

Abstract
Objective
Amyloid β (Aβ) depositions in plaques and cerebral amyloid angiopathy (CAA) represent common features of Alzheimer's disease (AD). Sequential deposition of post‐translationally modified Aβ in plaques characterizes distinct biochemical stages of Aβ maturation. However, the molecular composition of vascular Aβ deposits in CAA and its relation to plaques remain enigmatic.
Methods
Vascular and parenchymal deposits were immunohistochemically analyzed for pyroglutaminated and phosphorylated Aβ in the medial temporal and occipital lobe of 24 controls, 27 pathologically‐defined preclinical AD, and 20 symptomatic AD cases.
Results
Sequential deposition of Aβ in CAA resembled Aβ maturation in plaques and enabled the distinction of three biochemical stages of CAA. B‐CAA stage 1 was characterized by deposition of Aβ in the absence of pyroglutaminated Aβ N3pE and phosphorylated Aβ pS8. B‐CAA stage 2 showed additional Aβ N3pE and B‐CAA stage 3 additional Aβ pS8. Based on the Aβ maturation staging in CAA and plaques, three case groups for Aβ pathology could be distinguished: group 1 with advanced Aβ maturation in CAA; group 2 with equal Aβ maturation in CAA and plaques; group 3 with advanced Aβ maturation in plaques. All symptomatic AD cases presented with end‐stage plaque maturation, whereas CAA could exhibit immature Aβ deposits. Notably, Aβ pathology group 1 was associated with arterial hypertension, and group 2 with the development of dementia.
Interpretation
Balance of Aβ maturation in CAA and plaques defines distinct pathological subgroups of β‐amyloidosis. The association of CAA‐related Aβ maturation with cognitive decline, the individual contribution of CAA and plaque pathology to the development of dementia within the defined Aβ pathology subgroups, and the subgroup‐related association with arterial hypertension should be considered for differential diagnosis and therapeutic intervention.
Introduction
Alzheimer's disease (AD) represents the most common form of dementia, characterized by the accumulation of amyloid β (Aβ) in extracellular plaques and hyperphosphorylated tau in intracellular neurofibrillary tangles (NFTs).1 The majority of AD patients additionally develop amyloid lesions in the cerebrovasculature.2 Cerebral amyloid angiopathy (CAA) describes an AD‐associated vessel disorder, defined by the deposition of Aβ in leptomeningeal and/or parenchymal arteries, veins, and capillaries.3 Cerebrovascular Aβ structurally resembles Aβ fibrils in plaques of AD patients.4 The overall abundance of CAA and plaques in AD2, 5 and the spatio‐temporal relation between CAA and plaques in the pathogenesis of AD6, 7, 8 suggest a pathogenic link between both pathologies.
Recently, we revealed a hierarchical sequence for the deposition of different Aβ species in the pathogenesis of AD‐related amyloid plaques.9 Three biochemical stages of Aβ aggregate maturation in plaques (B‐Aβ plaque stages) were identified based on the immunohistochemical detection of Aβ and its modified species. B‐Aβ plaque stage 1 was defined by the parenchymal deposition of Aβ lacking N‐terminal truncated, pyroglutaminated Aβ (Aβ N3pE) and phosphorylated Aβ (Aβ pS8), whereas Aβ including Aβ N3pE was prevalent in B‐Aβ plaque stage 2. B‐Aβ plaque stage 3, finally, exhibited Aβ pS8 within the parenchymal Aβ aggregates in addition to other forms of Aβ including Aβ N3pE. 9
Post‐translational modification of Aβ by N‐terminal truncation and pyroglutamination as well as phosphorylation affect the aggregation, stability, and toxicity of Aβ.10, 11 In particular pyroglutamination at glutamate 3 and phosphorylation at serine 8 promote aggregation, thereby enhancing metabolic stability and toxicity of Aβ.12, 13, 14, 15, 16
Despite the abundance of Aβ N3pE and Aβ pS8 in plaques and CAA of AD patients,9, 15, 17, 18, 19, 20 the molecular composition of CAA lesions and its relation to the progression of AD remain enigmatic. To compare amyloid deposition in CAA and plaques, we analyzed the medial temporal and occipital lobe of control, pathologically‐defined preclinical AD (p‐preAD), and symptomatic AD cases for the presence of Aβ and its modified forms Aβ N3pE and Aβ pS8 in CAA‐affected blood vessels.
Materials and Methods
Neuropathology
Brain tissue originated from the brain bank of the “Laboratory of Neuropathology” at the “University of Ulm” (Germany) that collected brain tissue in accordance with German legal regulations. Collection of brain tissue and experimental analyses of this project were approved by the ethics committees of the Universities of Ulm (Germany), Bonn (Germany), and KU Leuven (Belgium) where experiments have been performed (Votes Nos. Bonn: 161/01, 238/04; Ulm: 238/07, 54/08, 57/12; Leuven: S‐58102, S‐59295).
The brain collection consists of hospital‐based autopsy cases that were included into the brain bank at the time point of autopsy. The clinical information, therefore, included only information from files that could be reviewed retrospectively in the respective hospital. Longitudinal data and data from neuropsychological tests were not available.
Morphological analysis of cerebrovascular and parenchymal Aβ lesions was performed in autopsy brains of 24 control, 27 p‐preAD, and 20 sporadic AD cases (Table 1). The diagnosis of AD was performed neuropathologically with consideration of clinical information about the cognitive status. Control cases were defined by absence of amyloid plaques including cases with primary age‐related tauopathy21 and occasionally CAA. Non‐demented cases with AD pathology, comprising Aβ plaques and NFTs, were designated as p‐preAD cases, whereas symptomatic AD cases were characterized by substantial AD pathology and impairment of cognition.1, 22
Table 1.
List of control, p‐preAD, and AD cases used for analysis of CAA and plaques
| No | Agea | Genderb | Diagnosisc | Diabetes mellitusd | Hyper‐tensiond | Alcohol abused | CDR scoree | Aβ‐MTL phasef | Braak‐NFT stageg | CERAD scoreh | NIA‐AA AD degreei | APOE genotypej | B‐Aβ plaque stagek | B‐CAA stage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 60 | M | Control | ‐ | + | ‐ | 0 | 0 | 0 | 0 | not AD | “2/3” | 0 | 0 |
| 2 | 69 | F | Control | ‐ | + | ‐ | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 3 | 66 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 4 | 71 | F | Control | ‐ | + | ‐ | 0 | 0 | 1 | 0 | not AD | “2/3” | 0 | 0 |
| 5 | 58 | F | Control | + | ‐ | ‐ | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 6 | 46 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 7 | 45 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 0 | 0 | not AD | “3/4” | 0 | 0 |
| 8 | 35 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 9 | 59 | M | Control | ‐ | ‐ | + | ‐ | 0 | 1 | 0 | not AD | “3/4” | 0 | 0 |
| 10 | 57 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “3/4” | 0 | 0 |
| 11 | 74 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “3/4” | 0 | 0 |
| 12 | 66 | M | Control | ‐ | ‐ | + | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 13 | 61 | M | Control | ‐ | + | ‐ | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 14 | 66 | M | Control | + | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “2/3” | 0 | 0 |
| 15 | 60 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 16 | 69 | F | Control | ‐ | + | ‐ | 0 | 0 | 0 | 0 | not AD | “2/3” | 0 | 0 |
