

Abstract
Thiol-ene/acrylate shape-memory polymers can be used as base substrates for neural electrodes to treat neurological dysfunction. Neural electrodes are implanted into the body to alter or record impulse conduction. This study characterizes thiol-ene/acrylate polymers to determine which synthesis methods constitute an ideal substrate for neural implants. To achieve a desired Tg between 50 and 56.5 °C, curing conditions, polymer thickness, monomer ratios, and water uptake were all examined and controlled for. Characterization with dynamic mechanical analysis and thermal gravimetric analysis reveals that thin, thiol-ene/acrylate polymers composed of at least 50 mol % acrylate content and cured for at least 1 h at 365 nm are promising as substrates for neural electrodes.
Introduction
Biotechnological advancements have created an emerging field for flexible implants that help reduce the burden of damaged body systems, such as neurological dysfunction. Although previous technologies provide promising solutions, they have their share of drawbacks, such as lack of long-term robustness and too much stiffness.1,2 For example, silicone-based neural electrodes are soft in vivo but may lack the stiffness to pierce soft tissue and have led to glial scar formation that reduces long-term stability.3 One reason is due to a chronic response from the body owing to the mechanical mismatch between the foreign, stiff electrode implants, and the significantly softer body tissue.4−8 Fortunately, flexible shape-memory polymers (SMPs) can address this problem and reduce the abiotic–biotic mismatch by being stiff during insertion, yet soft in vivo.2,9−12
A shape-memory polymer (SMP) is a polymer that can change its shape when exposed to an external stimulus, such as temperature, solvent, pH, or electricity.13−22 The temperature at which the polymer changes its shape is defined as the transition temperature (Ttrans). A shape change due to a change in environmental temperature is called a thermally induced shape-memory effect (SME). Meanwhile, a shape change due to the introduction of a solvent, typically water, is called a softening-induced shape-memory effect, which occurs due to the polymer’s molecular structure.14,23,24 When the polymer interacts with the solvent, a physical swelling effect or chemical plasticizing effect occurs between the solvent molecules and the polymer network. The solvent molecules become absorbed in the polymer and plasticize the polymer by reducing the interaction forces in the polymer network.25 This force depression makes the polymer chains more flexible and lowers the Ttrans.26 The thiol-ene/acrylate SMP system relies on both thermally induced and softening-induced shape-memory effects.
Although there has been extensive research on the SMP systems such as polyacrylate and polyurethane system, less is known about the thiol-ene/acrylate system. The thiol-ene/acrylate SMP system is a tunable system that allows for the creation of specific polymer formulas that are composed of different concentrations of monomers from thiol, alkene, and acrylate families.27,28 These monomers form a “click” reaction, meaning that the so-called mixed-mode reaction produces high-yield products with high reaction specificity and minimal byproducts while occurring in simple reaction conditions.29 In comparison to the acrylate system, the thiol-ene/acrylate system forms stable uniform networks, a low cure stress product, and is not inhibited by oxygen.10,28 Moreover, the thiol-ene/acrylate system benefits from strong interactions between the thiol groups and noble metals. This improved adhesion from the thiol-ene/acrylate polymer has proven to be successful in intracortical probes that used noble metals, such as gold, to record the neural activity while displaying long-term robustness.1,30 It has recently been demonstrated that thiol-ene/acrylate polymers are biocompatible12 and capable of sterilization with ethylene oxide,31 which makes them ideal candidates for biomedical applications.
SMPs have been extensively studied for their potential in biomedical devices, such as acting as vascular stents, self-tightening sutures, and neuroprosthetics.2,32−43 By synthesizing and characterizing a thiol-ene/acrylate SMP system, this study strives to determine how differences in the polymer structure and synthesis process affect its potential as a neural electrode substrate. We implement various fabrication methods and composition ratios to understand how to tune the glass transition temperature (Tg) so that the softening-induced SME results in the polymer being stiff near normal body temperature but soft after plasticization in bodily fluids. Dynamic mechanical analysis (DMA) test results show Tg shift based on different curing scenarios and varying monomer ratios. Meanwhile, the thermal gravimetric analysis (TGA) and swelling tests provide further valuable information regarding the degradation and expansion of the polymer, respectively. A careful examination of this combination of monomers will help advance further research on the thiol-ene/acrylate system’s potential as neuroprosthetic implants.
Results
Table 1 provides an overview of the variations in the polymer synthesis for each part of the study. In each part, one variable was altered, whereas all of the other variables were held constant, so the results would be solely due to the variable in question.
Table 1. Independent Variables in the Measurementsa.
| part I |
part II |
part III |
|||
|---|---|---|---|---|---|
| curing
conditions |
processing |
polymer
composition |
|||
| samples 1A | samples 1B | samples 2A | samples 2B | samples 3A | samples 3B |
| 31 mol % TCMDA | 31 mol % TCMDA | 31 mol % TCMDA | 31 mol % TCMDA | 10, 20, 31, 45, 50 mol % TCMDA | 10, 31, 50 mol % TCMDA |
| spin-coated | spin-coated | casted | spin-coated | spin-coated | casted |
| 365 nm | 254 nm | 365 nm | 365 nm | 365 nm | 365 nm |
| 30, 60, 120 min | 30, 60, 120 min | 120 min | 120 min | 60 min | 60 min |
Part I of the study tested how differences in the curing wavelength, as well as exposure to UV light during the polymerization process, affected the Tg. Part II tested the differences in the polymer characteristics when casted compared with when spin-coated. Part III examined how monomer mole variations affect Tg, the degradation process, and water uptake. Samples 3A and 3B were not tested against each other, but against themselves.
