

Abstract
Colon cancer has emerged as one of the most devastating diseases in the whole world. Mitogen-activated protein kinase (MAPK)-phosphatidylinsitol-3-kinase (PI3K) signaling hub has gained lots of attention due to its deregulation in colon cancer cells. However, selective targeting of oncogenic MAPK-PI3K hub in colon cancer has remained highly challenging, hence it has mostly been unexplored. To address this, we have engineered a hyaluronic acid layered lipid-based chimeric nanoparticle (HA-CNP) consisting of AZD6244 (MAPK inhibitor), PI103 (PI3K inhibitor), and cisplatin (DNA impairing drug) ratiometrically in a single particle. Electron microscopy (field emission scanning electron microscopy and atomic force microscopy) and dynamic light scattering were utilized to characterize the size, shape, morphology, and surface charge of the HA-CNPs. Fluorescent confocal laser scanning microscopy and flow cytometry analysis confirmed that HA-CNPs were taken up by HCT-116 colon cancer cells by merging of clathrin and CD44 receptor-mediated endocytosis along with macropinocytosis to home into acidic organelles (lysosomes) within 1 h. A gel electrophoresis study evidently established that HA-CNPs simultaneously inhibited MAPK-PI3K signaling hub with DNA damage in HCT-116 cells. These HA-CNPs stalled the cell cycle into G0/G1 phase, leading to induction of apoptosis (early and late) in colon cancer cells. Finally, these HA-CNPs exerted remarkable cytotoxicity in HCT-116 colon cancer cells at 24 h compared to that of the free triple drug cocktail as well as HA-coated dual drug-loaded nanoparticles without showing any cell death in healthy L929 fibroblast cells. These HA-coated CNPs have potential to be translated into clinics as a novel platform to perturb various oncogenic signaling hubs concomitantly toward next-generation targeted colon cancer therapy.
1. Introduction
Colon cancer has materialized as the third foremost malignancy in the whole world with 1.4 million new cases and 700 000 casualties per year.1,2 Several chemotherapeutic small molecule drugs (5-fluorouracil, oxaliplatin, capecitabin, and irinotecan) have already been approved by the FDA and are extensively used in clinics for the treatment of colon cancer patients.3−5 However, these traditional chemotherapeutic drugs extinguish noncancerous healthy cells along with rapidly growing cancer cells as collateral damage leading to severe off-target toxic side effects to the patients. However, in the last couple of decades, the advent of molecularly targeted therapy shifted the paradigm to reduce off-target toxicity.6−10 In this context, receptor tyrosine kinases (RTKs), downstream mitogen-activated protein kinase (MAPK) (involving RAS-RAF-MEK-ERK cascade), and phosphatidylinsitol-3-kinase (PI3K) (involving PI3K-Akt-mTOR cascade) signaling hubs remain highly dysfunctional in different types of cancer including colon cancer.11−13 As a result, components of MAPK and PI3K pathways served as potential targets for novel anticancer drug development.14−17 However, due to tumor heterogeneity, the emergence of drug resistance (intrinsic and extrinsic), and complex inter-/intracascade crosstalk, single pathway targeting strategies remained suboptimal and less effective.18−21 Subsequently, synchronized targeting of MAPK and PI3K signaling by polypharmacy evolved as an interesting strategy.22−27 Nevertheless, small molecule MAPK and PI3K inhibitors showed dose-limiting cardio- and immunotoxicity, developmental lethality, and hyperglycemia.28−30 Nanoscale toolkits have the promise to address these challenges.
