

Abstract
Nanopores are emerging as new single-molecule tools in the study of enzymes. Based on progress in nanopore sequencing of DNA, a tool called Single-molecule Picometer Resolution Nanopore Tweezers (SPRNT) was developed to measure the movement of enzymes along DNA in real time. In this new method, an enzyme is loaded onto a DNA (or RNA) molecule. A single stranded DNA end of this complex is drawn into a nanopore by an electrostatic potential that is applied across the pore. The single-stranded DNA passes through the pore’s constriction until the enzyme comes into contact with the pore. Further progression of the DNA through the pore is then controlled by the enzyme. An ion current that flows through the pore’s constriction is modulated by the DNA in the constriction. Analysis of ion current changes reveals the advance of the DNA with high spatiotemporal precision, thereby providing a real-time record of the enzyme’s activity. Using an engineered version of the protein nanopore MspA, SPRNT has spatial resolution as small as 40 pm at milliseconds time scales, while simultaneously providing the DNA’s sequence within the enzyme. In this chapter SPRNT is introduced and its extraordinary potential is exemplified using the helicase Hel308. Two distinct sub-states are observed for each one-nucleotide advance; one of these about half-nucleotide long steps is ATP-dependent and the other is ATP-independent. The spatiotemporal resolution of this low-cost single-molecule technique lifts the study of enzymes to a new level of precision, enabling exploration of hitherto unobservable enzyme dynamics in real-time.
1. Why are high resolution real-time measurements on enzymes interesting?
Enzymes are essential to the understanding of the molecular basis of life and our knowledge of them is rapidly growing. X-ray crystallography of enzymes provides atomistic detail about their structure; however, this picture is mostly static so that how the enzymes function in real time is mostly subject of prediction. Bulk assays reveal the outcome of enzymes’ function and how they depend on, for example, ATP concentration or temperature, but these ensemble measurements also leave much of the detailed enzymatic pathway to speculation.
Single-molecule measurements can elucidate the functional pathways that isolated enzymes take in real-time. In the past few decades, technological advances from a variety of areas that are unrelated to biology have led to single-molecule techniques such as TIRF FRET, optical tweezers, magnetic tweezers, etc.
These single-molecule tools have been tuned to their highest possible spatial and temporal resolution to study enzymes. However, in vivo enzyme activity often involves motion that is of sub-angstrom-length and occurs on sub-millisecond timescales. Therefore, direct in vitro observation of isolated single enzymes with matching spatial and temporal resolution are needed to provide the most realistic insights into how enzymes function.
2. Introduction to SPRNT
In this book chapter we describe a relatively new single-molecule technique that was born out of research in developing nanopore sequencing of DNA. We call the method SPRNT, which stands for Single-molecule Picometer Resolution Nanopore Tweezers (Derrington et al., 2015). This method is ideally suited to enzymes that process along DNA, RNA or other charged polymers. SPRNT’s big advantages are that it has (i) about ten times more spatial resolution than presently available single-molecule techniques, and (ii) simultaneously provides unprecedented temporal resolution. In addition to its spatiotemporal resolution SPRNT, (iii) provides the enzyme’s location in terms of the nucleotide sequence along the DNA, (iv) is hardware-inexpensive and, last- not-least, (v) is relatively simple. This chapter describes measurements with nanopores by first explaining how nanopore sequencing works and then describes SPRNT. We then show a typical SPRNT measurement that reveals molecular behavior that could not have been seen with other single-molecule measurements and compare SPRNT to other prominent single-molecule techniques.
3. Nanopore measurements
3.1. Nanopore sequencing of DNA (Principle)
Nanopores are emerging as powerful single-molecule tools (Squires, Gilboa, Torfstein, Varongchayakul, and Meller, 2016). Many nanopore techniques are extensions of the Coulter principle (Coulter, 1953), where an ionic flow through a small opening identifies a small object, e.g. a cell or a molecule, that is also passing through the opening. One of the most notable applications of this concept is nanopore sequencing of DNA. In nanopore sequencing an ion current carried by a salt solution is driven through a small pore. Single-stranded DNA, which is poly-negatively charged, is driven into the pore by the same electrostatic field that causes the ion current to flow.
In 1996 Kasianowicz et al. hypothesized that individual nucleotides in a DNA strand passing through the pore would modulate the ion current depending on nucleotide type; the ion current measurement could then be translated directly into the DNA’s sequence. In the seminal work (Akeson, Branton, Kasianowicz, Brandin, & Deamer, 1999; Kasianowicz, Brandin, Branton, & Deamer, 1996) a single biological pore, α- hemolysin (αHL), was prepared in a bilayer (Figure 1). When 120 mV was applied across the membrane, approximately 120 pA flowed through αHL. After RNA molecules were added, sub-millisecond reductions of the current were observed. The ion current obstructions from 100 nucleotide-long RNA molecules consisting of two homopolymer sections showed a small step in the ion current. While these results were inspirational for nanopore sequencing it became clear that individual nucleotides could not be resolved with αHL. ΑHL has an about 5-nm long β-barrel with nearly uniform 1.4-2.2 nm diameter (Song et al., 1996) that can encompass about 12 nucleotides. All these 12 nucleotides affect the ion current (Stoddart, Maglia, Mikhailova, Heron, & Bayley, 2010), prohibiting reading the DNA with single nucleotide precision. Furthermore, DNA passes through practically all types of nanopores at a speed greater than one nucleotide/μs which is too short to resolve picoampere current differences.
Figure 1. The nanopore system with a biological pore.
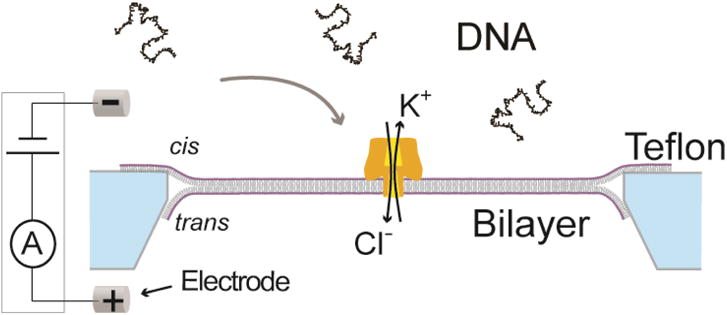
A phospholipid lipid bilayer (purple/grey) spans a Teflon (light blue) aperture, separating a KCl solution into two compartments, cis and trans. A single αHL nanopore (gold) is inserted into the bilayer. A voltage is applied via two Ag/AgCl electrodes and the resulting ion current through the pore is measured. The polynegatively charged DNA is attracted to the pore and moves through the pore. While the DNA moves through the pore it reduces the ion current. Modified from (A. H. Laszlo, Derrington, & Gundlach, 2016).
3.2. Making an ideal pore: MspA
In 2008 Butler et al. (Butler, Pavlenok, Derrington, Niederweis, & Gundlach, 2008) introduced Mycobacterium smegmatis porin A (MspA) (Faller, Niederweis, & Schulz, 2004) to nanopore sequencing. In contrast to αHL, MspA has a short constriction that is 1.2-nm wide and is only about 0.6-nm long which is closely matched to the size of and distance between nucleotides (Figure 2). In order for DNA to pass through MspA, its constriction was reengineered from being dominated by negatively charged aspartic acids to neutral asparagines. A few further mutations from negative to positive residues on MspA’s rim domain made the pore attractive to DNA and enhanced the rate of DNA entry into the pore 100-fold.
Figure 2. Mycobacterium smegmatis porin A (MspA).
