

Abstract
The transferrin receptor 1 (TfR1) is an attractive target for antibody-mediated cancer therapy. We previously developed a mouse/human chimeric IgG3 antibody (ch128.1) targeting human TfR1, which exhibits direct in vitro cytotoxicity against certain human malignant B cells through TfR1 degradation and iron deprivation. ch128.1 also demonstrates exceptional antitumor activity in two xenograft models of the B-cell malignancy multiple myeloma (MM): SCID-Beige mice bearing disseminated ARH-77 or KMS-11 cells in an early disease setting. Interestingly, this activity is observed even against KMS-11 cells, which show no sensitivity to the direct cytotoxic activity of ch128.1 in vitro. To understand the contributions of the Fc fragment, we generated a ch128.1 mutant with impaired binding to FcγRs and to the complement component C1q, which retains binding to FcRn. We now report that this mutant antibody does not show antitumor activity in these two MM models, indicating a crucial role of the Fc fragment in the antitumor activity of ch128.1, which can be attributed to effector functions (ADCC, ADCP, and/or CDC). Interestingly, in the KMS-11 model, complement depletion does not affect protection, while macrophage depletion does. Consistent with this observation, we found that ch128.1 induces ADCC and ADCP against KMS-11 cells in the presence of murine bone marrow-derived macrophages. Finally, we found that ch128.1 therapy effectively increases survival in a late MM disease setting. Our results suggest that macrophages play a major role in ch128.1-mediated antitumor protection in our models and that ch128.1 can be effective against human B-cell malignancies such as MM.
Keywords: multiple myeloma, transferrin receptor 1, antibody effector functions, macrophages, antitumor activity, antibody therapy
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy in the United States. In 2017, it is estimated that there were 30,280 new cases of MM with 12,590 estimated deaths (1). It is a B-cell cancer, composed of malignant plasma cells, and is characterized by the over production of a monoclonal immunoglobulin (M-protein), immunosuppression, and end-organ damage including osteolytic lesions (2). While novel treatment options have led to improved response rates and increased survival, the majority of patients acquire drug resistance and ultimately relapse with more aggressive tumors, developing treatment-refractory disease associated with shortened survival times (2–6). As MM is a heterogeneous disease (7), it is advantageous to take a broad-spectrum approach to address multiple pathways and targets for sustained efficacy.
Iron is important for multiple cellular processes including metabolism, respiration, and DNA synthesis (8). Iron in blood is bound by transferrin (Tf), which is transported into cells through interaction with its receptor, the transferrin receptor 1 (TfR1), also known as CD71 (8–10). TfR1 is constitutively internalized and recycled back to the cell surface (9, 10). TfR1 is ubiquitously expressed at relatively low levels on normal cells and at increased levels in rapidly proliferating cells and malignant cells, including hematological malignancies, in which overexpression has been associated with poor prognosis (9–12).
Interestingly, TfR1 has also been reported to be involved in regulating mitochondrial function (13). TfR1 can support mitochondrial respiration and reactive oxygen species (ROS) production, both of which are key players in tumor cell growth and survival (14). Furthermore, TfR1 mediates NF-κB signaling in malignant cells through interaction with the inhibitor of the NF-κB kinase (IKK) complex, increasing cancer cell survival (15). Importantly, NF-κB plays an important role in MM cell pathogenesis and inhibition of this pathway blocks MM cell growth and survival (16). In addition, MM cells are dependent on an increased influx of iron (17). Taken together, the high expression of TfR1 on malignant cells, its extracellular accessibility, and its central role in cancer cell pathology make this receptor an attractive target for antibody-mediated cancer therapy, including therapy for MM.
Targeting TfR1 on malignant cells with an antibody may result in direct cytotoxicity through interference with TfR1 function by inhibiting Tf binding, triggering receptor degradation, or blocking receptor internalization, all of which ultimately impair iron uptake leading to lethal iron starvation (10, 18). Since the TfR1 may also function in regulating mitochondrial respiration and mediating NF-κB signaling as mentioned above, it is possible that antibodies may also interfere with these TfR1-mediated functions as well. Furthermore, antibodies may induce cytotoxicity against cancer cells through the induction of Fc effector functions: antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cell-mediated phagocytosis (ADCP), and/or complement-dependent cytotoxicity (CDC) (19, 20). In addition to their direct use as anticancer agents, antibodies targeting TfR1 can be used as delivery systems for a variety of anticancer agents that are internalized into malignant cells by receptor-mediated endocytosis (9, 18).
ch128.1 is a mouse/human chimeric IgG3 that binds specifically to human TfR1 and does not interfere with binding of the TfR1 ligands Tf and hemochromatosis protein (HFE) to TfR1 (21). It promotes degradation of TfR1 (22, 23) and in vitro cytotoxicity against certain malignant B-cell lines such as ARH-77 and IM-9 (22, 24, 25). In addition, a single dose of ch128.1 resulted in significant protection in two disseminated human MM xenograft SCID-Beige mouse models bearing either ARH-77 or KMS-11 cells. Intriguingly, this in vivo antitumor activity was observed even against KMS-11 MM cells that are insensitive to the cytotoxic effects of ch128.1 in vitro (24). ch128.1 also prolonged survival in an AIDS-related human Burkitt lymphoma xenograft model of NOD-SCID mice bearing 2F7 tumors, although no sensitivity was observed in vitro (26). Using the disseminated models of MM, the present study aims to define the mechanism of antitumor activity exhibited by ch128.1 and explore its in vivo efficacy in different therapeutic settings.
Materials and Methods
Cell lines
The KMS-11 human MM cell line was a kind gift from Lawrence H. Boise (Emory University, Atlanta, GA) and were cultured in IMDM (Life Technologies, Inc., Carlsbad, CA). ARH-77, an Epstein-Barr virus-transformed human B lymphoblastoid cell line, was purchased from ATCC (Manassas, VA) and cultured in RPMI 1640 (Life Technologies, Inc.). All cell lines were cultured in media supplemented with penicillin, streptomycin (ThermoFisher Scientific Inc., Canoga Park, CA) and 10% heat-inactivated FBS (Atlanta Biologicals, Inc., Atlanta, GA) in 5% CO2 at 37°C.
Recombinant antibodies
ch128.1 (IgG3/κ) and the ch128.1 triple mutant L234A/L235A/P329S were produced in murine myeloma cells and affinity purified as described (27, 28). Mutations were previously generated on the ch128.1 heavy chain γ3 expression vector to disrupt binding to FcγRs and complement component C1q (27). An IgG3/κ isotype control antibody, specific for the hapten dansyl (5-dimethylamino naphthalene-1-sulfonyl chloride; referred to as IgG3) (22), was produced with the expression vectors and methods used to develop ch128.1.
Proliferation assay
ARH-77 cells were incubated with various concentrations of the antibodies for a total of 96 hours. Proliferation was monitored using the [3H]-thymidine incorporation assay as described (25). The cells were incubated with [3H]-thymidine for the final 16 hours of the treatment period.
In vivo antitumor activity
All experimental protocols were approved by the UCLA Institutional Animal Care and Use Committee, and all local and national guidelines on the care of animals were strictly adhered to. C.B-17 SCID-Beige mice were obtained and housed in the Defined-Flora Mouse Facility in the Department of Radiation Oncology at UCLA. Female mice (8–12 weeks old) were exposed to 3 gray total body, sublethal irradiation (MARK-1-30 irradiator 137Cs source, J.L. Shepherd & Associates, San Fernando, CA) one day before tumor challenge. To establish disseminated disease, 5 × 106 ARH-77 or KMS-11 cells were injected i.v. into the lateral tail vein, as described (24). Mice were randomized into treatment groups. A single dose of ch128.1, its triple mutant, the IgG3 isotype control, or buffer alone was injected i.v. 2 or 9 days after tumor challenge. All animals are littermates and animals in the same treatment groups were co-housed. Survival was based on the time from tumor challenge to development of hind limb paralysis. Survival plots were generated using GraphPad Prizm Version 5 (GraphPad Software, Inc., La Jolla, CA). Significant differences in survival were determined by the log-rank test using the same software. Results were considered significant if p < 0.05.