Part I: Impact of Curing Conditions
Part I of the study consisted of determining the most optimal curing wavelength of the polymers. All of the samples comprised of 31% tricyclodecanedimethanoldiacrylate (TCMDA), which was often used in previous studies, and were spin-coated to mimic the thinness of polymers that will be used in neural probes.44 Two categories of polymers were synthesized: 254 nm cured and 365 nm cured. The chosen wavelengths have been frequently studied in previous works.10,30,31,44 Each category contained three samples, which varied by the amount of time they were cured in their respective UV-curing chamber. Both categories’ samples were cured for 30 min, 1 h, or 2 h. The DMA was performed to determine how the curing method and time differences affect the Tg. The TGA was performed to understand differences in the polymer structure and decomposition.
The DMA measurements for the 365 versus 254 nm samples, as shown in Figure 1a, depict Tg, as denoted by the peaks of the tan δ curves, shifting for 254 nm cured polymers. As the curing time increased, the Tg increased for the 254 nm cured polymers from 58.8 to 69.3 °C. The spread of the tan δ curves also increased with curing time for polymers cured at 254 nm. In addition, the storage modulus in the rubbery regime for the 254 nm cured polymers increased with time and was greater than that in regime for the 365 nm cured polymers. Meanwhile, the samples cured at 365 nm showed no significant change in Tg as the time of curing increased. Furthermore, the TGA measurements, shown in Figure 1b, reveal that aside from the 30 min 365 nm cured sample, there were no significant differences among the curing times or curing wavelengths. The 30 min 365 nm cured sample started to degrade at a lower temperature than its counterparts.
Figure 1.

The thiol-ene/acrylate polymers cured under 365 or 254 nm UV bulbs and for various amounts of time, characterized by (a) dynamical mechanical analysis for Tg and (b) thermal gravimetric analysis for degradation characteristics.
Part II: Casting versus Spin-Coating
Part II of the study tested the differences between the casting method and spin-coating method. Thicker, casted samples are commonly used in polymer characterization studies.30,44,45 However, future applications of SMPs will use much thinner polymers as substrates for neural electrodes. The determined optimal curing method from part I of the study was held constant for the part II samples, as were the mole percentages of the monomers and the time cured in the UV-curing chamber. The DMA was performed on both the casted and spin-coated samples to determine how the methods affected the Tg. Meanwhile, the TGA was performed to reveal how the polymer structure may differ between the two methods.
The DMA measurements, as shown in Figure 2a, display the casted and spin-coated polymer with similar transition and modulus curve shapes but differing in Tg. The spin-coated sample displays a higher Tg of 39.4 °C than the casted sample, which had a Tg of 32.2 °C. As for the TGA measurements, shown in Figure 2b, the spin-coated sample starts degrading at a lower temperature than the casted sample.
Figure 2.
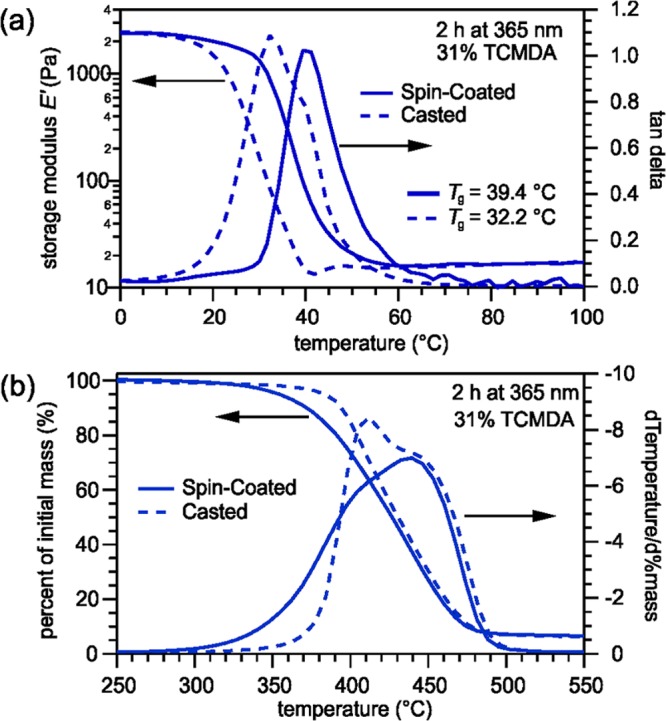
(a) DMA measurements indicating the differences in Tg between the spin-coated and casted polymers. (b) TGA measurements displaying differences in the degradation process between the spin-coated and casted polymers.