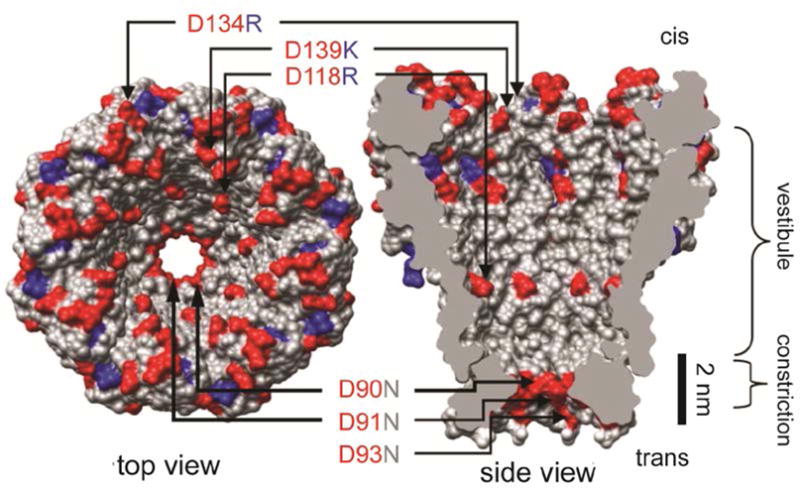
Wild type MspA contains negatively charged residues D90, D91, and D93. Butler et al. found that mutation of these residues to neutrally charged N allowed DNA to translocate through the MspA pore. Further mutation of the pore to include positively charged residues within the vestibule (D118R, D134R, and E139K) resulted in increased DNA interaction as the negatively charged DNA was attracted into the pore by the positive residues. Modified from (Butler et al., 2008).
Derrington et al. (Derrington et al., 2010) showed that DNA held stationary in MspA produces well-resolved nucleotide-specific ion current signals. With more experiments in which DNA was held stationary Manrao et al. (Manrao, Derrington, Pavlenok, Niederweis, & Gundlach, 2011) showed that single nucleotide substitutions in DNA generated significant current differences. She also found that the single nucleotide substitution produced a current different from the DNA without the substitution, when the substituted nucleotide was held up to two nucleotide positions from either side of MspA’s constriction, i.e. about four adjacent nucleotide positions were involved in controlling the measured ion current. Given the short constriction of MspA, this was at first surprising, but it was quickly realized that the springiness of the DNA between where it is held and where it controls the current allowed for Brownian motion. Since the measured ion current is the time-average over all the DNA positions accessed by the Brownian motion within the time scale of the measurement, this effectively widens the width of MspA’s read head.
Use the following procedure to establish a single MspA pore in an unsupported bilayer.
Build a single-channel patch-clamp system as described in (Akeson et al., 1999; Butler et al., 2008).
Pre-clean the Teflon™ setup with Folch solution (1:2 ratio of methanol:chloroform) or pirhana solution (3:1 ratio of concentrated sulfuric acid:30% hydrogen peroxide). Use proper personal protective equipment (see Laszlo et al., 2016 for further details on cleaning).
Use a syringe to pull subsequent rinses of water, ethanol, and hexane through the u-tube aperture and let dry.
Prime the aperture surface by depositing and drying 1μl of DPhPC or DOPC lipid (Avanti Lipids product no. 850356C or 999984C) dissolved in hexanes on the aperture surface (0.3% by weight lipid). Let dry 15 min and repeat one more time.
Fill the trans volume of the “u-tube” and trans well with the desired trans well buffer, then fill the cis volume with the same trans-well buffer (the cis buffer will be replaced once a bilayer is established). Filling in this order ensures no bubbles are trapped within the u-tube.
With a single bristle brush, mix together a ~0.1 μl drop of hexadecane or hexadecene with some dried down DPhPC or DOPC lipid until the mixture takes on a consistency slightly wetter than toothpaste. If it is too dry the bilayers made will be leaky, if too wet, the bilayers will turn into oil clogs of the aperture.
With the single-bristle brush, paint the lipid-oil mixture on and near the aperture on the u-tube. If the aperture becomes clogged, clear it by pushing buffer out through the u-tube from the trans side with a syringe.
Apply 180 mV across the aperture using the patch-clamp amplifier, the current measured by the amplifier should rail at its maximum value.
Using a pipette, blow a ~5 μl bubble across the surface of the aperture, then retract the bubble. This should form a bilayer, if successful the current measured on the patch-clamp amplifier should read zero. Verify the presence of a bilayer by applying a 5 ms, 1 V voltage pulse which should pop the bilayer. If the current still displays zero pA, you instead have a clog that can be cleared as described above.
Once a bilayer has been established, add 1 μl of 1 μg/ml MspA protein to the cis well and wait several minutes looking for pore insertions. A single MspA pore will have an open pore current of ~115 pA if 180 mV is applied across the bilayer in 300 mM KCl while two MspA pores will have a current of ~230 pA. (It is important that the patch-clamp amplifier be zeroed such that when no voltage is applied across the bilayer, zero current is measured).
Once a single MspA pore has inserted into the bilayer, rinse the excess MspA from the cis well by perfusing fresh buffer into the cis well with one syringe, while pulling off excess buffer with a second waste syringe.
This procedure is as much an art as it is a science; indeed it is called the ‘painting method’ of bilayer formation. With practice, an operator can go from a primed setup to running an experiment in as little as 10 minutes.
3.3. Translocation speed control
Next, the fast translocation speed had to be addressed. Polymerases and helicases are molecular motors that move along DNA slowly enough to read the small nanopore currents. Gyarfas et al. published work using the Klenow fragment of Escherichia coli DNA polymerase (DNAP) I or the bacteriophage T7 DNA polymerase in conjunction with αHL (Gyarfas et al., 2009). Instead of attaching the DNAP to the pore, the DNAP was loaded on the DNA and then the single-stranded end of the complex was drawn into the pore until the DNAP came in contact with the pore; further motion of the DNA was limited by the motion of the DNA through the DNAP. In these early studies the DNAP- DNA complex was static, i.e. the DNAP was prevented from moving on the DNA by using a dideoxy terminated primer. This was necessary so that the DNAP would not process in bulk and DNAP-DNA complexes could be captured. In 2012, Cherf et al. refined this technique using the highly processive Bacilllus subtilis phage phi29 DNAP (Cherf et al., 2012). In this case an unextendable primer, called the blocking oligo, stalled the DNAP. Once the DNAP came into contact with the pore, the force exerted by the pore rim on the DNAP was big enough to push the DNAP to unzip the blocking oligo off the template strand. Once the blocking oligo was out of the way, the DNAP encountered a second primer on which extension was possible. In the presence of dNTPs and Mg2+ the DNAP then began synthesizing double-stranded DNA, while reeling the 5′ end of the template strand that had filed through the pore, out of the pore. In the synthesis mode, the force the DNAP exerted on the pore was bigger than the force the electrostatic field applied on the DNA. Once the DNAP pulled the 5′ end of the DNA out of the pore, the DNAP-DNA complex dissociated from the pore and the current of the unblocked pore resumed. We call the time interval during which the ion current is reduced by the presence of DNA in the pore an “event.”
3.4. First nanopore sequencing
Cherf et al.’s technique was then combined with the engineered MspA nanopore of Butler et al. The result, reported by Manrao et al. (Manrao et al., 2012), was stunning as it showed discrete and very distinct current levels ranging from 20 pA to 60 pA in 300 mM KCl at 180 mV (Figure 3b). These current levels were repeatable from one event to the next to within less than +/− 1 pA, but the durations of the levels were randomly distributed. There were more than four current levels which was to be expected based on the four nucleotides that are involved in determining the ion current (described above).
Figure 3. Phi29 DNAP controls DNA translocation via the blocking oligomer technique.