Binding to neonatal Fc receptor (FcRn) via surface plasmon resonance (SPR)
The interaction of ch128.1 and its mutant with murine FcRn was monitored by SPR detection on a BIAcore 3000 instrument using a CM5 sensor chip (BIAcore, GE Healthcare Life Sciences, Pittsburgh, PA), as described (29), with modifications. Recombinant mouse FcRn (100 µg/ml, R&D Systems, Inc., Minneapolis, MN) was amine-coupled to flow cell 2 of the sensor chip and flow cells were blocked with 1M ethanolamine-HCl, pH 8.5. Flow cell 1 without FcRn was used as a control surface. ch128.1 or its mutant (10 to 400 nM) were flowed over FcRn in PBS/Tween-20 (50 mM sodium phosphate pH 6.0, 150 mM NaCl, 0.02% NaN3, 0.01% Tween-20) at 25°C, 20 µl/min for 10 minutes. Flow cells were regenerated using PBS, pH 8.0 containing 0.05% Tween-20. Sensograms were generated and analyzed, and equilibrium KD values determined using the steady state affinity model included in the BIAevaluation software v4.1 (29). Murine FcRn, which binds human IgG (30), was used to reflect binding in the in vivo model.
Evaluation of serum bioavailability
ch128.1 or its mutant (100 µg) were injected i.v. into the lateral tail vein of SCID-Beige female mice 8–12 weeks old. Blood was collected from the lateral tail vein at 2, 24, 48, 72, and 168 hours after injection from 2 alternating groups of mice. Serum antibody levels were quantified by ELISA and antibody integrity by immunoblotting. Briefly, ELISA Immulon 2HB plates (Thermo Fisher Scientific Inc., Waltham, MA) were coated with 1 µg/ml anti-human IgG (Southern Biotech, Birmingham, AL). Pooled serum from 4–5 mice was diluted 1:500 into 1% FBS/PBS. Two-fold serial dilutions were tested for all sera. Recombinant ch128.1 was used to generate a standard curve. Binding was detected with alkaline phosphatase (AP)-conjugated goat anti-human κ (MilliporeSigma, St. Louis, MO) and AP substrate (MilliporeSigma) dissolved in diethanolamine buffer (9.6% diethanolamine (v/v), 0.24 mM MgCl2 in water, pH 9.8). Plates were read at absorbance 405 nm using a FilterMax F5 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). Immunoblot analysis was performed with pooled serum (diluted 1:500) separated by SDS-PAGE under non-reducing conditions and transferred onto nitrocellulose membranes (GE Healthcare Life Sciences). The human IgG3 antibodies were detected using rabbit anti-human IgG (1:1000, Bethyl laboratories, Inc., Montgomery, TX) and HRP-conjugated donkey anti-rabbit IgG (1:5000, GE Healthcare Life Sciences). The ChemiGlow West Chemiluminescent Subtrate Kit (ProteinSimple, San Jose, CA) was used for detection. Recombinant antibodies, ch128.1 or its mutant were used as positive controls, while serum from untreated mice was used as a negative control. Digital images of blots were obtained using a FluorChem Megapixel High Performance Fluorescence, Chemiluminescence, and Visible Imaging System (ProteinSimple).
Macrophage depletion and confirmation of depletion by immunohistochemistry (IHC)
Macrophages in female SCID-Beige mice 8–12 weeks old were depleted by i.p. injection of 200 µl Clodrosome® (clodronate-encapsulated liposomes, 5 mg/ml, Encapsula NanoSciences LLC., Brentwood, TN) 4 hours after tumor inoculation, followed by a second dose (100 µl) 5 days later. Clodronate phagocytosed by macrophages inhibits mitochondrial ATP/ADP translocase (31) and induces death by apoptosis (32). PBS-encapsulated liposomes were used as a negative control. Depletion was confirmed by IHC of the spleen using F4/80 as a marker (33). Spleens of mice injected with the same doses of Clodrosome® were excised 5 days after the second dose. Briefly, spleen tissue specimens were fixed in 10% buffered formalin, and embedded in paraffin. Antigen retrieval was performed using citrate buffer and macrophages were visualized using rat anti-mouse F4/80 antibody (Bio-Rad Laboratories, Inc., Hercules, CA), rabbit anti-rat secondary antibody (Vector Laboratories, Burlingame, CA), Dako EnVision+ System-HRP-labelled polymer anti-rabbit kit (Dako, Carpenteria, CA), and 3,3’-diaminobenzidine (DAB; BioCore Medical Technologies, Inc., Elkridge, MD). Tissue sections were counterstained with Harris hematoxylin. Serial sections were stained with H&E. Images were obtained using an Olympus BX51 light microscope at 20× original magnification. Staining intensity was quantified using the reciprocal intensity method and the Fiji Software (ImageJ) as described (34). The reciprocal intensity is derived in the image after color deconvolution and is directly proportional to the amount of chromogen present in the image.
Complement depletion and confirmation via ELISA
For complement depletion, female SCID-Beige mice 8–12 weeks old were given 25 U cobra venom factor (CVF; Quidel, San Diego, CA) i.p. on the day of tumor challenge and at 3, 6, and 9 days post-challenge. CVF is a structural and functional analog of the complement component C3 that forms a stable C3/C5 convertase resistant to the regulatory complement proteins, resulting in exhausted complement activation and ultimately complement depletion (35). Depletion was assessed via detection of C3a in mouse serum by ELISA as described (36) with modifications. Immulon-H2B plates (Thermo Fisher Scientific, Inc.) were coated overnight at 4°C with 4 µg/ml rat anti-mouse C3a (BD Biosciences, San Jose, CA) in PBS, pH 6.5. Plates were washed with PBS then blocked with 3% BSA in PBS. Mouse serum samples from the same time point were pooled and diluted 1:50–1:500 in 1% BSA in PBS. Diluted mouse serum and purified mouse C3a (used as a positive control, BD Biosciences) were incubated overnight at 4°C. Plates were washed and incubated with biotinylated rat anti-mouse C3a (BD Biosciences) for 1 hour at room temperature. The presence of C3a was detected with streptavidin-AP and AP substrate (MilliporeSigma) dissolved in diethanolamine buffer and read at absorbance 405 nm using a FilterMax F5 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA).
Isolation of bone marrow-derived macrophages (BMDM)
Macrophages were derived from bone marrow (BM), as described (37) with modifications. Briefly, BM was flushed from femurs and tibias of BALB/c mice bred in-house (6–12 weeks) in RPMI-1640 supplemented with 10% FBS, glutamine, and antibiotics. Cells were passed through a 40 µm cell strainer (Corning Life Sciences, Tewksbury, MA). RBC were lysed with ammonium chloride for 30 minutes. Cells were cultured in Petri dishes in RPMI with 10% FBS and 20–30 ng/ml of recombinant M-CSF (Peprotech, Rocky Hill, NJ) for 7 days with replenishment every 3 days. Cells were confirmed to be macrophages via flow cytometry using fluorescence-conjugated antibodies against CD11b (Bio-Rad Laboratories, Inc.) and F4/80 on an LSRII cytometer (BD Biosciences).