Part III: Altered TCMDA Content
In part III of the study, we wanted to understand how the mole percentages of TCMDA, the acrylate component, affected the characteristics of the polymer. In the samples 3A, the mole percentages of TCMDA varied at 10, 20, 31, 40, and 50%, whereas the spin-coating process, curing wavelength, and curing time were all held constant. The DMA was performed to determine how the acrylate percentage alters the Tg. The TGA was performed to understand differences in the degradation temperature between the varied TCMDA mole percentages and the structural character of the polymer.
The DMA performance in part III, as shown in Figure 3a, on the thiol-ene/acrylate polymers with varied ratios of TCMDA revealed an increase in Tg and a spread in the transition curve with increase in the mole percentage of TCMDA. The Tg varied from 28.0 °C for the 10% TCMDA polymer to 45.9 °C for the 50% TCMDA polymer. In addition, the TGA data on the polymers with varied TCMDA content, as shown in Figure 3b, reveal unique characteristics when taking the derivative of the degradation percentage curve. The 10 and 20% TCMDA degradation rate curves showed similar symmetrical curves that degraded uniformly, whereas the 40 and 50% TCMDA both had a front-shoulder that degraded before the rest of the polymer. The 31% TCMDA results displayed a mixture of the low-TCMDA percentage and high-TCMDA percentage curve characteristic.
Figure 3.

(a) DMA measurements displaying an increase in Tg as the percentage of TCMDA increases. (b) TGA measurements illustrating the degradation behaviors of mole-variated thiol-ene/acrylate polymers.
Despite differences between the casted and spin-coated polymers, the samples for swelling tests were made with the casting method to create larger, thicker samples to the decrease marginal errors in the accuracy-focused water uptake measurements. Swelling tests were performed to mimic physiological conditions and to understand how well the polymers withstand water uptake. Three polymers were created with the compositions of 10, 31, and 50%. Samples were casted between two glass slides with a 0.5 mm thick spacer and cured in a 365 nm UV oven for 1 h. The samples were soaked in phosphate-buffered saline (PBS) and measured at desired time periods to determine if the mole percentages of TCMDA significantly affect the water uptake.
Swelling tests results (Figure 4) from the three chosen TCMDA percentages revealed favorable results regarding fluid uptake. The water uptake percentages of the polymers were calculated as an average of five samples of each polymer. As the percentage of TCMDA decreased, the amount of swelling increased. Each of the three polymers reached a water uptake limit and leveled off within the first day. All of the polymers remained below 1.11% water uptake by volume.
Figure 4.
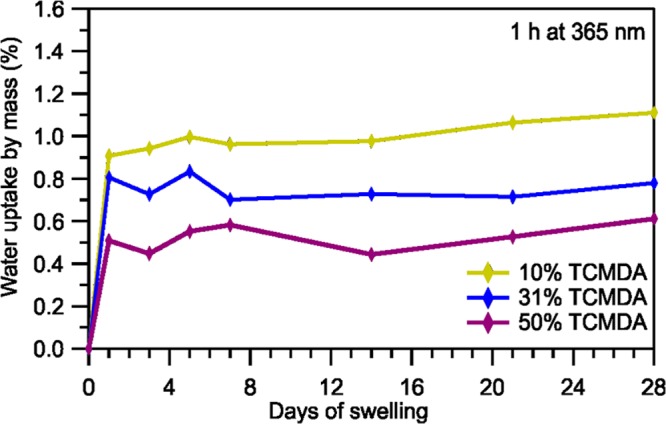
Percentage of water uptake for 10, 31, and 50% TCMDA samples over a period of 28 days.
Discussion
Determination of a suitable candidate for the polymers to act as substrates for neural electrodes requires understanding which synthesis methods are optimal and tuning Tg to the desired temperature range. The normal body temperature ranges from 36.5 to 37.5 °C but, depending on the patient, may range from 34 °C for hypothermic patients to 38 °C for those with fever. However, during the surgical process, both anesthesia and implant location have an effect on the desired range and must be taken into account. Body temperature in the limbs is generally lower than that in the core, which houses the central nervous system and is the intended implant location of the neural probes. Nonetheless, anesthesia can reduce core body temperature by 0.5–1.5 °C, thus suggesting an ideal temperature range between 35 and 36.5 °C.46 In a study by Ecker et al., Tg of the polymer decreased by 15–20 °C after being soaked in PBS, when compared with dry polymers.31 Because of the softening-induced shape-memory effect when soaking the polymer, ideal Tg values should range between 50 and 56.5 °C.
Every step of the synthesis process has the potential to alter the polymer’s Tg. Previous studies have cured the polymer under either 254 or 365 nm UV light for curing times ranging from 15 min to 2 h.10,31 Part I of the study was conducted to determine whether one UV wavelength was more optimal for curing, or if they had no effect. In part I, DMA data in Figure 1a reveal sharper tan δ curves for the 365 nm samples, when compared with the 254 nm samples, indicating a more homogeneous polymer that changes shape at approximately the same temperature.31 On the other hand, the 254 nm cured curves possess more spread, suggesting a less homogeneous polymer that slowly changes shape over a range of temperatures. Explanations for the nonhomogeneous-like characteristics may include the strength of the UV length. The 254 nm light exhibits a high energy that may have damaged the intramolecular and intermolecular bonds among the monomers, creating radicals that can react in unintended ways, such as with oxygen. As a result, the surface of the polymer has a different monomer structure than the deeper, less-exposed areas of the polymer. The minor bond-linkage differences between different regions of the polymer lead to a nonhomogeneous sample. As the length of time of exposure to the high-energy 254 nm UV light increases, more damages occur on the bonds, thus creating more nonhomogeneity and a tan δ curve with a greater spread.