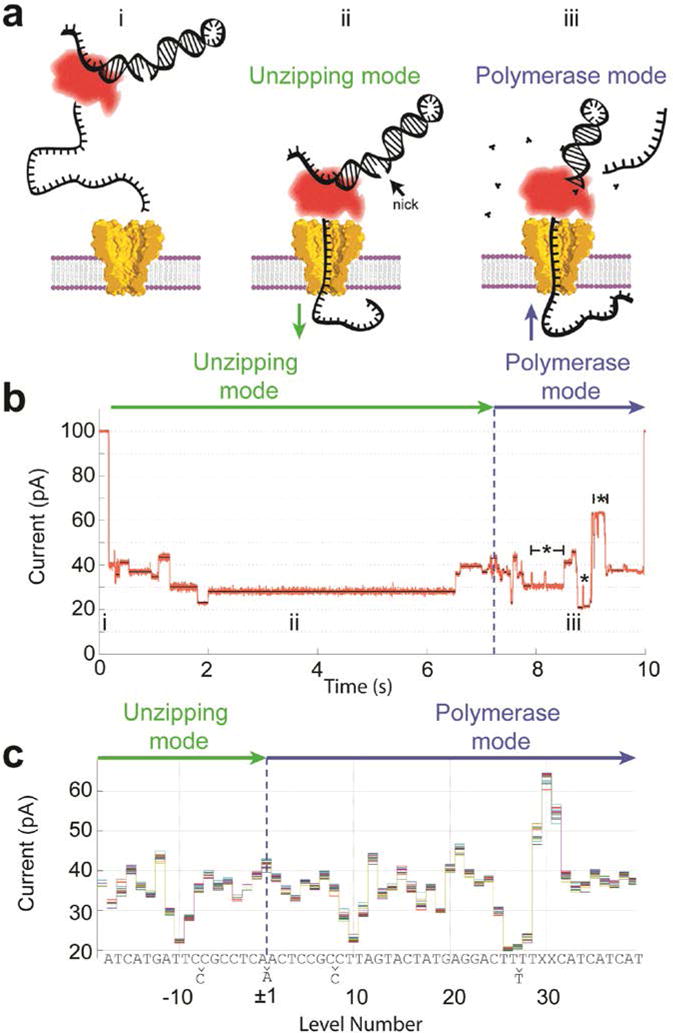
a) In bulk, phi29 DNAP loads onto the target DNA but is prevented from making double- stranded DNA by an unextendable oligomer (i). The single-stranded 5′-end inserts into the pore until the DNAP come to rest on the top of the pore. The electric force on the DNA pulls it through the phi29 DNAP unzipping the frayed end of the duplex DNA like a zipper (ii). The DNA moves nucleotide by nucleotide toward trans. Once the blocking oligomer is unzipped and dissociated (iii), the DNAP encounters the extendable end of the primer, which can be formed by a hairpinned 3′-end of the template strand. If proper buffer conditions are present, the phi29 DNAP begins extending the primer, thereby pulling the template strand back out of the pore from trans to cis. b) As phi29 DNAP steps DNA through the pore, a series of current levels are observed. Green and blue arrows indicate the current levels that correspond to unzipping and polymerase mode, respectively. Regions of the current trace that contain backsteps are indicated with a ‘*’. c) Because the durations of the observed current levels have stochastic durations, we depict observed current level amplitudes without duration information to form a “level plot.” Here we show a level plot of 20 separate single-molecule reads of identical DNA strands. The level plot displays a symmetry about the change-over from unzipping mode to polymerase mode. Each nucleotide sequence of the DNA can be associated with the current levels. In some places, adjacent levels are indistinguishable, thus we observe slightly fewer levels than there are DNA bases within the sequence. To compensate for well evaporation and electrode offsets a linear scale and offset is applied to the measured current values. Modified from (Manrao et al., 2012).
The data analysis proceeded by using an algorithm to find the beginning and end of each ion current level and determining each level’s average current amplitude. The time-ordered average current amplitudes of each level, but not their duration, were compiled into a “level plot.” Figure 3c shows the overlay of 20 level plots collected with the same DNA sequence. Occasionally the DNA skips a level or it moves back to a previous level; the level plots in Figure 3c were adjusted with local expansion or omission of levels to produce a consensus level plot.
After this step in the data analysis, a reflection symmetry in the level plot becomes apparent. When the DNAP displaces the blocking oligo in the forced unzipping mode, it walks (or better is forced to walk) along the DNA and pauses its motion at practically each nucleotide. When the DNAP begins synthesis, it walks along the DNA in the opposite direction and pauses at exactly the same positions, causing ion current levels with exactly the same current amplitudes but in opposite time order compared to the levels observed in the unzipping mode.
Figure 3b shows the current trace and level plot with a DNA that contains two abasic sites (denoted by “X” in the DNA sequence shown in Fig. 3c). These abasic sites were introduced to the DNA to produce a very large current, which can be used as a marker to align the level plot DNA sequence. With the additional knowledge that thymine (T) causes low currents the known letter sequence of the DNA can be aligned to the level plot. Nearly every nucleotide corresponded to a level in the consensus level plot with the only exception being nucleotides in homopolymer sections; nucleotides in the center homopolymer sections with more than four nucleotides produce degenerate current levels.
The 2012 Manrao et al. paper was the first demonstration that nanopore sequencing is viable and formed the basis for industrial nanopore sequencing platforms. In subsequent work, it was shown that this system is sensitive not only to the A, C, G and T, but also has exquisite sensitivity to 5-methycytosine, 5-hydroxymethylcytosine, 5- formylcytosine, and 5-carboxylcytosine (A. H. Laszlo et al., 2013; Schreiber et al., 2013; Wescoe, Schreiber, & Akeson, 2014). These epigenetically modified bases have relevance in gene expression. Each of these cytosine variants differ from C by only a few atoms, yet produce remarkably different ion current patterns.
To develop the new sequencing technology further, it was important to determine automatically the DNA sequence from the nanopore ion current reads. Since each current level is generated from contributions of four nucleotides, Laszlo et al. (Andrew H. Laszlo et al., 2014) measured the ion current of all 44= 256 four-nucleotide combinations (quadromers) to produce the “quadromer map.” This was achieved by using the 256 nt- long de Bruijn sequence (de Bruijn, 1946), which contains all 256 quadromers. Laszlo et al. then went on to measure long sections of DNA from the genome of the phage phiX 174. The 5000 nt-long phiX 174 genome was adapted with oligomers to promote loading of phi29 DNAP and insertion of single-stranded sections into the pore. Furthermore, the DNA adapters contained a cholesterol that intercalates into the bilayer, thereby increasing the density of DNA near the pore, improving the event rate (Adam & Delbruck, 1968). The measured level plots were compared with ion current level plots generated using the known sequence of phiX 174 and the ion currents of the quadromer map. Special alignment tools were developed that could align the measured level plots in the presence of significant rates of backsteps and skips. Long and highly specific alignments were found that showed a read length over a few thousand nucleotides.
De novo sequencing using hidden Markov models and the quadromer map suffered from low base calling accuracy of 60-65% for a single read. This low accuracy was dominated by enzyme behavior: (1) skipped levels (deletes), where the phi29 DNAP either made a step with too short a duration to be found by the level finding algorithm, or where the enzyme in the unzipping mode failed to pause and went forward by one or more levels, or (2) backsteps, where the enzyme reversed direction and repeated levels. Other sources of error arose due to homopolymer sections and successions of adjacent degenerate current levels.
4. Nanopore measurements turned into SPRNT
With the basic concepts of nanopore sequencing explained, we are now able to describe SPRNT. From nanopore sequencing it is already clear that a nanopore is a very useful single-molecule tool to study the properties of the enzyme that controls the DNA. Gyarfas et al. demonstrated enzyme functionality at single-nucleotide step resolution (Gyarfas et al., 2009), but their sensitivity was in part limited by using αHL instead of the much more sensitive engineered MspA porin. However, SPRNT is distinct since it goes much beyond the precision of ~6Å single nucleotide steps.
4.1. The ion current is a smooth function of position
In nanopore sequencing the DNA stops temporarily in one-nucleotide intervals. During the pauses of the DNA motion, the current is measured. From one stop to the next the ion current can change considerably. If the current change for a full nucleotide displacement of ~6 Å is ΔI, would the current for the DNA being displaced by only 2 Å be ΔI′= ΔI 2Å/6Å= ΔI/3? In other words, can one interpolate between ion current levels to find the DNA’s position with sub-nucleotide resolution? Or more generally, is the level plot the result of sampling a smoothly varying ion current curve as a function of position so that a nonlinear interpolation can be used? The hypothesis of the ion current varying smoothly with position seemed likely to be correct since up to four nucleotides were involved in determining each current level, effectively employing a low-pass filter that smoothes the underlying current function.