ADCC/ADCP analysis via flow cytometry
A three-color flow cytometric assay was performed as described (38) with modifications. Briefly, KMS-11 target cells were labeled with CFSE at 250 nM/106 cells/ml (Life Technologies, Inc), coated with antibodies (10 µg/ml) for 30 minutes on ice, and incubated with BMDM effector cells (E:T ratio of 3:1) at 37°C for 2 hours in duplicates. Cells were washed in buffer (5% FBS in PBS), and incubated for 30 minutes on ice with a PE-conjugated anti-F4/80 antibody (Bio-Rad Laboratories, Inc.) to visualize macrophages. DAPI (1 µg/ml) was added to identify dead cells. At this concentration only dead cells are stained with DAPI. Cells treated with saponin (0.3% final concentration, MilliporeSigma) were used as a positive control for dead cells. 50,000 events were acquired by flow cytometry. Acquisition and measurement of single cell events were monitored by forward scatter versus side scatter dot plots and compared to control single- and mixed-population samples. ADCC and ADCP were calculated as described (38) with CFSE+/PE+ cells classified as ADCP, and CFSE+/DAPI+ cells classified as ADCC. Results were considered significant if p < 0.05 as determined by the Student’s t-test.
Visualization of ADCP by ImageStream imaging flow cytometry
The ADCC/ADCP assay was performed as described above. After the 2-hour incubation, the cells were fixed using 2% paraformaldehyde in PBS. Samples were run on a ImageStream®X Mark II (Amnis, part of MilliporeSigma) Imaging Flow Cytometer using the INSPIRE acquisition software (Version 2, Amnis). Brightfield and fluorescent images were acquired at 40× magnification. During event acquisition and excluding cell debris, 10,000 events were recorded with the following settings: Brightfield LED intensity 25.81 mW; 405 nm laser 10 mW; 488 nm laser 2.0 mW. The IDEAS software (Version 6.2; Amnis) was used for data analysis. Single stained samples were used to construct a compensation matrix which was then applied to subsequent flow analyses as well to draw positive and negative signal gates. Events that were best in focus (determined using the Gradient RMS value of 40 or greater on the brightfield channel) and single cells (determined from the brightfield area versus aspect ratio) were selected via gating for further analysis. From this population gate, a scatterplot was drawn graphing fluorescence intensity of the CFSE signal versus the fluorescence intensity of the PE signal. Gates were drawn containing PE+/CFSE− (macrophage population), PE−/CFSE+ (KMS-11 tumor cell population), and PE+/CFSE+ subpopulations using the single stained controls.
Results
The Fc region of ch128.1 is important for its antitumor activity
To examine the mechanism of protection conferred by ch128.1, we used a previously generated mutant with impaired binding to FcγRs and C1q (27). The triple mutant (L234A/L235A/P329S) retains the antigen-binding activity of ch128.1 and exhibits a lack of ADCC and CDC activity in vitro (27). We now show that this mutant also retains the in vitro cytotoxic activity of ch128.1. Exposure to the ch128.1 mutant led to the reduced proliferation of ARH-77 cells, to levels comparable to those observed in cells treated with ch128.1 (Fig. 1A). ARH-77 cells were used because they previously showed sensitivity to ch128.1 in vitro and in vivo, in contrast to KMS-11 cells, which only showed sensitivity in vivo (24). These results show that, as expected, these mutations do not alter the in vitro anti-proliferative effect of ch128.1.
Figure 1. The antitumor activity of ch128.1.

(A) ch128.1 and its mutant inhibit the proliferation of malignant B cells. ARH-77 cells were incubated with the specified concentrations of the antibodies for a total of 96 hours. [3H]-thymidine was added for the final 16 hours of the treatment period. Values are the mean of triplicate samples expressed as the percent of the [3H]-thymidine incorporation in cells treated with buffer alone control. Error bars show SD. (B–C) In vivo efficacy of the ch128.1 triple mutant (L234A/L235A/P329S) in two disseminated models of MM. Kaplan-Meier plots indicate survival of SCID-Beige mice challenged i.v. with 5 × 106 ARH-77 (B) or KMS-11 cells (C). 100 µg of ch128.1 or its mutant was administered i.v. 2 days after tumor challenge. *p < 0.0001 as determined by the log-rank test. Data in all panels are representative of 2 independent experiments.
Interestingly, the in vivo protective effect of ch128.1 was not observed with the mutant in the two disseminated xenograft models of MM. ARH-77 B lymphoblastoid cells have been used as a model of MM, as its i.v. injection in SCID mice leads to the development of a disease that mimics human MM (39, 40). KMS-11 human MM cells have been used to evaluate the in vivo efficacy of MM therapeutics (41, 42). In both models, significant prolonged survival was observed with ch128.1-treated animals (p < 0.0001 compared to animals treated with buffer alone; Fig. 1 B, C). In contrast, the mutant did not show significant antitumor activity and mice succumbed to disease at rates similar to control animals. An isotype control antibody tested previously showed no antitumor effect in these models (24), confirming that TfR1 targeting was necessary for antitumor protection.
FcRn, also known as the Brambell receptor or “salvage receptor”, is critical to maintain the bioavailability of IgG antibodies in blood preventing their degradation (20, 43). Thus, we determined if binding of the ch128.1 triple mutant to mouse FcRn was impaired, as its lack of antitumor activity might be due to less bioavailability. However, SPR analysis showed similar binding affinity between mouse FcRn and ch128.1 or its mutant (Table I). To determine if the blood bioavailability of the mutant antibody was affected, antibody levels in the serum of SCID-Beige mice were examined after i.v. administration. The mutant ch128.1 antibody did not show decreased serum levels, in fact, the levels found were slightly higher than those of wild type ch128.1 (Fig. 2A). Additionally, we found that both antibodies remain intact in mouse serum (Fig. 2B), showing bands consistent with the m.w. of human IgG3 (165 kDa) (20). Together these data indicate that the Fc region of ch128.1 is required for the antitumor activity in these models and that the lack of antitumor activity of the mutant is not due to a decrease in blood bioavailability (or increased clearance) in vivo.
Table I.
Antibody binding to mouse FcRn receptor via SPR analysis
| Antibody | KD (M) |
|---|---|
| ch128.1 | 3.58E-08 |
| ch128.1 mutant | 3.37E-08 |
Data are representative of 2 independent experiments.
Figure 2. Serum bioavailability of ch128.1 and its mutant.

Female SCID-Beige mice were injected i.v. with 100 µg of ch128.1 or its mutant (L234A/L235A/P329S). Blood was collected at 2, 24, 48, 72, and 168 hours after injection. (A) The amount of human IgG3 in pooled serum, was determined by ELISA performed in triplicate. Data are representative of 2 independent experiments using 2 different sets of animals. Error bars show SD. (B) Immunoblotting was performed using pooled serum diluted 1:500. Purified ch128.1 or the L234A/L235A/P329S mutant (labelled “Ab”) was used as a positive control. Mouse serum from untreated animals (labelled “N”) was used as a negative control. Blots are representative of 2 independent experiments.
Macrophages mediate ch128.1 antitumor activity
The SCID-Beige model used is deficient in B and T cells (44), and has impaired NK cell and neutrophil activity (45–47). However, this model has macrophages capable of eliminating tumor cells (48–50) and an active complement pathway (51). To further define the importance of the Fc region in ch128.1-mediated antitumor activity, the role of macrophages was examined. Mice challenged with KMS-11 cells were treated with Clodrosome® to deplete macrophages. The depletion of macrophages in ch128.1-treated mice significantly reduced the antitumor effect (p < 0.001 compared to that of ch128.1 administered with the liposomal control; Fig. 3A). However, a statistically significant survival benefit was still observed in these mice, compared with similarly depleted mice without ch128.1 treatment (p < 0.01). Depletion of macrophages in the absence of ch128.1 did not affect tumor growth and mice succumbed to disease at the same rate as the buffer control group. Depletion of macrophages (F4/80+ cells) was confirmed in the spleen of mice using IHC (Fig. 3B). Even though macrophage depletion was not complete (approximately a 63% reduction in macrophages was observed), it was sufficient to significantly decrease ch128.1-induced antitumor activity. These results suggest that macrophages play a crucial role in the antitumor activity of ch128.1 in the KMS-11 xenograft model.