| 17 | 66 | F | Control | ‐ | + | + | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 18 | 62 | M | Control | ‐ | ‐ | + | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 19 | 72 | F | Control | + | + | ‐ | 0 | 0 | 1 | 0 | not AD | “3/3” | 0 | 0 |
| 20 | 62 | M | Control | ‐ | ‐ | ‐ | 0 | 0 | 0 | 0 | not AD | “3/3” | 0 | 0 |
| 21 | 72 | F | p‐preAD | + | + | ‐ | 0 | 1 | 2 | 0 | low | “3/3” | 2 | 0 |
| 22 | 71 | M | p‐preAD | + | ‐ | ‐ | 0 | 3 | 2 | 1 | low | “2/4” | 3 | 3 |
| 23 | 68 | F | p‐preAD | ‐ | ‐ | ‐ | 0 | 2 | 2 | 0 | low | “2/3” | 3 | 3 |
| 24 | 73 | F | p‐preAD | ‐ | + | ‐ | 0 | 1 | 2 | 0 | low | “3/3” | 2 | 0 |
| 25 | 77 | F | p‐preAD | ‐ | ‐ | ‐ | 0 | 3 | 2 | 0 | low | “3/4” | 3 | 0 |
| 26 | 78 | F | p‐preAD | ‐ | ‐ | ‐ | 0 | 3 | 2 | 0 | low | “3/4” | 3 | 3 |
| 27 | 71 | F | p‐preAD | ‐ | + | ‐ | 0 | 3 | 2 | 1 | low | “3/3” | 2 | 3 |
| 28 | 77 | F | p‐preAD, VD | + | + | ‐ | 3 | 2 | 3 | 1 | low | “3/3” | 2 | 3 |
| 29 | 73 | F | p‐preAD | ‐ | ‐ | ‐ | 0 | 1 | 2 | 0 | low | “2/3” | 2 | 0 |
| 30 | 74 | M | p‐preAD | + | + | ‐ | 0 | 2 | 2 | 0 | low | “3/3” | 2 | 0 |
| 31 | 64 | M | p‐preAD, brain infarction | ‐ | + | ‐ | ‐ | 2 | 1 | 0 | low | “3/3” | 2 | 0 |
| 32 | 74 | M | p‐preAD | + | + | ‐ | 0 | 4 | 3 | 1 | intermediate | “3/4” | 3 | 1 |
| 33 | 53 | M | p‐preAD | ‐ | ‐ | ‐ | 0 | 1 | 1 | 0 | low | “3/3” | 2 | 0 |
| 34 | 78 | F | p‐preAD, VD, CBD | ‐ | + | ‐ | 3 | 1 | 1 | 0 | low | “3/3” | 2 | 0 |
| 35 | 68 | F | p‐preAD | ‐ | + | ‐ | 0 | 2 | 1 | 0 | low | “3/3” | 2 | 0 |
| 36 | 67 | F | p‐preAD | + | ‐ | ‐ | 0 | 2 | 2 | 0 | low | “3/3” | 2 | 0 |
| 37 | 82 | F | p‐preAD, microinfarcts | ‐ | + | ‐ | ‐ | 2 | 1 | 1 | low | “3/3” | 2 | 3 |
| 38 | 87 | M | p‐preAD | ‐ | + | ‐ | 0 | 3 | 3 | 1 | intermediate | “3/4” | 3 | 3 |
| 39 | 84 | F | p‐preAD | ‐ | + | ‐ | 0 | 3 | 2 | 0 | low | “2/3” | 3 | 0 |
| 40 | 84 | F | p‐preAD, brain infarction | ‐ | ‐ | ‐ | 0 | 3 | 3 | 0 | intermediate | “3/3” | 3 | 3 |
| 41 | 88 | M | p‐preAD, AGD | ‐ | + | ‐ | 2 | 3 | 2 | 1 | low | “2/3” | 3 | 2 |
| 42 | 83 | F | p‐preAD | + | ‐ | ‐ | 0 | 3 | 3 | 1 | intermediate | “2/3” | 3 | 1 |
| 43 | 72 | M | p‐preAD | + | ‐ | ‐ | 0 | 2 | 3 | 0 | low | “3/3” | 2 | 0 |
| 44 | 64 | M | p‐preAD | ‐ | ‐ | ‐ | 0 | 2 | 1 | 0 | low | “3/3” | 2 | 0 |
| 45 | 63 | F | p‐preAD, brain infarction | ‐ | ‐ | ‐ | 0 | 4 | 3 | 1 | intermediate | “3/3” | 2 | 0 |
| 46 | 85 | F | p‐preAD | + | ‐ | ‐ | 0 | 4 | 3 | 1 | intermediate | “3/3” | 2 | 0 |
| 47 | 83 | M | p‐preAD, brain infarction | + | + | ‐ | 0 | 2 | 3 | 0 | low | “3/3” | 1 | 0 |
| 48 | 79 | F | AD | ‐ | ‐ | ‐ | ‐ | 3 | 4 | 2 | intermediate | “3/3” | 3 | 3 |
| 49 | 64 | F | AD | ‐ | + | ‐ | ‐ | 4 | 6 | 3 | high | “3/4” | 3 | 2 |
| 50 | 62 | F | AD | ‐ | ‐ | ‐ | 3 | 4 | 6 | 3 | high | “3/4” | 3 | 3 |
| 51 | 84 | M | AD | ‐ | ‐ | ‐ | 3 | 4 | 6 | 3 | high | “3/4” | 3 | 2 |
| 52 | 72 | F | AD | ‐ | ‐ | ‐ | 1 | 4 | 4 | 2 | intermediate | “3/3” | 3 | 2 |
| 53 | 83 | M | AD | ‐ | ‐ | ‐ | 1 | 4 | 4 | 2 | intermediate | “3/4” | 3 | 3 |
| 54 | 78 | M | AD | ‐ | ‐ | ‐ | 3 | 4 | 4 | 1 | intermediate | “3/4” | 3 | 2 |
| 55 | 75 | F | AD | ‐ | ‐ | ‐ | 0.5 | 4 | 3 | 1 | intermediate | “4/4” | 3 | 3 |
| 56 | 84 | M | AD, AGD, ALS, VD | ‐ | ‐ | + | 3 | 3 | 4 | 2 | intermediate | “3/3” | 3 | 3 |
| 57 | 68 | F | AD | ‐ | ‐ | ‐ | 1 | 4 | 6 | 3 | high | “3/3” | 3 | 1 |
| 58 | 82 | M | AD | + | + | ‐ | 2 | 3 | 3 | 2 | intermediate | “3/4” | 3 | 3 |
| 59 | 86 | F | AD, AGD | ‐ | ‐ | ‐ | 3 | 4 | 6 | 3 | high | “3/3” | 3 | 3 |
| 60 | 83 | M | AD | ‐ | ‐ | ‐ | 3 | 4 | 4 | 2 | intermediate | “3/3” | 3 | 3 |
| 61 | 78 | F | AD | ‐ | + | ‐ | 3 | 4 | 5 | 3 | high | “3/4” | 3 | 3 |
| 62 | 89 | F | AD | + | ‐ | ‐ | 2 | 4 | 4 | 3 | intermediate | “3/4” | 3 | 3 |
| 63 | 87 | F | AD | ‐ | ‐ | ‐ | 3 | 4 | 4 | 1 | intermediate | “3/3” | 3 | 2 |
| 64 | 78 | M | AD | + | ‐ | ‐ | 1 | 3 | 4 | 1 | intermediate | “3/3” | 3 | 3 |
| 65 | 89 | F | AD | + | ‐ | ‐ | 3 | 4 | 5 | 2 | high | “3/4” | 3 | 2 |
| 66 | 81 | F | AD | + | ‐ | ‐ | 3 | 4 | 5 | 1 | intermediate | “3/3” | 3 | 2 |
| 67 | 83 | M | AD | ‐ | + | ‐ | 3 | 4 | 5 | 3 | high | “4/4” | 3 | 3 |
| 68 | 68 | F | Control, pure CAA | ‐ | + | ‐ | 0 | 0 | 2 | 0 | not AD | “3/3” | 0 | 3 |
| 69 | 64 | F | Control, pure CAA, LBD | ‐ | + | ‐ | 0.5 | 0 | 3 | 0 | not AD | ‐ | 0 | 2 |
| 70 | 63 | F | Control, pure CAA | ‐ | ‐ | ‐ | ‐ | 0 | 2 | 0 | not AD | ‐ | 0 | 2 |
| 71 | 63 | F | Control, pure CAA | + | + | ‐ | 1 | 0 | 1 | 0 | not AD | ‐ | 0 | 2 |
age [years].
F = female, M = male.
AD = Alzheimer's disease, AGD = argyrophilic grain disease, ALS = amyotrophic lateral sclerosis, CAA = cerebral amyloid angiopathy, CBD = corticobasal degeneration, LBD = Lewy body disease, p‐preAD = pathologically‐defined preclinical Alzheimer's disease, VD = vascular dementia.
diabetes mellitus/hypertension/alcohol abuse: present (+), absent (‐).
clinical dementia rating (CDR) score.64
Aβ ‐ medial temporal lobe (MTL) phase.27
Consortium to Establish a Registry for Alzheimer's disease (CERAD) score.26
National Institute on Aging‐Alzheimer's Association (NIA‐AA) degree of Alzheimer's disease pathology.1, 65
APOE = apolipoprotein E.
B‐Aβ plaque stage.9
Following autopsy, brains were fixed in a 4% aqueous solution of formalin and tissue from both the medial temporal and occipital lobe was embedded in paraffin and cut into sections of 12 μm. Neuropathological diagnosis of AD was performed according to established guidelines of the “National Institute of Aging‐Alzheimer's Association” (NIA‐AA AD degree)1 and included (1) the assessment of NFT distribution (Braak‐NFT stage)23, 24 on the basis of Gallyas' silver impregnation24 and/or immunohistochemical staining of abnormal phosphorylated tau (AT8, 1:1000, Pierce Endogen; Rockford, USA),25 (2) the assignment of neuritic plaque density (Consortium to Establish a Registry for AD (CERAD) score)26 on the basis of Gallyas' silver impregnation24 and/or immunohistochemical staining of abnormal phosphorylated tau (AT8, 1:1000, Pierce Endogen; Rockford, USA),1 and (3) the evaluation of amyloid plaque distribution in the medial temporal lobe (Aβ‐MTL phase)27 on the basis of immunohistochemical staining for Aβ 17‐24 (4G8, 1:5000, Covance; Princeton, USA). Apolipoprotein E (APOE) genotypes were determined by restriction isotyping of unfixed brain tissue with HhaI28 (Table 1).
The severity of CAA‐related vessel wall destruction (CAA severity) was graded according to Vonsattel et al.29; and the stage of the anatomical expansion of CAA throughout the brain (CAA stage) was rated according to Thal et al.30 CAA with affection of capillaries was referred to as CAA type 1, and CAA without capillary Aβ deposits was designated as CAA type 2.31
Medical examination of control, p‐preAD, and AD cases was performed one to four weeks prior to death according to standardized protocols and included the assessment of (1) cognition (including short term and long term memory), (2) speech, writing, and reading, (3) self‐dependence and self‐care, (4) habit of eating, (5) bladder and bowel continence, and (6) orientation within the hospital setting.8 Clinical data were used to retrospectively assess clinical dementia rating scores (CDR scores)32 and for information about arterial hypertension, diabetes mellitus, and alcohol abuse. Cases with CDR scores ≥ 0.5 in conjunction with intermediate to high NIA‐AA AD degrees were considered as symptomatic AD cases.1 Due to missing clinical data, CDR scores could not be obtained for 5 of 24 control, 2 of 27 p‐preAD, and 2 of 20 AD cases (Table 1).