In the last couple of decades, nanomedicine has changed the course of cancer treatment by packing multiple therapeutic entities (small molecule drugs, siRNA, microRNA, antibodies, and proteins) of different physicochemical properties in a single nano-platform.31−35 Nanocarriers have unique properties that enable them to be accumulated into malignant tissues by dysfunctional blood vessels (passive targeting).36−38 However, much improved accumulation of therapeutics into cancerous tissues in a more specific manner can be achieved by surface decoration of nanoplatforms with targeting moieties that can recognize specific marker overexpressed on tumor tissues (active targeting).39,40 Several small molecules (biotin, folic acid), nucleic acids (aptamers), proteins (antibodies), and biopolymers (carbohydrates) have been used to surface wrap the nanoplatforms for improved therapeutic efficacy of nanomedicine.41−45 In this context, recently, bio-polysaccharide hyaluronic acid (HA) has been widely explored to coat different nanoplatforms to actively target overexpressed CD44 receptors in different types of cancer cells, especially colon cancer cells.46−48 Despite having immense improvements in developing numerous nanocarriers for drug delivery into cancer tissues, targeting of the therapeutically relevant oncogenic signaling hub (MAPK-PI3K) selectively in colon cancer cells remains in its infancy for next-generation cancer treatment.49−52
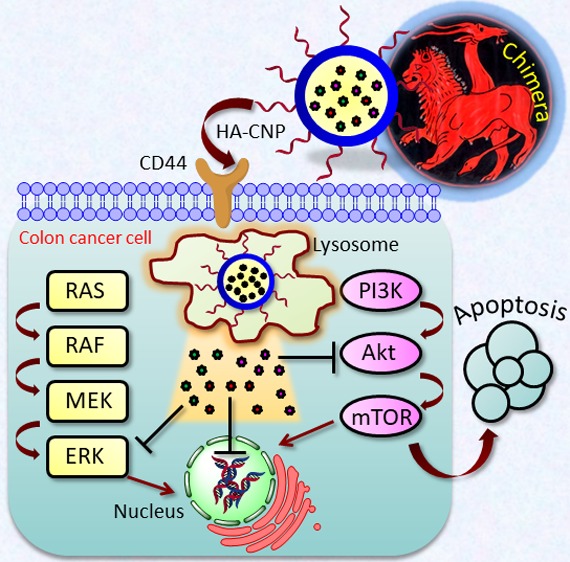
Encouraged by this less explored space, in this article, we have engineered hyaluronic acid-coated chimeric nanoparticles (HA-CNPs) (inspired by a Greek mythological creature called “Chimera” having lion, goat, and serpent in the same body) comprising AZD6244 (MAPK inhibitor), PI103 (PI3K inhibitor), and cisplatin (DNA-damaging FDA-approved drug) in a ratiometric manner. These HA-CNPs were hypothesized to be internalized more efficiently through CD44 receptor-mediated endocytosis into colon cancer cells to target MAPK-PI3K signaling hub along with cellular DNA for improved efficacy (Scheme 1c). These HA-CNPs were found to be internalized into the colon cancer cells (HCT-116) through a combination of CD44 receptor and clathrin-mediated endocytosis mechanisms followed by localization into acidic lysosomal compartments within 1 h. Simultaneous inhibition of MAPK and PI3K signaling was achieved along with DNA damage by these HA-CNPs in HCT-116 cells leading to cell cycle arrest in the G0/G1 phase, followed by induction of apoptosis (early and late stage). Finally, these HA-CNPs demonstrated remarkably improved cell killing ability compared to that of the free drug combinations in HCT-116 colon cells, without showing toxicity to the healthy fibroblast cells. This hyaluronic acid-coated chimeric nanoparticle-mediated inhibition of MAPK-PI3K signaling hub has the potential to usher in a novel therapeutic strategy toward colon cancer patients in the future.
Scheme 1. (a) Synthesis of Cholesterol–AZD6244 Conjugate and (b, c) Schematic Representations.

(b) Schematic representation of chimeric nanoparticle synthesis by blending cholesterol–drug conjugates. (c) Schematic representation of engineering of hyaluronic acid-coated chimeric nanoparticles and their mode of action into colon cancer cells.
