScan is a web-server for rapid scanning of large genomes for high-scoring matches to a user-supplied or server-resident PWM. Compared to other web-based PWM scanning tools, PWMScan is unique in that it scans server-resident whole genomes rather than user-uploaded DNA sequences. Other key features are: (i) menu-driven access to genomes of >30 model organisms; (ii) menu-driven access to >300 public PWM libraries; (iii) support of various PWM representations and formats; (iv) cut-off values can be specified as match scores or P-values; (v) output in BEDdetail format with match scores and P-values; (vi) links to UCSC genome browser for visualization of results; and (vii) action buttons to transfer match lists to analysis tools.
A short description of the PWMScan server follows. Technical details about algorithms, programs and data are provided under Supplementary Material.
2 Data and methods
Genome sequences were downloaded from the NCBI in FASTA format. Indexed versions for rapid scanning were generated for Bowtie (Langmead et al., 2009). The motif databases offered by PWMScan have been downloaded from the MEME Suite website (Bailey et al., 2009). LPMs have been converted to integer PWMs (see Supplementary Material).
The input form of the PWMScan server is shown in Figure 1. The user chooses a genome assembly from a menu. Optionally, a BED file may be uploaded to restrict the search to genomic regions of particular interest, e.g. open chromatin regions. The right side of the form offers several ways to specify a DNA motif. PWMs from a server-resident database are chosen from a pull-down menu. Alternatively, matrices can be entered into a text area or uploaded. Accepted motif types are: PFMs, LPMs, real or integer PWMs and IUPAC consensus sequences. PFMs can be entered in several formats, including TRANSFAC and JASPAR.
Fig. 1.
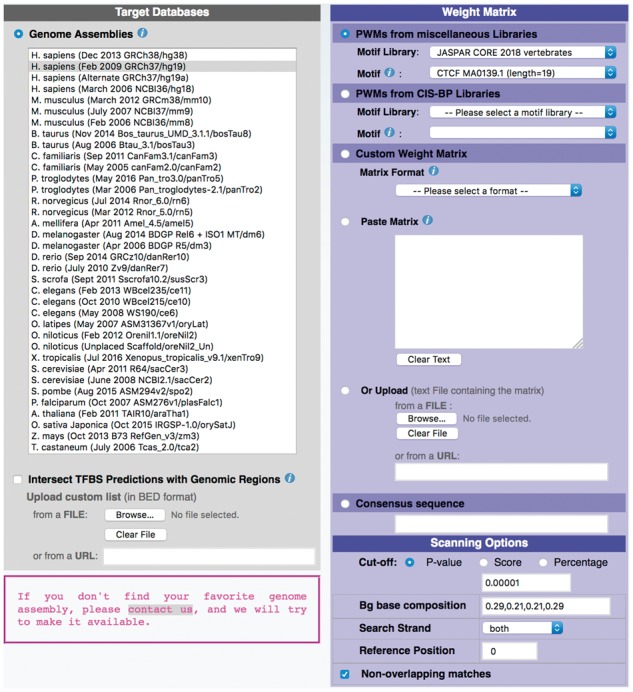
Screen shot of the PWMScan graphical user interface
All motif types have to be converted into integer PWMs for input to the genome search engines (see Supplementary Material). Default conversion parameters are proposed and can be changed by the user. For instance, real PWMs can be rescaled on input by a multiplication factor to ensure sufficient resolution after integer conversion. IUPAC consensus sequences are converted into binary matrices consisting of 0 and 1.
For all matrix formats, the cut-off value can be specified as PWM score, as P-value or as percentage of the score range (0% = minimal score and 100% = maximal score). For IUPAC consensus sequences, the cut-off value is specified as a maximal number of mismatches allowed.
The P-value of a PWM score x is defined as the probability that a random k-mer sequence of the length of the PWM has a binding score ≥ x given the base composition of the genome.
