

Abstract
Disorganization of nodes of Ranvier is associated with motor and sensory dysfunctions. Mechanisms that allow nodal recovery during pathological processes remain poorly understood. A highly enriched nodal cytoskeletal protein βIV spectrin anchors and stabilizes the nodal complex to actin cytoskeleton. Loss of murine βIV spectrin allows the initial nodal organization, but causes gradual nodal destabilization. Mutations in human βIV spectrin cause auditory neuropathy and impairment in motor coordination. Similar phenotypes are caused by nodal disruption due to demyelination. Here we report on the precise timelines of nodal disorganization and reorganization by following disassembly and reassembly of key nodal proteins in βIV spectrin mice of both sexes before and after βIV spectrin re-expression at specifically chosen developmental time points. We show that the timeline of nodal restoration has different outcomes in the PNS and CNS with respect to nodal reassembly and functional restoration. In the PNS, restoration of nodes occurs within 1 month regardless of the time of βIV spectrin re-expression. In contrast, the CNS nodal reorganization and functional restoration occurs within a critical time window; after that, nodal reorganization diminishes, leading to less efficient motor recovery. We demonstrate that timely restoration of nodes can improve both the functional properties and the ultrastructure of myelinated fibers affected by long-term nodal disorganization. Our studies, which indicate a critical timeline for nodal restoration together with overall motor performance and prolonged life span, further support the idea that nodal restoration is more beneficial if initiated before any axonal damage, which is critically relevant to demyelinating disorders.
SIGNIFICANCE STATEMENT Nodes of Ranvier are integral to efficient and rapid signal transmission along myelinated fibers. Various demyelinating disorders are characterized by destabilization of the nodal molecular complex, accompanied by severe reduction in nerve conduction and the onset of motor and sensory dysfunctions. This study is the first to report in vivo reassembly of destabilized nodes with sequential improvement in overall motor performance. Our study reveals that nodal restoration is achievable before any axonal damage, and that long-term nodal destabilization causes irreversible axonal structural changes that prevent functional restoration. Our studies provide significant insights into timely restoration of nodal domains as a potential therapeutic approach in treatment of demyelinating disorders.
Keywords: axonal health, motor coordination, myelination, nerve conduction, nodal restoration, nodes of Ranvier
Introduction
Nodes of Ranvier are the myelin-free regions that are indispensable for fast action potential propagation along myelinated axons. The nodal region is established during myelination and involves clustering of cell-adhesion molecules—the neuronal isoform of Neurofascin 186 (NfascNF186) and NrCAM, voltage-gated sodium (NaV) channels, and axonal cytoskeletal scaffolding proteins ankyrin G (AnkG) and βIV spectrin (Kordeli et al., 1995; Davis et al., 1996; Lambert et al., 1997; Jenkins and Bennett, 2001, 2002; Dzhashiashvili et al., 2007; Feinberg et al., 2010; Zhang et al., 2012; Nelson and Jenkins, 2017). While many studies have provided valuable insights into the mechanisms regulating nodal clustering and long-term stability (Buttermore et al., 2013; Susuki et al., 2013; Saifetiarova et al., 2017a; Taylor et al., 2017), the question of whether nodes disorganized due to loss of key nodal proteins or under pathological circumstances can be reorganized and their functions restored remains unknown. It has been well documented that under pathological conditions, such as central and peripheral demyelinating disorders, molecular interactions within the nodal complex are highly compromised, leading to redistribution of the core nodal proteins along the damaged axons (Craner et al., 2004). As a result of such perturbations, patients with disintegrated nodes acquire motor, cognitive, and sensory dysfunctions (Frohman et al., 2006; Waxman, 2006; Huang et al., 2017). In addition, considering chronic progressive development of demyelinating disorders and possibility of key irreversible changes in the nervous system after a certain time point, it is important to determine and identify critical time windows in which restoration of the nodes is achievable.
βIV spectrin is a cytoskeletal scaffolding protein that anchors the nodal molecular complex to the actin cytoskeleton through its interaction with AnkG (Jenkins and Bennett, 2002; Lacas-Gervais et al., 2004). βIV Spectrin mutants show progressive destabilization of the nodes, which is accompanied by chronic motor dysfunctions (Komada and Soriano, 2002). Mutations in the human βIV spectrin (SPTBN4) locus are associated with congenital myopathy, neuropathy, and central deafness, further underscoring the vital functions of βIV spectrin (Knierim et al., 2017).
The βIV spectrin mutants were created by insertion of ROSAβgeo* (referred to here as βgeo) flanked by LoxP sequences, which could be removed by Cre-mediated recombination (Komada and Soriano, 2002). This unique genetic situation provided us with an exciting experimental opportunity to restore βIV spectrin expression. Using these animals, we were able to study the time course of nodal destabilization and appearance of motor symptoms. In the current study we addressed the following questions: (1) Can nodes of Ranvier be restored in myelinated axons? If yes, what is the timeline of this restoration? (2) Would it be possible to obtain full functional recovery of the phenotypes resulting from destabilized nodes of Ranvier? and (3) Is there a critical time window for the best rescue outcome and beyond which rescue is not efficient or achievable?
Here we report that upon Cre-mediated recombination βIV spectrin is re-expressed in βIV spectrin mutants and clusters at the destabilizing nodes, allowing other nodal proteins to recluster at the nodes after they had been disorganized. Interestingly, the nodal rescue and reorganization reveals that nodes in the PNS myelinated axons are restored faster than in the CNS myelinated axons. Most importantly, PNS nodes were restored in the same timeframe independent of the timing of βIV spectrin re-expression, whereas the CNS nodal recovery occurs slowly and, if the rescue was initiated at the later stages, nodes failed to reassemble. Furthermore, timely nodal reorganization led to restoration of nerve conduction and motor performance and prevented further progression of motor paralysis. Together, our data shed new light on how nodal reorganization occurs in the PNS and CNS after nodes have been destabilized for extended periods of time. Furthermore, our data uncover specific differences in nodal reorganization and functional restoration in the PNS and CNS myelinated axons. Our studies provide insights into demyelinating disorders and into the timeline in which reorganization of destabilized nodes would become necessary to improve disease symptoms in patients. Our studies also add weight to the notion that early restoration will lead to better outcomes.
Materials and Methods
Animals.
Sptbn4geo (βIV spectrin) mutant animals used in these studies were characterized previously (Komada and Soriano, 2002). The actin-CreER transgenic mouse line has been described previously (Hayashi and McMahon, 2002; Taylor et al., 2017). Sptbn4geo/+ heterozygous mice were mated with the actin-CreER strain to obtain Sptbn4geo/+; actin-CreER animals, which were crossed to Sptbn4geo/+ to obtain actin-CreER;Sptbn4geo animals for further analysis. Mouse lines used in these studies were on a mixed C57BL/6 and 129/Sv genetic background. Equal numbers of males and females were used for all experiments and were analyzed at various ages of postnatal development from 4 to 10 months old. Humane endpoints were selected for survival studies. The end stage was defined as the point when mice exhibited inability to reach for food and water due to progressive weakness. Animals at that stage were euthanized using 95% CO2. All animal experiments were performed according to guidelines for ethical treatment of laboratory animals approved by the National Institutes of Health and the Institutional Animal Care and Use Committee the University of Texas Health Science Center at San Antonio.
Tamoxifen injections.
For re-expression of βIV spectrin, actin-CreER;Sptbn4geo mice received 1 mg of tamoxifen by intraperitoneal injections for 5 consecutive days at 4 or 7 months of age (Pillai et al., 2009). The actin-CreER;Sptbn4geo mice receiving tamoxifen are referred as Sptbn4res mice.
Antibodies.
βIV spectrin, AnkG, pan-NaV, NfascNF186, and Caspr antibodies used in the current studies were described previously (Bhat et al., 2001; Thaxton et al., 2011; Saifetiarova et al., 2017a; Taylor et al., 2017). Other primary antibodies used in this study were mouse anti-Caspr (75-001, NeuroMab), mouse anti-αtubulin (12G10, Developmental Studies Hybridoma Bank), mouse anti-ankyrin R (75-380, NeuroMab), and rabbit anti-βI spectrin (obtained from M. Stankewich). Fluorophore-conjugated secondary antibodies used for immunofluorescence were purchased from Invitrogen. Infrared-conjugated secondary antibodies used for immunoblotting were purchased from LI-COR.
Tissue preparation and immunostaining.
The tissue preparation and immunostaining technique has been described previously (Saifetiarova et al., 2017a). Briefly, animals were anesthetized with Avertin (400 mg/kg mouse body weight; T48402, Sigma-Aldrich) and transcardially perfused with PBS, pH 7.2–7.4, followed by a mixture of ice-cold 1% paraformaldehyde (PFA) and 1% sucrose in 0.1 m phosphate buffer (PB), pH 7.2–7.4. The spinal cords (SCs) were postfixed in the same fixative for 2 h at 4°C and then immersed in 30% sucrose in 0.1 m PB. Afterward, the tissue was frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek USA). Longitudinal 14 μm sections were cut with a cryostat (Leica), mounted on slides, and processed for immunostaining. Sciatic nerves (SNs) were dissected out from anesthetized animals, fixed in 4% PFA for 30 min, teased into individual nerve fibers, dried overnight at room temperature, and stored at −80°C before immunostaining.
Transmission electron microscopy.
The transmission electron microscopy (TEM) procedure has been described previously (Saifetiarova et al., 2017a). Briefly, anesthetized animals were transcardially perfused with freshly prepared normal saline followed by 2.5% glutaraldehyde/4% PFA EM fixative. After perfusion, entire mouse carcasses were postfixed for another 2 weeks in the same EM fixative. SNs and SCs were dissected out and incubated overnight in 0.1 m sodium cacodylate buffer. This was followed by incubation in 2% OsO4 solution and gradient ethanol dehydration. Samples were incubated in propylene oxide PolyBed resin and embedded in flat molds at 55°C for 36 h. After embedding, the molds were processed and imaged on a JEOL 1230 electron microscope at the University of Texas Health Science Center at San Antonio Electron Microscopy Laboratory.