Immunohistochemistry
Following deparaffinization, hydration, and blocking, sections of the medial temporal and occipital lobe were incubated for 24 h at room temperature with anti‐Aβ 17‐24 (4G8, 1:5000, Covance; Princeton, USA; formic acid pretreatment), anti‐Aβ N3pE 20 (1:100, IBL; Hamburg, Germany; formic acid/microwave pretreatment), or anti‐Aβ pS8 12, 33 (SA5434/1E4E11, 1:5; formic acid/microwave pretreatment) antibodies. The antibodies used in this study to detect phosphorylated or pyroglutaminated Aβ were raised specifically against synthetic Aβ peptides carrying the phosphorylation or pyroglutamate modification and recognize phosphorylated serine or pyroglutamate residues selectively in the context of the Aβ amino acid sequence. Primary antibodies were detected with biotinylated secondary antibodies and visualized with the ABC method (Vector Laboratories; Burlingame, USA) and 3,3′‐diaminobenzidine (DAB; brown color) as chromogen. Sections were counterstained with hematoxylin. Positive and negative controls were performed.
Sensitivity and specificity of phosphorylation‐state specific polyclonal (SA5434) and monoclonal (1E4E11) antibodies were examined by preabsorption with synthetic Aβ peptides followed by western immunoblotting and/or immunohistochemistry using brain tissue from transgenic mouse models and human AD cases.12, 17, 33 Additional staining with antibodies against Aβ 1‐17 (6E10; 1:1,250, Covance; Princeton, USA; formic acid pretreatment) was performed on selected sections as described previously.9
Analysis of Aβ, AβN3pE, and AβpS8 deposition in CAA
Analysis of CAA was conducted in the medial temporal lobe of control, p‐preAD, and AD cases stained for Aβ, Aβ N3pE, or Aβ pS8 (Table 1). Cases that exhibited Aβ deposits in leptomeningeal and/or parenchymal vessels were considered positive for CAA independent of the severity and extent of CAA pathology. Vascular Aβ deposition was determined independently for Aβ, Aβ N3pE, and Aβ pS8 for each case. Cases that did not exhibit vascular Aβ, Aβ N3pE, and Aβ pS8 deposition in the medial temporal lobe were additionally analyzed for CAA in the occipital lobe.
Quantification of Aβ, AβN3pE, and AβpS8 plaque loads
Aβ, Aβ N3pE, and Aβ pS8 plaque loads were quantified in the temporal cortex (Brodmann area 36) of control, p‐preAD, and AD cases (Table 1) stained with anti‐Aβ 17‐24 (Aβ plaque load), anti‐Aβ N3pE (Aβ N3pE plaque load), or anti‐Aβ pS8 (Aβ pS8 plaque load) antibodies as previously published.9 ImageJ 1.46 (National Institutes of Health; Bethesda, USA) was used to delineate the temporal cortex and, similarly, to delineate the plaques at morphological identification. The area covered by the plaques was calculated and related to the area of the temporal cortex to assess the plaque load.
Statistical analysis
Statistical analysis was performed using SPSS Statistics 22 (IBM; Chicago, USA). Partial Spearman's rank correlation (control for age/gender) was used to evaluate the association amongst CAA‐ and AD‐related parameters. Multinomial logistic regression (control for age/gender) was applied to compare Aβ pathology groups for their association with CAA‐ and AD‐related parameters, vascular risk factors, and alcohol abuse. Linear regression (control for age/gender) was used to determine the effect of B‐CAA and B‐Aβ plaque stages on NIA‐AA AD degree, CDR score, vascular risk factors, and alcohol abuse.
B‐Aβ plaque stages and amyloid plaque loads used for statistical analysis were obtained from a previous publication in which the present cases were analyzed for the biochemical composition of Aβ aggregates in plaques.9
Results
Molecular differentiation of amyloid deposition in CAA
To analyze amyloid composition of CAA, brains of control (including cases with CAA in the absence of plaques), p‐preAD, and AD cases were stained with antibodies against non‐modified epitopes of Aβ (Aβ 17‐24, Aβ 1‐17) or the post‐translationally modified species Aβ N3pE and Aβ pS8. Deposition of Aβ, including Aβ N3pE and Aβ pS8, was detected in arteries, veins, and/or capillaries of the leptomeninges and/or the parenchyma (Fig. 1). Of the 35 cases with CAA detected through anti‐Aβ 17‐24 staining, 32 cases (91.4%) also showed vascular Aβ N3pE, whereas vascular Aβ pS8 was limited to 21 cases with CAA (60%). Thereby, Aβ pS8 was exclusively detected in cases also exhibiting Aβ N3pE. Triple label immunofluorescence revealed that non‐modified (6E10‐positive) Aβ, Aβ N3pE, and Aβ pS8 could colocalize in the same vessels (Fig. S1). Moreover, all cases in which CAA could be detected with antibodies against Aβ 17‐24 also showed a positive staining for Aβ 1‐17.
Figure 1.
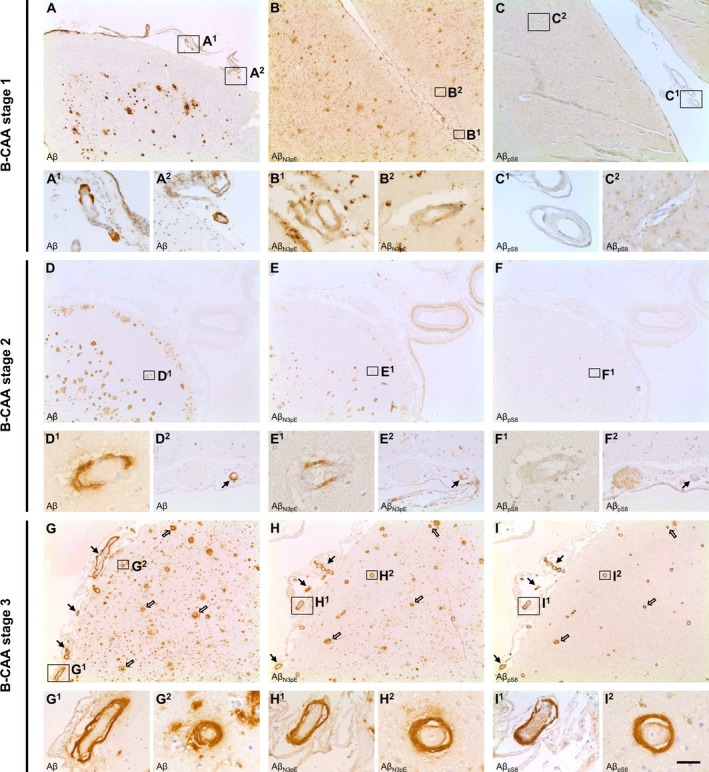
Stages of amyloid maturation in CAA. Detection of Aβ, Aβ N3pE, and Aβ pS8 in leptomeningeal (arrow) and parenchymal (open arrow) vessels of AD cases enabled the differentiation of three biochemical stages of amyloid deposition in the pathogenesis of CAA. B‐CAA stage 1 (A–C) was characterized by initial deposition of Aβ in the vessel (A) in the absence of Aβ N3pE (B) and Aβ pS8 (C) deposition. B‐CAA stage 2 (D–F), however, corresponded to the additional deposition of Aβ N3pE (E) whereas the vessels were still devoid of Aβ pS8 deposits (F, the intravascularly stained material in one vessel in F and F2 (no arrow) is related to insufficient peroxidase blocking in the erythrocytes and does not correspond to positivity for Aβ pS8 as demonstrated in I, I1, and I2). Co‐deposition of Aβ (G), Aβ N3pE (H), and Aβ pS8 (I) in the vessel could be detected in B‐CAA stage 3 (G–I). The figure displays representative images of the temporal cortex of AD cases stained with DAB for Aβ (A, D, G), Aβ N3pE (B, E, H), and Aβ pS8 (C, F, I). Scale bar: (A, B, C, D, E, F, G, H, I) 350 μm, (A1, A2, C1, C2, D2, E2, F2, G1, H1, I1) 70 μm, (B1, B2, D1, E1, F1, G2, H2, I2) 35 μm.
Our findings indicate the sequential deposition of distinct post‐translationally modified Aβ species in CAA analogous to the biochemical stages of Aβ deposition in amyloid plaques (B‐Aβ plaque stages),9 defining three biochemical (immunohistochemical) stages of CAA (B‐CAA stages). B‐CAA stage 1 was characterized by deposition of Aβ in the absence of Aβ N3pE and Aβ pS8; B‐CAA stage 2 was defined by the additional deposition of Aβ N3pE; and B‐CAA stage 3 corresponded to the deposition of Aβ including Aβ N3pE and Aβ pS8 (Fig. 1). In the combined cohort of control, p‐preAD, and AD cases, three of 35 cases with CAA (8.6%) presented with B‐CAA stage 1, whereas B‐CAA stage 2 was prevalent in 11 of 35 cases with CAA (31.4%). Twenty one of 35 cases with CAA (60%) exhibited B‐CAA stage 3 (Table 1).