Figure 3. Role of macrophages in ch128.1-induced antitumor activity.

(A) Kaplan-Meier plots indicate survival of SCID-Beige mice challenged i.v. with 5 × 106 KMS-11 human MM cells. 100 µg of ch128.1 or buffer was injected i.v. 2 days after tumor challenge. Macrophages were depleted with Clodrosome® i.p. 4 hours (200 µl) and 5 days (100 µl) after tumor challenge. PBS-encapsulated liposome was used as a negative control. *p, < 0.01, **p, < 0.001, (B) Spleens of mice were analyzed for macrophage depletion via IHC. Tissue sections were stained with rat anti-mouse F4/80 antibody or isotype control and counterstained with Harris hematoxylin. Serial sections were stained with H&E. Images were obtained at 20× original magnification. Scale bar =100 µm. Staining intensity was quantified and the reciprocal intensities for each of the images are shown. Data are representative of 2 independent experiments.
The complement pathway is not involved in ch128.1 antitumor activity
The role of complement in ch128.1-induced antitumor activity was assessed using CVF to deplete C3. Mice challenged with KMS-11 cells were treated with CVF or buffer. ch128.1 treatment alone or combined with CVF prevented MM in over 80% of animals with no significant difference in survival between the two groups (Fig. 4A). In contrast, CVF or buffer treated mice that did not receive the therapeutic antibody succumbed to disease at the same rate and were both significantly different compared to ch128.1 treatment alone (p < 0.0001). A substantial decrease of C3a levels in the sera of CVF-treated mice was observed within 24 hours and persisted up to 8 days, confirming complement inactivation (Fig 4B). The Fc region of human IgG1 and IgG3 is able to bind mouse complement and induce CDC, as shown with rituximab, a mouse/human chimeric anti-CD20 IgG1 (52), as well as with mouse/human chimeric anti-glucuronoxylomannan IgG1 and IgG3 antibodies (53). Even though ch128.1 is capable of activating complement in vitro (27), our studies indicate that this pathway is not relevant in the mouse model used.
Figure 4. Role of the complement pathway in ch128.1-induced antitumor activity.

(A) Kaplan-Meier plot indicates survival of SCID-Beige mice challenged i.v. with 5 × 106 KMS-11 human MM cells. Mice were injected i.v. 2 days after tumor challenge with 100 µg ch128.1. Complement depletion was achieved by injecting 25 U CVF i.p. on the day of tumor challenge and on days 3, 6, and 9. PBS was used as negative control. *p < 0.0001. (B) Confirmation of complement depletion. Blood from mice was collected before CVF injection (day 0) and on days 1 and 8. Serum C3a levels were examined by ELISA. Pooled sera collected at designated time points (n = 3–5, 1:500 dilution) were added to an ELISA plate coated with a rat anti-mouse C3a antibody. Binding was detected using biotinylated rat anti-mouse C3a, AP-conjugated streptavidin antibody and AP substrate. Values were normalized to the concentration of C3a in serum, where percent at initial time point was set at 100. Error bars show SD of samples in triplicate. Data are representative of 2 independent experiments.
Effector functions mediated by macrophages
To confirm the ability of ch128.1 to induce effector functions mediated by macrophages, an in vitro assay to assess ADCC and ADCP was performed. Murine macrophages were used for these studies since human IgG3 has been shown to bind all 4 murine FcγRs (54) and these cells were identified as a major player in the in vivo mechanism of action of the antibody. Significant ADCC and ADCP were exhibited in the presence of 10 µg/ml ch128.1 (Fig. 5A) (p < 0.05 compared to negative control), which was also observed with 5 µg/ml (data not shown). As expected, the levels of ADCC and ADCP induced by the ch128.1 mutant were similar to those of buffer control. In order to confirm the occurrence of phagocytosis induced by ch128.1 treatment, ImageStream imaging flow cytometry was performed. This method has previously been used to visualize macrophage phagocytosis of tumor cells mediated by rituximab (55). Phagocytosis was observed in the CFSE+/PE+ population, as indicated by the presence of tumor cells within macrophages (Fig. 5B). Together, these results show that murine macrophages can mediate tumor cell death via ADCC and ADCP.
Figure 5. Macrophages mediate ch128.1-induced ADCC and ADCP in vitro.
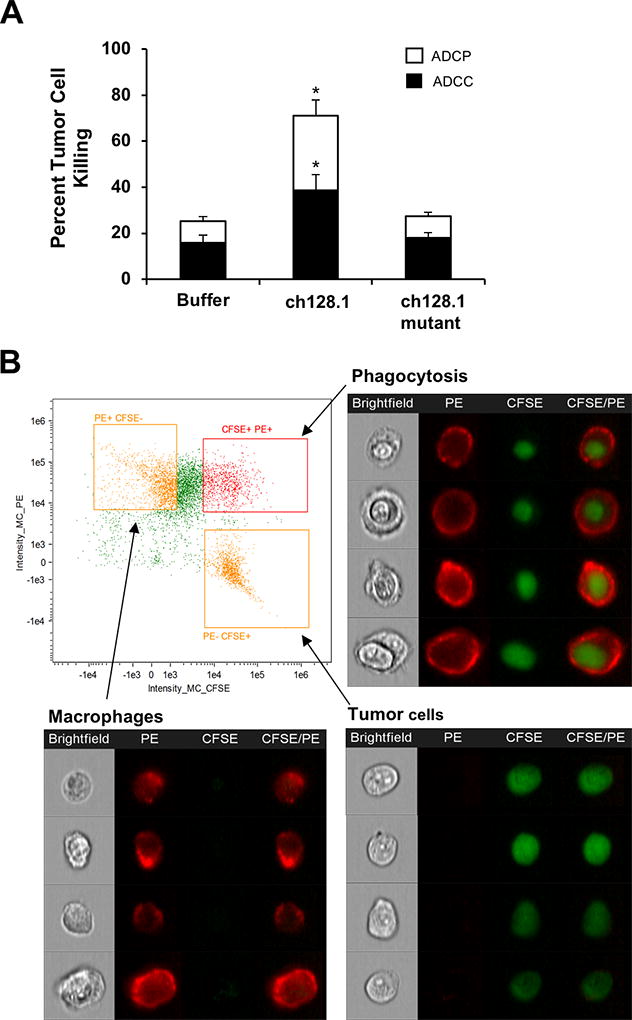
Macrophages were generated from progenitor cells from murine BM via differentiation with M-CSF for 7 days. CFSE-labeled KMS-11 cells were incubated with antibodies prior to addition of BMDM (E:T ratio of 3:1) at 37°C for 2 h. (A) Macrophages were visualized with a PE-conjugated anti-F4/80 antibody and all cells were stained with DAPI and analyzed by flow cytometry. CFSE+/PE+ cells designate occurrence of phagocytosis, while CFSE+/DAPI+ cells designate ADCC as described in the methods section. Error bars show SEM of samples in duplicates. Data are averages of 3 independent experiments. *p < 0.05 as determined by Student’s t-test. (B) Visualization of ch128.1-treated cells via ImageStream analysis focusing on CFSE and PE staining. In the dot plot, quadrants are drawn around the macrophage population (CFSE−/PE+), KMS-11 tumor cell population (CFSE+/PE−), and the double positive population (CFSE+/PE+). Representative bright field and fluorescent images of each population are shown. Images are obtained at original magnification X40.
ch128.1 prolongs the survival of SCID-Beige mice bearing different stages of MM
The in vivo antitumor activity of ch128.1 in an early disease setting has been examined in disseminated MM models using ARH-77 and KMS-11 cell lines as shown previously (24) and in Fig. 1B and C. We now show, for the first time, that a single dose of 50 µg ch128.1 (~2.5 mg/kg), administered 2 days after tumor inoculation (early-stage), is sufficient for protection against MM in SCID-Beige mice, with over 85% of tumor free mice at the termination of the experiment (120 days), compared to mice treated with buffer alone (p < 0.0005; Fig. 6A). This is similar to studies using a 100 µg dose in the KMS-11 disseminated disease model, as shown previously (24) and in Fig. 1C. Importantly, statistically significant survival benefit was still observed when treatment was delayed to 9 days after tumor cell challenge (p ≤ 0.001 compared to buffer treated mice; Fig. 6A). A higher dose of ch128.1 (400 µg) was also tested in this late-stage disease model, but increased protection, compared to the 100 µg dose, was not observed (Fig. 6B; p < 0.05 compared to buffer treated mice). The antitumor effects observed in these studies require targeting of the TfR1 since, as expected, an isotype control antibody did not confer protection in either the early-stage or late-stage disease setting (Fig. 6C). In both cases the isotype control, whether given at day 2 or day 9, did not show an improvement of survival. In this study, ch128.1 showed significant tumor protection, when compared to IgG3 isotype control or buffer alone, when animals were treated 2 days after tumor challenge. Mice treated with ch128.1 in the late-stage disease setting (9 days post tumor challenge) also showed significant protection when compared to the IgG3 isotype control.