In vivo nerve conduction measurements.
Subdermal electrodes were used for SN stimulation with 0.02 ms impulses using the Nicolet Teca Synergy neurological system (Natus Neurology). The method has been previously described (Taylor et al., 2017). Briefly, for each trace, amplitude was measured as the difference in millivolts from the onset to the peak of the compound action potential (CAP). The nerve conduction velocity (NCV) was determined by estimating the distance between the notch and ankle, and dividing that estimate by the difference between the notch and ankle latencies.
Catwalk analysis.
The Catwalk automated gait analysis system (Noldus) was used to assess the gait and motor coordination in control, mutant, and rescue animals. The apparatus consists of a glass-plate walkway with a fluorescent light beaming into the glass from the side. As the mouse crosses the walkway, fluorescent light illuminates the animal's paws and when the paw touches the glass a bright print image of it is produced (Hamers et al., 2001). Catwalk XT 10.6 software (Noldus) was used to record and process the position of footprints, allowing quantitative analysis of gait. All mice were trained to cross the walkway for at ≥3 times a day over 4 successive days. On day 5, the final day, data were collected from three replicate crossings by each mouse. A run was defined as successful when the animal crossed the runway without interruption. The average speed for animals through the walkway floor was calculated (cm/s).
RNA extraction and quantitative RT-PCR analyses.
Harvested dorsal root ganglia (DRGs) and SC tissue were immediately frozen in liquid nitrogen and stored at −80°C until further processing. Total RNA from tissues was isolated by using PureLinkTM RNA Minikit (Ambion) with Trizol reagents (Invitrogen). cDNA reverse transcription was performed with a High Capacity cDNA Reverse Transcription Kit (Applied Bio System). Sequences of primers spanning exons 17/18, 20/21, and 31/32 were designed using the National Institutes of Health primer tool and synthesized by Eurofins Genomics. Sequence for primers were as follows: exon 17/18, forward, 5′-TCACCACGATGGAGCTGAAC-3′, reverse, 5′-CAGCTGATTTTCTTGGCTCTTCT-3′; exon 20/21, forward, 5′-ACCAGCTAGTGCAGAGCTTCG-3′, reverse, 5′-CACCACTCC TCCACCTGAGA-3′; exon 31/32, forward, 5′-TATCAGCCAGAGTGGCCTTC-3′, reverse, RP 5′-TATCAGCCAGAGTGGCCTTC-3′. Quantitative RT-PCR (qRT-PCR) was performed with Power SYBR PCR Master Mix (Applied Bio System) on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Data were normalized to β-actin and control mRNA levels using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Immunoblotting.
Samples were processed as described previously (Saifetiarova et al., 2017a). Briefly, mouse brains, SCs, and SNs were dissected out and homogenized on ice in lysis buffer. Homogenized samples were incubated at 4°C followed by centrifugation at 20,000 × g at 4°C for 30 min. Supernatant was collected as a final lysate, heated for 5 min at 37°C, loaded on SDS-PAGE, transferred to nitrocellulose, and probed with primary antibodies. Afterward membranes were incubated in infrared-conjugated secondary antibodies followed by detection using the Odyssey CLx Imaging System.
Quantification and statistics: intensities of nodal proteins and dot plot charts.
For each experiment, SC sections and teased SNs from at ≥3 animals per group were immunostained. Consistently, throughout the study, lateral tracts of the cervical region of the SCs and distal SNs were analyzed. The number of independent measurements (n) represents the total number of nodes from three animals per genotype. Images were acquired by a Zeiss LSM 710 confocal microscope with a 40× magnification objective using identical settings. Optical fields were randomly selected throughout the slide and three independent images for each control and mutant animal were acquired. On average, ≥100 nodes for the PNS and 200 nodes for the CNS from each animal were collected for analysis. To have the same amount of βIV spectrin-positive nodes in the Sptbn4res group, we increased the number of acquired images to 5–6 for SNs and 8–10 for SCs. All nodes flanked by Caspr immunostaining from both sides were quantified within each field of view. Heminodes or nodes with partially captured profiles were not included in the analysis. Data measurements were performed by one examiner in a nonblinded manner.
Protein intensities were measured over the entire nodal area, which was defined by Caspr-positive paranodal immunostaining and selected manually using the freeform drawing tool in ImageJ software. To calculate corrected values of the nodal fluorescence, mean background readings were taken and subtracted from the integrated density values for each node. The following formula was used for calculations: corrected nodal fluorescence = integrated density − (area of selected node × mean fluorescence of background readings) (Burgess et al., 2010; Gavet and Pines, 2010). Mean corrected nodal fluorescence values were averaged from ∼300 nodes for SNs and 600 nodes for SCs from three animals per genotype (≥100 and 200 nodes/mouse for SNs and SCs, respectively) and final results were standardized to same-age control values. All animals and samples within the same-age group were processed at the same time (perfused, sectioned, immunostained, and imaged). For Sptbn4res mice, only βIV spectrin-positive nodes were included in the statistical analysis.
Percentage of βIV spectrin-positive nodes were calculated over the total number of nodal gaps (defined by Caspr immunostaining) in the entire field of view and the mean values from three animals (≥100 and 200 nodes per animal for PNS and CNS, respectively) were plotted into the graphs. One-way ANOVA with Bonferroni's post hoc analysis test was used for comparison of groups between each other. Unless otherwise stated, all data are represented as mean ± SEM and analyzed using MatLab software. Statistical significance between groups is represented by *p < 0.05; **p < 0.01; ***p < 0.001 using two-way ANOVA with Bonferroni's post hoc analysis test, where time and experimental groups were considered as two variable factors.
Results
Removal of the ROSAβgeo* insertion element from the Sptbn4geo locus restores expression of βIV spectrin at nodes of Ranvier
βIV spectrin is encoded by the Sptbn4 locus. In the following sections, the wild-type animals are referred as Sptbn4 (+/+), the βIV spectrin mutant mice are referred as Sptbn4geo, and the mutants in which the ROSAβgeo* insertion has been removed are referred as Sptbn4res. Sptbn4geo mutants were previously generated by gene-trap mutagenesis, where expression of the βIV spectrin protein was disrupted by ROSAβgeo* insertion, leading to a null allele (Komada and Soriano, 2002). To determine the precise site of the ROSAβgeo* insertion within the Sptbn4 gene for our studies, we first mapped its exact location by screening genomic sequences around exons 19 and 22 using a series of primer sets that spanned this genomic region (data not shown). Based on the PCR amplification with a specific set of primers, we determined that the ROSAβgeo* insertion was located in the intron between exons 20 and 21 (Fig. 1A). We used the following sets of primers to amplify a 715 bp fragment in the wild-type (+/+) and a 450 bp fragment in Sptbn4geo mutants from genomic tail DNA: common forward primer, 5′-AAA CTG GGC GTC TTC CTT AG-3′; reverse wild-type primer, 5′-GTT TGA TTC TCC TGC TGA CCT C-3′; reverse mutant primer, 5′-CTG AGT GAT TGA CTA CCC GT-3′ (Fig. 1B). Interestingly, the original design of the ROSAβgeo*, which includes flanking LoxP sites, gave us an opportunity to test whether re-expression of the βIV spectrin would be possible after excision of the ROSAβgeo* construct by tamoxifen-inducible Cre-recombinase in actin-CreER;Sptbn4geo mice. A single injection of tamoxifen to postnatal day (P) 24, P120, or P210 actin-CreER;Sptbn4geo mice was able to induce re-expression of the βIV spectrin and its accumulation at the nodes. As shown in Figure 1C,H, wild-type (+/+) nodes showed normal localization of βIV spectrin (red, red arrowheads) flanked by the paranodal Caspr (green) in both the PNS (SN; Fig. 1C) and CNS (SC; Fig. 1H) myelinated axons. No βIV spectrin was detected at the nodes from Sptbn4geo mice in the SNs (Fig. 1D, white arrowheads) and SCs (Fig. 1I, white arrowheads). In tamoxifen-injected actin-CreER;Sptbn4geo (Sptbn4res) mice, βIV spectrin expression was restored and the protein localized at the nodes in myelinated axons from both the SNs (Fig. 1E–G, red arrowheads) and the SCs (Fig. 1J–L, red arrowheads). Note for comparison, nodes that have not restored βIV spectrin are also shown (Fig. 1E–G, J–L, white arrowheads). No βIV spectrin protein was detected in actin-CreER;Sptbn4geo mice without tamoxifen administration, thus excluding the possibility of leaky expression of βIV spectrin (data not shown). Moreover, to determine whether there were age-specific differences in the recombination efficiency and rescue rates, we analyzed percentage of βIV spectrin-positive nodes flanked by Caspr-positive paranodal immunostaining in control, mutant, and various-aged rescue SN and SC samples (Fig. 1M,N). Our quantification data revealed that percentage of nodes with re-expressed βIV spectrin in rescue samples was slightly reduced. However, that percentage was not significantly different among 2-month-old, 5-month-old, and 8-month-old animals 1 month after tamoxifen injection (mpi) in both the PNS and the CNS (Fig. 1M,N). To determine the levels of βIV spectrin protein expression in Sptbn4res animals compared with +/+ and Sptbn4geo animals, we performed immunoblot analysis of SNs, SCs, and brain lysates using antibodies generated against the C terminus of βIV spectrin (Saifetiarova et al., 2017a). Immunoblot analysis revealed a minor ∼300 kDa isoform and a major ∼160 kDa spectrin isoform (Komada and Soriano, 2002) in control samples. These were absent in Sptbn4geo mutant tissues (Fig. 1O–Q) for both PNS and CNS. In the Sptbn4res samples, the major ∼160 kDa βIV spectrin isoform and the minor ∼300 kDa isoform were both restored in actin-CreER;Sptbn4geo mice upon tamoxifen injection. Given the low level of recombination in actin-CreER;Sptbn4geo mice upon tamoxifen injection, the protein samples loaded were at levels 2.5X (SN), 6X (SC), and 4X (brains) the levels loaded for control samples (Fig. 1O–Q). Immunoblots against tubulin served as loading controls. Moreover, we did not observe differences in the levels of re-expressed protein between younger (2 months old) and older (8 months old) 1 mpi animals (data not shown). These data show that removal of ROSAβgeo* from the Sptbn4 locus restores expression of both isoforms of βIV spectrin. Additionally, the degree of rescue was not Cre-dependent or tamoxifen-dependent, as the Slick-H-CreER;Sptbn4geo line revealed similar percentages of βIV spectrin-positive nodes in both the PNS and CNS (data not shown). Next, to confirm that there was no leaky expression of Sptbn4 gene in our model, we performed qPCR analysis of relative mRNA levels in control, Sptbn4geo, and Sptbn4res animals with primers specific to exon/intron sites in the Sptbn4 locus. As the ROSAβgeo* insertion, which disrupts gene expression, is located in the intron between exons 20 and 21, we designed three sets of qPCR primers: (1) before insertion (located in exons 17/18); (2) flanking insertion (located in exons 20/21), and (3) after the insertion (located in exon 31/32; Fig. 1A). As expected, expression of the Sptbn4 locus before ROSAβgeo* insertion was detected by primers in exons 17/18 in DRG and SC samples from all three groups (control, mutant, and rescue; Fig. 1R,S). However, the second primer set, which is flanking the insertion site and is located in exons 20/21, as well as the third primer set, which is located in exons 31/32 after the insertion, showed no detectable mRNA in DRGs and barely detectable (<1%) mRNA in SC samples from Sptbn4geo mutant animals (Fig. 1R,S). In contrast, increased levels of mRNA expression was detected with exons 20/21 and exons 31/32 primer sets in DRG (∼25% of control) and SC (∼10% of control) Sptbn4res samples, indicating successful re-expression of the Sptbn4 gene after Cre-mediated recombination of the ROSAβgeo* insertion (Fig. 1R,S). Together, our data demonstrate that removal of the ROSAβgeo* insertion from the Sptbn4geo locus allows restoration and re-expression of βIV spectrin isoforms from the Sptbn4 locus and that re-expressed βIV spectrin clusters normally at the nodes.