The whole genome scan takes as input an integer PWM and a corresponding cut-off. The output is a list of sequence regions that match the PWM with a match score higher or equal to the cut-off value. Depending on the length of the PWM and the cut-off, one of the following search strategies is chosen: (i) Bowtie, a fast memory-efficient short read aligner using indexed genomes and (ii) matrix_scan, a C program developed by our group using a conventional search algorithm.
The first strategy is more efficient for short PWMs and high cut-off values. It requires as a first step the generation of a list of all k-mers that match the PWM with the given cut-off. The list of k-mers is then mapped to the genome using Bowtie.
The second strategy takes genome sequences in FASTA format as input. Individual chromosomes are processed in parallel and distributed to multiple cores by a Python script. We empirically found that this approach becomes more efficient if the number of k-mers exceeds 105 sequences. matrix_scan was benchmarked for speed together with five other matrix scanners and was found to be the fastest (see Supplementary Material).
The basic search step outputs a list of PWM matches, including the genomic coordinates, the DNA sequence and the match score. Post-processing of this list involves computation of the corresponding P-values, addition of the matrix name and, optionally, elimination of overlapping matches. The final match list is provided in BEDdetail format. The output page further shows the total number of PWM matches and a sequence logo reflecting the letter-probabilities of the input matrix. Action buttons are provided for: (i) sending the match lists to analysis tools of the ChIP-Seq and SSA servers (Ambrosini et al., 2016), (ii) extracting DNA sequences around the matches, (iii) sending the output to the UCSC genome browser for visualization and (iv) liftover of the match coordinates to other assemblies of the same or related species.
PWMScan is meant to support many types of genomic data analysis and designed to be interoperable with other tools from our group and elsewhere. An example of a typical workflow involving ChIP-seq data is presented in Supplementary Material.
PWMScan is also available as a command-line software package from SourceForge, including a master script scheduling all computational steps running during a web job.
3 Benchmark
The runtime of PWMScan was measured by scanning the human genome (UCSC assembly hg19) with two different PWMs from JASPAR, STAT1 with length 11 bp and CTCF with length 19 bp, and different cut-off values expressed as P-values. Results are shown in Table 1. Note that for longer motifs and higher P-values, the Bowtie-based approach becomes inefficient, whereas matrix_scan remains reasonably fast.
Table 1.
Benchmark results with different PWMs and P-values
| PWM/P-value | Bowtie speed | Matrix_scan speed |
|---|---|---|
| STAT1(len = 11 bp)/10−5 | 3 | 30/5a |
| STAT1(len = 11 bp)/10−4 | 8 | 40/8a |
| STAT1(len = 11 bp)/10−3 | 60 | 65/30a |
| CTCF(len = 19 bp)/10−5 | 12 | 40/6a |
| CTCF(len = 19 bp)/10−4 | 90 | 50/10a |
| CTCF(len = 19 bp)/10−3 | 720 | 90/35a |
Note: Speed is expressed in seconds. The benchmarking tests have been run on a Linux/CentOS7/x86_64 workstation with 48 CPU-cores and 256 GB of DRAM.
Performance measurements using matrix_scan in parallel over 10 CPU-cores.
Supplementary Material
Acknowledgements
The PWMScan server is hosted by Vital-IT, the SIB Swiss Institute of Bioinformatics’ Competence Centre in Bioinformatics and Computational Biology.
Funding
This work was supported by the Swiss Institute of Bioinformatics and Swiss Federal Institute of Technology Lausanne (EPFL).
Conflict of Interest: none declared.
References
- Ambrosini G. et al. (2016) The ChIP-Seq tools and web server: a resource for analyzing ChIP-seq and other types of genomic data. BMC Genomics, 17, 938.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. et al. (2009) MEME Suite: tools for motif discovery and searching. Nucleic Acids Res., 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. et al. (2017) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res., 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy I.V. et al. (2017) HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res., 46, D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.