Figure 6. In vivo efficacy of ch128.1 in early and late-stage models of MM.

Kaplan-Meier plots indicate survival of SCID-Beige mice challenged i.v. with 5 × 106 KMS-11. (A) 50 µg of ch128.1 was adminsitered i.v. 2 or 9 days after tumor challenge, (B) 100 or 400 µg of ch128.1 was injected i.v. 9 days after tumor challenge, or (C) 100 µg of either ch128.1 or the isotype control IgG3 were administered 2 or 9 days after tumor challenge. Buffer control was administered 2 days after tumor challenge. *p < 0.05, **p < 0.001. Data in panel B are representative of 2 independent experiments.
Discussion
We have previously shown that ch128.1 exhibits cytotoxic activity against certain hematopoietic malignant cell lines in vitro, including ARH-77 cells, through TfR1 degradation and iron deprivation (22, 24). However, KMS-11 cells were not sensitive to this direct in vitro activity of ch128.1 (24). We have also shown that ch128.1 induces ADCC and CDC in vitro against Ramos human non-Hodgkin lymphoma B cells (27) and that it confers similar antitumor protection in two disseminated MM xenograft models, ARH-77 and KMS-11, in an early-stage disease setting (24). This serves as a cautionary note that in vitro models may not always predict in vivo outcomes, at least in the case of antibodies targeting TfR1. In this report, we examined the mechanism of the in vivo antitumor activity of ch128.1. For these studies, mutations were generated at sites important for FcγR and complement C1q binding (L234, L235, and P329), which resulted in disrupted interaction with effector cells and the inability to fix complement, leading to impaired ADCC and CDC (27). As expected, this triple mutant antibody retains binding to the antigen (27), as well as the in vitro anti-proliferative activity against sensitive malignant B cells (ARH-77) as we now report. It was used in this study to evaluate the relevance of the ch128.1 Fc region in its in vivo antitumor activity.
The ch128.1 mutant antibody failed to provide a survival benefit in vivo, indicating the relevance of the Fc fragment in ch128.1-induced antitumor activity. We explored the possibility that this loss of activity was due to a loss of bioavailability in mouse serum. Importantly, ch128.1 and its mutant showed similar binding affinity to mouse FcRn. Thus, the mutations at residues L234, L235, and P329 did not affect the binding of ch128.1 to this receptor, consistent with a human IgG1 mutated at the same sites, which similarly exhibited abolished FcγR and C1q interactions but retained binding to FcRn (56). The residues mutated are located at sites distinct from those residues that are known to play a crucial role in binding of human IgG antibodies to mouse or human FcRn (especially S254, H310, H435, I253, and Y436) (57, 58). Since the FcRn protects IgG from degradation, and in so doing is crucial to maintain the antibody bioavailability in blood (20, 43), and binding to this receptor is not affected in the ch128.1 triple mutant, its lack of antitumor activity is not expected to be due to a decrease in bioavailability. In fact, we showed that the mutations introduced in ch128.1 did not result in a decrease in its serum bioavailability.
Importantly the ch128.1 mutant retains the direct in vitro cytotoxic effect against sensitive (ARH-77) cells. However, both sensitive (ARH-77) and resistant (KMS-11) cells respond similarly in vivo to ch128.1 treatment; thus, TfR1 degradation followed by iron deprivation does not appear to be the mechanism of action in these mouse models. Macrophages are important for this activity, while the complement pathway did not appear to contribute to ch128.1-mediated antitumor activity in our model, even though it binds C1q and induces CDC in vitro (27). There are two possible and non-mutually exclusive ways in which the Fc fragment and interaction with macrophages could potentially elicit antitumor activity. One is through Fc effector functions including ADCC and/or ADCP. Even though TfR1 is constitutively internalized, significant ADCC has been observed in vitro by us and others (27, 59, 60) and tumor growth inhibition induced by antibodies targeting TfR1 in oral squamous cell carcinoma and adult T-cell leukemia/lymphoma murine xenograft models has been reported (59, 60). Here we also show that murine BMDM mediate ADCC and ADCP in the presence of ch128.1, but not its mutant. The other possibility is that ch128.1 simultaneously binds TfR1 on cancer cells and FcγRs on the surface of effector cells such as macrophages, leading to extensive cross-linking of the TfR1, resulting in inhibition of receptor internalization and iron uptake. This has been observed with rat IgM anti-TfR1 antibodies RI7 208 and REM 17.2, which as multivalent antibodies induce crosslinking of TfR1, interference with TfR1internalization, and growth inhibition of AKR1 thymic lymphoma cells and SL-2 leukemic cells in vitro (61, 62). RI7 208 also exhibited antitumor activity against SL-2 cells in an AKR/J syngeneic model (62).
SCID-Beige mice are deficient in B and T cells (44), have impaired NK cell and neutrophil activity (45–47), but retain functional macrophages and complement activity (45, 48–50). Even though the depletion of macrophages is incomplete in our studies, the antitumor activity induced by ch128.1 was significantly reduced. We have also shown previously that ch128.1 prolonged the survival of NOD-SCID mice bearing xenograft tumors of 2F7 AIDS-related NHL cells (26), tumors that are infiltrated with F4/80+ cells (63). NOD-SCID mice are deficient in B and T cells and have impaired NK cell and complement activity (64), supporting the importance of macrophages for ch128.1-induced antitumor activity. Macrophages are a particularly relevant component of the stroma in MM, providing support to the malignant plasma cells and vice versa (65). Such cross-talk protects the malignant cells from drug-induced apoptosis (66). However, it is expected that the presence of an antibody targeting the malignant cells in the MM microenvironment would result in macrophage-mediated MM cell killing as our studies suggest. It is also possible that the in vivo antitumor activity of ch128.1 may be magnified in the presence of the complete repertoire of functional immune effector cells such as NK cells and neutrophils.
Consistent with our results, phagocytosis by human monocyte-derived macrophages of human MM cell lines and MM cells isolated from patients has been observed in vitro in the presence of the anti-CD38 antibody daratumumab (67). Additionally, macrophages contribute to the in vivo anti-tumor activity of daratumumab in SCID-Beige mice bearing Daudi Burkitt lymphoma (67). Furthermore, the recruitment of macrophages and mediation of ADCP by an antibody specific for the B-cell maturation antigen in subcutaneous and disseminated MM models in SCID-Beige mice has also been demonstrated (68). Together, these studies support the notion that macrophages play an important role in mediating the antitumor effects of various therapeutic antibodies in mice bearing human malignant B cells.