Additionally, in Figure 1a, the higher storage modulus in the 254 nm cured polymers indicates more cross-linking among the monomers than that in the 365 nm cured polymers, which may create more mesh-like structures leading to less bond rotation. In addition, oxidized sulfur–oxygen double bonds may result between trimethylolpropanetris(3-mercaptopropionate) (TMTMP) and other monomers, creating less rotational freedom between the bonds. As a result of the two explanations, the more exposure to the UV light the polymer has, the less internal freedom there is in the polymer to change shape, and more energy is needed to rotate the bonds. Therefore, the Tg increases as the curing time increases.
In the TGA measurements in part I of the study, shown in Figure 1b, the 30 min 365 nm cured sample degraded at a lower temperature than the other samples because the lower-energy 365 nm UV light did not have enough time to fully polymerize the monomers. This created weaker bonds compared with those in the samples that had either more time to polymerize or were polymerized with more energy. Polymers cured at 365 nm for at least 1 h are more suitable candidates for neuroprosthetic implants compared with 254 nm cured samples because their Tg was independent of the curing time and the samples displayed greater structural homogeneity. For the 365 nm samples, there were no significant differences in the polymer characteristics when cured for 1 h than when cured for 2 h. In addition, the onset of thermal degradation in TGA measurements is vital for cleanroom processing, during which procedures may require temperatures of 300 °C or higher. A polymer with a later onset of degradation, in this case samples cured at 1 h or more, would be more suitable for future neuroprosthetic devices.
After understanding how the curing wavelength and time affected the polymer, casted and spin-coated samples were tested to determine any difference. Thin, spin-coated polymers will be used in neural probe devices, but they are often difficult to test with DMA and TGA. Previous studies have admitted to using thick, casted samples rather than thin, spin-coated samples due to mechanical testing limitations.10 Part II of the study was conducted to determine if there were significant differences between casted and spin-coated samples for future experiments. DMA results, shown in Figure 2a, revealed the spin-coated sample having a higher Tg than the casted sample due to differences in UV light energy absorption, which is consistent with the findings of Ecker et al., who studied the mechanical characteristics of thiol-ene/acrylate SMPs upon sterilization.31 Because the casted sample, 0.5 mm, is thicker than the spin-coated sample, 30 μm, the energy from the UV light may damage the surface layers more than the internal layers, thus altering its mechanical characteristics.31 Furthermore, the thickness of the casted sample may result in the UV energy being more distributed between the monomers compared with that in the spin-coated sample, which has fewer overall monomers. As a result, there may not be as many or as strong bonds in the casted samples than in the thinner samples. The fewer or weaker bonds allow for more flexibility and require less energy to rotate. So, the casted sample changes shape at a lower temperature than does the spin-coated samples and the difference between the two synthesis methods is significant.
When performing the TGA in part II of the study, the casted samples were cut with a hole-puncher and placed as a disk on the tray. On the other hand, the thin spin-coated samples were cut and folded into a zig-zag shape into a tray because a single disk-shaped sample’s mass is too minimal to provide reliable data. So, the greater surface area in the spin-coated samples allows for the greater exposure of intermolecular bonds to break due to temperature. As a result of the surface area differences, the spin-coated samples degraded before the casted samples and had a higher rate of degradation until 390 °C. Despite the casted samples being less complicated to perform with, the spin-coated and casted samples have different characteristics, and future studies should be directed more toward samples with the same thinness as those used in prospective biomedical devices.
Thin films were subsequently used to determine how monomer composition affects the polymer. Previous works have studied the thermomechanical properties of the polymer at 31% TCMDA.44 Ware et al. determined that varying the mole percentage of TCMDA in a different thiol-ene/acrylate polymer between 0 and 31% alters Tg.10 Therefore, mole variations of TCMDA can be used to precisely tune Tg to the desired physiological temperature under anesthesia to activate the polymer upon implantation.14Tg of the polymers ranging from 10 to 50% TCMDA were measured to determine how to tune the thiol-ene/acrylate polymer to have a dry Tg above 36.5 °C for stiffness during insertion but a soaked Tg below 35 °C for flexibility with tissues. A future step would comprise of conducting DMA measurements in PBS to mimic in vivo conditions to verify the transition temperature in the soaked state.