Figure 1.
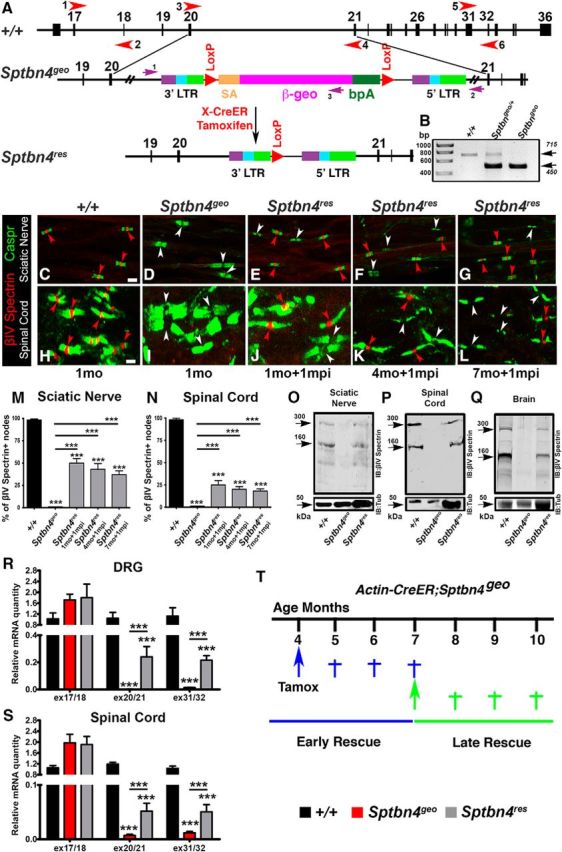
Removal of Rosaβgeo insertion allows re-expression of βIV spectrin. A, Partial genomic map of the Sptbn4 locus and the location of the gene trap Rosa βgeo* insertion, which is removed by Cre-mediated recombination. Red arrowheads represent location of primers that were used for qRT-PCR amplification of Sptbn4 transcripts. B, PCR amplification of genomic tail DNA isolated from wild-type (+/+), heterozygous (Sptbn4geo/+), and homozygous floxed (Sptbn4geo) mice. C–L, Immunostaining of teased SN fibers (C–G) and SCs (H–L) from control (+/+) (C, H), Sptbn4geo mutant (D, I), and tamoxifen-injected 2-month-old, 5-month-old, and 8-month-old actin-CreER;Sptbn4geo mice after 1 mpi (E–G, J–L, Sptbn4res) with antibodies against nodal βIV spectrin (red) and paranodal Caspr (green). Red arrowheads point to βIV spectrin-positive nodes and white arrowheads indicate βIV spectrin-negative nodes. M, N, Percentage of βIV spectrin-positive nodes in SNs (M) and SCs (N) of control, Sptbn4geo mutant, and Sptbn4res rescue animals depicted in C–L. Data are represented as mean ± SEM. n = 3 mice per genotype, with ≥100 and 200 nodes/animal for SNs and SCs, respectively. ***p < 0.001, one-way ANOVA, Bonferroni's post hoc analysis. Scale bar, 4 μm. O–Q, Immunoblot analysis of SN (O), SC (P), and brain (Q) lysates from control, Sptbn4geo mutant, and Sptbn4res rescue animals 1 mpi with antibodies against βIV spectrin (top) and tubulin (bottom). R, S, qPCR analysis of relative mRNA quantity in DRG (R) and SC (S) samples from control, Sptbn4geo mutant, and Sptbn4res rescue animals 1 mpi using primers specific to various exon/intron junctions of the Sptbn4 locus (exons 17/18, 20/21, and 31/32, depicted in the schematic in A as 1-2, 3-4, and 5-6, respectively). Data are represented as mean ± SEM. n = 4–7 mice per genotype. ***p < 0.001, one-way ANOVA, Bonferroni's post hoc analysis. T, Schematic representing the design of rescue strategies. Tamoxifen injection times in actin-CreER;Sptbn4geo animals and the timeline for phenotypic analysis of the two rescue groups.
Previous phenotypic analysis of Sptbn4geo mutants revealed disrupted clustering of AnkG and NaV channels at the nodes of Ranvier in adult 3-month-old mice and complete paralysis of the mutants by 6–10 months and their inevitable demise (Komada and Soriano, 2002). To address the precise timeline of nodal disorganization and also address whether there is a critical time window for the βIV spectrin expression that allows full recovery of the nodes in myelinated axons and improvement of motor symptoms in Sptbn4geo mutants, we designed an experimental strategy and created two groups of mice (Fig. 1T) correlated with the severity of the motor symptoms. In Group 1 (early rescue), re-expression of βIV spectrin in actin-CreER;Sptbn4geo mice was induced at 4 months, when they exhibit moderate motor phenotype. In Group 2 (late rescue), re-expression of βIV spectrin in actin-CreER;Sptbn4geo mice was induced at 7 months, when motor dysfunctions had progressed to eventual paralysis. After induction of βIV spectrin re-expression, phenotypic analyses of each group were performed for 3 consecutive months.
Nodal restoration in the PNS occurs independent of the timeline of βIV spectrin re-expression
Since Sptbn4geo mutants are characterized by nodal destabilization in the PNS, our next step was to evaluate whether nodes of Ranvier will be able to recluster after βIV spectrin re-expression. For this purpose, we performed immunofluorescent staining of SN fibers from wild-type (+/+), Sptbn4geo, and Sptbn4res animals followed by quantification of intensities of core nodal proteins, such as NfascNF186, AnkG, and NaV channels. Group 1 (early rescue) Sptbn4res animals receiving tamoxifen at 4 months were analyzed at 5, 6, and 7 months; and Group 2 (late rescue) Sptbn4res animals receiving tamoxifen at 7 months were analyzed at 8, 9, and 10 months. Wild-type, Sptbn4geo, and Sptbn4res animals were identically processed at the same time points. Evaluation of βIV spectrin at SN nodes showed that within 1 mpi its intensity in rescued animals reached control levels and further remained stable for the next 3 months that the animals were observed (Fig. 2A–S). As shown in Figure 2A–F, immunostaining against AnkG (red), paranodal Caspr (green), and βIV spectrin (blue) revealed that Sptbn4geo mutants showed a drastic decrease in AnkG levels with barely detectable traces of AnkG at the mutant nodes. At 4 months there was close to 25% reduction in AnkG intensity in Sptbn4geo mutant animals, which further decreased to 65% at 7 months and dropped further to 75% at 10 months (Fig. 2T). Moreover, dot plots of individual AnkG intensities from three animals per each genotype revealed that in Sptbn4geo mutants the distribution of the majority of nodal population progressively shifted toward decreased intensities from 4 to 10 months (Fig. 2W,W′). After tamoxifen injections, the levels of AnkG at the nodes quickly returned to control levels within 1 month after βIV spectrin re-expression (Fig. 2T). Additionally, the dot plot distribution showed recovery of AnkG intensities in the βIV spectrin-positive nodal population of Sptbn4res mice from early-rescue and late-rescue groups (Fig. 2W,W′). These data indicate that loss of βIV spectrin has a direct impact on the levels of AnkG at the nodes, and that re-expression of βIV spectrin allows quick restoration of AnkG at the nodes.