The first test of the hypothesis was to run a piece of DNA that has a level plot with large current changes. The level plot was measured at 180 mV and then at 140 mV. The 180 mV potential pulls the DNA with a greater force toward the trans chamber than at 140 mV. Since the DNA acts like a spring, the measurements at 180 mV place the DNA further toward trans than at 140 mV. Figure 4c shows the level plots for these two voltages fitted with smoothly varying spline interpolations. Both splines look similar in shape with the 180 mV current curve being scaled up because of the higher voltage, but most importantly, the positions of the two curves are shifted by a fraction of a nucleotide relative to each other. In Figure 4d the 140 mV data points were scaled upwards and shifted horizontally by δ= 0.29 nucleotide positions. This demonstration shows that the level plots are sampled from a common smooth curve and that the ion currents through nanopores can resolve positional changes of the DNA that are much smaller than one nucleotide.
Figure 4. Transduction of current to distance.
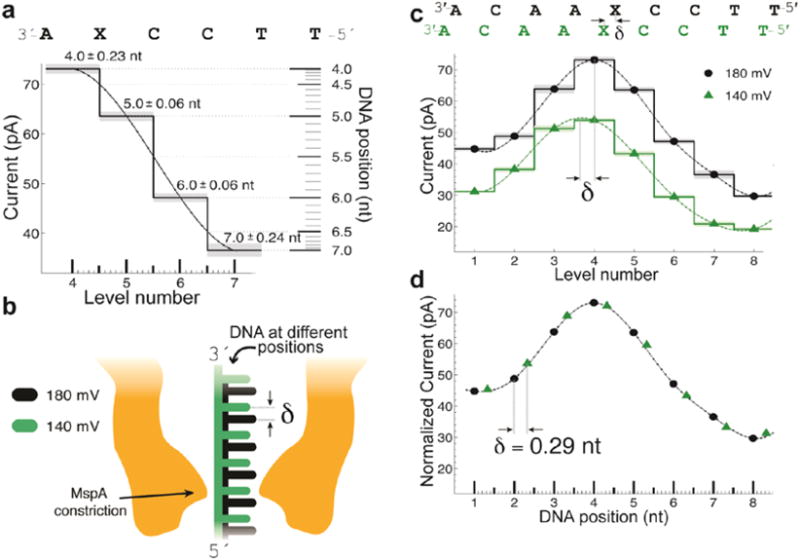
a) Regions of high current contrast can be used to measure DNA position precisely. This shows how small uncertainties in measured current translate to small positional uncertainty. b) Schematic depiction of DNA position within the pore at two different voltages; differences in the applied electric force result in different DNA extensions. c) Current levels observed for phi29 DNAP controlled motion of DNA through MspA at 180 mV and 140 mV of applied potential. A cubic spline interpolant has been applied to each set of current steps. Note that, apart from a scaling factor, the shape of the spline is identical but the peak of the spline has shifted a distance δ. d) after a linear scale and offset is applied to the two splines a horizontal displacement δ = 0.29 nt brings the two splines in line with one another. This experiment has two important results: 1) The current levels observed during single-nucleotide stepping of DNA through MspA lie along an underlying smooth curve that is well-approximated by a spline. 2) This spline provides a direct mapping from current to DNA position and we can use it to measure sub-nucleotide movement of DNA. Figure modified from (Derrington et al., 2015).
The variance of the ion current levels is typically less than one pA. In some regions with large ion current changes (Figure 4a) this variance translates to a DNA position uncertainty of ~0.06 nucleotides. With an inter-nucleotide spacing of ~6 Å this means that DNA position changes as small as ~0.4 Å (40 pm) are resolvable with each event. Such extreme position precision seems impossible, given that the DNA, the enzyme position and the pore length are subject to Brownian motion with significantly larger amplitudes. However, these fluctuations happen at much shorter time scales so that the time average (mean position) over typical level durations remains precisely determined.
SPRNT’s temporal resolution is remarkable also (see discussion in section 6 of this chapter) with large level transitions being resolvable after ~50 μs. As in all precision measurements, the time resolution and position resolution are anti-correlated. Both the time resolution and position resolution are optimal if the magnitude of current changes is large. In places along the DNA where the underlying smooth current curve is flat, e.g. at minima and maxima, SPRNT’s resolution is less good. Using the quadromer map, discussed above, specific DNA constructs can be designed that optimize SPRNT’s sensitivity.
5. Application of SPRNT: Helicase Hel308
Derrington et al. (Derrington et al., 2015) applied SPRNT to study the Hel308 helicase from Thermococcus gammatolerans (hereafter referred to as Hel308). Hel308 is an ATP-dependent Ski2-like superfamility II (SF2) helicase/translocase that unwinds duplex DNA in the 3′ to 5′ direction. The crystal structure of Hel308 is known, making it a good model system for studying SF2 helicases (Büttner, Nehring, & Hopfner, 2007; Woodman & Bolt, 2011).
A typical Hel308 experiment is performed via the following procedure:
Establish a single MspA nanopore in an unsupported lipid bilayer as described above, also see (Akeson et al., 1999; Butler et al., 2008; Manrao et al., 2012).
Perfuse the experimental buffer into the cis well (400 mM KCl, 10 mM HEPES, 1000 μM ATP, 10 mM MgCl2, 1 mM DTT, pH 8.00 ± 0.05)
Add 1 μl ~500 nM annealed experimental DNA construct (template: <m>TACTACTACTACTACTACXXTTTTTCAGGAGTATCATGATTCCCGCCTCAAA ATCAGATCTCACTATCGCATTCTCATGCAGGTCGTAGCC</m> complement: <m>CCTGCATGAGAATGCGATAGTGAGATTTTTTTTTTTTTTTTTTTTZ</m>; ‘X’ denotes an abasic residue, ‘Z’ a 3′ cholesterol tag. To anneal DNA mix template and complement at a 1:1.1 ratio and heat to 95°C for 3 min. and then slowly cool to below 30°C over 10 minutes.)
Add 1μl of 5 μM Hel308 protein to the experimental volume. Hel308 self- assembles with the DNA in vitro to form SPRNT-ready experimental constructs.
Apply 180 mV across the bilayer and record the data. The applied voltage will pull the template DNA strand into the pore initiating a SPRNT measurement. The operator should observe a few events per minute, which appear similar to the data shown in Figure 3b. If the pore clogs, it can be cleared by reversing the voltage.
Since Hel308 was expected to operate differently than the phi29 DNAP, it required a different strategy to load Hel308 onto the template DNA strand wherein a complement primer was annealed to the template strand such that an eight-base 3′ overhang remained onto which the helicase loads. The 5′ end of the primer had a (non- complementary) poly-T overhang with cholesterol at its end. In solution, Hel308 binds to the 3′-overhang on the template strand and may begin unwinding the dsDNA towards the 5′ direction. The 5′-end of the template DNA strand is drawn into the pore causing the complement strand to dissociate quickly. The Hel308 bound to the template prevents further translocation toward trans. Hel308 then becomes a ssDNA translocase, drawing the template strand out of the nanopore in the 3′ to 5′ direction, back into the cis well. Since the core of the template strand had the same DNA sequence as the DNA used in phi29 DNAP experiments, Derrington et al. expected the Hel308 level plot (Figure 5b) to look similar to that seen with phi29 DNAP in synthesis mode (Figure 5a).
Figure 5. Sub-nucleotide steps in helicase movement.