The B‐CAA stages highly correlated with the overall anatomical expansion of CAA as represented by the CAA stage30 and the severity of CAA‐related vessel wall damage according to the Vonsattel grading29 (P < 0.001, Table 2).
Table 2.
Partial Spearman's rank correlations (control for age/gender)
| Correlation coefficient | P‐value | |
|---|---|---|
| B‐CAA stage | ||
| CAA stage | 0.910 | <0.001 |
| CAA severity | 0.911 | <0.001 |
| Aβ plaque load | 0.163 (0.007)* | 0.264 (0.967)* |
| Aβ N3pE plaque load | 0.216 (0.055)* | 0.137 (0.761)* |
| Aβ pS8 plaque load | 0.469 (‐0.044)* | 0.001 (0.807)* |
| CTRL/p‐preAD/AD | 0.442 | <0.001 |
| CDR score | 0.535 + | <0.001 + |
| NIA‐AA AD degree | 0.403 | 0.001 |
| Aβ‐MTL phase | 0.394 | 0.001 |
| Braak‐NFT stage | 0.509 | <0.001 |
| CERAD score | 0.599 | <0.001 |
| B‐Aβ plaque stage | 0.500 | <0.001 |
| APOE ε4 allele | ||
| B‐CAA stage | 0.357 | 0.003 |
| CAA severity | 0.372 | 0.002 |
| B‐Aβ plaque stage | 0.379 | 0.002 |
| Aβ‐MTL phase | 0.387 | 0.001 |
Aβ = amyloid β, AD = Alzheimer's disease, APOE = apolipoprotein E, B‐CAA stage = biochemical CAA stage, B‐Aβ plaque stage = biochemical Aβ plaque stage, CAA = cerebral amyloid angiopathy, CDR = clinical dementia rating, CERAD = Consortium to Establish a Registry for Alzheimer's disease, CTRL = control, MTL = medial temporal lobe, NFT = neurofibrillary tangle, NIA‐AA = National Institute on Aging‐Alzheimer's Association, p‐preAD = pathologically‐defined preclinical Alzheimer's disease; Aβ/Aβ N3pE/Aβ pS8 plaque load9 [* numbers display the correlation coefficients and P‐values when restricting the correlation analysis to cases with Aβ pathology, whilst numbers in brackets correspond to the correlation coefficients and P‐values when restricting the correlation analysis to cases with CAA pathology (B‐CAA stage ≥1)], Aβ‐MTL phase27, B‐Aβ plaque stage9, Braak‐NFT stage25, CAA severity29, CAA stage30, CDR score32 [+ correlation analysis of CDR scores was restricted to cases without VD, CBD, LBD, and/or AGB], CERAD score26, NIA‐AA AD degree1, 65. Values in bold represent statistically significant results.
Heterogeneous amyloid deposition in CAA and plaques
Analysis of Aβ deposition in CAA and plaques revealed heterogeneity between B‐Aβ plaque and B‐CAA stages. On the one hand, B‐CAA stage 1 and 2 could be detected in cases with B‐Aβ plaque stage 3. Two cases with B‐Aβ plaque stage 3 even presented without CAA. On the other hand, cases with B‐CAA stage 3 could exhibit initial stages of amyloid deposition in plaques (B‐Aβ plaque stage 2) or no amyloid plaques at all (Table 1). The distribution of the distinct B‐Aβ plaque stages within the different B‐CAA stages of control, p‐preAD, and AD cases indeed supported the heterogeneous deposition of modified Aβ in CAA and plaques (Table 3). Accordingly, analysis of control, p‐preAD, and AD cases with Aβ pathology revealed no significant correlation between the B‐CAA stages and the Aβ/Aβ N3pE plaque load (P ≥ 0.137, Table 2). However, the B‐CAA stages weakly correlated with the Aβ pS8 plaque load (P = 0.001, Table 2). The dissociation of the B‐CAA stages from the plaque load became particularly obvious when restricting the correlation analysis to cases with CAA pathology (≥ B‐CAA stage 1) (P ≥ 0.761, Table 2).
Table 3.
Distribution of cases within different B‐Aβ plaque and B‐CAA stages
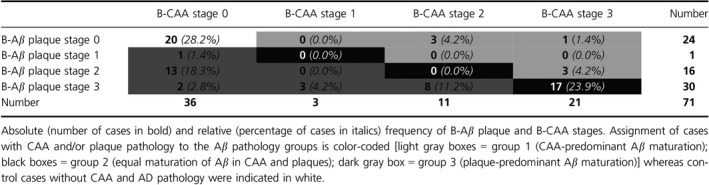
Based on the distribution of the B‐Aβ plaque and B‐CAA stages, cases with Aβ pathology could be subclassified into three groups of Aβ aggregate maturation: group 1 corresponded to cases with biochemically more advanced maturation of CAA pathology (CAA‐predominant group: B‐CAA stage > B‐Aβ plaque stage; this group included CAA cases without plaque pathology); group 2 comprised cases with equal biochemical maturation of Aβ aggregates in CAA and plaques (equal maturation group: B‐CAA stage = B‐Aβ plaque stage; this group contained only cases with end‐stage Aβ pathology); and group 3 referred to cases with biochemically more advanced maturation of plaque pathology (plaque‐predominant group: B‐CAA stage < B‐Aβ plaque stage; this group included 16 cases without CAA). Seven of 51 cases with Aβ pathology (13.7%) were assigned to group 1 whereas group 3 comprised 27 of 51 cases with Aβ pathology (53.0%). Notably, 17 of 51 cases (33.3%) exhibited equal maturation of Aβ in CAA and plaques, thus being classified into group 2 (Table 3).
Association of Aβ pathology groups with CAA‐ and AD‐related pathology
Comparison of the CAA‐predominant (group 1), plaque‐predominant (group 3), and equally maturating (group 2) cases by multinomial logistic regression (controlled for age and gender) revealed no association with the CAA stage and severity when comparing groups 1 and 2 (P ≥ 0.535, Table 4). However, group 3 exhibited lower levels of CAA severity and expansion throughout the brain than the other two groups (P ≤ 0.016, Table 4; Fig. 2A).
Table 4.
Multinomial logistic regression models (control for age/gender)
| Aβ pathology group 1 vs Aβ pathology group 2 | Aβ pathology group 1 vs Aβ pathology group 3 | Aβ pathology group 2 vs Aβ pathology group 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| P‐value | odds ratio | 95% confidence interval | P‐value | odds ratio | 95% confidence interval | P‐value | odds ratio | 95% confidence interval | |
| CAA severity | 0.583 | 0.520 | 0.050–5.376 | 0.016 | 0.060 | 0.006–0.597 | 0.001 | 0.031 | 0.004–0.240 |
| CAA stage | 0.535 | 0.543 | 0.079–3.734 | 0.014 | 0.072 | 0.009–0.588 | 0.002 | 0.133 | 0.037–0.481 |
| CAA type 1 | 0.452 | 2.812 | 0.190–41.587 | 0.689 | 1.729 | 0.119–25.179 | 0.035 | 4.861 | 1.115–21.183 |
| CAA type 2 | 0.249 | 4.385 | 0.355–54.137 | 0.040 | 12.106 | 1.123–130.467 | 0.162 | 2.761 | 0.665–11.452 |
| APOE ε4 allele | 0.998 | 2.678 × 10‐9 | ‐ | 0.998 | 1.176 × 10‐8 | ‐ | 0.021 | 0.228 | 0.064–0.804 |
| Arterial hypertension | 0.010 | 0.028 | 0.002–0.426 | 0.042 | 0.080 | 0.007–0.918 | 0.160 | 2.898 | 0.657–12.788 |
| Diabetes mellitus | 0.282 | 0.272 | 0.025–2.914 | 0.726 | 0.687 | 0.084–5.606 | 0.207 | 2.530 | 0.597–10.713 |
| Alcohol abuse | 1.000 | 1.398 | ‐ | 1.000 | 1.172 x 10‐8 | ‐ | 0.998 | 2.279 × 10‐8 | ‐ |
| Aβ‐MTL phase | 0.008 | 5.279 | 1.530–18.210 | 0.042 | 2.867 | 1.039–7.907 | 0.134 | 1.841 | 0.829–4.089 |
| Braak‐NFT stage | 0.037 | 3.008 | 1.069–8.466 | 0.137 | 2.095 | 0.790–5.555 | 0.131 | 1.436 | 0.897–2.298 |
| CERAD score | 0.034 | 3.322 | 1.092–10.103 | 0.433 | 1.517 | 0.535–4.296 | 0.021 | 2.190 | 1.128–4.253 |
| Aβ plaque load | 0.048 | 1.328 | 1.002–1.759 | 0.124 | 1.222 | 0.947–1.577 | 0.303 | 1.087 | 0.928–1.273 |
| Aβ N3pE plaque load | 0.009 | 2.613 | 1.271–5.374 | 0.057 | 1.816 | 0.982–3.357 | 0.106 | 1.439 | 0.926–2.237 |
| Aβ pS8 plaque load | 0.000 | 2.514 × 10 77 | 1.034 × 10 77–6.112 × 10 77 | ‐ | ‐ | ‐ | 0.099 | 2.110 | 0.868–5.128 |
| CDR score | 0.000 | 3.733 × 10 10 | 2.096 × 10 10 –6.650 × 10 10 | 0.997 | 6.324 × 10‐13 | ‐ | 0.389 | 1.289 | 0.724–2.296 |
Aβ = amyloid β, APOE = apolipoprotein E, CAA = cerebral amyloid angiopathy, CDR = clinical dementia rating, CERAD = Consortium to Establish a Registry for Alzheimer's disease, MTL = medial temporal lobe, NFT = neurofibrillary tangle; Aβ/Aβ N3pE/Aβ pS8 plaque load,9 Aβ‐MTL phase,27 Braak‐NFT stage,25 CAA severity,29 CAA stage,30 CAA type,31 CDR score,32 CERAD score.26 Bold values represent statistically significant results.