Figure 2.
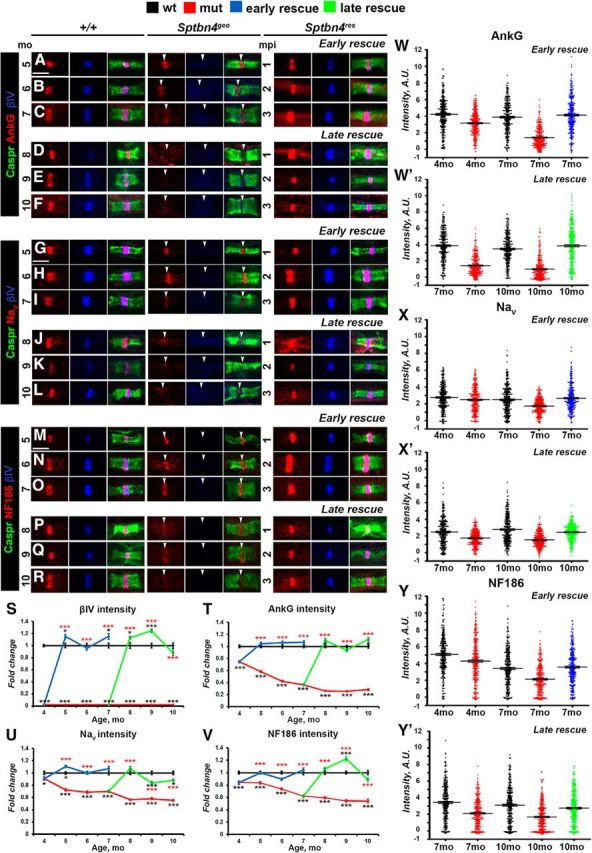
Timeline of disorganization and reorganization of the nodes in the PNS. A–R, Immunostaining of SN fibers from 5–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice with antibodies against βIV spectrin (blue) and Caspr (green) in combination with antibodies against either of the following proteins: AnkG (A–F), pan-NaV (G–L), or NfascNF186 (M–R; red). Arrows indicate βIV spectrin-negative nodes. Scale bar, 4 μm. S–V, Quantification of average fluorescence intensities of βIV spectrin (S), AnkG (T), pan-NaV (U), and NfascNF186 (V) in the SN nodes standardized to the same age control values from 4–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice (n = 300 nodes from 3 mice per genotype; ≥100 nodes per animal, all data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; 2-way ANOVA with Bonferroni's post hoc analysis; red stars indicate rescue group statistical significance compared with the age-matched mutants; black stars indicate rescue and mutant group statistical significance compared with the age-matched controls). W–Y′, Distribution of AnkG (W, W′), NaV (X, X′), and NfascNF186 (Y, Y′) nodal fluorescence intensities in SNs in control (+/+), Sptbn4geo mutant, and Sptbn4res mice at the initial prerescue stage and at the latest rescue time points (n = 300 nodes from 3 mice per genotype; ≥100 nodes per animal). Fluorescence intensity, arbitrary units (A.U.) × 100.
Next, we followed the consequences of absence of βIV spectrin on the stability of nodal NaV channels. As shown in Figure 2G–L,U, starting from 4 months, there was a significant difference in average NaV intensity between control and Sptbn4geo mutants with a ∼12% reduction; by 7 months the NaV channel levels showed 30% decrease with further decline to 55% by 10 months. Arrangement of individual nodal NaV intensities into a dot plot chart revealed differential distribution between genotypes, with a higher percentage of nodes with lower intensities in the mutant group (Fig. 2X,X′). After tamoxifen injection, re-expression of βIV spectrin within 1 month resulted in a proper localization and recovery of nodal NaV channels levels in the early-rescue group (Fig. 2G–L,T). In the late-rescue group, within 1 month after βIV spectrin re-expression, levels of NaV at the node reached control levels in Sptbn4res mice. However, within the next 2 months it showed slight decline from initial values, but remained significantly higher than in Sptbn4geo mutant mice. The dot plot charts showed recovery in distribution of individual nodal NaV intensities both in early-rescue and late-rescue groups similar to that observed in controls (Fig. 2X,X′).
Immunostaining against NfascNF186 revealed that Sptbn4geo mutants undergo a gradual decrease of nodal NfascNF186 intensity over time (Fig. 2M–R), where at 4 months, NfascNF186 intensity had decreased by 18% and continued to decrease by 46% of the control levels at 10 months (Fig. 2V). Tamoxifen injections to early and late rescue groups resulted in the restoration of NfascNF186 intensities in βIV Spectrin-positive nodes, where they remained at control levels within the next 3 months in early rescue groups and slightly decreased at 10 months in late-rescue animals (Fig. 2M–R,V). In addition, βIV spectrin restoration resulted in a redistribution of intensities leading to a higher percentage of nodes with increased levels of NfascNF186 in both rescue groups compared with mutant animals (Fig. 2Y,Y′). These data show that re-expression of βIV spectrin restores NfascNF186 to normal levels within 1 month after its expression. Together, our results indicate that loss of βIV spectrin leads to a sequential loss of nodal proteins with AnkG getting destabilized first followed by NaV channels and NfascNF186. On the other hand, restoration of βIV spectrin at the nodes allows a quick reassembly of the nodal proteins beginning with AnkG and full restoration within 1 month after βIV spectrin re-expression.
Timeline of βIV spectrin re-expression in the CNS is critical for optimal nodal reorganization
To establish whether the CNS nodal proteins followed a similar disorganization pattern as the PNS, and whether the βIV spectrin re-expression timeline had different effects on the CNS nodes, we performed immunofluorescent staining of SCs from two rescue time points in all genotypes for the core nodal proteins AnkG, NaV channels, and NfascNF186 at 4–10 months. Efficiency of our rescue models in the CNS was evaluated by quantification of nodal βIV spectrin intensities, which showed that re-expressed βIV spectrin intensities reached and remained at control levels (Fig. 3A–S). At the Sptbn4geo nodes, which lack βIV spectrin, AnkG levels dramatically decrease over time, starting from a 54.1% reduction at 4 months and reaching close to 85% reduction by 10 months (Fig. 3A–F,T), where most nodes either had no AnkG or had barely trace amounts of AnkG. After tamoxifen injections to 4-month-old actin-CreER;Sptbn4geo animals, the level of AnkG intensity started to increase and within 3 months reached close to control levels (65, 83, and 89% intensity at 5, 6, and 7 months; Fig. 3T). Surprisingly, in the late-rescue actin-CreER;Sptbn4geo animals, AnkG levels did not return to control levels. Even though βIV spectrin was re-expressed, levels of AnkG remained slightly higher than in Sptbn4geo mutants, but not significantly higher than in mutant animals, reaching 47.3, 47.1, and 38.5% at the maximum at 8, 9, 10 months, respectively. Additionally, we observed that distribution of AnkG nodal intensities were recovered only in the early-rescue group, with only moderate improvement in the late-rescue animals (Fig. 3W,W′). These data suggest that as nodal disorganization continued for 7 months in Sptbn4geo mutants, re-expression of βIV spectrin was not able to fully restore AnkG at the CNS nodes.
Figure 3.
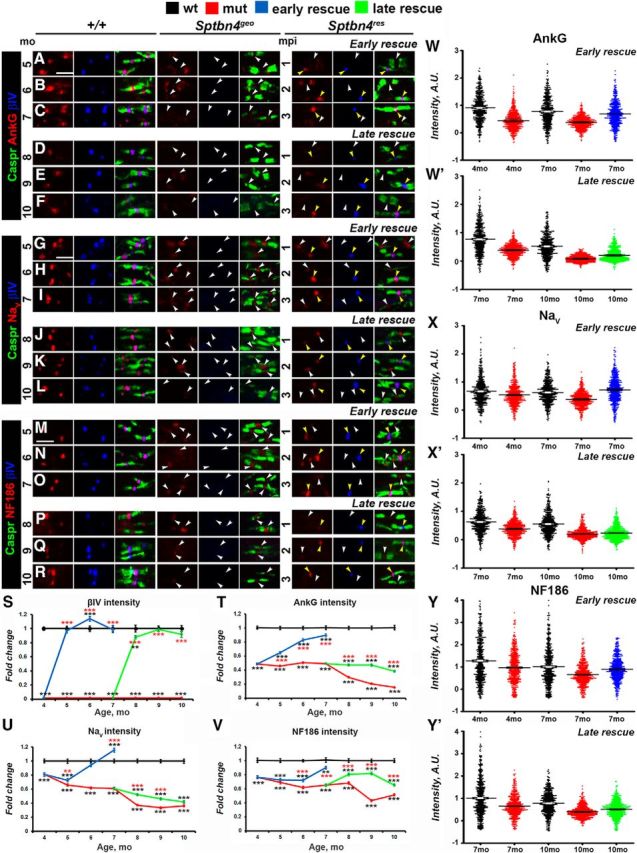
Timeline of disorganization and reorganization of the nodes in the CNS. A–R, Immunostaining of SCs from 5–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice with antibodies against βIV spectrin (blue) and Caspr (green) in combination with antibodies against either of the following proteins: AnkG (A–F), NaV (G–L), or NfascNF186 (M–R; red). White and yellow arrows indicate βIV spectrin-negative and βIV spectrin-positive nodes, respectively. Scale bar, 4 μm. S–V, Quantification of average fluorescence intensities of βIV spectrin (S), AnkG (T), pan-NaV (U), and NfascNF186 (V) in the SC nodes standardized to the same age control values from 4–7-month-old and 7–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice (n = 600 nodes from 3 mice per genotype; ≥200 nodes per animal; all data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; 2-way ANOVA with Bonferroni's post hoc analysis; red stars indicate rescue group statistical significance compared with the age-matched mutants; black stars indicate rescue and mutant group statistical significance compared with the age-matched controls). W–Y′, Distribution of the nodal population by their intensities of AnkG (W, W′), NaV (X, X′), and NfascNF186 (Y, Y′) in control (+/+), Sptbn4geo mutant, and Sptbn4res mice at the initial prerescue and at the latest rescue time point (n = 600 nodes from 3 mice per genotype; ≥200 nodes per animal). Fluorescence intensity: arbitrary units (A.U.) × 100.