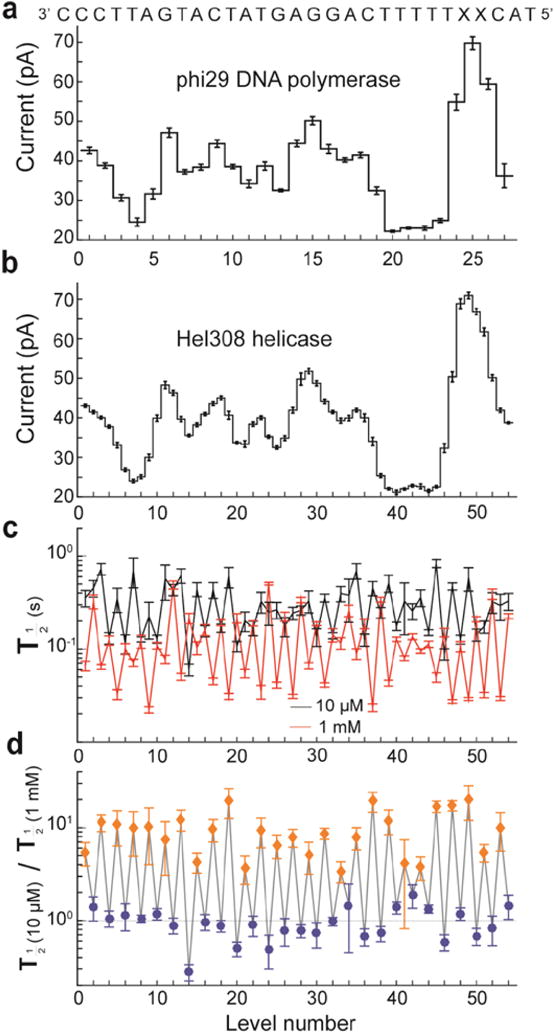
a) Levels observed when stepping the indicated DNA strand through MspA with phi29 DNAP controlling the DNA motion and b) with helicase T. gammatolerans Hel308 controlling the DNA motion. The observed current patterns are similar; however the helicase takes twice as many steps. c) Comparison of the half-lives of each level at two different concentrations of ATP. d) Ratio of observed half-lives from c). Interestingly, the duration of odd-numbered levels (orange diamonds) varies as [ATP] changes, while even-numbered levels (blue diamonds) remain unchanged. With the ability to resolve such small substates and observe their chemical dependencies, SPRNT has the potential to shed light on how such motors actually do their work. Modified from (Derrington et al., 2015).
Hel308′s consensus level plot overall structure of current peaks and troughs was similar to that with phi29 DNAP, but Hel308 had twice as many levels. Therefore, Hel308 took two steps to advance by one nucleotide. As one would expect, Hel308 level plots did not start in the same region where phi29 DNAP began synthesis.
Enzymatic steps with sub-nucleotide length had never been observed heretofore because single-molecule tools lacked the resolution. However, Büttner et al. had predicted (Büttner et al., 2007) a second kinetic step in the motion of Hel308. Placing the steps observed with Hel308 onto the phi29 DNAP spline curve revealed that the odd- and even-numbered steps corresponded to DNA being held 0.14 ± 0.03 nt toward cis and 0.41 ± 0.03 nt toward trans of the closest observed phi29 DNAP level, with the Hel308 steps being about 0.5 nt long.
To understand better the origin of these two steps, Derrington et al. measured the median level durations at 10 μM and 1 mM ATP (Fig. 5c). Taking the ratio of these rates at these two ATP concentrations (Fig. 5d) made the effect of ATP clearer: the stepping rate for one of the levels was ATP dependent but the other step was unaffected by ATP concentration. While the phi29 DNAP moved along the DNA strand in single-nucleotide steps via a Brownian ratchet model (Lieberman et al., 2013; Morin et al., 2015), Hel308 moved along the DNA strand in two steps per nucleotide, which seemed to support a power stroke model proposed by Büttner et al. (Büttner et al., 2007).
To examine the nature of the two states in Hel308′s hydrolysis cycle further Derrington et al. systematically varied the ATP concentration from 10 μM to 3 mM. Figure 6 shows the Lineweaver-Burk plot with the reaction times, averaged over all levels. Data points from ATP-dependent steps are in orange and ATP-independent ones in blue. The ATP-dependent step follows Michaelis-Menten kinetics: rate = V*[ATP] / (K + [ATP]), with V = 15.5 ± 3.5 s−1 and K = 92 ± 22 μM.
Figure 6. ATP dependence of Hel308 sub-nucleotide steps.
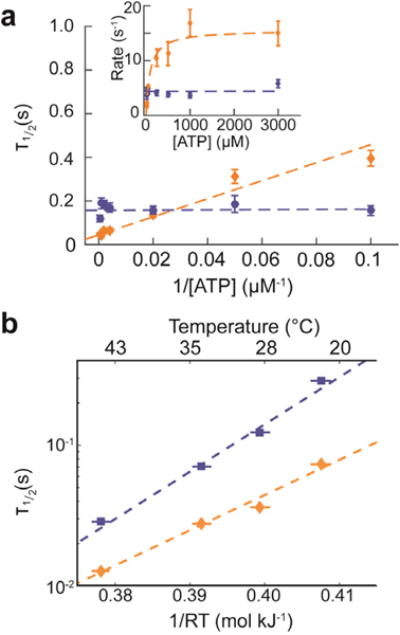
a) The half-lives for the ATP- dependent step, averaged over all ATP-dependent levels (gold) and the ATP-independent step, averaged over all ATP-independent levels (blue) as a function of 1/[ATP], with best fit lines drawn as dashed lines over both. The inset shows the inverse plot of rate (rate = ln(2) / τ1/2) vs. [ATP]. b) Temperature dependence of the ATP-dependent (gold) and ATP- independent (blue) steps and resulting linear fit assuming Arrhenius kinetics. Panel a) modified from (Derrington et al., 2015).
The duration distribution functions for each individual ATP-independent levels are well- described by an exponential distribution. Interestingly, the levels’ half-lives vary from level to level in a statistically significant way, possibly due to sequence dependence. To analyze the ATP-dependent levels, Derrington et al. first examined the high [ATP] data, where the rate-limiting step dominates. These distributions are also well described by a single exponential.
Many other experimental parameters are accessible with SPRNT. For instance, in Figure 6b we show the temperature dependence of averaged ATP-dependent and ATP- independent steps; both levels adhere to Arrhenius kinetics.
6. Advantages and limitations of SPRNT
SPRNT has many intrinsic advantages that allow for very detailed studies of enzymes that process DNA or other charged polymers. The foremost feature of SPRNT is the spatiotemporal resolution.
6.1. Spatial sensitivity
SPRNT can monitor the progression of DNA through enzymes with ~40 pm precision at enzyme state durations of ~10 ms. This resolution may become even better as the understanding of the nanopore system will be improved. To our knowledge such spatial resolution is not available with any other single-molecule tool.
Because SPRNT’s exquisite spatial sensitivity, it becomes important to define exactly the reference point on the enzyme relative to which the DNA motion is measured. This reference point is given by the contact points where the enzyme touches the rim domain of MspA. To determine these contact points, knowledge of the enzyme’s crystal structure is important.
Since SPRNT uses ssDNA as a ruler, its distance calibration is given by the inter- nucleotide spacing. For enzyme kinetic studies, nucleotide spacing (nt) is the most relevant natural unit. For applications that require reduction to absolute metric units the nucleotide spacing of ssDNA under the tension of SPRNT’s force is used for this conversion. For our experiments at 180 mV, we found 630+/−20 pm/nt for this conversion (Bosco, Camunas-Soler, & Ritort, 2013; Smith, Cui, & Bustamante, 1996) to be adequate, but it should be pointed out that this spacing depends non-linearly on voltage, on salt type, on salt concentration, as well as DNA composition.
SPRNT’s spatial resolution to DNA motion is best when measured in regions of large current changes and when measuring steps that are about one or two nucleotides long or shorter. When measuring distances that are several nucleotides long the effect of secondary structure of the DNA that stretches between the enzyme and MspA’s constriction can introduce additional uncertainty. At the forces, i.e. voltages, we use in standard measurements (180 mV), two different random 14-nucleotide DNA sections cause position uncertainty of less than ~1/10 of a nucleotide. This uncertainty can be minimized by properly selecting the DNA that is used as a ruler. For very long sections of DNA the fractional position uncertainty of SPRNT is excellent because SPRNT simultaneously sequences the DNA that is passed through the pore (see section 6.3).
6.2. Temporal sensitivity
SPRNT’s direct electronic readout allows for high bandwidth recordings of enzyme behavior. Figure 7 shows a level transition using the raw unfiltered data taken at 50 kHz sampling rate (10 kHz 8-pole Bessel filtered). Upon inspection, it is clear that some of the apparent “noise” visible in current traces is not due to instrumental and fundamental noise sources such as current amplifier noise, coupling to external noise, electrode drift or shot noise. Instead, the apparent noise is in part due to the complexities of the pore-DNA interaction and the enzyme acting on the DNA. While the former noise can be considered as a type of sensor noise, the latter noise actually is not noise, but rather contains sought after biophysically relevant information about enzyme behavior. For example, starving phi29 DNAP of one of the four dNTPs generated not only longer levels but caused the polymerase to backstep numerous times until the proper dNTP was incorporated (Cherf et al., 2012; Manrao et al., 2012). These backsteps can be very short, appearing as noise that rapidly toggles between two states.