In part III, the increasing Tg with increasing TCMDA content of the study, shown in Figure 3a, is attributed to the quantity and structure of acrylates in the samples. TCMDA consist of a bridged tricyclic component that may offer less structural freedom than TMTMP, the thiol monomer, and 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO), the acrylate monomer. As a result of the greater stiffness, it takes more energy or a higher temperature, for the bonds to rotate and let the polymer change shape. In addition, the increase in the curve spread with increase in the TCMDA content is presumably due to the chemical nature of the acrylate. The acrylates have the ability to react with other acrylates, thus causing small regions containing more acrylates than others. The result is less homogeneity with the increasing TCMDA content, which may explain the wider curves. Furthermore, the thiol-ene/acrylate polymer with 50% TCMDA shows promise due to its Tg of 45.9 °C, as shown in Figure 3. This suggests it will be stiff during insertion around 35–36.5 °C while being soft in vivo with a soaked Tg presumably around 25–30 °C. Fine-tuning the Tg of the polymer to be between 50 and 56.5 °C can be achieved by studying the thiol-ene/acrylate polymer with increasing variations of TCMDA above 50%. Likewise, future DMA tests with the polymer soaked in PBS will need to be performed to confirm the degree of softening.
As for the TGA measurements in part III of the study, shown in Figure 3b, the differences between the degradation rate curves are attributed to the quantity of the monomers themselves as well as the bonds between the monomers because each sample has different absolute amounts of each monomer. Furthermore, variations may be attributed to carbon dioxide being randomly released from TMTMP and TCMDA monomers during the degradation process. Additionally, the fact that none of the samples had more than 1.11% water uptake by volume in the swelling tests, shown in Figure 4, is greatly beneficial for the neural electrodes that will be fabricated onto the polymers. Too much swelling in vivo leads to polymer expansion that could potentially destroy the neural devices due to breakage or delamination of the electronics.
Conclusions
Future application for the thiol-ene/acrylate SMP system is directed toward a base substrate for neural electrodes. In other words, neural electronics are fabricated onto the polymer and then inserted into the body. Within the body, these neural electrodes can record, stimulate, or block neural conduction. In this study, we have identified characteristics pertaining to the thiol-ene/acrylate SMP system that could lead to the development of the ideal implant with a Tg between 50 and 56.5 °C. Curing wavelength and time, polymer thickness, monomer composition ratios, and water uptake were all examined with all of the other factors controlled for. An analysis of DMA and TGA results indicate that thin thiol-ene/acrylate polymers cured at 365 nm for at least 1 h with at least 50% TCMDA show promise as polymer substrates for neural devices. This study, in combination of future work, will help pave the path for manipulating nerve conduction that has the potential to treat neurological dysfunction.
Methods
Materials
1,3,5-Triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO), trimethylolpropanetris(3-mercaptopropionate) (TMTMP), tricyclodecanedimethanoldiacrylate (TCMDA), and 2,2-dimethoxy-2-phenyl-acetophenone (DMPA) were purchased from Sigma-Aldrich and used without further purification.
Polymer Synthesis
Varying the concentration of TCMDA, while keeping the stoichiometric ratios of TATATO and TMTMP equal, has been shown to alter Tg.44 The monomer solution consisted of equal stoichiometric ratios of TATATO and TMTMP, 0.1 wt % DMPA, and TCMDA concentrations at mole percentages of 10, 20, 31, 40, and 50%. The monomers used are depicted in Figure 5a, whereas Table 1 shows which mole percentages were used for the different tests.
Figure 5.

a) Monomers used to make the thiol-ene/acrylate polymer. (b) Graphic of the casting process. Monomer solution was pipetted in between two glass slides separated by spacers of the desired thickness at two ends. (c) Two-part diagram of the spin-coating process. Monomer solution was added to the top of a spin-coater, which rotated to evenly spread the monomer solution over the surface at the desired thickness.
First, TATATO, TCMDA, and DMPA were mixed in proportionate amounts to create the appropriate mole ratios of the monomers in each sample. Immediately after the addition of the photoinitiator (DMPA), the vial was wrapped with aluminum foil to prevent polymerization from external light. A vortexing step followed to homogenize the monomer solution at 2000 rpm for 5 min in a speedmixer. TMTMP was subsequently added in the foil-wrapped vial and the solution was vortexed at 2000 rpm for 5 min for a second homogenizing step.
The sample was then either casted between two glass slides to create thick samples or spin-coated, as illustrated in Figure 5b. During the casting process, two glass slides were first cleaned with acetone and sprayed with a hydrophobic spray to allow for later removal of the polymer from the slide. Two plastic spacers 0.5 mm thick were placed on the edges between the two glass slides and clamped with a standard binder clip. The monomer solution was subsequently pipetted between the glass slides, creating a 0.5 mm thick product.10 For samples undergoing the spin-coating process instead, the monomer solution was poured onto a glass slide. A Laurell WS-650-23B spin-coater spun at 600 rpm with an acceleration of 2000 m s–2 for 25 s to create uniform 30 μm samples.31
After either casting or spin-coating the sample, the sample on the glass slide was polymerized by photoinitiating the sample in an UV cross-linker oven with either 365 or 254 nm UV bulbs for desired periods of time as depicted in Table 1. For the casted samples, the top glass slide was removed after 5 min in the oven to prevent it from blocking the UV light from reaching the polymer.31 Finally, the sample was placed in a postcuring chamber at 120 °C and 0.17 bar for 24 h. The spin-coated polymer was removed from the glass slide by soaking the sample in deionized water for 5 min and separating the polymer with a razor.44
Dynamic Mechanical Analysis (DMA)
A Mettler Toledo DMA/SDTA861e was used to perform dynamic mechanical analysis on the polymers. The 30 μm thick polymer samples were laser-cut with CO2 using a Gravograph LS100 to create rectangles 10.5 mm in length and 2.8 mm in width. Samples were heated from −10 to 150 °C at a heating rate of 2 °C min–1 in a nitrogen environment. The polymers were measured with tension at a frequency of 1 Hz, force amplitude of 2 N, and a displacement amplitude of 20 μm.10,30
Thermal Gravimetric Analysis (TGA)
A Mettler Toledo TGA/DSC 1 was used to perform thermal gravimetric analysis on the 30 μm spin-coated samples. The polymer samples were heated from 25 to 700 °C at a heating rate of 20 °C min–1 and a flow of nitrogen gas of 50 mL min–1.30 Samples were approximately 4 mg each.