Similarly, the intensity levels of Nav channels at the nodes were decreasing progressively in Sptbn4geo mutants (Fig. 3G–L,U) starting from a 19% reduction at 4 months and reaching a 64% reduction by 10 months (Fig. 3U). Tamoxifen injections at 4 months to actin-CreER;Sptbn4geo animals led to gradual increase in Nav intensity and within 2 months it reached control levels (72, 94, and slightly >100% intensity levels of control over 5, 6, and 7 months). Nevertheless, the levels did not increase after injection of tamoxifen to 7-month-old actin-CreER;Sptbn4geo animals and the levels continued to decline reaching 52, 46, and 42% of control levels in 8, 9, and 10 months, respectively. Moreover, NaV intensities in the late-rescue group remained distributed similarly to mutant 10-month-old animals (Fig. 3X′), in contrast to recovered distribution in the early-rescue group (Fig. 3X), further indicating that late expression of βIV spectrin is unable to rescue Nav-channel restoration at the CNS nodes.
Next, we followed NfascNF186 intensity changes and recovery at the CNS nodes. As shown in Figure 3M–R,V, NfascNF186 levels declined over time in the Sptbn4geo mutants, compared with controls with 24% reduction at 4 months to 49% at 10 months. In addition, loss of βIV spectrin affected distribution of nodal NfascNF186 levels in the mutants, especially at 10 months, shifting them toward a range of lower intensities (Fig. 3Y,Y′). After, tamoxifen injections at 4 months to actin-CreER;Sptbn4geo animals, NfascNF186 levels began to increase and within 3 months reached from 73, to 78, to 90% of control levels over 5, 6, and 7 months, respectively (Fig. 3V). However, tamoxifen injections at 7 months to actin-CreER;Sptbn4geo animals resulted in initial elevation of NfascNF186 intensity in βIV-positive nodes with 81 and 82% intensity at 8 and 9 months, respectively, but by 10 months levels of NfascNF186 dropped and were close to mutant levels (Fig. 3V). Similarly, distribution of NfascNF186 intensities was recovered in the early-rescue group, in contrast to the late-rescue group (Fig. 3Y,Y′). Together, these data indicate that CNS nodes lacking βIV spectrin for extended periods develop a limited ability for reorganization and restoration, and that re-expression of βIV spectrin after just 7 months fails to allow full reorganization of the CNS nodes, highlighting key differences between CNS and PNS nodal reorganization.
βI spectrin and ankyrin R occupying βIV spectrin-deficient nodes get extruded after βIV spectrin re-expression
Recent studies have shown that loss of nodal cytoskeletal scaffolding proteins βIV spectrin and AnkG leads to aberrant compensatory increase of βI spectrin and ankyrin R (AnkR) at the nodes (Ho et al., 2014; Saifetiarova et al., 2017a; Taylor et al., 2017). To determine the timeline of how βI spectrin and AnkR nodal levels change in Sptbn4geo mutants and whether re-expression of βIV spectrin leads to changes in the levels of βI spectrin and AnkR at the reorganizing nodes, we performed immunofluorescent staining of SNs and SCs followed by quantification of intensities of βI spectrin (Fig. 4A–R) and AnkR (Fig. 5A–R) at 4–10 months in control, Sptbn4geo mutant, and actin-CreER;Sptbn4geo animals. The levels of βI spectrin remained high in the SNs (Fig. 4A–F,M) and SCs (Fig. 4G–L,N) of Sptbn4geo mutants. Similarly, AnkR levels were also significantly high at most nodes in SNs (Fig. 5A–F,Q) and SCs (Fig. 5G–L,R) of Sptbn4geo mutants. At later time points there were nodes that lacked both βI spectrin and AnkR, suggesting that acute nodal destabilization affects the stability of both βI spectrin and AnkR at the nodes (Fig. 5E,F, Sptbn4geo panels). In addition, distribution of nodal βI spectrin and AnkR levels was significantly shifted toward increased intensity levels in mutant animals compared with controls (Figs. 4O–R, 5M–P). After tamoxifen injection to actin-CreER;Sptbn4geo animals at 4 and 7 months, re-expression of βIV spectrin caused a decrease of βI spectrin and AnkR intensities within 1 month in both early-rescue and late-rescue groups in the PNS (Figs. 4A–F,M, 5A–F,Q). In the CNS, the levels of both proteins in the early-rescue group gradually decreased to control levels (Figs. 4G–L,N, 5G–L,R). Even though intensities of βI spectrin and AnkR decreased in the late-rescue group, their levels remained between control and Sptbn4geo mutant levels at 10 months (Figs. 4N, 5R). βI spectrin and AnkR dot plot charts of individual nodal intensities showed their redistribution in rescue groups toward profiles similar to those of controls, except for the SCs in the late-rescue group, where redistribution remained similar to that observed at the mutant nodes (Figs. 4O–R, 5M–P). These data suggest that as soon as βIV spectrin is re-expressed and the core nodal proteins are reassembled, βI spectrin and AnkR are extruded from the reorganizing nodes. However, as disorganization of nodes continues, as in the later-rescue group, re-expression of βIV spectrin either fails to properly reorganize nodal components, allowing βI spectrin and AnkR to stay, or the older mutant nodes continue to become destabilized with βIV spectrin unable to restore them.
Figure 4.

Re-expression of βIV spectrin displaces βI spectrin. A–L, Immunostaining of SNs (A–F) and SCs (G–L) from 5–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice with antibodies against βI spectrin (red), βIV spectrin (blue), and Caspr (green). White and yellow arrows indicate βIV spectrin-negative and βIV spectrin-positive nodes, respectively. Scale bar, 4 μm. M, N, Quantification of βI spectrin average fluorescence intensity in the SN (M) and SC (N) nodes standardized to the same age control values from 4–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice (n = 300 from 3 mice, with ≥100 nodes per animal in SNs; n = 600 from 3 mice, with ≥200 nodes from each animal in SCs; all data are represented as mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; 2-way ANOVA with Bonferroni's post hoc analysis; red stars indicate rescue group statistical significance compared with the age-matched mutants; black stars indicate rescue and mutant group statistical significance compared with the age-matched controls). O–R, Distribution of the nodal βI spectrin fluorescence intensities in SNs (O, P) and SCs (Q, R) at the initial prerescue stage and at the latest rescue time point (n = 300 nodes from 3 mice per genotype in SNs; n = 600 nodes from 3 mice per genotype in SCs). Fluorescence intensity: arbitrary units (A.U.) × 100.
Figure 5.
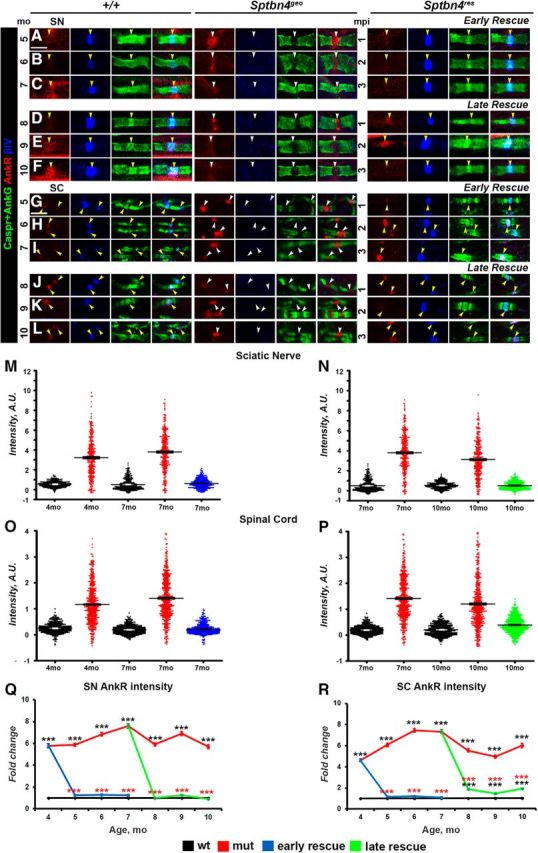
Newly expressed βIV spectrin causes decrease in AnkR levels at the nodes. A–L, Immunostaining of SNs (A–F) and SCs (G–L) from 4–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice with antibodies against AnkR (red), βIV spectrin (blue), and Caspr (green). Yellow and white arrows indicate βIV spectrin-negative and βIV spectrin-positive nodes, respectively. Scale bar, 4 μm. M–P, Distribution of the nodal population by their nodal AnkR fluorescence intensities in SNs (M, N) and SCs (O, P) at the initial prerescue stage and at the latest rescue time point (n = 300 nodes from 3 mice per genotype in SNs; n = 600 nodes from 3 mice per genotype in SCs). Fluorescence intensity: arbitrary units (A.U.) × 100. Q, R, Quantification of average AnkR fluorescence intensity in the SN (Q) and SC (R) nodal area standardized to the same age control values from 4–10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice (n = 300, with ≥100 nodes from each animal's SNs; n = 600, with ≥200 nodes from each animal's SCs. All data are represented as mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; 2-way ANOVA with Bonferroni's post hoc analysis).