Figure 2.
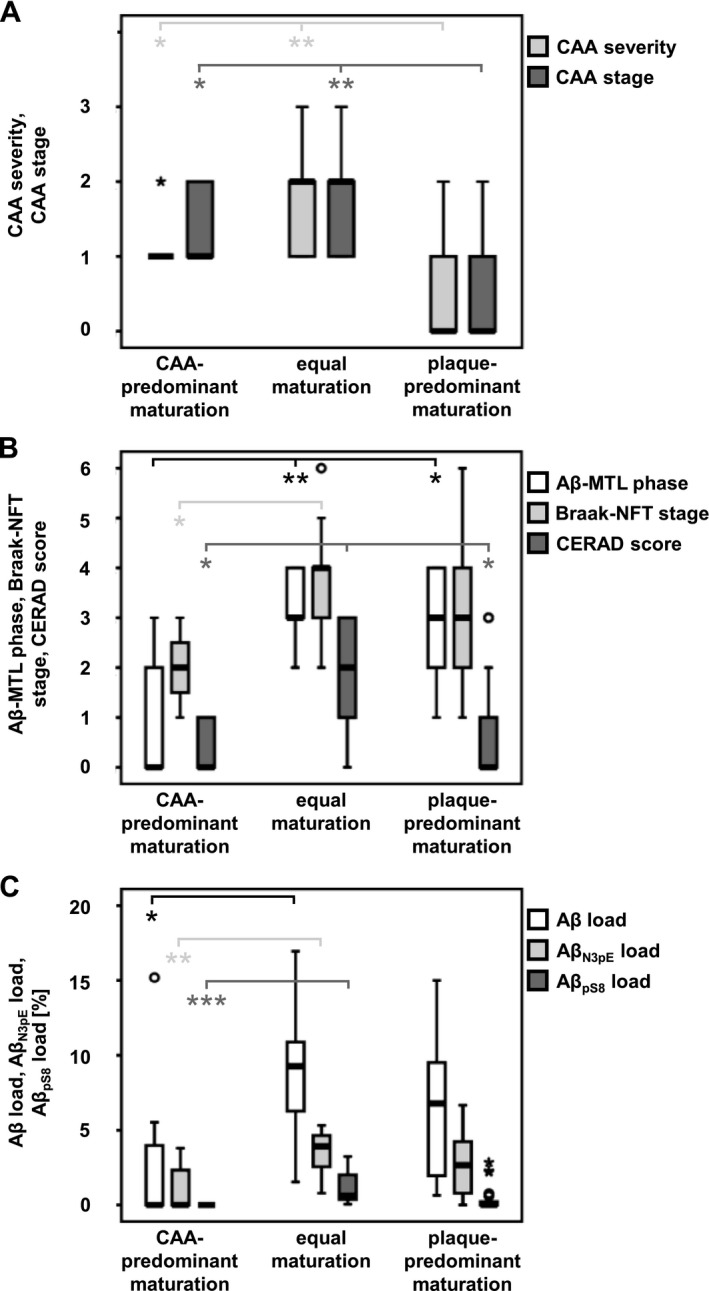
Neuropathologic associations of Aβ pathology groups. Relation of case groups for Aβ pathology to CAA stage (of CAA distribution)30 and CAA severity29 (A), to Aβ‐MTL phases,27 Braak‐NFT stages,23 and CERAD scores26 (B), and to Aβ, Aβ N3pE, and Aβ pS8 plaque loads (C).
Notably, cases with capillary CAA (CAA type 1) more likely belonged to group 2 than to group 3 compared to cases without capillary Aβ deposits (CAA type 2) or without CAA (P = 0.035, Table 4). No significant difference became obvious between groups 1 and 2, or groups 1 and 3 (P ≥ 0.452, Table 4). In contrast, the presence of CAA type 2 significantly increased the probability of a case for belonging to group 1 compared to group 3 (P = 0.040, Table 4) that could not be observed for group 2 (P ≥ 0.162, Table 4). The APOE ε4 allele frequency was higher in group 2 compared to group 3 (P = 0.021, Table 4). Furthermore, group 1 (P ≤ 0.042, Table 4), but not groups 2 and 3 (P = 0.160, Table 4), was associated with arterial hypertension. The differential prevalence of arterial hypertension within the Aβ pathology groups indeed supported the association with CAA‐related Aβ maturation (Table 5). None of the groups showed an association with diabetes mellitus or alcohol abuse (P ≥ 0.207, Table 4).
Table 5.
Prevalence of arterial hypertension within Aβ pathology groups
| Aβ pathology group 1 | Aβ pathology group 2 | Aβ pathology group 3 | |
|---|---|---|---|
| Arterial hypertension | 6 (85.7%) | 4 (23.5%) | 11 (40.7%) |
| No arterial hypertension | 1 (14.3%) | 13 (76.5%) | 16 (59.3%) |
Absolute (number of cases in bold) and relative (percentage of cases in italics) frequency of (no) arterial hypertension within Aβ pathology groups. Of the 20 control cases without Aβ pathology that could not be assigned to either of the Aβ pathology groups, seven cases (35%) exhibited arterial hypertension, whilst arterial hypertension was not observed in 13 cases (65%).
Cases in groups 2 or 3 presented with higher Aβ‐MTL phases compared to group 1 (P ≤ 0.042, Table 4). However, no significant difference was detected between groups 2 and 3 (P = 0.134, Table 4). Higher Braak‐NFT stages were observed in group 2 (equal maturation) compared to group 1 with CAA‐predominant Aβ pathology (P = 0.037, Table 4) whereas no significant difference became obvious between groups 1 and 3, or groups 2 and 3 (P ≥ 0.131, Table 4). Likewise, CERAD scores for neuritic plaque pathology were higher in group 2 than in groups 1 and 3 (plaque‐predominant maturation) (P ≤ 0.034, Table 4). Groups 1 and 3 did not differ significantly (P = 0.433, Table 4; Fig. 2B).
Notably, cases of groups 1 and 2 showed significant differences in the Aβ, Aβ N3pE, and Aβ pS8 plaque load (P ≤ 0.048, Table 4). Comparison of group 3 with groups 1 or 2, however, revealed no association with the Aβ, Aβ N3pE, or Aβ pS8 plaque load (P ≥ 0.057, Fig. 2C).
Since six cases presented with vascular dementia (VD), corticobasal degeneration (CBD), Lewy body disease (LBD), and/or argyrophilic grain disease (AGD) additional to AD pathology that might contribute to cognitive decline, multinomial logistic regression of CDR scores was restricted to cases with “pure” CAA and/or AD pathology, thereby preventing the distortion of statistics through the contribution of these co‐morbidities to dementia. Notably, group 2 exhibited higher CDR scores compared to group 1 (P < 0.001, Table 4). No significant difference was detected between groups 1 and 3, or groups 2 and 3 (P ≥ 0.389, Table 4).
Association of B‐CAA stages with AD‐related pathology, risk factors, and clinical progression
CAA was prevalent in 4 of 24 control cases (16.7%), 11 of 27 p‐preAD cases (40.7%), and 20 of 20 AD cases (100%). All B‐CAA stages could be detected in p‐preAD and AD cases, whereby two of 11 p‐preAD cases with CAA (18.2%) and one of 20 AD cases with CAA (5%) exhibited B‐CAA stage 1. B‐CAA stage 2 was prevalent in one of 11 p‐preAD (9.1%) and seven of 20 AD (35%) cases with CAA. B‐CAA stage 3, however, became obvious in eight of 11 p‐preAD cases with CAA (72.7%) but only in 12 of 20 AD cases with CAA (60%). In this context, it is important to re‐note that 16 of 27 p‐preAD cases (59.3%) did not exhibit CAA. Three of four control cases with CAA (75%) exhibited B‐CAA stage 2, whereas B‐CAA stage 3 was prevalent in one of four control cases with CAA (25%) (Table 1).
Partial Spearman's rank correlation (controlled for age and gender) revealed a moderate correlation between the B‐CAA stages and (1) the progression of AD pathogenesis (control, p‐preAD, AD) or (2) the degree of dementia, provided by the CDR score (P < 0.001, Table 2). Correlation of the B‐CAA stages with the CDR score was restricted to cases without VD, CBD, LBD, and/or AGD to avoid bias caused by these co‐morbidities. Furthermore, the anatomical expansion of amyloid plaques (Aβ‐MTL phases) as well as the B‐Aβ plaque stages, Braak‐NFT stages, CERAD scores, and NIA‐AA AD degrees showed a weak to moderate correlation with the B‐CAA stages (P ≤ 0.001, Table 2). Interestingly, the B‐CAA stages also correlated with the APOE ε4 allele frequency similar to the CAA severity, the B‐Aβ plaque stages, and the Aβ‐MTL phases (P ≤ 0.003, Table 2). Linear regression (controlled for age and gender) furthermore revealed a negative association of arterial hypertension with the B‐Aβ plaque stages (P = 0.022, Table 6) but not with the B‐CAA stages (P = 0.319, Table 6). Diabetes mellitus and alcohol abuse did not affect the B‐CAA or B‐Aβ plaque stages (P ≥ 0.064, Table 6).