Swelling Tests
The 0.5 mm casted samples were cut and soaked in a PBS solution and stored at 37 °C for a 4 week period in enclosed glass vials to simulate physiological conditions. The samples were weighed on a balance with 0.01 mg precision before the soaking period. During certain days throughout the soaking process, the samples were removed from the PBS vials and dried using a lint-free cloth. The swollen sample was weighed on the balance with 0.01 mg precision. Polymers consisting of 10, 31, and 50% TCMDA polymerized under UV light for 1 h were tested. Samples were weighed after 1, 3, 4, 5, 7, 14, 21, and 28 days of soaking. The amount of swelling was calculated as a mass change percent from samples when dry using the following formula
where ms is the weight of the swollen samples and mi is the weight of the samples when dry.30 The mass change percentage of five samples from the three different polymers were calculated and averaged to determine the final swelling percentage of the respective polymer.
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH-15-1-0607. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. Additionally, the contents do not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government. The authors want to thank Romil Modi for providing the graphics of neural interfaces for the graphical abstract.
The authors declare no competing financial interest.
References
- Ware T.; Simon D.; Hearon K.; Kang T. H.; Maitland D. J.; Voit W. Thiol-Click Chemistries for Responsive Neural Interfaces. Macromol. Biosci. 2013, 13, 1640–1647. 10.1002/mabi.201300272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware T.; Simon D.; Rennaker R. L. II; Voit W. Smart Polymers for Neural Interfaces. Polym. Rev. 2013, 53, 108–129. 10.1080/15583724.2012.751924. [DOI] [Google Scholar]
- Zhong Y.; Bellamkonda R. V. Dexamethasone Coated Neural Probes Elicit Attenuated Inflammatory Response and Neuronal Loss Compared to Uncoated Neural Probes. Brain Res. 2007, 1148, 15–27. 10.1016/j.brainres.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. P.; Capadona J. R.; Miller R. H.; Healy B. C.; Shanmuganathan K.; Rowan S. J.; Weder C.; Tyler D. J. Mechanically adaptive intracortical implants improve the proximity of neuronal cell bodies. J. Neural Eng. 2011, 8, 066011 10.1088/1741-2560/8/6/066011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capadona J. R.; Tyler D. J.; Zorman C. A.; Rowan S. J.; Weder C. Mechanically adaptive nanocomposites for neural interfacing. MRS Bull. 2012, 37, 581–589. 10.1557/mrs.2012.97. [DOI] [Google Scholar]
- Nguyen J. K.; Park D. J.; Skousen J. L.; Hess-Dunning A. E.; Tyler D. J.; Rowan S. J.; Weder C.; Capadona J. R. Mechanically-compliant intracortical implants reduce the neuroinflammatory response. J. Neural Eng. 2014, 11, 056014 10.1088/1741-2560/11/5/056014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan A.; Nguyen J. K.; Capadona J. R.; Muthuswamy J. Compliant intracortical implants reduce strains and strain rates in brain tissue in vivo. J. Neural Eng. 2015, 12, 036002 10.1088/1741-2560/12/3/036002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikov V. S.; Tresco P. A.; Reichert W. M. Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods 2005, 148, 1–18. 10.1016/j.jneumeth.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Ware T.; Simon D.; Hearon K.; Liu C.; Shah S.; Reeder J.; Khodaparast N.; Kilgard M. P.; Maitland D. J.; Rennaker R. L. II; Voit W. E. Three-Dimensional Flexible Electronics Enabled by Shape Memory Polymer Substrates for Responsive Neural Interfaces. Macromol. Mater. Eng. 2012, 297, 1193–1202. 10.1002/mame.201200241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware T.; Simon D.; Liu C.; Musa T.; Vasudevan S.; Sloan A.; Keefer E. W.; Rennaker R. L. II; Voit W. Thiol-ene/acrylate substrates for softening intracortical electrodes. J. Biomed. Mater. Res., Part B 2014, 102, 1–11. 10.1002/jbmb.32946. [DOI] [PubMed] [Google Scholar]