A similar survival benefit in the KMS-11 model is achieved even with less than half of the previously reported dose of ch128.1 (125 µg) (24), indicating that this antibody is particularly effective against disseminated B-cell malignancies. We also examined the efficacy of ch128.1 in a late-stage disease setting. As expected, less protection was detected compared to treatment at an earlier time point. However, significant prolongation of survival was still observed using a single dose of ch128.1. Interestingly, both 100 and 400 µg of ch128.1 comparably prolonged survival in this setting, suggesting that saturating conditions were reached.
Normal cells expressing high levels of TfR1 may be targeted by anti-TfR1 antibodies including ch128.1. Cells that are of particular concern include activated immune cells that are rapidly dividing, as well as hematopoietic progenitor cells, including erythroblasts that require iron for heme synthesis (10). The xenograft models used in this study do not adequately address the toxicity of ch128.1 to these cell populations, since this antibody does not cross-react with the murine TfR1 (69). However, this is a common problem for many antibodies developed for clinical use, and despite this fact, these models have still shown predictive value for antibodies that are currently used in the clinic (70–72). Toxicity of anti-human TfR1 antibodies to normal cells has previously been evaluated. Toxicity to normal erythroblasts and PHA-activated T cells has been reported in vitro upon treatment with the fully human anti-TfR1 IgG1 antibody JST-TFR09, while no significant cytotoxicity was observed with this antibody in resting T cells (60). Similar effects against activated cells were observed with the mouse/human chimeric IgG1 antibody D2C (73). Two studies in cynomolgus monkeys (Macaca fascicularis) have been reported using anti-TfR1 antibodies and are consistent with these in vitro findings. Monkeys given the murine IgG2b A24 antibody did not show significant toxicity; however, they showed decreased hemoglobin levels, decreased serum iron levels, and increased apoptosis in lymph node germinal centers that contain highly proliferative B and T cells (74). In the second report, monkeys given multiple doses of 30 mg/kg of the JST-TFR-09 (also known as PPMX-T003) human IgG1 antibody demonstrated moderate anemia, but no other toxicities were observed (60, 75). Even though committed, hematopoietic progenitor cells express high levels of the TfR1 and are vulnerable to the effects of anti-TfR1 antibodies; hematopoietic pluripotent stem cells lack TfR1 expression (10, 76–78). If fact, we have shown that this cell population is not affected by an immunotoxin consisting of an antibody-avidin fusion protein composed of ch128.1 genetically fused to chicken avidin conjugated to the plant toxin saporin (79). One Phase I clinical trial using an anti-TfR1 antibody has been reported (80). The 42/6 murine IgA antibody was generally well-tolerated in this study; however an allergic-type response that was associated with the human anti-mouse antibody (HAMA) response was observed in one patient that received two doses of the IgA (80). Further studies using antibodies with human constant regions are needed to fully define their toxic effects and therapeutic potential. Due to the expression of the TfR1 on a variety of normal cells, toxicity must be carefully monitored.
In summary, we show that the Fc fragment of ch128.1 is crucial for its antitumor activity in the disseminated MM xenograft models examined. We have shown that macrophages can mediate both ADCC and ADCP in vitro in the presence of ch128.1. Additionally, our studies are consistent with the conclusion that macrophages play a major role in ch128.1-induced in vivo antitumor activity in our mouse models. Importantly, a single low dose of ch128.1 is protective in models of different stages of MM disease. Our results suggest that ch128.1 is a promising therapeutic against human B-cell malignancies such as MM.
Acknowledgments
We thank Dr. Fu Li (Seattle Genetics, Inc.) for his technical advice on Clodrosome® depletion of macrophages in SCID mice and Dr. Donald M. Lamkin (University of California, Los Angeles) for his technical advice on BMDM isolation and differentiation. We also thank Salem Haile [UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility] for technical assistance running the ImageStream®X Mark II and Dr. Tim C. Chang (Amnis, part of MilliporeSigma) for assistance with the analysis of these data.
Abbreviations used in this article
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cell-mediated phagocytosis
- AP
alkaline phosphatase
- BMDM
bone marrow-derived macrophages
- CDC
complement-dependent cytotoxicity
- CVF
cobra venom factor
- FcRn
neonatal Fc receptor (also known as the Brambell receptor)
- IHC
immunohistochemistry
- MM
multiple myeloma
- SPR
surface plasmon resonance
- Tf
transferrin
- TfR1
transferrin receptor 1
Footnotes
Conflict of Interest Statement: Manuel L. Penichet is a shareholder of Klyss Biotech, Inc. The Regents of the University of California are in discussions with Klyss to license Dr. Penichet’s technology to this firm. The other authors declare no competing financial interests.
This work was supported in part by NIH grants R01CA107023, R01CA196266, P30AI028697, and P30CA016042. Flow cytometry and ImageStream Analysis was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30CA016042 and P30AI028697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor's Office, and the UCLA Vice Chancellor's Office of Research. Histology was performed by the UCLA Translational Pathology Core Laboratory.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. 2015;12:42–54. doi: 10.1038/nrclinonc.2014.200. [DOI] [PubMed] [Google Scholar]
- 3.Lonial S, Durie B, Palumbo A, San-Miguel J. Monoclonal antibodies in the treatment of multiple myeloma: current status and future perspectives. Leukemia. 2016;30:526–535. doi: 10.1038/leu.2015.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orlowski RZ, Lonial S. Integration of Novel Agents into the Care of Patients with Multiple Myeloma. Clin Cancer Res. 2016;22:5443–5452. doi: 10.1158/1078-0432.CCR-16-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majithia N, Rajkumar SV, Lacy MQ, Buadi FK, Dispenzieri A, Gertz MA, Hayman SR, Dingli D, Kapoor P, Hwa L, Lust JA, Russell SJ, Go RS, Kyle RA, Kumar SK. Early relapse following initial therapy for multiple myeloma predicts poor outcomes in the era of novel agents. Leukemia. 2016;30:2208–2213. doi: 10.1038/leu.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laubach J, Garderet L, Mahindra A, Gahrton G, Caers J, Sezer O, Voorhees P, Leleu X, Johnsen HE, Streetly M, Jurczyszyn A, Ludwig H, Mellqvist UH, Chng WJ, Pilarski L, Einsele H, Hou J, Turesson I, Zamagni E, Chim CS, Mazumder A, Westin J, Lu J, Reiman T, Kristinsson S, Joshua D, Roussel M, O'Gorman P, Terpos E, McCarthy P, Dimopoulos M, Moreau P, Orlowski RZ, Miguel JS, Anderson KC, Palumbo A, Kumar S, Rajkumar V, Durie B, Richardson PG. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia. 2016;30:1005–1017. doi: 10.1038/leu.2015.356. [DOI] [PubMed] [Google Scholar]
- 7.de Mel S, Lim SH, Tung ML, Chng WJ. Implications of heterogeneity in multiple myeloma. Biomed Res Int. 2014;2014:232546. doi: 10.1155/2014/232546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823:1468–1483. doi: 10.1016/j.bbamcr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820:291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Yeh CJ, Taylor CG, Faulk WP. Transferrin binding by peripheral blood mononuclear cells in human lymphomas, myelomas and leukemias. Vox Sang. 1984;46:217–223. doi: 10.1111/j.1423-0410.1984.tb00078.x. [DOI] [PubMed] [Google Scholar]