Timely βIV spectrin re-expression allows nodal restoration and prevents axonal degeneration
The nodal destabilization observed in Sptbn4geo mutants over time could lead to ultrastructural changes in the myelinated axons. This, in combination with the sequential full recovery of nodes upon βIV spectrin re-expression in the PNS and partial reorganization of the CNS nodes in both rescue groups, prompted us to analyze the ultrastructure of myelinating axons and assess the health of the axons before and after βIV spectrin re-expression. In addition, the differences seen in the CNS and PNS nodal recovery in early-rescue and late-rescue groups could potentially reveal irreversible changes in axonal morphology by or after the time the rescue was initiated. We performed TEM of PNS and CNS myelinated axons at the final stages of rescue at 7 months (early-rescue group) and 10 months (late-rescue group) together with corresponding age-matched control and Sptbn4geo mutants. At 7 and 10 months, SNs (Fig. 6A,G) and SCs (Fig. 6D,J) from control groups showed normal morphology of axons ensheathed by tight compact myelin. In contrast, ultrastructural morphology of Sptbn4geo mutant SNs (Fig. 6B,H) and SCs (Fig. 6E,K) revealed shrunken axons (Fig. 6B,H, arrowheads) or axons containing abnormal cytoskeletal deformities and inclusions (Fig. 6E,K, arrowheads), which is generally taken as an indication of axonal degeneration (Fig. 6M–R, higher-magnification images). Moreover, quantification of the degenerated axons revealed that there is a higher degree of degeneration in 10-month-old Sptbn4geo mutants compared with 7-month-old Sptbn4geo mutants (34 vs 22% in PNS; 40 vs 23.33% in CNS; Fig. 6S,T). The degree of degeneration was significantly lower in both rescue groups in SNs (Fig. 6C,I). However, in SCs, the 10-month-old animals (late-rescue group) showed the same amount of pathology as Sptbn4geo mutants at 10 months (Fig. 6, compare L, K). The 7-month-old samples from the early-rescue group showed healthy axons with overall morphology close to that seen in age-matched control animals (Fig. 6, compare D, F). There was clear correlation between the level of degeneration and nodal recovery between PNS and CNS axons. The PNS axons exhibited a slightly different level of degeneration in Sptbn4geo mutants at 7 and 10 months, whereas the CNS axons at 10 months showed significantly higher levels of pathology and, after a certain time point, the axonal degeneration reached profound levels, which may prevent restoration of axonal domains. Together, the ultrastructural analyses indicate that timely restoration of axonal domains in myelinated axons that have undergone nodal domain disorganization is necessary to prevent axonal degeneration.
Figure 6.

Timely reorganization of nodes prevents axonal degeneration. A–L, TEM of cross sections from 7-month-old (A–F) and 10-month-old (G–L) age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice SNs (A–C, G–I) and SCs (D–F, J–L). M–R, Axonal pathology seen in Sptbn4geo mutants. TEM images at higher magnification showing different stages of axonal degeneration, starting with the accumulation of cytoskeletal inclusions (M, N, Q, R), which eventually results in axon and myelin structural disintegration (O, P). S, T, Quantification of axonal degeneration in 7-month-old and 10-month-old age-matched control (+/+), Sptbn4geo mutant, and Sptbn4res mice in the PNS and CNS, respectively (n = 3 mice/genotype, 2-way ANOVA, Bonferroni's post hoc analysis). All data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001, two-way ANOVA, Bonferroni's post hoc analysis. Scale bars: A–L, 4 μm; M–P, 400 nm; Q, R, 1 μm.
Timely βIV spectrin re-expression allows functional recovery and prevents motor paresis
As has been reported, Sptbn4geo mutants develop progressive motor dysfunctions accompanied by significantly shortened life span (Komada and Soriano, 2002). Since disorganization of axonal domains is one of the primary causes underlying these phenotypes, we aimed to determine whether re-expression of the βIV spectrin and sequential reassembly of the nodal components will restore functional properties and motor performance of the Sptbn4geo mutant animals deficient in βIV spectrin. As shown in Figure 7A–C, the control (+/+) animals at 10 months show normal body posture and motor coordination (Fig. 7A) compared with Sptbn4geo mutants, which show small body stature with weakened motor coordination and severe hindlimb paralysis at 10 months (Fig. 7B). Upon tamoxifen injection at 7 months, Sptbn4res animals show a remarkable recovery of body posture and do not display severe hindlimb weakness or paralysis (Fig. 7, compare B, C), indicating significant recovery of motor functions. Next, we wanted to analyze motor coordination and improvements in walking patterns of Sptbn4res mice before and after tamoxifen injection using the Catwalk gait analysis system (Noldus), which monitors digital paw impressions as animals touch the surface. The pattern of paw impressions is consistent as animals walk in the path. At 4 months of age, in addition to acquiring profound tremor and demonstrating clenching of hindlimbs to the body when suspended by the tail, Sptbn4geo mutants became significantly slower in crossing Catwalk compared with control animals. Also, they lost the normal left–right walking pattern. Moreover, mutant animals display reduced hindpaw footprint area, indicating their inability to properly use rear limbs while moving (Fig. 7D,E, compare green, pink footprints). At 6–10 months of age, most animals were paralyzed and incapable of moving around or completing Catwalk measurements. Re-expression of the βIV spectrin prevented motor paralysis in those animals ≤10 months old. The rescue animals regained their walking pattern and were able to move and performed better on Catwalk than mutant groups (Fig. 7F). However, measurements of the walking speed revealed that this parameter did not completely return to control levels (Fig. 7G). The motor tremor remained persistent in both rescue groups, but tremors were significantly less severe than those of mutants. Next, we performed body-weight measurements at two rescue stages. Sptbn4geo mutants at 7 months were already low in body weight, which continued to decline at 10 months (21.06 ± 0.8201 vs 17.13 ± 1.12 g at 7 and 10 months, respectively (Fig. 7H). Upon tamoxifen injections at 4 and 7 months, the body weight was significantly restored in the early-rescue group, but remained lower than control levels in the late-rescue group, indicating that timely restoration of nodal function is necessary for full body-weight recovery in Sptbn4geo mutants (Fig. 7H). We also monitored animal survival for up to 10 months and quantified survival rates in both rescue groups. As shown in Figure 7I, 44% of Sptbn4geo mutants died within 6 months and barely 10% of Sptbn4geo mutants survived to 10 months. Upon tamoxifen injection, the survival rate increased significantly in both rescue groups. However, the early-rescue group animals had a much better survival rate and their general health conditions were much better than those of the late-rescue groups (25 and 19% survived by 10 months in early-rescue and late-rescue group, respectively; Fig. 7I). Loss of βIV spectrin and NaV-channel destabilization at the nodes are also accompanied by altered conductive properties in myelinated axons, which is characterized by decreased amplitudes of the CAPs and decreased conduction velocities. Sptbn4geo mutants were severely impaired in both the NCV and the action potential amplitudes (Fig. 7J). Upon re-expression of βIV spectrin, Sptbn4res animals showed improvements in both parameters compared with Sptbn4geo mutants in the early-rescue group. However, the parameters in rescue animals did not reach control levels (Fig. 7J–L). The early-rescue group again showed much better restoration of nerve conduction measurements compared with the late-rescue group, further highlighting the importance of the axonal health and timely rescue in Sptbn4res mice. Together, our data demonstrate that reassembly of the nodes in myelinated axons improves their functional properties and restores motor performance in a time-dependent manner, and that if nodal domains continue to destabilize, it will irreversibly harm the health of the myelinated axons, thereby preventing restoration of their function and motor performance.
Figure 7.
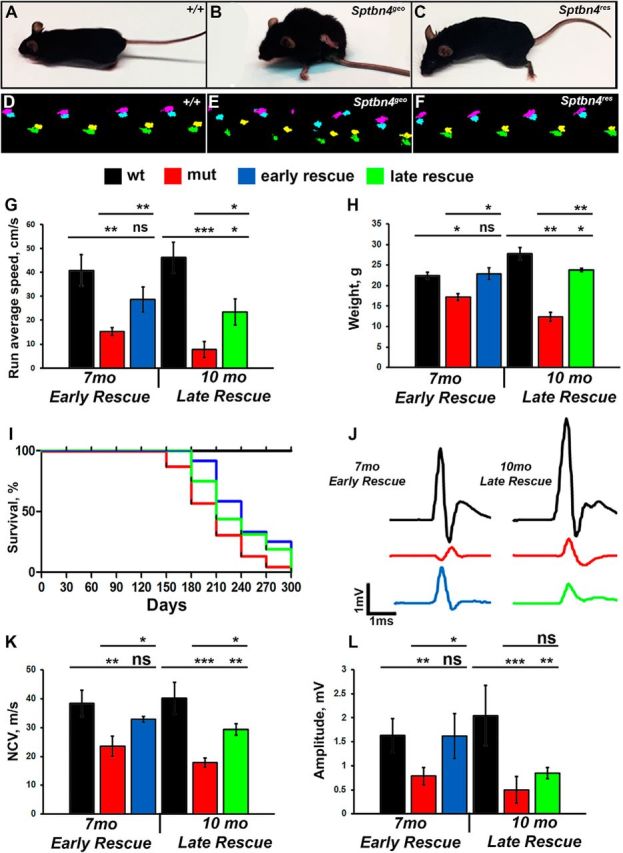
Motor function and nerve conduction restoration after βIV spectrin re-expression. A–C, Photographs of 10-month-old control (+/+), Sptbn4geo mutant, and Sptbn4res mice at 3 mpi. D–F, Representative Catwalk footprints of 10-month-old control (+/+), Sptbn4geo mutant, and Sptbn4res mice at 3 mpi. G, Quantifications of the average running speed from Catwalk gait recordings of early-rescue and late-rescue mice (n = 7–10 mice/genotype). H, Body-mass change in control (+/+), Sptbn4geo mutant, and Sptbn4res mice at early-rescue and late-rescue stages. Equal numbers of males and females were included in each control (+/+), Sptbn4geo mutant, and Sptbn4res early-rescue and late-rescue groups (n = 7–10 mice/genotype). I, Survival curve for control (+/+), Sptbn4geo mutant, and Sptbn4res early-rescue and late-rescue groups. J, Representative electrophysiological profiles of CAPs from 7-month-old and 10-month-old SNs of control (+/+), Sptbn4geo mutant, and Sptbn4res early-rescue and late-rescue groups. K, L, Quantification of the NCV (K) and amplitude (L) in control (+/+), Sptbn4geo mutant, and Sptbn4res early-rescue and late-rescue groups (n = 7–10 mice/genotype). All data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001, two-way ANOVA, Bonferroni's post hoc analysis.