- Reit R.; Abitz H.; Reddy N.; Parker S.; Wei A.; Aragon N.; Ho M.; Weittenhiller A.; Kang T.; Ecker M.; Voit W. E. Thiol–epoxy/maleimide ternary networks as softening substrates for flexible electronics. J. Mater. Chem. B 2016, 4, 5367–5374. 10.1039/C6TB01082B. [DOI] [PubMed] [Google Scholar]
- Simon D. M.; Charkhkar H.; St. John C.; Rajendran S.; Kang T.; Reit R.; Arreaga-Salas D.; McHail D. G.; Knaack G. L.; Sloan A.; Grasse D.; Dumas T. C.; Rennaker R. L.; Pancrazio J. J.; Voit W. E. Design and demonstration of an intracortical probe technology with tunable modulus. J. Biomed. Mater. Res., Part A 2017, 105, 159–168. 10.1002/jbm.a.35896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.; Zhang X.; Wang M.; Ma D.; Xu M.; Li F. Thermally Stimulated Shape-Memory Behavior of Ethylene Oxide–Ethylene Terephthalate Segmented Copolymer. J. Appl. Polym. Sci. 1997, 64, 2433–2440. . [DOI] [Google Scholar]
- Lendlein A.; Kelch S. Shape-Memory Polymers. Angew. Chem., Int. Ed. 2002, 41, 2034–2057. . [DOI] [PubMed] [Google Scholar]
- Maitland D. J.; Wilson T.; Metzger M.; Schumann D. L.. Laser-Activated Shape Memory Polymer Microactuators for Treating Stroke. In Biomedical Nanotechnology Architectures and Applications; Bornhop D. J., Dunn D. A., Mariella R. P., Murphy C. J., Nicolau D. V., Nie S., Palmer M., Raghavachari R., Eds.; Spie-Int Soc Optical Engineering: Bellingham, 2002; Vol. 4626, pp 394–402. [Google Scholar]
- Beloshenko V. A.; Beigelzimer Y. E.; Borzenko A. P.; Varyukhin V. N. Shape-memory effect in polymer composites with a compactible filler. Mech. Compos. Mater. 2003, 39, 255–264. 10.1023/A:1024522211390. [DOI] [Google Scholar]
- Huang W. M.; Yang B.; An L.; Li C.; Chan Y. S. Water-driven programmable polyurethane shape memory polymer: Demonstration and mechanism. Appl. Phys. Lett. 2005, 86, 114105 10.1063/1.1880448. [DOI] [Google Scholar]
- Lendlein A.; Jiang H.; Jünger O.; Langer R. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. 10.1038/nature03496. [DOI] [PubMed] [Google Scholar]
- Buckley P. R.; McKinley G. H.; Wilson T. S.; Small W.; Benett W. J.; Bearinger J. P.; McElfresh M. W.; Maitland D. J. Inductively Heated Shape Memory Polymer for the Magnetic Actuation of Medical Devices. IEEE Trans. Biomed. Eng. 2006, 53, 2075–2083. 10.1109/TBME.2006.877113. [DOI] [PubMed] [Google Scholar]
- Mohr R.; Kraftz K.; Weigel T.; Lucka-Gabor M.; Moneke M.; Lendlein A. Initiation of shape-memory effect by inductive heating of magnetic nanoparticles in thermoplastic polymers. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 3540–3545. 10.1073/pnas.0600079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Hu J. L. Shape Memory Studies of Functionalized MWNT-reinforced Polyurethane Copolymers. Iran. Polym. J. 2006, 15, 135–142. [Google Scholar]
- Dietsch B.; Tong T. A review - Features and benefits of shape memory polymers (SMPs). J. Adv. Mater. 2007, 39, 3–12. [Google Scholar]
- Sun L.; Huang W. M. Thermo/moisture responsive shape-memory polymer for possible surgery/operation inside living cells in future. Mater. Des. 2010, 31, 2684–2689. 10.1016/j.matdes.2009.11.036. [DOI] [Google Scholar]
- Salvekar A. V.; Huang W. M.; Xiao R.; Wong Y. S.; Venkatraman S. S.; Tay K. H.; Shen Z. X. Water-Responsive Shape Recovery Induced Buckling in Biodegradable Photo-Cross-Linked Poly(ethylene glycol) (PEG) Hydrogel. Acc. Chem. Res. 2017, 50, 141–150. 10.1021/acs.accounts.6b00539. [DOI] [PubMed] [Google Scholar]
- Levine H.Water as a Plasticizer: Physico-Chemical Aspects of Low-Moisture Polymeric Systems. In Water Science Reviews 3; Franks F., Ed.; Cambridge University Press: Cambridge, U.K., 1988; Vol. 3, pp 79–185. [Google Scholar]
- Safranski D. L.; Griffis J. C.. Mechanical Properties of Shape-Memory Polymers for Biomedical Applications. In Shape Memory Polymers for Biomedical Applications, 1st ed.; Yahia L., Ed.; Elsevier Science: Cambridge, U.K., 2015; Vol. 97, pp 9–33. [Google Scholar]