- 12.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, P F. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 13.Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK, Vidal-Puig A, Teleman AA. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature. 2015;525:124–128. doi: 10.1038/nature14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong SM, Hwang S, Seong RH. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem Biophys Res Commun. 2016;471:373–379. doi: 10.1016/j.bbrc.2016.02.023. [DOI] [PubMed] [Google Scholar]
- 15.Kenneth NS, Mudie S, Naron S, Rocha S. TfR1 interacts with the IKK complex and is involved in IKK-NF-kappaB signalling. Biochem J. 2013;449:275–284. doi: 10.1042/BJ20120625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hideshima T, Chauhan D, Kiziltepe T, Ikeda H, Okawa Y, Podar K, Raje N, Protopopov A, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Biologic sequelae of I{kappa}B kinase (IKK) inhibition in multiple myeloma: therapeutic implications. Blood. 2009;113:5228–5236. doi: 10.1182/blood-2008-06-161505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanderWall K, Daniels-Wells TR, Penichet M, Lichtenstein A. Iron in multiple myeloma. Crit Rev Oncog. 2013;18:449–461. doi: 10.1615/critrevoncog.2013007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniels-Wells TR, Penichet ML. Transferrin receptor 1: a target for antibody-mediated cancer therapy. Immunotherapy. 2016;8:991–994. doi: 10.2217/imt-2016-0050. [DOI] [PubMed] [Google Scholar]
- 19.Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front Immunol. 2018;8:1–19. doi: 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez JA, Helguera G, Daniels TR, Neacato II, Lopez-Valdes HE, Charles AC, Penichet ML. Binding specificity and internalization properties of an antibody-avidin fusion protein targeting the human transferrin receptor. J Control Release. 2007;124:35–42. doi: 10.1016/j.jconrel.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 22.Ng PP, Helguera G, Daniels TR, Lomas SZ, Rodriguez JA, Schiller G, Bonavida B, Morrison SL, Penichet ML. Molecular events contributing to cell death in malignant human hematopoietic cells elicited by an IgG3-avidin fusion protein targeting the transferrin receptor. Blood. 2006;108:2745–2754. doi: 10.1182/blood-2006-04-020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez JA, Luria-Perez R, Lopez-Valdes HE, Casero D, Daniels TR, Patel S, Avila D, Leuchter R, So S, Ortiz-Sanchez E, Bonavida B, Martinez-Maza O, Charles AC, Pellegrini M, Helguera G, Penichet ML. Lethal iron deprivation induced by non-neutralizing antibodies targeting transferrin receptor 1 in malignant B cells. Leuk Lymphoma. 2011;52:2169–2178. doi: 10.3109/10428194.2011.596964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniels TR, Ortiz-Sanchez E, Luria-Perez R, Quintero R, Helguera G, Bonavida B, Martinez-Maza O, Penichet ML. An antibody-based multifaceted approach targeting the human transferrin receptor for the treatment of B-cell malignancies. J Immunother. 2011;34:500–508. doi: 10.1097/CJI.0b013e318222ffc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daniels TR, Ng PP, Delgado T, Lynch MR, Schiller G, Helguera G, Penichet ML. Conjugation of an anti transferrin receptor IgG3-avidin fusion protein with biotinylated saporin results in significant enhancement of its cytotoxicity against malignant hematopoietic cells. Mol Cancer Ther. 2007;6:2995–3008. doi: 10.1158/1535-7163.MCT-07-0330. [DOI] [PubMed] [Google Scholar]
- 26.Daniels-Wells TR, Widney DP, Leoh LS, Martinez-Maza O, Penichet ML. Efficacy of an Anti-transferrin Receptor 1 Antibody Against AIDS-related Non-Hodgkin Lymphoma: A Brief Communication. J Immunother. 2015;38:307–310. doi: 10.1097/CJI.0000000000000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leoh LS, Daniels-Wells TR, Martinez-Maza O, Penichet ML. Insights into the effector functions of human IgG3 in the context of an antibody targeting transferrin receptor 1. Mol Immunol. 2015;67:407–415. doi: 10.1016/j.molimm.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leoh LS, Morizono K, Kershaw KM, Chen IS, Penichet ML, Daniels-Wells TR. Gene delivery in malignant B cells using the combination of lentiviruses conjugated to anti-transferrin receptor antibodies and an immunoglobulin promoter. J Gene Med. 2014;16:11–27. doi: 10.1002/jgm.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monnet C, Jorieux S, Urbain R, Fournier N, Bouayadi K, De Romeuf C, Behrens CK, Fontayne A, Mondon P. Selection of IgG Variants with Increased FcRn Binding Using Random and Directed Mutagenesis: Impact on Effector Functions. Front Immunol. 2015;6:39. doi: 10.3389/fimmu.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 31.Lehenkari PP, Kellinsalmi M, Napankangas JP, Ylitalo KV, Monkkonen J, Rogers MJ, Azhayev A, Vaananen HK, Hassinen IE. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol. 2002;61:1255–1262. doi: 10.1124/mol.61.5.1255. [DOI] [PubMed] [Google Scholar]
- 32.van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193:93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 33.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95:272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen DH, Zhou T, Shu J, Mao J. Quantifying chromogen intensity in immunohistochemistry via reciprocal intensity. Cancer InCytes. 2013;2:e. [Google Scholar]
- 35.Vogel CW, Finnegan PW, Fritzinger DC. Humanized cobra venom factor: structure, activity, and therapeutic efficacy in preclinical disease models. Mol Immunol. 2014;61:191–203. doi: 10.1016/j.molimm.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 36.Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, Haas KM, Tedder TF. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. 2008;112:1205–1213. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manzanero S. Generation of mouse bone marrow-derived macrophages. Methods Mol Biol. 2012;844:177–181. doi: 10.1007/978-1-61779-527-5_12. [DOI] [PubMed] [Google Scholar]
- 38.Bracher M, Gould HJ, Sutton BJ, Dombrowicz D, Karagiannis SN. Three-colour flow cytometric method to measure antibody-dependent tumour cell killing by cytotoxicity and phagocytosis. J Immunol Methods. 2007;323:160–171. doi: 10.1016/j.jim.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Alsina M, Boyce B, Devlin RD, Anderson JL, Craig F, Mundy GR, Roodman GD. Development of an in vivo model of human multiple myeloma bone disease. Blood. 1996;87:1495–1501. [PubMed] [Google Scholar]
- 40.Gado K, Silva S, Pálóczi K, Domján G, Falus A. Mouse plasmacytoma: an experimental model of human multiple myeloma. Haematologica. 2001;86:227–236. [PubMed] [Google Scholar]
- 41.Paton-Hough J, Chantry AD, Lawson MA. A review of current murine models of multiple myeloma used to assess the efficacy of therapeutic agents on tumour growth and bone disease. Bone. 2015;77:57–68. doi: 10.1016/j.bone.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Stein R, Smith MR, Chen S, Zalath M, Goldenberg DM. Combining milatuzumab with bortezomib, doxorubicin, or dexamethasone improves responses in multiple myeloma cell lines. Clin Can Res. 2009;15:2808–2817. doi: 10.1158/1078-0432.CCR-08-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J Immunol. 2015;194:4595–4603. doi: 10.4049/jimmunol.1403014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 45.Roder J, Duwe A. The beige mutation in the mouse selectively impairs natural killer cell function. Nature. 1979;278:451–453. doi: 10.1038/278451a0. [DOI] [PubMed] [Google Scholar]
- 46.Takechi Y, Hara I, Naftzger C, Xu Y, Houghton AN. A melanosomal membrane protein is a cell surface target for melanoma therapy. Clin Cancer Res. 1996;2:1837–1842. [PubMed] [Google Scholar]