Figure 7. Close-up view of an example Hel308-controlled current transition.
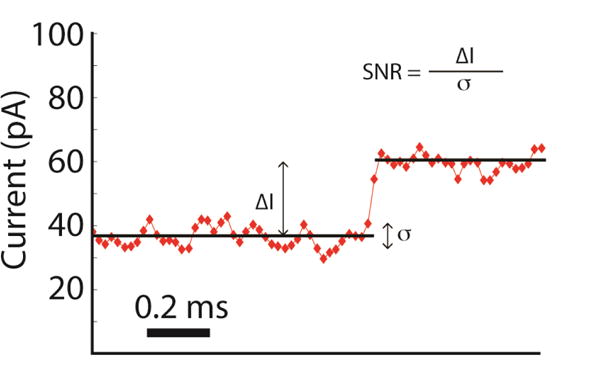
The observed step is a transition of the ATP independent state going into the ATP dependent state and therefore represents the motion of the DNA through the pore by just 0.45 ±0.04 nt. This data was acquired at a sampling frequency of 50 kHz (10 kHz Bessel filtered). The exquisite signal to noise ratio available in SPRNT enables resolution of enzymatic states as short as 200 μs with precision in the timing of such transitions to within 40 μs.
6.3. Simultaneous sequencing/absolute position
A significant bonus of SPRNT is that it simultaneously reveals the DNA sequence located within the enzyme. For example, this permits direct study of enzyme pausing (Adelman et al., 2002; Herbert et al., 2006; Neuman, Abbondanzieri, Landick, Gelles, & Block, 2003) in vitro. Pausing during transcription is particularly interesting because of expression regulation. SPRNT permits direct sequence-specific identification of pausing sites as well as observing the mechanistic behavior while the enzyme encounters the pause site.
The DNA sequence read is not the sequence located in the enzyme’s active site, but it is offset by the 10-20 nucleotides, depending on the location of the active site within the enzyme. To measure this offset exactly, one can use dNTP starvation experiments or perform mutual information (MI) analysis (Ross, 2014), correlating ion current and level duration. Our MI analysis with phi29 DNAP in the synthesis mode found this distance to be 14.5 nt, consistent with starvation experiments.
6.4. Ease of implementation
SPRNT also offers practical advantages. SPRNT requires only very limited hardware setup and the experiments can be performed with a relatively moderate amount of training and cost. In our experiments we use bilayer setups similar to those described in (Akeson et al., 1999; Butler et al., 2008). Our experimental setup is constructed from Teflon™ with PTFE heat-shrink tubing (heat shrunken around a needle and cut off to form an aperture ~20 μm in diameter) used to form the apertures. We use an Axopatch 200B integrating patch clamp amplifiers (Axon Instruments) although several other viable alternatives are available. For those not wishing to build their setups up from scratch, commercial bilayer setups with integrated electronics such as those from Nanion can be used. Some commercial bilayer recording platforms allow parallelization. Furthermore, enzymes do not need to be modified as required for attachment to beads or tethers in optical or magnetic tweezers, or by introducing fluorophores into the enzyme as required for FRET.
Because of the wealth and detail of the information uncovered with SPRNT, data interpretation can be most challenging, but once sorted out will provide a trove of new biological understanding.
7. Comparison to other single-molecule techniques
Common single-molecule techniques that are being used to study enzymes are optical tweezers (OT), magnetic tweezers (MT) and TIRF-FRET. Data analysis for each of these techniques, (as well as SPRNT) essentially boils down to looking for steps in noisy data. For experiments that are statistically limited (as opposed to being limited by systematic errors or experimental bandwidth), the position and temporal resolution limits of many single-molecule techniques are related: the precision in the position of a state (or level) that is established for a time Δt is proportional to (Δt)−½ (Moffitt, Chemla, Smith, & Bustamante, 2008).
In such cases, the signal-to-noise ratio (SNR) can be a useful metric for the spatiotemporal resolution. The SNR is given by the equation shown in Figure 7. It is the ratio of the observable step size to the standard deviation of independently sampled data points from a given state. However, direct comparison of SNR for various experiments is complicated by the fact that the data from different experiments are acquired at different bandwidths and measured step sizes of varying size, of which Figure 8 is a prime example. The SPRNT data depicted in Figure 8 is median filtered at 200 Hz and shows ~half-nucleotide steps along ssDNA, while the OT data shown is filtered at 20 Hz (pink in Fig. 8d) and depicts full nucleotide steps along dsDNA. Thus, for direct comparison of various experiments it is often useful to compute the scaled SNR by multiplying the SNR by the square root of the bandwidth and dividing by the physical step size in nm. Such scaling converts the raw SNR into the scaled SNR (sSNR), the SNR that would be observed for a 1 nm step at 1 Hz bandwidth (Moffitt, Chemla, Izhaky, & Bustamante, 2006). Using comparisons of sSNR from various experiments (Abbondanzieri, Greenleaf, Shaevitz, Landick, & Block, 2005; Derrington et al., 2015; Dulin et al., 2015; Holden et al., 2010) we generated Figure 9, which shows a log-log sensitivity plot, highlighting the large new phase-space available to SPRNT experiments. Specifically figure 9 was constructed as follows:
The diagonal line representing the spatiotemporal resolution limit for OT comes from (Moffitt et al., 2008).
Using the sSNR described by (Moffitt et al., 2006) we calculated scaled SNR values for each technique shown for representative data from Abbondanzieri et al. Derrington et al., Dulin et al. and Holden et al. Comparison of each of these sSNR values (sSNROT = 41.6, sSNRFRET = 41.6, sSNRMT = 24.3, and sSNRSPRNT = 2360) to one another yields the relative resolving power of each technique.
Taking the ratio of sSNROT/sSNRMT = 1.7, we see OT would have ~twice the signal to noise ratio of MT for observing a 1 nm step at 1 Hz bandwidth, thus the line representing the resolution limit of MT lies a factor of 1.7 higher than the line for OT. (On the time scales shown, the noise in SPRNT data is Gaussian distributed; therefore the SPRNT noise floor also follows the same (Δt)−½ power law as seen with Brownian-limited OT.)
Figure 8. Conversion of current trace to DNA displacement and comparison to OT.
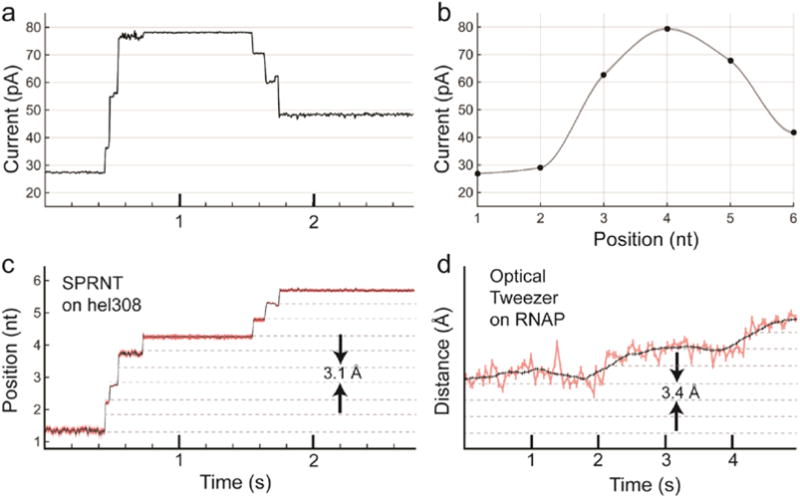
a) Raw current trace from SPRNT measurement of Hel308 helicase median filtered at 200 Hz (5 ms). b) A piecewise cubic hermite interpolating polynomial (PCHIP) interpolation of current values observed for translocation of the same DNA through an MspA pore using phi29 DNAP. c) The interpolation in b) is used to translate the currents observed in a) to DNA position. Note that the relatively uniform noise seen in the current trace is either enhanced or suppressed due to the slope of the interpolant. Regions of high slope yield precise measurements of position on short time-scales while regions of low slope yield less precise position measurements. Nanopore data is displayed at 200 Hz. d) Example OT data of single nucleotide RNAP steps from Abbondanzieri, et al. (Abbondanzieri et al., 2005) for comparison. Note, OT data in pink is filtered at 20 Hz (50 ms window) and the black is filtered at 1.33 Hz (750 ms window), while the black trace in the nanopore data is median filtered at 200 Hz (5 ms window). Figure originally appeared in (A. H. Laszlo et al., 2016).