Table 6.
Linear regression models (control for age/gender)
| Model | B‐CAA stage | B‐Aβ plaque stage | |||
|---|---|---|---|---|---|
| β‐coefficient | P‐value | β‐coefficient | P‐value | ||
| 1 | NIA‐AA AD degree | 0.087 | 0.248 | 0.823 | <0.001 |
| 2 | CDR score | 0.401 | 0.005 | 0.216 | 0.173 |
| 3 | Arterial hypertension | −0.105 | 0.319 | ‐ | ‐ |
| 4 | ‐ | ‐ | −0.205 | 0.022 | |
| 5 | Diabetes mellitus | −0.133 | 0.213 | ‐ | ‐ |
| 6 | ‐ | ‐ | −0.053 | 0.569 | |
| 7 | Alcohol abuse | −0.068 | 0.522 | ‐ | ‐ |
| 8 | ‐ | ‐ | −0.169 | 0.064 | |
Aβ = amyloid β, AD = Alzheimer's disease, CAA = cerebral amyloid angiopathy, CDR score = clinical dementia rating score,32 B‐CAA stage = biochemical CAA stage, B‐Aβ plaque stage = biochemical Aβ plaque stage,9 NIA‐AA AD degree = National Institute on Aging‐Alzheimer's Association Alzheimer's disease degree1, 65; model 1 ‐ model 2: dependent variables: NIA‐AA AD degree, CDR score; independent variables: B‐CAA stage, B‐Aβ plaque stage; confounding variables: age, gender; model 3 ‐ model 8: dependent variables: B‐CAA stage, B‐Aβ plaque stage; independent variables: arterial hypertension, diabetes mellitus, alcohol abuse; confounding variables: age, gender; – = variable is not included in the model. Values in bold represent statistically significant results.
To clarify the impact of plaque‐ and CAA‐related Aβ maturation on the development of AD according to the NIA‐AA AD criteria and the degree of dementia as described by the CDR score, we calculated two linear regression models (controlled for age and gender) including both B‐CAA and B‐Aβ plaque stages. Plaque maturation (B‐Aβ plaque stages; P < 0.001, Table 6) but not CAA maturation (B‐CAA stages; P = 0.248, Table 6) correlated with the progression of AD (NIA‐AA AD degree). Accordingly, all symptomatic AD cases exhibited B‐Aβ plaque stage 3 but only 60% of symptomatic AD cases presented with B‐CAA stage 3. Additional linear regression (restricted to cases without VD, CBD, LBD, and/or AGD) indicated that CAA maturation significantly contributed to the degree of cognitive decline (CDR score) (P = 0.005, Table 6). As expected from the finding that all cases with symptomatic AD exhibited B‐Aβ plaque stage 3, no additional impact of plaque‐related Aβ maturation on cognitive decline was detected (P = 0.173, Table 6).
Discussion
Biochemical stages of CAA‐related Aβ maturation (B‐CAA stages)
The combined detection of different Aβ variants revealed a hierarchical sequence of Aβ deposition in CAA that could be differentiated into three distinct stages (Fig. 3A): B‐CAA stage 1 corresponded to the deposition of Aβ not modified by pyroglutamination and/or phosphorylation; B‐CAA stage 2 was characterized by the additional deposition of Aβ N3pE; and B‐CAA stage 3, finally, included Aβ pS8. This sequential deposition of Aβ in CAA corresponds to the previously observed hierarchical sequence for the deposition of modified Aβ in plaques,9 suggesting that vascular and parenchymal Aβ deposition represent two aspects of a common biochemical process of Aβ maturation. A common sequence of Aβ deposition in CAA and plaques is indeed supported by the correlation of the B‐CAA stages with the B‐Aβ plaque stages and with the anatomical expansion of amyloid plaques and CAA‐affected blood vessels throughout the brain. Although studies with transgenic mouse models expressing the amyloid precursor protein in neurons34 and on the drainage of parenchymal Aβ 35 suggest a neuronal origin of Aβ in CAA, it remains to be determined whether modified Aβ species detected in the vasculature originate from the parenchyma or whether modification can occur within vessels. However, despite the common sequence of Aβ deposition in CAA and plaques, the segregation of CAA and plaque pathology became particularly obvious through the case‐by‐case analysis of B‐CAA and B‐Aβ plaque stages and the absent correlation of the B‐CAA stages with amyloid plaque load.
Figure 3.
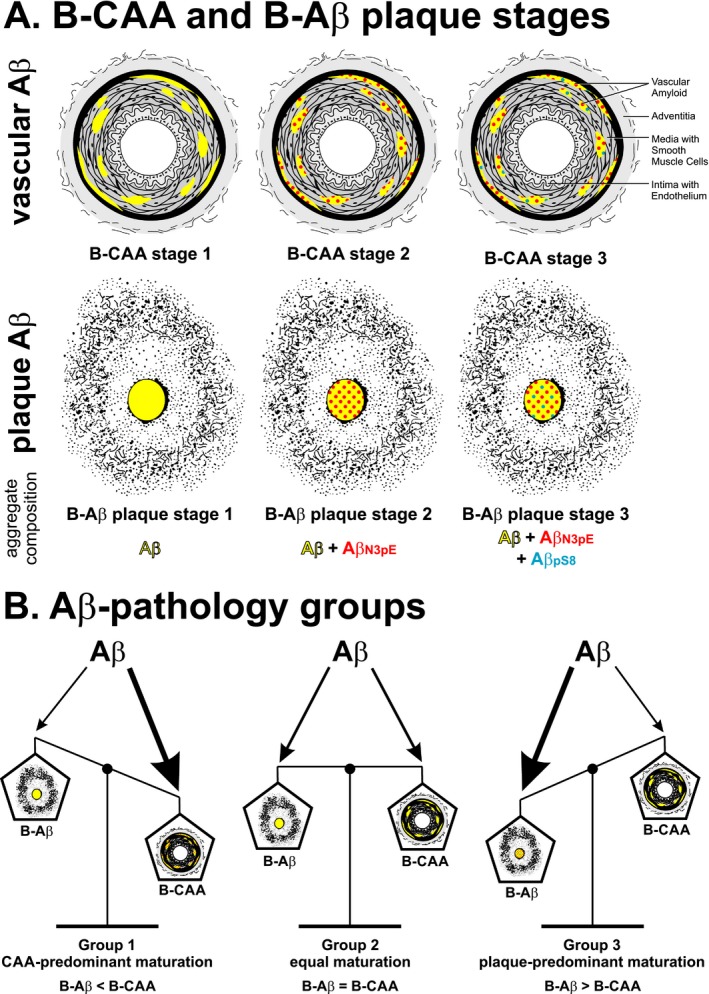
Amyloid maturation within Aβ pathology groups. Schematic representation of the biochemical (immunohistochemical) stages of CAA‐ (B‐CAA stage) and plaque‐ (B‐Aβ plaque stage) related Aβ maturation (A) and their balance in distinct Aβ pathology groups (B).
Cross‐sectional autopsy studies cannot prove the sequential deposition of Aβ and its modified forms. However, the following arguments strongly support a sequential process: (1) none of the cases showed deposition of Aβ pS8 in the absence of Aβ N3pE, (2) the B‐CAA stages correlate with the sequential expansion of plaques throughout the MTL27 which correlates with increased amyloid PET‐tracer retention,36, 37, 38 and (3) the sequential occurrence of Aβ and its modified forms in line with the B‐Aβ plaque stages within the human brain has been confirmed in a mouse model for AD.39 Thus, there is at least indirect evidence that the B‐CAA stages indeed represent a sequential process of Aβ, Aβ N3pE, and Aβ pS8 deposition.
Case groups for plaque‐ and CAA‐related Aβ maturation (Aβ pathology groups)
Despite the correlation of the B‐CAA stages with the B‐Aβ plaque stages and the Aβ‐MTL phases, significant variations between the B‐CAA and B‐Aβ plaque stages existed within individual cases. The specific composition of vascular and parenchymal Aβ deposits indicated three distinct case groups for Aβ pathology (Fig. 3B, Table 3): group 1 was defined by predominant Aβ maturation in CAA (B‐CAA stage > B‐Aβ plaque stage); group 2 included cases with equal Aβ maturation in CAA and plaques (B‐CAA stage = B‐Aβ plaque stage); and group 3 was characterized by predominant Aβ maturation in plaques (B‐CAA stage < B‐Aβ plaque stage), including cases without CAA. Interestingly, these case groups showed additional neuropathological associations. Cases with CAA‐predominant pathology showed less advanced Aβ‐MTL phases for the anatomical expansion of plaque pathology, whereas cases with plaque‐predominant pathology exhibited less widespread and severe CAA. Previous neuropathological studies revealed that CAA cases could differ in their relation between CAA severity and AD‐related plaque pathology.3, 7, 40, 41, 42 Thus, our finding of differences in the balance of Aβ maturation between CAA and plaques might provide an explanation at the molecular level for the well‐known variation in CAA in relation to plaque pathology.3, 7, 40, 41, 42
The association of the CAA‐predominant pathology group with arterial hypertension furthermore indicates that this condition could affect the leading site of Aβ maturation and, thereby, might act as additional risk factor for Aβ seeding and maturation. This is indeed supported by the development of CAA in a rat model for hypertension.43 Thus, arterial hypertension should be taken into account for therapeutic intervention because it might modify the pathological picture of AD to the CAA‐predominant pattern.