Discussion
The emergence of myelination during evolution became coupled with saltatory conduction, which allowed species to perform many neuronally controlled processes faster than those without myelination. The primary driver of this fast conduction are the nodes of Ranvier, assembled with a unique set of proteins that include transmembrane cell-adhesion molecules and voltage-gated Nav channels anchored by cytoskeletal scaffolding proteins. The reorganization of nodes after their disorganization and the timeline in which restoration may occur remain unknown. Here, we reported on the reorganization of the nodes in βIV spectrin-deficient nodes, which undergo destabilization in a protein-specific sequential manner. We uncovered key differences in the timeline of nodal reorganization and functional restoration after re-expression of βIV spectrin in the peripheral and central myelinated axons. Our studies highlight that nodal disorganization for extended periods critically harms axonal health, thus impeding complete functional restoration.
Cytoskeletal scaffolding proteins in nodal organization and maintenance
The initial organization of the nodal domain coincides with the onset of myelination followed by nodal maturation to ensure long-term stability and maintenance. Recent studies from several groups have provided insights into how nodal proteins are recruited to the nodal region and the specific role that individual nodal proteins play in not just the initial assembly but also in its maturation and maintenance (Jenkins and Bennett, 2001; Komada and Soriano, 2002; Thaxton et al., 2011; Zhang et al., 2012; Susuki et al., 2013; Saifetiarova et al., 2017a; Taylor et al., 2017). The flanking paranodal protein complex does not seem to directly aid in this initial assembly but plays a role in the long-term stability, as when the paranodal disruption is combined with loss of nodal βIV spectrin and nodal destabilization occurs (Susuki et al., 2013; Taylor et al., 2017). Thus, several mechanisms have been postulated that rely on the contributions of the nodal and paranodal cytoskeletal-scaffolding proteins, including the spectrins and ankyrins in nodal organization and maintenance (Susuki et al., 2013; Saifetiarova et al., 2017a; Taylor et al., 2017). Given the intermolecular interactions between node-specific proteins and the presence of flanking axoglial paranodal molecular complexes, it is still unclear how timely interplay between these complexes causes the initial nucleation of the nodal site. Recent studies using spatiotemporal ablation methods showed that loss of NfascNF186 after nodal organization causes slow but progressive nodal destabilization (Taylor et al., 2017) and loss of AnkG at early or late postnatal stages affects the maturation and maintenance of the nodes but not their initial organization (Thaxton et al., 2011; Saifetiarova et al., 2017a; Taylor et al., 2017). Interestingly, combined loss of nodal NfascNF186 and paranodal NfascNF155 after nodal organization led to exacerbated destabilization of the remaining nodal components (Taylor et al., 2017). Disruption of the paranodal and/or juxtaparanodal regions did not affect nodal organization or maintenance, but severely affected nerve function and neuromuscular health of the mutants (Bhat et al., 2001; Saifetiarova et al., 2017b).
Like AnkG, βIV spectrin is highly enriched at the nodes and loss of AnkG or βIV spectrin does not affect the initial nodal assembly, as NfascNF186 and Nav channels, along with either βIV spectrin or AnkG, respectively, still cluster at the node (Komada and Soriano, 2002; Susuki et al., 2013; Saifetiarova et al., 2017a). Prenatal loss of βIV spectrin leads to a slow but progressive destabilization of the node over a period of 6–8 months (this study), however, loss of AnkG prenatally causes nodal destabilization within 1 month; and, if AnkG is lost after nodal organization, the remaining nodal components stay stable for almost 1 year after ablation (Saifetiarova et al., 2017a). These observations suggest that nodal component assembly and disassembly may follow a sequential order in which individual nodal components act either as major organizers, like e.g., NfascNF186, which clusters at the nodal axolemma and interacts with both the extracellular and intracellular proteins, or as stabilizers like AnkG and βIV spectrin to allow the transmembrane nodal proteins to get anchored with the axonal nodal cytoskeleton and thus stabilize this complex (Jenkins and Bennett, 2001). Loss of βIV spectrin affects in increasing severity the localization of AnkG, Nav channel, and NfascNF186, as AnkG is the first protein to diffuse out from the nodes, followed by Nav channel and last NfascNF186, as revealed by precise intensity measurements reported here. To add to the complexity of nodal destabilization, absence of βIV spectrin allows enrichment of βI spectrin and AnkR, which are present at the nodes but at reduced levels (Saifetiarova et al., 2017a). However, the presence of βI spectrin and AnkR is not able to prevent progressive destabilization of the βIV spectrin-deficient nodes, suggesting that their molecular interactions with nodal proteins may be much weaker compared with AnkG and βIV spectrin.
Reorganization and restoration of disorganized nodes
The mechanisms that govern in vivo reassembly of the nodes of Ranvier have not been addressed. Also, and most importantly, we do not understand at what state of destabilization nodal reorganization is still possible; nor do we know at what stage axons can still tolerate nodal destabilization before they become too unhealthy to recover. Previous studies reported nodal restoration by selective expression of either NfascNF186 or NfascNF155 in Nfasc-null background using transgenic expression (Zonta et al., 2008). These studies showed re-establishment of axo-glial paranodal junctions together with the rescue of nodal molecular complexes in the CNS. Since the expression of the Nfasc rescue constructs was occurring during prenatal and postnatal stages in Nfasc mutants, the nodes formed normally and were never disorganized or destabilized. Our model of nodal restoration provides several advantages. First, Sptbn4geo mutant mice, in contrast to other nodal mutant animals deficient in NfascNF186, AnkG, or Nav, are unique in that they are postnatally viable and undergo gradual destabilization of the nodal components over extended periods. Second, progressive loss of nodal functions causes motor phenotypes in Sptbn4geo mice mimicking disease course in patients with demyelinating disorders. Third, our model allows us to induce nodal restoration in a spatiotemporal manner in both the PNS and CNS at any stage of phenotypic progression. Our data show that the PNS myelinated axons respond better with full recovery of the nodes accompanied with the ultrastructural and functional improvements, independent of time when rescue is initiated. Meanwhile, the CNS myelinated axons performed better when restored at early stages but failed to fully restore nodes and continued toward axonal health decline and progressive motor dysfunction. While the level of βIV spectrin in the rescued nodes was comparable to those in controls, the percentage of actually positive nodes in the PNS and CNS reached 50 and 25%, respectively. The level of rescue was neither Cre-dependent nor age-dependent, as after the rescue initiation in slick-H-CreER; Sptbn4geo mice showed the same level of rescue as in the actin-CreER; Sptbn4geo mice. It remains to be established whether re-expression of βIV spectrin that allowed rescue of a greater percentage of nodes could lead to better motor outcomes. Collectively, these observations suggest that urgency is required to achieving nodal reorganization and functional restoration, and that long-term disorganization of nodes in the CNS myelinated axons may be associated with irreversible axonal damage, even when the nodal protein components are reassembled. These observations also highlight differences in the molecular mechanisms that may be operating in PNS and CNS myelinated axons.
Nodal disorganization and myelinated axon pathologies
Numerous mouse and human studies about nodal disorganization have linked that disorganization with auditory, motor, and nerve conduction impairments. Autoantibodies against βIV spectrin isoforms were found in the serum of a cancer patient with the paraneoplastic lower motor neuron syndrome (Berghs et al., 2001). It had been suggested that disruption of the nodal regions and the axon initial segments by βIV spectrin autoantibodies altered neuronal functions, leading to death of the motor neurons. Moreover, mutations in the human SPTBN4 locus have been recently linked to congenital myopathy, neuropathy, and central deafness (Knierim et al., 2017). Altered nodal morphologies have also been observed in demyelinating disorders (Hahn et al., 2001; Craner et al., 2004; Howell et al., 2006; Arancibia-Carcamo and Attwell, 2014), in immune-mediated neuropathies (Santoro et al., 1990; Cifuentes-Diaz et al., 2011), and in spinal cord and diffuse brain injuries (Ouyang et al., 2010; Reeves et al., 2010), as well as under normal aging conditions (Hinman et al., 2006). Thus, changes in the structure and function of the nodes affects electrical properties of myelinated axons, eventually resulting in severe ataxia and paralysis. Sequence analyses across human populations have also uncovered an association of mutations in loci that encode nodal proteins to psychiatric disorders, such as autism, schizophrenia, bipolar disorders, and personality disorders (Davis et al., 2003; Ahn et al., 2004; Aoki et al., 2013). In addition, microarray-based gene-expression profiling also revealed downregulation of Nfasc, NrCAM, Nav1.6, and Ank3 genes in neuropsychiatric disorders. Ank3, which encodes AnkG, was identified as a susceptibility gene in bipolar disorders, attention deficit hyperactivity disorder, intellectual disability, and epilepsy (Jia et al., 2011; Bi et al., 2012; Iqbal et al., 2013). While these genetic associations suggest a link between nodal proteins, no direct evidence is available to link these proteins to human diseases. Our studies with βIV spectrin, which provide a window into nodal disorganization and into axonal pathology and dysfunction, aim for functional restoration by nodal reorganization in a spatiotemporal manner. Mouse mutants carrying human mutations could be created to establish direct functional links with nodal dysfunction. Then rescue strategies could be used to determine the time course of reversing associated pathologies. Together, our studies provide significant novel insights into the processes of in vivo nodal disorganization and reorganization associated with functional restoration. These strategies may serve as a great therapeutic tool for understanding the molecular basis of slowing down disease progression in myelinated axon nodal pathologies and thus for improving patients' quality of life.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences Grant GM063074, the National Multiple Sclerosis Society, the Owens Foundation, the Morrison Trust, and the Zachry Foundation (M.A.B). We thank members of the Bhat laboratory for technical assistance and valuable discussions. We also thank J. Gelfond (Department of Epidemiology and Biostatistics, University of Texas Health San Antonio), for guidance and assistance with statistical data analysis, and M. Stankewich (Yale University), for sharing βI spectrin antibodies. All electron microscopy was performed at the University of Texas Health San Antonio electron microscopy facility.