Figure 9.
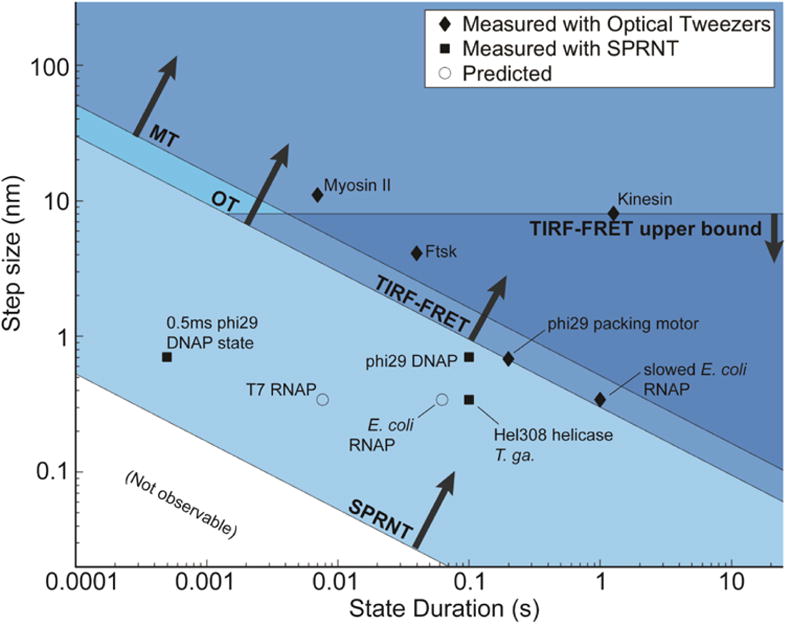
Spatiotemporal resolution of SPRNT (Derrington et al., 2015) as compared to OT (Abbondanzieri et al., 2005; Moffitt et al., 2006; Moffitt et al., 2008), MT (Dulin et al., 2015), and TIRF-FRET (Holden et al., 2010). Many nucleic acid processing enzymes work on time- scales and step sizes that are too fast and too small to be resolved with current techniques. The spatiotemporal resolution provided by SPRNT may enable study of previously unobservable kinetic steps. SPRNT has already shown its ability to measure sub-nucleotide steps on time scales that exceed the spatiotemporal resolution of OT. To provide context, black diamonds indicate average step-sizes and durations for various enzyme steps observed with OT (Abbondanzieri et al., 2005; Finer, Simmons, & Spudich, 1994; Moffitt et al., 2009; Moffitt et al., 2008; Saleh, Perals, Barre, & Allemand, 2004; Svoboda, Schmidt, Schnapp, & Block, 1993), black squares indicate enzymes measured with SPRNT (Derrington et al., 2015; Manrao et al., 2012), while open circles represent estimated step-sizes and step durations for several other enzymes based on known stepping rates and suspected step size (Seidel & Dekker, 2007). While an average step may be resolvable with a given technique, it can be useful to have spatiotemporal resolution exceeding the average stepping rate of an enzyme so that the entire stepping distribution can be observed. The square labeled “0.5ms phi29 DNAP state” shows the position of a fast, yet resolved single-step measurement (Derrington et al., 2015) that falls far closer to the noise limit than the average observed step duration. Figure originally appeared in (A. H. Laszlo et al., 2016).
In essence, SPRNT owes its high spatiotemporal resolution to the scale on which the experiment is performed. In SPRNT the distance between the enzyme and MspA’s constriction is only ~12 nt (~8 nm) which is much closer than the distance between beads in state-of-the-art dual bead optical traps (Abbondanzieri et al., 2005). In these optical traps, which can achieve 300 pm positional resolution, the length of DNA (often referred to as a tether) spanning the distance between the beads is kilobases long (~1 μm). The DNA (or other polymer) between the enzyme and the sensor forms a spring that becomes softer with length, allowing for larger Brownian motion amplitude at the sensor. Figure 8 is a direct comparison of SPRNT with an optical tweezers measurement. The SPRNT position measurement was linearized using methods above described involving a third order spline. The optical tweezers data are from Abbondanzieri et al. (Abbondanzieri et al., 2005).
All single-molecule techniques have a host of applications. Here we only discuss applications to enzymes that process along DNA, where the OT, MT, TIRF-FRET and SPRNT overlap. For example, it is clear that OT have many other applications that are not accessible with SPRNT, such as measuring processive cytoskeletal motors, e.g. kinesin. On the other hand, nanopores also have other applications such as DNA sequencing which is not feasible with OT.
8. Outlook
SPRNT is a relatively new technology ready to be used for single-molecule enzyme studies. Many improvements to SPRNT can be envisioned.
8.1 Backwards pore
In the work described here, MspA has been oriented with the periplasmic end toward trans (“forwards pore”). It is however possible to reverse the orientation of MspA in the lipid bilayer (“backwards pore”). With enzymes and DNA presented to the backwards pore, the constriction is significantly closer to the enzyme. This reduces the stretchiness of the DNA and enhances ion current contrast between adjacent nucleotides/quadromers, compared to forwards pore setups. Our recent nanopore sequencing experiments indicate that the backwards pore setup is feasible and that it has significant advantages.
The backwards pore setup can also be used with enzymes that are too small to stay on MspA’s rim domain or where MspA’s rim interferes with enzyme function. Furthermore the backwards pore provides an independent set of contact points; this can help with understanding conformational dynamics.
Since the current modulation of DNA in the constriction depends on the orientation of the DNA relative to the pore, a new quadromer map for the backwards pore is being built. In the backwards setups the presence of the enzyme near the constriction interferes with the current generation. This access resistance, which is due to the enzyme, is not observed (negligible) with the forwards pore. Therefore, backwards pore analysis is somewhat more complicated.
8.2 Asymmetric salt
Most enzymes do not function well in high salt conditions desired for high precision SPRNT measurements. We are presently studying cis-trans asymmetric salt conditions in which the cis side salt concentration is minimal. Serendipitously, MspA with DNA in the constriction becomes cation selective. Therefore, with positive voltage applied to trans (as in SPRNT), the conductance of anions from cis becomes unimportant, allowing for low salt concentration in the cis compartment near the enzyme. We have collected phi29 DNAP data with <20 mM KCl in cis and 500 mM KCl in trans and found signal-to-noise nearly equivalent to symmetric salt conditions.
8.3 Need for direct force calibration
Depending on the applied voltage, SPRNT produces 10 to 50 pN of constant force on the DNA (in the absence of access resistance). While the electrostatic force on the DNA can be estimated from the charge per nucleotide or from the DNA stretching at various voltages, the uncertainties on these force estimates are large. This necessitates a precise and model-independent force calibration. For example, such a force calibration can be established by using optical tweezers to hold DNA in MspA. Since MspA is atomistically reproducible, this calibration needs to be carried out only once and is then transferable to all SPRNT experiments using MspA.
8.4 Combination with other techniques
Combining single-molecule techniques can substantially increase information. In principle, SPRNT is amenable to be combined with optical techniques such as FRET.
8.5 Adaptation to non-DNA-translocating enzymes
Many interesting enzymes do not translocate DNA (or other polymers). In such cases SPRNT can still be used to monitor enzyme activity. DNA tethers can be attached to the domains of the enzyme of which motion is of interest. These tethers can be drawn into the pore and will modulate the ion current through MspA as the domain moves. With the high spatiotemporal resolution of SPRNT, domain motion or conformational change can be studied with substantially better precision than with TIRF-FRET.
9. Summary
SPRNT is a new single-molecule tool with many desirable properties for studying enzymes that walk on DNA. For this class of measurements, SPRNT has already been used successfully and has provided insights that could not have been obtained with other single-molecule tools. SPRNT is experimentally simple to carry out. The hardware and analysis tools capitalize on vast progress in nanopore sequencing.