2. Results and Discussion
2.1. Engineering Hyaluronic Acid-Coated Chimeric Nanoparticles
To target MAPK-PI3K signaling hub, we have chosen AZD6244 (MEK inhibitor) and PI103 (dual Akt-mTOR inhibitor) as both of them are currently under clinical trials.53−56 Moreover, inhibition of MAPK-PI3K signaling hub along with downstream nuclear DNA damage augmented therapeutic efficacy in cancer therapy.57−60 Therefore, we have chosen cisplatin (FDA-approved anticancer drug) as the DNA-damaging agent in combination with AZD6244 and PI103. Biocompatible and biodegradable cholesterol was used as the vector for developing the chimeric nanoparticles. Furthermore, our group recently developed cholesterol-based nanoparticles for targeting PI3K signaling to overcome drug resistance in breast cancer cells.49 Hyaluronic acid, a negatively charged polysaccharide, was applied to surface decorate the chimeric nanoparticles to target overexpressed CD44 receptors on colon cancer cells.61,62
Cholesterol (1) was first reacted with succinic anhydride to append a linker to afford the cholesterol−succinic acid conjugate (2) in 77% yield,49 followed by conjugation with AZD6244 through an ester linkage in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI)/4-dimethylaminopyridine (DMAP) to achieve the cholesterol–AZD6244 conjugate (3) in 78% yield (Scheme 1a). The cholesterol–AZD6244 conjugate was characterized by 1H, 13C, and 19F NMR spectroscopy and matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) (Figures S1–S4). We further conjugated PI103 and cisplatin (CDDP) with cholesterol through ester and Pt–O–carboxylato linkages to acquire cholesterol–PI103 (4) and cholesterol–CDDP (5) conjugates.49 To impart positive charge on the chimeric nanoparticles for further coating with negatively charged HA, we synthesized the cholesterol–ethylenediamine conjugate (6) from cholesteryl chloroformate.63
Chimeric nanoparticles (CNPs) were engineered by blending conjugates 3, 4, 5, and 6 with phosphatidylcholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polythylene glycol)] (DSPE-PEG) in a ratiometric manner (Scheme 1b).49 Size and surface charge of non-HA-coated bare CNPs were determined by dynamic light scattering (DLS). The mean hydrodynamic diameter and ζ potential were found to be 143.4 nm (polydispersity index (PDI) = 0.078) and +36.4 mV, respectively (Figure S5). These triple drug-loaded CNPs were further surface layered by incubation with hyaluronic acid to obtain HA-CNPs (Scheme 1b).50 Mean hydrodynamic diameter of the HA-CNPs was found to be increased to 176.5 nm (PDI = 0.139) (Figure 1a). On the other hand, the surface charge of the HA-CNPs was also found to be −13.9 mV (Figure 1b). This increment in hydrodynamic diameter and reversal of surface charge from positive to negative clearly confirmed that the surface of the CNPs was successfully covered by hyaluronic acid. The shape and morphology of the HA-CNPs were further visualized by electron microscopy. The field emission scanning electron microscopy (FESEM) and atomic force microscopy (AFM) images (Figure 1c,d) evidently revealed that HA-CNPs had a spherical shape and sub-200 nm size for potential accumulation into tumor tissues by passive targeting through dysfunctional vascularization.36 To confirm the presence of AZD6244 and cisplatin in the same particle, energy-dispersive X-ray spectroscopy (EDX) was performed. The elemental analysis from EDX spectra (Figure S6) confirmed the presence of Br and Pt atoms as components of AZD6244 and cisplatin. This EDX assay exhibited that HA-CNP contained inhibitors and DNA damaging drug in the same nanoparticle.
Figure 1.

Characterization of HA-CNPs by (a) DLS, (b) ζ potential, (c) FESEM, and (d) AFM.
Triple drug loading into HA-CNPs was subsequently evaluated by UV–vis spectroscopy through concentration versus absorbance calibration curves at characteristic λmax = 273, 296, and 706 nm for AZD6244, PI103, and cisplatin, respectively (Figure S7a–c). The mean loading of AZD6244, PI103, and cisplatin in the HA-CNPs was found to be 313.4 ± 4.5 μM (loading efficiency = 29.0%), 286.9 ± 7.5 μM (loading efficiency = 23.5%), and 201.9 ± 6.9 μM (loading efficiency = 16.8%), respectively (Figure S7d). HA-CNPs with this triple drug loading were used for further biological studies.