The length of Aβ could also play an important role in the balance between vascular and parenchymal Aβ deposition. Aβ 40 predominantly occurs within vascular Aβ deposits whereas Aβ 42 predominates in plaques.44 Likewise, mouse models producing mainly Aβ 40 develop a CAA‐predominant Aβ pathology and mice exhibiting augmented amounts of Aβ 42 show negligible CAA pathology but abundant plaques.45 The identification of B‐CAA stages with the same Aβ maturation sequence as plaques suggests that post‐translational modifications could occur in both Aβ 40‐ and Aβ 42‐predominant aggregates in blood vessels and parenchymal plaques. However, since vascular deposits in the human brain contain both Aβ 40 and Aβ 42 even in early stages of pathogenesis,31, 44 it might require transgenic animal models to specifically address the question whether the ratio of Aβ 40 and Aβ 42 influences the site of Aβ aggregate maturation.
Furthermore, the site of leading Aβ maturation might attract further proteins to accumulate. This interpretation corresponds to the finding that seeds of Aβ aggregates could induce further Aβ deposition46, 47, 48 and argues in favor of the view that the presence of local seeds determines the aggregation pattern. However, it will be important to investigate whether the apparent maturation of Aβ in individual deposits results from addition of modified species to existing Aβ aggregates or whether already aggregated Aβ within deposits undergoes post‐translational modification. Previously, we demonstrated Aβ N3pE and Aβ pS8 within non‐detergent extracts of human and APP transgenic mouse brains,9, 12, 17 indicative for the presence of these Aβ variants in monomeric or soluble oligomeric form. Whether already aggregated and deposited Aβ is amenable to pyroglutamination and/or phosphorylation is not known. Own preliminary studies suggest that Aβ aggregates can be phosphorylated at serine 8 in vitro but further work would be required to proof that this could occur in vivo. It has also been shown that plaques and CAA in human and transgenic mouse brains contain N‐terminally truncated Aβ species19, 49, 50, 51, 52, 53 that would also be detected by the anti‐Aβ 17‐24 antibody used in this study. Since the cerebrovascular deposits were detected with antibodies raised against Aβ 17‐24 and Aβ 1‐17, it is quite likely that these deposits contain non‐truncated Aβ. Only some early plaque types, such as fleecy amyloid, appeared to contain N‐terminal truncated Aβ with a non‐identified N‐terminus as reported previously.54, 55 The simultaneous presence of truncated forms of Aβ other than Aβ N3pE and Aβ pS8 in B‐CAA stage 1 cerebrovascular Aβ deposits, however, could not be excluded. Additional to the aggregation‐promoting effect, phosphorylation, and pyroglutamination also affect the proteolytic degradation of Aβ monomers and the stability of Aβ aggregates.13, 56, 57, 58 Thus, pyroglutamination could potentially favor further phosphorylation and, thereby, increase the stability of Aβ aggregates, and exaggerate Aβ aggregation.
Contribution of CAA‐related Aβ maturation to cognitive decline
The B‐CAA stages significantly correlated with the CDR score representing cognitive decline. However, only cases with equal maturation of Aβ in CAA and plaques showed an association with cognitive decline, suggesting that Aβ maturation in CAA represents only one of numerous factors that contribute to dementia. The mere presence of end‐stage CAA maturation (that was observed both in CAA‐predominant and equal maturation cases) seems to be insufficient to determine the development of dementia on its own. Rather end‐stage plaque maturation has to be present. Accordingly, all symptomatic AD cases presented with end‐stage plaque maturation. In contrast, only 60% of symptomatic AD cases exhibited full CAA maturation, supporting the importance of plaque maturation for the development of dementia. However, a multiple linear regression model revealed only the selective impact of CAA maturation on the CDR score, indicating that the impact of plaques on the development of dementia might only partially be ascribed to Aβ maturation but that instead the high level of other AD neuropathologic changes that correlate with end‐stage plaque maturation, such as NFT pathology,59 might contribute to the course of dementia in AD.
Molecular characterization of amyloid pathology, based on the composition of CAA and plaques, might not only help to identify pathological subgroups of β‐amyloidosis but also to understand the effect of therapeutic interventions. Aβ immunization, for example, could reduce the amyloid load within plaques but simultaneously exacerbate CAA pathology and CAA‐related hemorrhages.60, 61 The association of CAA‐predominant Aβ maturation with arterial hypertension argues for a higher risk for therapy‐related bleedings in these patients as both CAA and arterial hypertension represent risk factors for intracerebral hemorrhages.62, 63
Author Contribution
Conception and design of study: JW, DRT, JG; acquisition and analysis of data: (i) neuropathology: DRT, (ii) production and characterization of non‐commercial antibodies: SK, JG, JW, (iii) immunohistochemistry: JG, DRT, ARU, SK, JW, (iv) APOE genotyping: EG, JG, (v) clinical assessment: CAFVA, (vi) statistical analysis: JG, DRT; draft of figures and manuscript: JG, DRT, JW.
Conflicts of Interest
DRT received consultancies from Covance Laboratories (UK) and GE‐Healthcare (UK), a speaker honorarium from GE‐Healthcare (UK), and collaborated with Novartis Pharma Basel (Switzerland), Probiodrug (Germany) and Janssen Pharmaceutical Companies (Belgium). CAFVA received honoraria from serving on the scientific advisory board of Nutricia GmbH and Hongkong University Research council, received funding for travel and speaker honoraria from Nutricia GmbH, Novartis Pharma GmbH, Lilly Deutschland GmbH, Desitin Arzneimittel GmbH, Biogen, and Dr. Willmar Schwabe GmbH & Co. KG, and collaborated with Roche Diagnostics GmbH, Biologische Heilmittel Heel GmbH, and ViaMed GmbH. All other authors declare no conflicts of interest with the content of the publication.
Supporting information
Figure S1: Vascular colocalization of modified Aβ.
Acknowledgment
All authors thank Irina Kosterin, Christine Schneider, Kathrin Pruy, Alice Yates, Daniela Demharter, and Sandra Theil for technical help. We are also grateful to Drs. Stephan Schilling and Inge Lues (Probiodrug AG) for providing antibodies against Aβ N3pE, and for critically reading the manuscript. The work was supported by the “Deutsche Forschungsgemeinschaft” (WA1477/6 (JW), TH624/6‐1 (DRT)), “Alzheimer Forschung Initiative” (grant #13803, #10810 (DRT); #12854 (SK)), “KU Leuven starting grant” (DRT), “Fonds Wetenschappelijk Onderzoek Vlaanderen” (FWO‐G0F8516N Odysseus (DRT)), and “Vlaamse Impulsfinanciering voor Netwerken voor Dementie‐Onderzoek” (IWT 135043 (DRT)).
Funding Information
The work was supported by the “Deutsche Forschungsgemeinschaft” (WA1477/6 (JW), TH624/6‐1 (DRT)), “Alzheimer Forschung Initiative” (grant #13803, #10810 (DRT); #12854 (SK)), “KU Leuven starting grant” (DRT), “Fonds Wetenschappelijk Onderzoek Vlaanderen” (FWO‐G0F8516N Odysseus (DRT)), and “Vlaamse Impulsfinanciering voor Netwerken voor Dementie‐Onderzoek” (IWT 135043 (DRT)).
Funding Statement
This work was funded by Deutsche Forschungsgemeinschaft grants TH624/6‐1 and WA1477/6; Alzheimer Forschung Initiative grants 10810, 12854, and 13803; KU Leuven grant ; Fonds Wetenschappelijk Onderzoek grant ; Vlaamse Impulsfinanciering voor Netwerken voor Dementie‐Onderzoek grant IWT 135043.
Contributor Information
Dietmar R. Thal, Email: Dietmar.Thal@kuleuven.be.
Jochen Walter, Email: Jochen.Walter@ukbonn.de.