Acknowledgments
This work was initiated with funding from the National Institutes of Health, National Human Genome Research Institute (NHGRI) $1000 Genome Program Grant R01HG005115.
References
- Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438(7067):460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam G, Delbruck M. Reduction of dimensionality in biological diffusion processes. San Francisco: W.H: Freeman & Co; 1968. [Google Scholar]
- Adelman K, La Porta A, Santangelo TJ, Lis JT, Roberts JW, Wang MD. Single molecule analysis of RNA polymerase elongation reveals uniform kinetic behavior. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13538–13543. doi: 10.1073/pnas.212358999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeson M, Branton D, Kasianowicz JJ, Brandin E, Deamer DW. Microsecond time-scale discrimination among polycytidylic acid, polyadenylic acid, and polyuridylic acid as homopolymers or as segments within single RNA molecules. Biophysical Journal. 1999;77(6):3227–3233. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Camunas-Soler J, Ritort F. Elastic properties and secondary structure formation of single-stranded DNA at monovalent and divalent salt conditions. Nucleic Acids Research. 2013 doi: 10.1093/nar/gkt1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TZ, Pavlenok M, Derrington IM, Niederweis M, Gundlach JH. Single-molecule DNA detection with an engineered MspA protein nanopore. Proc Natl Acad Sci U S A. 2008;105(52):20647–20652. doi: 10.1073/pnas.0807514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14(7):647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- Cherf GM, Lieberman KR, Rashid H, Lam CE, Karplus K, Akeson M. Automated forward and reverse ratcheting of DNA in a nanopore at 5- angstrom precision. Nature Biotechnology. 2012;30(4):344–348. doi: 10.1038/Nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter WH. U S P Office 1953 [Google Scholar]
- de Bruijn NG. A Combinatorial Problem. Koninklijke Netherlandse Akademie v Wetenschappen. 1946;49:758–764. [Google Scholar]
- Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Nanopore DNA sequencing with MspA. Proc Natl Acad Sci U S A. 2010;107(37):16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrington IM, Craig JM, Stava E, Laszlo AH, Ross BC, Brinkerhoff H, Gundlach JH. Subangstrom single-molecule measurements of motor proteins using a nanopore. Nat Biotechnol. 2015;33(10):1073–1075. doi: 10.1038/nbt.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin D, Cui TJ, Cnossen J, Docter MW, Lipfert J, Dekker NH. High Spatiotemporal-Resolution Magnetic Tweezers: Calibration and Applications for DNA Dynamics. Biophys J. 2015;109(10):2113–2125. doi: 10.1016/j.bpj.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller M, Niederweis M, Schulz GE. The Structure of a Mycobacterial Outer-Membrane Channel. Science. 2004;303(5661):1189–1192. doi: 10.1126/science.1094114. [DOI] [PubMed] [Google Scholar]
- Finer JT, Simmons RM, Spudich JA. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 1994;368(6467):113–119. doi: 10.1038/368113a0. [DOI] [PubMed] [Google Scholar]
- Gyarfas B, Olasagasti F, Benner S, Garalde D, Lieberman KR, Akeson M. Mapping the Position of DNA Polymerase-Bound DNA Templates in a Nanopore at 5 angstrom Resolution. ACS Nano. 2009;3(6):1457–1466. doi: 10.1021/nn900303g. [DOI] [PubMed] [Google Scholar]
- Herbert KM, La Porta A, Wong BJ, Mooney RA, Neuman KC, Landick R, Block SM. Sequence-resolved detecton of pausing by single RNA polymerase molecules. Cell. 2006;125(6):1083–1094. doi: 10.1016/j.cell.2006.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden SJ, Uphoff S, Hohlbein J, Yadin D, Le Reste L, Britton OJ, Kapanidis AN. Defining the limits of single-molecule FRET resolution in TIRF microscopy. Biophys J. 2010;99(9):3102–3111. doi: 10.1016/j.bpj.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci U S A. 1996;93(24):13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Gundlach JH. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(47):18904–18909. doi: 10.1073/Pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo AH, Derrington IM, Gundlach JH. MspA nanopore as a single- molecule tool: From sequencing to SPRNT. Methods. 2016 doi: 10.1016/j.ymeth.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Gundlach JH. Decoding long nanopore sequencing reads of natural DNA. Nat Biotech. 2014;32(8):829–833. doi: 10.1038/nbt.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman KR, Dahl JM, Mai AH, Cox A, Akeson M, Wang H. Kinetic mechanism of translocation and dNTP binding in individual DNA polymerase complexes. J Am Chem Soc. 2013;135(24):9149–9155. doi: 10.1021/ja403640b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Gundlach JH. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nature Biotechnology. 2012;30(4):349–U174. doi: 10.1038/Nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrao EA, Derrington IM, Pavlenok M, Niederweis M, Gundlach JH. Nucleotide Discrimination with DNA Immobilized in the MspA Nanopore. PLoS One. 2011;6(10):e25723. doi: 10.1371/journal.pone.0025723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Aathavan K, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457(7228):446–450. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Izhaky D, Bustamante C. Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc Natl Acad Sci U S A. 2006;103(24):9006–9011. doi: 10.1073/pnas.0603342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Smith SB, Bustamante C. Recent advances in optical tweezers. Annu Rev Biochem. 2008;77:205–228. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- Morin JA, Cao FJ, Lazaro JM, Arias-Gonzalez JR, Valpuesta JM, Carrascosa JL, Ibarra B. Mechano-chemical kinetics of DNA replication: identification of the translocation step of a replicative DNA polymerase. Nucleic Acids Res. 2015;43(7):3643–3652. doi: 10.1093/nar/gkv204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman KC, Abbondanzieri EA, Landick R, Gelles J, Block SM. Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell. 2003;115(4):437–447. doi: 10.1016/S0092-8674(03)00845-6. [DOI] [PubMed] [Google Scholar]
- Ross BC. Mutual Information between Discrete and Continuous Data Sets. PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0087357. doi:ARTN e87357 10.1371/journal.pone.0087357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh OA, Perals C, Barre FX, Allemand JF. Fast, DNA-sequence independent translocation by FtsK in a single-molecule experiment. Embo J. 2004;23(12):2430–2439. doi: 10.1038/sj.emboj.7600242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber J, Wescoe ZL, Abu-Shumays R, Vivian JT, Baatar B, Karplus K, Akeson M. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc Natl Acad Sci U S A. 2013;110(47):18910–18915. doi: 10.1073/pnas.1310615110. 1310615110 [pii]10.1073/pnas.1310615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel R, Dekker C. Single-molecule studies of nucleic acid motors. Curr Opin Struct Biol. 2007;17(1):80–86. doi: 10.1016/j.sbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Smith SB, Cui YJ, Bustamante C. Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science. 1996;271(5250):795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- Song LZ, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274(5294):1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Squires AH, Gilboa T, Torfstein C, Varongchayakul N, Meller A. Single-Molecule Characterization of DNA-Protein Interactions Using Nanopore Biosensors. Methods in Enzymology. 2017;582:353–385. doi: 10.1016/bs.mie.2016.08.010. [DOI] [PubMed] [Google Scholar]
- Stoddart D, Maglia G, Mikhailova E, Heron AJ, Bayley H. Multiple Base-Recognition Sites in a Biological Nanopore: Two Heads are Better than One. Angewandte Chemie International Edition. 2010;49(3):556–559. doi: 10.1002/anie.200905483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Schmidt CF, Schnapp BJ, Block SM. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 1993;365(6448):721–727. doi: 10.1038/365721a0. [DOI] [PubMed] [Google Scholar]
- Wescoe ZL, Schreiber J, Akeson M. Nanopores discriminate among five C5-cytosine variants in DNA. J Am Chem Soc. 2014;136(47):16582–16587. doi: 10.1021/ja508527b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman IL, Bolt EL. Winged helix domains with unknown function in Hel308 and related helicases. Biochemical Society Transactions. 2011;39:140–144. doi: 10.1042/Bst0390140. [DOI] [PubMed] [Google Scholar]