- 47.Jones-Carson J, Vazquez-Torres A, Balish E. Defective killing of Candida albicans hyphae by neutrophils from beige mice. J Infect Dis. 1995;171:1664–1667. doi: 10.1093/infdis/171.6.1664. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z, Taylor PR, Locklin RM, Gordon S, Cui Z, Triffitt JT. Innate immune response to human bone marrow fibroblastic cell implantation in CB17 scid/beige mice. J Cell Biochem. 2006;98:966–980. doi: 10.1002/jcb.20730. [DOI] [PubMed] [Google Scholar]
- 49.Elvington M, Huang Y, Morgan BP, Qiao F, van Rooijen N, Atkinson C, Tomlinson S. A targeted complement-dependent strategy to improve the outcome of mAb therapy, and characterization in a murine model of metastatic cancer. Blood. 2012;119:6043–6051. doi: 10.1182/blood-2011-10-383232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roder JC, Lohmann-Matthes ML, Domzig W, Wigzell H. The beige mutation in the mouse. II. Selectivity of the natural killer (NK) cell defect. J Immunol. 1979;123:2174–2181. [PubMed] [Google Scholar]
- 51.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–2743. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 52.Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 53.Beenhouwer DO, Yoo EM, Lai CW, Rocha MA, Morrison SL. Human immunoglobulin G2 (IgG2) and IgG4, but not IgG1 or IgG3, protect mice against Cryptococcus neoformans infection. Infect Immun. 2007;75:1424–1435. doi: 10.1128/IAI.01161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, Parren PW. Crosstalk between human IgG isotypes and murine effector cells. J Immunol. 2012;189:3430–3438. doi: 10.4049/jimmunol.1200356. [DOI] [PubMed] [Google Scholar]
- 55.Church AK, VanDerMeid KR, Baig NA, Baran AM, Witzig TE, Nowakowski GS, Zent CS. Anti-CD20 monoclonal antibody-dependent phagocytosis of chronic lymphocytic leukaemia cells by autologous macrophages. Clin Exp Immunol. 2016;183:90–101. doi: 10.1111/cei.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umana P, Mossner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016 doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 57.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 58.Kim JK, Firan M, Radu CG, Kim CH, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29:2819–2825. doi: 10.1002/(SICI)1521-4141(199909)29:09<2819::AID-IMMU2819>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 59.Nagai K, Nakahata S, Shimosaki S, Tamura T, Kondo Y, Baba T, Taki T, Taniwaki M, Kurosawa G, Sudo Y, Okada S, Sakoda S, Morishita K. Development of a complete human anti-human transferrin receptor C antibody as a novel marker of oral dysplasia and oral cancer. Cancer Med. 2014;3:1085–1099. doi: 10.1002/cam4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimosaki S, Nakahata S, Ichikawa T, Kitanaka A, Kameda T, Hidaka T, Kubuki Y, Kurosawa G, Zhang L, Sudo Y, Shimoda K, Morishita K. Development of a complete human IgG monoclonal antibody to transferrin receptor 1 targeted for adult T-cell leukemia/lymphoma. Biochem Biophys Res Commun. 2017;485:144–151. doi: 10.1016/j.bbrc.2017.02.039. [DOI] [PubMed] [Google Scholar]
- 61.Lesley JF, Schulte RJ. Inhibition of cell growth by monoclonal anti-transferrin receptor antibodies. Mol Cell Biol. 1985;5:1814–1821. doi: 10.1128/mcb.5.8.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sauvage CA, Mendelsohn JC, Lesley JF, Trowbridge IS. Effects of monoclonal antibodies that block transferrin receptor function on the in vivo growth of a syngeneic murine leukemia. Cancer Res. 1987;47:747–753. [PubMed] [Google Scholar]
- 63.Widney DP, Olafsen T, Wu AM, Kitchen CM, Said JW, Smith JB, Pena G, Magpantay LI, Penichet ML, Martinez-Maza O. Levels of murine, but not human, CXCL13 are greatly elevated in NOD-SCID mice bearing the AIDS-associated Burkitt lymphoma cell line, 2F7. PLoS One. 2013;8:e72414. doi: 10.1371/journal.pone.0072414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–191. [PubMed] [Google Scholar]
- 65.Ribatti D, Moschetta M, Vacca A. Macrophages in multiple myeloma. Immunol Lett. 2014;161:241–244. doi: 10.1016/j.imlet.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 66.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114:3625–3628. doi: 10.1182/blood-2009-05-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, Groen RW, Breij E, Martens AC, Bleeker WK, Parren PW. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7:311–321. doi: 10.1080/19420862.2015.1007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, Craigen J, Yates J, Gliddon L, Fieles W, Hoang B, Tunstead J, Christie AL, Kung AL, Richardson P, Munshi NC, Anderson KC. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–3138. doi: 10.1182/blood-2013-10-535088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helguera G, Jemielity S, Abraham J, Cordo SM, Martinez MG, Rodriguez JA, Bregni C, Wang JJ, Farzan M, Penichet ML, Candurra NA, Choe H. An antibody recognizing the apical domain of human transferrin receptor 1 efficiently inhibits the entry of all new world hemorrhagic fever arenaviruses. J Virol. 2012;86:4024–4028. doi: 10.1128/JVI.06397-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pegram M, Ngo D. Application and potential limitations of animal models utilized in the development of trastuzumab (Herceptin): a case study. Adv Drug Deliv Rev. 2006;58:723–734. doi: 10.1016/j.addr.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin) Semin Oncol. 1999;26:60–70. [PubMed] [Google Scholar]
- 72.Wang M, Han XH, Zhang L, Yang J, Qian JF, Shi YK, Kwak LW, Romaguera J, Yi Q. Bortezomib is synergistic with rituximab and cyclophosphamide in inducing apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leukemia. 2008;22:179–185. doi: 10.1038/sj.leu.2404959. [DOI] [PubMed] [Google Scholar]
- 73.Lei P, He Y, Ye Q, Zhu HF, Yuan XM, Liu J, Xing W, Wu S, Dai W, Shen X, Wang GB, Shen GX. Antigen-binding characteristics of AbCD71 and its inhibitory effect on PHA-induced lymphoproliferation. Acta Pharmacol Sin. 2007;28:1659–1664. doi: 10.1111/j.1745-7254.2007.00623.x. [DOI] [PubMed] [Google Scholar]
- 74.Moura IC, Lepelletier Y, Arnulf B, Bazarbachi A, Monteiro RC, Hermine O. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Retrovirology. 2011;8:A60. doi: 10.1182/blood-2003-07-2440. [DOI] [PubMed] [Google Scholar]
- 75.Zhang L, Nomura F, Aikawa Y, Kurosawa Y, Morishita K, Sudo Y. PPMX-T003, a fully human anti-TfR1 antibody with potent efficacy against hematologic malignancies. Proceedings of the American Association for Cancer Research 108th Annual Meeting. 2017;58 Abstract #5586. [Google Scholar]
- 76.Gross S, Helm K, Gruntmeir JJ, Stillman WS, Pyatt DW, Irons RD. Characterization and phenotypic analysis of differentiating CD34+ human bone marrow cells in liquid culture. Eur J Haematol. 1997;59:318–326. doi: 10.1111/j.1600-0609.1997.tb01693.x. [DOI] [PubMed] [Google Scholar]
- 77.Knaan-Shanzer S, van der Velde-van Dijke I, van de Watering MJ, de Leeuw PJ, Valerio D, van Bekkum DW, de Vries AA. Phenotypic and functional reversal within the early human hematopoietic compartment. Stem Cells. 2008;26:3210–3217. doi: 10.1634/stemcells.2007-0117. [DOI] [PubMed] [Google Scholar]
- 78.Lansdorp PM, Dragowska W. Long-term erythropoiesis from constant numbers of CD34+ cells in serum-free cultures initiated with highly purified progenitor cells from human bone marrow. J Exp Med. 1992;175:1501–1509. doi: 10.1084/jem.175.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daniels-Wells TR, Helguera G, Rodriguez JA, Leoh LS, Erb MA, Diamante G, Casero D, Pellegrini M, Martinez-Maza O, Penichet ML. Insights into the mechanism of cell death induced by saporin delivered into cancer cells by an antibody fusion protein targeting the transferrin receptor 1. Toxicol In Vitro. 2013;27:220–231. doi: 10.1016/j.tiv.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brooks D, Taylor C, Dos Santos B, Linden H, Houghton A, Hecht TT, Kornfeld S, Taetle R. Phase Ia trial of murine immunoglobulin A antitransferrin receptor antibody 42/6. Clin Cancer Res. 1995;1:1259–1265. [PubMed] [Google Scholar]