References
- 1. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 2001;81:741–766. [DOI] [PubMed] [Google Scholar]
- 3. Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011;37:75–93. [DOI] [PubMed] [Google Scholar]
- 4. Wisniewski H, Johnson AB, Raine CS, et al. Senile plaques and cerebral amyloidosis in aged dogs. A histochemical and ultrastructural study. Lab Invest 1970;23:287–296. [PubMed] [Google Scholar]
- 5. Love S, Miners S, Palmer J, et al. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front Biosci (Landmark Ed) 2009;14:4778–4792. [DOI] [PubMed] [Google Scholar]
- 6. Kovari E, Herrmann FR, Hof PR, Bouras C. The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer's disease. Neuropathol Appl Neurobiol 2013;39:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thal DR, Griffin WST, De Vos RAI, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol 2008;115:599–609. [DOI] [PubMed] [Google Scholar]
- 8. Thal DR, Rüb U, Orantes M, Braak H. Phases of Abeta‐deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 9. Rijal Upadhaya A, Kosterin I, Kumar S, et al. Biochemical stages of amyloid β‐peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically‐preclinical Alzheimer's disease. Brain 2014;137:887–903. [DOI] [PubMed] [Google Scholar]
- 10. Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid‐beta (Abeta): a hatchet man in Alzheimer disease. J Biol Chem 2011;286:38825–38832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kummer MP, Heneka MT. Truncated and modified amyloid‐beta species. Alzheimers Res Ther 2014;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar S, Rezaei‐Ghaleh N, Terwel D, et al. Extracellular phosphorylation of the amyloid beta‐peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease. EMBO J 2011;30:2255–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar S, Singh S, Hinze D, et al. Phosphorylation of amyloid‐beta peptide at serine 8 attenuates its clearance via insulin‐degrading and angiotensin‐converting enzymes. J Biol Chem 2012;287:8641–8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Russo C, Violani E, Salis S, et al. Pyroglutamate‐modified amyloid beta‐peptides–AbetaN3(pE)–strongly affect cultured neuron and astrocyte survival. J Neurochem 2002;82:1480–1489. [DOI] [PubMed] [Google Scholar]
- 15. Saido TC, Yamao‐Harigaya W, Iwatsubo T, Kawashima S. Amino‐ and carboxyl‐terminal heterogeneity of beta‐amyloid peptides deposited in human brain. Neurosci Lett 1996;215:173–176. [DOI] [PubMed] [Google Scholar]
- 16. Schlenzig D, Manhart S, Cinar Y, et al. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 2009;48:7072–7078. [DOI] [PubMed] [Google Scholar]
- 17. Ashby EL, Miners JS, Kumar S, et al. Investigation of abeta phosphorylated at serine 8 (pAbeta) in Alzheimer's disease, dementia with Lewy bodies and vascular dementia. Neuropathol Appl Neurobiol 2015;41:428–444. [DOI] [PubMed] [Google Scholar]
- 18. Frost JL, Le KX, Cynis H, et al. Pyroglutamate‐3 amyloid‐beta deposition in the brains of humans, non‐human primates, canines, and Alzheimer disease‐like transgenic mouse models. Am J Pathol 2013;183:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iwatsubo T, Saido TC, Mann DM, et al. Full‐length amyloid‐beta (1‐42(43)) and amino‐terminally modified and truncated amyloid‐beta 42(43) deposit in diffuse plaques. Am J Pathol 1996;149:1823–1830. [PMC free article] [PubMed] [Google Scholar]
- 20. Saido TC, Iwatsubo T, Mann DM, et al. Dominant and differential deposition of distinct beta‐amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995;14:457–466. [DOI] [PubMed] [Google Scholar]
- 21. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. The National Institute on Aging . Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on aging, and reagan institute working group on diagnostic criteria for the neuropathological assessment of alzheimer's disease. Neurobiol Aging 1997;18:S1–S2. [PubMed] [Google Scholar]
- 23. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 24. Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1991;1:213–216. [DOI] [PubMed] [Google Scholar]
- 25. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 27. Thal DR, Rüb U, Schultz C, et al. Sequence of Abeta‐protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol 2000;59:733–748. [DOI] [PubMed] [Google Scholar]
- 28. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–548. [PubMed] [Google Scholar]
- 29. Vonsattel JP, Myers RH, Hedley‐Whyte ET, et al. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991;30:637–649. [DOI] [PubMed] [Google Scholar]
- 30. Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer's disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003;62:1287–1301. [DOI] [PubMed] [Google Scholar]
- 31. Thal DR, Ghebremedhin E, Rüb U, et al. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 2002;61:282–293. [DOI] [PubMed] [Google Scholar]
- 32. Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 33. Kumar S, Wirths O, Theil S, et al. Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer's disease. Acta Neuropathol 2013;125:699–709. [DOI] [PubMed] [Google Scholar]
- 34. Calhoun ME, Burgermeister P, Phinney AL, et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A 1999;96:14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weller RO, Subash M, Preston SD, et al. Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008;18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curtis C, Gamez JE, Singh U, et al. Phase 3 Trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol 2015;72:287–294. [DOI] [PubMed] [Google Scholar]
- 37. Thal DR, Beach TG, Zanette M, et al. [18F]flutemetamol amyloid PET in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of Aβ pathology. Alzheimers Dement 2015;11:975–985. [DOI] [PubMed] [Google Scholar]
- 38. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a prospective cohort study. Lancet Neurol 2012;11:669–678. [DOI] [PubMed] [Google Scholar]
- 39. Balakrishnan K, Rijal Upadhaya A, Steinmetz J, et al. Impact of amyloid beta aggregate maturation on antibody treatment in APP23 mice. Acta Neuropathol Commun 2015;3:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983;14:924–928. [DOI] [PubMed] [Google Scholar]
- 41. Jellinger KA, Attems J. Prevalence and impact of cerebrovascular pathology in Alzheimer's disease and parkinsonism. Acta Neurol Scand 2006;114:38–46. [DOI] [PubMed] [Google Scholar]
- 42. Olichney JM, Hansen LA, Hofstetter CR, et al. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol 2000;57:869–874. [DOI] [PubMed] [Google Scholar]
- 43. Held F, Morris AWJ, Pirici D, et al. Vascular basement membrane alterations and beta‐amyloid accumulations in an animal model of cerebral small vessel disease. Clin Sci (Lond) 2017;131:1001–1013. [DOI] [PubMed] [Google Scholar]
- 44. Roher AE, Lowenson JD, Clarke S, et al. beta‐Amyloid‐(1‐42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:10836–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004;7:954–960. [DOI] [PubMed] [Google Scholar]
- 46. Kane MD, Lipinski WJ, Callahan MJ, et al. Evidence for seeding of beta ‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta ‐amyloid precursor protein‐transgenic mice. J Neurosci 2000;20:3606–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 2006;313:1781–1784. [DOI] [PubMed] [Google Scholar]
- 48. Xu F, Fu Z, Dass S, et al. Cerebral vascular amyloid seeds drive amyloid beta‐protein fibril assembly with a distinct anti‐parallel structure. Nat Commun 2016;7:13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Masters CL, Simms G, Weinman NA, et al. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A 1985;82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Antonios G, Saiepour N, Bouter Y, et al. N‐truncated Abeta starting with position four: early intraneuronal accumulation and rescue of toxicity using NT4X‐167, a novel monoclonal antibody. Acta Neuropathol Commun 2013;1:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guntert A, Dobeli H, Bohrmann B. High sensitivity analysis of amyloid‐beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 2006;143:461–475. [DOI] [PubMed] [Google Scholar]
- 52. Harigaya Y, Saido TC, Eckman CB, et al. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer's disease brain. Biochem Biophys Res Commun 2000;276:422–427. [DOI] [PubMed] [Google Scholar]
- 53. Tekirian TL, Saido TC, Markesbery WR, et al. N‐terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol 1998;57:76–94. [DOI] [PubMed] [Google Scholar]
- 54. Thal DR, Capetillo‐Zarate E, Schultz C, et al. Apolipoprotein E co‐localizes with newly formed amyloid beta‐protein (Abeta)‐deposits lacking immunoreactivity against N‐terminal epitopes of Abeta in a genotype‐dependent manner. Acta Neuropathol 2005;110:459–471. [DOI] [PubMed] [Google Scholar]
- 55. Thal DR, Sassin I, Schultz C, et al. Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N‐terminal truncated fragments of Abeta. J Neuropathol Exp Neurol 1999;58:210–216. [DOI] [PubMed] [Google Scholar]
- 56. Rezaei‐Ghaleh N, Amininasab M, Kumar S, et al. Phosphorylation modifies the molecular stability of beta‐amyloid deposits. Nat Commun 2016;7:11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wulff M, Baumann M, Thummler A, et al. Enhanced fibril fragmentation of N‐terminally truncated and pyroglutamyl‐modified abeta peptides. Angew Chem Int Ed Engl 2016;55:5081–5084. [DOI] [PubMed] [Google Scholar]
- 58. Kumar S, Wirths O, Stüber K, et al. Phosphorylation of the amyloid β‐peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol 2016;131:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42:631–639. [DOI] [PubMed] [Google Scholar]
- 60. Racke MM, Boone LI, Hepburn DL, et al. Exacerbation of cerebral amyloid angiopathy‐associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci 2005;25:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boche D, Nicoll JA. The role of the immune system in clearance of abeta from the brain. Brain Pathol 2008;18:267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jellinger K. Cerebrovascular amyloidosis with cerebral hemorrhage. J Neurol 1977;214:195–206. [DOI] [PubMed] [Google Scholar]
- 63. Yamori Y, Horie R, Handa H, et al. Pathogenetic similarity of strokes in stroke‐prone spontaneously hypertensive rats and humans. Stroke 1976;7:46–53. [DOI] [PubMed] [Google Scholar]
- 64. Morris JC, Heyman A, Mohs RC, et al. The consortium to establish a registry for Alzheimer's Disease (CERAD). Part I. clinical and neuropsychological assessment of Alzheimer's disease. Neurology 1989;39:1159–1165. [DOI] [PubMed] [Google Scholar]
- 65. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Vascular colocalization of modified Aβ.