To be successfully translated to clinical applications, the nanoparticles should be stable in biological milieu for a prolonged time for effective accumulation in tumor tissues by active and passive targeting.64,65 Hence, we evaluated the stability of HA-CNPs in cell culture media for 7 days at body temperature. HA-CNPs were incubated in Dulbecco’s modified Eagle’s medium (DMEM) cell culture media containing 10% fetal bovine serum (FBS) at 37 °C, and the hydrodynamic diameter, polydispersity index (PDI), and surface charge were evaluated by DLS. It was observed that HA-CNPs showed an increase in hydrodynamic diameter from 160.4 ± 1.2 to 173.3 ± 1.0 nm at day 6 (Figure S8a). However, the size increased to 182.2 ± 3.0 nm on day 7. On the other hand, the PDI value increased marginally from 0.13 ± 0.01 to 0.0.19 ± 0.05 over 6 days with a sudden increase to 0.38 ± 0.02 on day 7 (Figure S8b). Finally, the surface charge of the HA-CNPs decreased continuously from −17.5 ± 0.1 to −8.2 ± 0.9 mV over 6 days and decreased to −6.2 ± 0.9 mV on the 7th day (Figure S8c). From this stability assay, it was evident that HA-CNPs remained stable in cell culture media for 6 days, which is enough for them to be accumulated into tumor tissues from blood circulation. However, the rapid increase in hydrodynamic diameter, PDI, and surface charge after 7 days indicated that serum proteins were absorbed on the nanoparticle surface leading to aggregation after a week.
2.2. Cellular Internalization
We hypothesized that HA-CNPs will be internalized into the acidic organelles like lysosomes through endocytosis (Scheme 1c). To evaluate the subcellular localization of HA-CNPs, we tagged the nanoparticles with a green fluorescent fluorescence isothiocyanate (FITC). We treated FITC with the cholesterol–ethylenediamine conjugate (6) in the presence of diisopropylethylamine (DIPEA) as base to obtain the cholesterol–FITC conjugate (8) (Figure S9). 1H and 13C NMR spectroscopy and MALDI-TOF spectra confirmed the structure of the cholesterol–FITC conjugate (Figures S10–S12). FITC-labeled HA-CNPs were synthesized by blending conjugates 3, 4, 5, 6, and 7 with PC and DSPE-PEG in a ratiometric manner, followed by hyaluronic acid coating to obtain HA-FITC-CNPs. The size and ζ potential of FITC-CNP and HA-FITC-CNP were determined by DLS study. The mean diameter and ζ potential of FITC-CNP were found to be 142.4 nm and +34.6 mV (Figure S13a,b). Whereas the mean diameter and surface charge of HA-FITC-CNP were determined to be 162.5 nm and −19.8 mV (Figure S13c,d). The size and surface charge of HA-FITC-CNP and HA-CNP were found to be similar, which confirmed our choice of HA-FITC-CNP for the cellular internalization study. To evaluate the cellular internalization of nanoparticles, we treated CD44 receptor overexpressed HCT-116 colon cancer cells with green fluorescent HA-FITC-CNPs at different time points (1, 3, and 6 h) followed by staining the acidic compartments by LysoTracker Red DND-99 (red fluorescent). The cellular nucleus was subsequently stained with blue fluorescent DAPI. High-resolution fluorescence confocal laser scanning microscopy (CLSM) was used to visualize the cellular localization of HA-FITC-CNPs. From Figure 2, we observed that HA-FITC-CNPs were internalized into HCT-116 cells and localized into red fluorescently labeled acidic organelles (lysosomes) within 1 h. However, over 3 and 6 h, a gradual decrease of HA-FITC-CNP internalization and localization in lysosomes was visualized. To further validate the above observation, we quantified the overlapping of green (HA-FITC-CNPs) and red fluorescence (LysoTracker Red) signals from confocal images through Pearson’s and Mander’s coefficients. Table S1 demonstrates that 77.3% volume colocalization was found within 1 h. On the other hand, the % volume colocalization of green and red fluorescence gradually decreased to 40.6 and 23.4% at 3 and 6 h, respectively. This quantification indeed supported the fluorescence confocal imaging study conclusion that HA-FITC-CNPs internalized and homed into acidic organelles within 1 h, with a gradual decrease over 6 h. We anticipated that over 6 h exposure of HCT-116 cells with HA-CNPs, all HA-binding receptors were saturated by hyaluronic acid on the nanoparticle surface, leading to reduced cellular uptake of further nanoparticles.
Figure 2.