The authors declare no competing financial interests.
References
- Ahn KH, Lyoo IK, Lee HK, Song IC, Oh JS, Hwang J, Kwon J, Kim MJ, Kim M, Renshaw PF (2004) White matter hyperintensities in subjects with bipolar disorder. Psychiatry Clin Neurosci 58:516–521. 10.1111/j.1440-1819.2004.01294.x [DOI] [PubMed] [Google Scholar]
- Aoki Y, Abe O, Nippashi Y, Yamasue H (2013) Comparison of white matter integrity between autism spectrum disorder subjects and typically developing individuals: a meta-analysis of diffusion tensor imaging tractography studies. Mol Autism 4:25. 10.1186/2040-2392-4-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia-Carcamo IL, Attwell D (2014) The node of Ranvier in CNS pathology. Acta Neuropathol 128:161–175. 10.1007/s00401-014-1305-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghs S, Ferracci F, Maksimova E, Gleason S, Leszczynski N, Butler M, De Camilli P, Solimena M (2001) Autoimmunity to beta IV spectrin in paraneoplastic lower motor neuron syndrome. Proc Natl Acad Sci U S A 98:6945–6950. 10.1073/pnas.121170798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30:369–383. 10.1016/S0896-6273(01)00294-X [DOI] [PubMed] [Google Scholar]
- Bi C, Wu J, Jiang T, Liu Q, Cai W, Yu P, Cai T, Zhao M, Jiang YH, Sun ZS (2012) Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat 33:1635–1638. 10.1002/humu.22174 [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A (2010) Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A 107:12564–12569. 10.1073/pnas.0914191107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttermore ED, Thaxton CL, Bhat MA (2013) Organization and maintenance of molecular domains in myelinated axons. J Neurosci Res 91:603–622. 10.1002/jnr.23197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Dubourg O, Irinopoulou T, Vigny M, Lachkar S, Decker L, Charnay P, Denisenko N, Maisonobe T, Léger JM, Viala K, Hauw JJ, Girault JA (2011) Nodes of Ranvier and paranodes in chronic acquired neuropathies. PLoS One 6:e14533. 10.1371/journal.pone.0014533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A 101:8168–8173. 10.1073/pnas.0402765101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V (1996) Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain−) and NrCAM at nodal axon segments. J Cell Biol 135:1355–1367. 10.1083/jcb.135.5.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR, Buxbaum J, Haroutunian V (2003) White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry 60:443–456. 10.1001/archpsyc.60.5.443 [DOI] [PubMed] [Google Scholar]
- Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL (2007) Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol 177:857–870. 10.1083/jcb.200612012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, Peles E (2010) A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 65:490–502. 10.1016/j.neuron.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med 354:942–955. 10.1056/NEJMra052130 [DOI] [PubMed] [Google Scholar]
- Gavet O, Pines J (2010) Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell 18:533–543. 10.1016/j.devcel.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM (2001) Pathological findings in the x-linked form of Charcot–Marie–Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol 101:129–139. [DOI] [PubMed] [Google Scholar]
- Hamers FP, Lankhorst AJ, van Laar TJ, Veldhuis WB, Gispen WH (2001) Automated quantitative gait analysis during overground locomotion in the rat: its application to spinal cord contusion and transection injuries. J Neurotrauma 18:187–201. 10.1089/08977150150502613 [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244:305–318. 10.1006/dbio.2002.0597 [DOI] [PubMed] [Google Scholar]
- Hinman JD, Peters A, Cabral H, Rosene DL, Hollander W, Rasband MN, Abraham CR (2006) Age-related molecular reorganization at the node of Ranvier. J Comp Neurol 495:351–362. 10.1002/cne.20886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell OW, Palser A, Polito A, Melrose S, Zonta B, Scheiermann C, Vora AJ, Brophy PJ, Reynolds R (2006) Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 129:3173–3185. 10.1093/brain/awl290 [DOI] [PubMed] [Google Scholar]
- Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, Bennett V, Rasband MN (2014) A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci 17:1664–1672. 10.1038/nn.3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WJ, Chen WW, Zhang X (2017) Multiple sclerosis: pathology, diagnosis and treatments. Exp Ther Med 13:3163–3166. 10.3892/etm.2017.4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA, Roca LT, Vulto-van Silfhout AT, Nijhof B, Kramer JM, Van der Aa N, Ansar M, Peeters H, Helsmoortel C, Gilissen C, Vissers LE, Veltman JA, de Brouwer AP, Frank Kooy R, Riazuddin S, et al. (2013) Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet 22:1960–1970. 10.1093/hmg/ddt043 [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001) Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155:739–746. 10.1083/jcb.200109026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci U S A 99:2303–2308. 10.1073/pnas.042601799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Ewers JM, Zhao Z (2011) Prioritization of epilepsy associated candidate genes by convergent analysis. PLoS One 6:e17162. 10.1371/journal.pone.0017162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knierim E, Gill E, Seifert F, Morales-Gonzalez S, Unudurthi SD, Hund TJ, Stenzel W, Schuelke M (2017) A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum Genet 136:903–910. 10.1007/s00439-017-1814-7 [DOI] [PubMed] [Google Scholar]
- Komada M, Soriano P (2002) [Beta]IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 156:337–348. 10.1083/jcb.200110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359. 10.1074/jbc.270.5.2352 [DOI] [PubMed] [Google Scholar]
- Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, De Camilli P, Moser T, Rasband MN, Solimena M (2004) BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol 166:983–990. 10.1083/jcb.200408007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V (1997) Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci 17:7025–7036. 10.1523/JNEUROSCI.17-18-07025.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Nelson AD, Jenkins PM (2017) Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front Cell Neurosci 11:136. 10.3389/fncel.2017.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H, Sun W, Fu Y, Li J, Cheng JX, Nauman E, Shi R (2010) Compression induces acute demyelination and potassium channel exposure in spinal cord. J Neurotrauma 27:1109–1120. 10.1089/neu.2010.1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA (2009) Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res 87:1773–1793. 10.1002/jnr.22015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Greer JE, Vanderveer AS, Phillips LL (2010) Proteolysis of submembrane cytoskeletal proteins ankyrin-G and alphaII-spectrin following diffuse brain injury: a role in white matter vulnerability at nodes of Ranvier. Brain Pathol 20:1055–1068. 10.1111/j.1750-3639.2010.00412.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifetiarova J, Taylor AM, Bhat MA (2017a) Early and late loss of the cytoskeletal scaffolding protein, ankyrin G reveals its role in maturation and maintenance of nodes of Ranvier in myelinated axons. J Neurosci 37:2524–2538. 10.1523/JNEUROSCI.2661-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifetiarova J, Liu X, Taylor AM, Li J, Bhat MA (2017b) Axonal domain disorganization in Caspr1 and Caspr2 mutant myelinated axons affects neuromuscular junction integrity, leading to muscle atrophy. J Neurosci Res 95:1373–1390. 10.1002/jnr.24052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M, Thomas FP, Fink ME, Lange DJ, Uncini A, Wadia NH, Latov N, Hays AP (1990) IgM deposits at nodes of Ranvier in a patient with amyotrophic lateral sclerosis, anti-GM1 antibodies, and multifocal motor conduction block. Ann Neurol 28:373–377. 10.1002/ana.410280312 [DOI] [PubMed] [Google Scholar]
- Susuki K, Chang KJ, Zollinger DR, Liu Y, Ogawa Y, Eshed-Eisenbach Y, Dours-Zimmermann MT, Oses-Prieto JA, Burlingame AL, Seidenbecher CI, Zimmermann DR, Oohashi T, Peles E, Rasband MN (2013) Three mechanisms assemble central nervous system nodes of ranvier. Neuron 78:469–482. 10.1016/j.neuron.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Saifetiarova J, Bhat MA (2017) Postnatal loss of neuronal and glial neurofascins differentially affects node of Ranvier maintenance and myelinated axon function. Front Cell Neurosci 11:11. 10.3389/fncel.2017.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA (2011) Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron 69:244–257. 10.1016/j.neuron.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. (2006) Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 7:932–941. 10.1038/nrn2023 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bekku Y, Dzhashiashvili Y, Armenti S, Meng X, Sasaki Y, Milbrandt J, Salzer JL (2012) Assembly and maintenance of nodes of Ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron 73:92–107. 10.1016/j.neuron.2011.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta B, Tait S, Melrose S, Anderson H, Harroch S, Higginson J, Sherman DL, Brophy PJ (2008) Glial and neuronal isoforms of neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol 181:1169–1177. 10.1083/jcb.200712154 [DOI] [PMC free article] [PubMed] [Google Scholar]