- Hoyle C. E.; Lee T. Y.; Roper T. Thiol–enes: Chemistry of the past with promise for the future. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. 10.1002/pola.20366. [DOI] [Google Scholar]
- Hoyle C. E.; Bowman C. N. Thiol–Ene Click Chemistry. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Simon D.; Ware T.; Marcotte R.; Lund B.; Smith D. Jr.; Prima M.; Rennaker R.; Voit W. A comparison of polymer substrates for photolithographic processing of flexible bioelectronics. Biomed. Microdevices 2013, 15, 925–939. 10.1007/s10544-013-9782-8. [DOI] [PubMed] [Google Scholar]
- Ecker M.; Danda V.; Shoffstall A. J.; Mahmood S. F.; Joshi-Imre A.; Frewin C. L.; Ware T. H.; Capadona J. R.; Pancrazio J. J.; Voit W. E. Sterilization of Thiol-ene/Acrylate Based Shape Memory Polymers for Biomedical Applications. Macromol. Mater. Eng. 2017, 302, 1600331 10.1002/mame.201600331. [DOI] [Google Scholar]
- Feninat F. E.; Laroche G.; Fiset M.; Mantovani D. Shape Memory Materials for Biomedical Applications. Adv. Eng. Mater. 2002, 4, 91–104. . [DOI] [Google Scholar]
- Gall K.; Yakacki C. M.; Liu Y. P.; Shandas R.; Willett N.; Anseth K. S. Thermomechanics of the shape memory effect in polymers for biomedical applications. J. Biomed. Mater. Res., Part A 2005, 73A, 339–348. 10.1002/jbm.a.30296. [DOI] [PubMed] [Google Scholar]
- Sokolowski W.; Metcalfe A.; Hayashi S.; Yahia L.; Raymond J. Medical applications of shape memory polymers. Biomed. Mater. 2007, 2, S23–S27. 10.1088/1748-6041/2/1/S04. [DOI] [PubMed] [Google Scholar]
- Mano J. F. Stimuli-Responsive Polymeric Systems for Biomedical Applications. Adv. Eng. Mater. 2008, 10, 515–527. 10.1002/adem.200700355. [DOI] [Google Scholar]
- Lendlein A.; Behl M.. Shape-Memory Polymers for Biomedical Applications, In CIMTEC 2008 - Proceedings of the third International Conference on Smart Materials, Structures and Systems - Smart Materials and Micro/Nanosystems, 2008; Vol. 54, pp 96–102.
- Yakacki C. M.; Gall K. Shape-Memory Polymers for Biomedical Applications. Adv. Polym. Sci. 2009, 226, 147–175. 10.1007/12_2009_23. [DOI] [Google Scholar]
- Lendlein A.; Behl M.; Hiebl B.; Wischke C. Shape-memory polymers as a technology platform for biomedical applications. Expert Rev. Med. Devices 2010, 7, 357–379. 10.1586/erd.10.8. [DOI] [PubMed] [Google Scholar]
- Small W. IV; Singhal P.; Wilson T. S.; Maitland D. J. Biomedical applications of thermally activated shape memory polymers. J. Mater. Chem. 2010, 20, 3356–3366. 10.1039/b923717h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M. C.; Ameer G. A. Recent Insights Into the Biomedical Applications of Shape-memory Polymers. Macromol. Biosci. 2012, 12, 1156–1171. 10.1002/mabi.201200097. [DOI] [PubMed] [Google Scholar]
- Singhal P.; Small W.; Cosgriff-Hernandez E.; Maitland D. J.; Wilson T. S. Low density biodegradable shape memory polyurethane foams for embolic biomedical applications. Acta Biomater. 2014, 10, 67–76. 10.1016/j.actbio.2013.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudis S.; Behl M.; Lendlein A. Smart Polymers for Biomedical Applications. Macromol. Chem. Phys. 2014, 215, 2399–2402. 10.1002/macp.201400561. [DOI] [Google Scholar]
- Wong Y.; Kong J.; Widjaja L. K.; Venkatraman S. S. Biomedical applications of shape-memory polymers: how practically useful are they?. Sci. China: Chem. 2014, 57, 476–489. 10.1007/s11426-013-5061-z. [DOI] [Google Scholar]
- Gaj M. P.; Wei A.; Fuentes-Hernandez C.; Zhang Y.; Reit R.; Voit W.; Marder S. R.; Kippelen B. Organic light-emitting diodes on shape memory polymer substrates for wearable electronics. Org. Electron. 2015, 25, 151–155. 10.1016/j.orgel.2015.06.029. [DOI] [Google Scholar]
- Reit R.; Zamorano D.; Parker S.; Simon D.; Lund B.; Voit W.; Ware T. H. Hydrolytically Stable Thiol–ene Networks for Flexible Bioelectronics. ACS Appl. Mater. Interfaces 2015, 7, 28673–28681. 10.1021/acsami.5b10593. [DOI] [PubMed] [Google Scholar]
- Sessler D. I. Temperature Monitoring and Perioperative Thermoregulation. Anesthesiology 2008, 109, 318–338. 10.1097/ALN.0b013e31817f6d76. [DOI] [PMC free article] [PubMed] [Google Scholar]