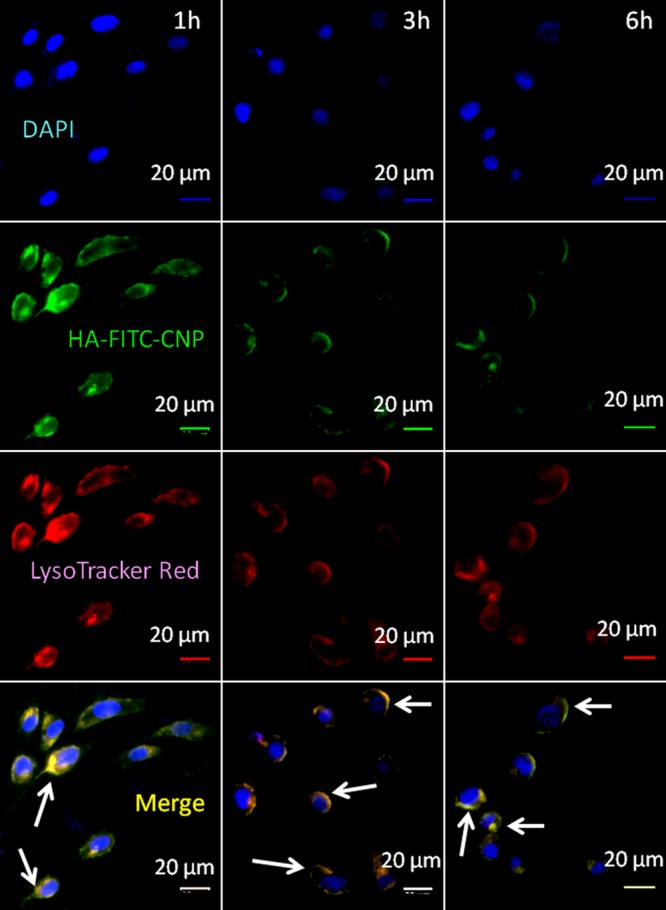
Confocal laser scanning microscopy images of HCT-116 colon cancer cells after treatment with FITC-labeled HA-CNPs (green) at 1, 3, and 6 h. Lysosomes and nuclei were stained with LysoTracker Red DND-99 (red) and DAPI (blue), respectively. Yellow regions are indicating the colocalization of HA-FITC-CNPs into lysosomes. Scale bar = 20 μm.
After localization into acidic lysosomes, the HA-CNPs should release their drug payload through cleavage of acid labile ester and Pt–O coordinate bonds.49,50 To evaluate the drug release in the acidic environment, we incubated HA-CNPs in pH = 5.5 solution (lysosome mimic), and the released drugs were quantified in predetermined time points through concentration versus absorbance calibration graphs in UV–vis spectroscopy (Figure S7a–c). At pH = 5.5, it was found that nearly 81.3 ± 3.8% of Akt-mTOR inhibitor PI103 was released after 72 h (Figure S14a). However, a slightly smaller amount (69.9 ± 8.6%) of MEK inhibitor AZD6244 was released after 72 h. On the other hand, only 58.5 ± 4.9% of DNA damaging drug cisplatin was found to be released after 72 h. We attributed the different amount of inhibitor and cytotoxic drug released in the acidic environment to the relatively labile nature of the chemical bonds through which they are linked with the cholesterol moiety. The phenolic ester linkage is highly labile in acidic media leading to a higher amount of free PI103 release from the HA-CNPs. On the other hand, the aliphatic ester linkage in the cholesterol–AZD6244 conjugate made it a little less labile in acidic media compared to the phenolic ester causing less release of AZD6244 even at 72 h compared to that of PI103. Finally, the Pt–O coordinate bond is much less labile compared to the phenolic and aliphatic ester linkages in acidic milieu. Hence, cisplatin release was much less compared to that of PI103 and AZD6244 after the same time interval (72 h). We expect that over prolonged exposure in a tumor acidic environment, HA-CNPs would constantly release the three drugs in a continuous manner.49 However, both the signaling inhibitors and the cytotoxic drug showed controlled and continuous release over 3 days. To demonstrate that the release of inhibitors and drug from HA-CNPs is pH dependent, we incubated HA-CNPs in physiological medium (pH = 7.4) and quantified drug release in a time dependent manner. It was found that only 25.0 ± 0.6, 31.8 ± 4.4, and 19.7 ± 2.9% of PI103, AZD6244, and cisplatin were released, respectively, even after 72 h (Figure S14b). It is also noteworthy that HA-CNPs released negligible amounts of inhibitors and drugs (less than 10%) within 1 h (before reaching the acidic lysosomes) at pH = 7.4, indicating that the nanoparticles can safe-guard the loaded drugs before reaching the drug release site. This drug release study evidently confirmed that HA-CNPs released their payloads by a much improved amount in an acidic environment compared to that in physiological medium. With excellent stability in cell culture medium, we anticipate that HA-CNPs would only release the drugs inside tumor tissues (acidic milieu) and show the least premature drug release in blood circulation (physiological milieu) before reaching the tumor tissues by passive and active targeting.
2.3. Mechanism of Cellular Internalization
Nanoparticles can home into the lysosomal compartments through endocytosis under different mechanisms (clathrin/caveolin-mediated endocytosis and macropinocytosis).66 The exact mechanism of endocytosis of HA-CNPs was evaluated. HCT-116 cells were pretreated with specific endocytosis inhibitors (chlorpromazine, genistein, and amiloride) followed by green fluorescent FITC-labeled HA-FITC-CNPs. As a control, HCT-116 cells were treated with only HA-FITC-CNPs without any pretreatment with endocytosis inhibitors. Cellular internalization of HA-FITC-CNPs was visualized by CLSM. Confocal microscopy images in Figure 3a evidently show that genistein-treated cells engulfed HA-FITC-CNPs in a similar way to the control cells. However, chlorpromazine pretreated cells showed remarkable reduction in HA-FITC-CNP internalization. Interestingly, on the other hand, amiloride pretreated cells demonstrated moderate uptake of HA-FITC-CNPs leading to more nanoparticle internalization compared to that of chlorpromazine-treated cells, but less compared to that of genistein-treated cells. We further evaluated the cellular uptake of HA-FITC-CNPs by quantifying the green fluorescence intensity inside the cells from confocal images. It was assessed that genistein pretreated cells showed only a 6% reduction in fluorescent intensity compared to that of no-inhibitor treated cells (Figure S15). However, chlorpromazine and amiloride pretreated cells demonstrated nearly 76 and 50% reduced fluorescent intensity compared to that of control cells. The mechanism of endocytosis was further evaluated by flow cytometric analysis. HCT-116 cells were pretreated with endocytosis inhibitors (chlorpromazine, genistein, and amiloride) followed by treatment with HA-FITC-CNPs. The fluorescently labeled cells were quantified by flow cytometry assay. The flow cytometry data clearly demonstrated that genistein-treated cells internalized HA-FITC-CNPs as well as control cells (treated with HA-FITC-CNPs only without any inhibitor pretreatment). However, interestingly, chlorpromazine- and amiloride-treated cells reduced HA-FITC-CNP uptake remarkably up to 94.5 and 47.2%, respectively (Figure 3b). As chlorpromazine, genistein, and amiloride are inhibitors for clathrin-mediated endocytosis, caveolin-mediated endocytosis, and macropinocytosis, respectively, these microscopy and cell sorting analyses confirmed that HCT-116 cells took HA-FITC-CNPs up by a combination of clathrin-mediated endocytosis and macropinocytosis mechanisms and homed them into acidic organelles like lysosomes.
Figure 3.

HCT-116 colon cancer cells were pretreated with endocytosis inhibitors (chlorpromazine, amiloride, and genestein) followed by treatment with HA-FITC-CNPs (green fluorescent) to evaluate the mechanism of endocytosis by (a) confocal microscopy. Nucleus was stained with DAPI (blue fluorescent). Scale bar = 35 μm. (b) Flow cytometry analysis.
We hypothesized that the hyaluronic acid coating over the chimeric nanoparticles containing MAPK-PI3K signaling inhibitors will help them to internalize by the interaction with the CD44 receptor overexpressed on colon cancer cells. In our previous study, we screened a panel of colon cancer cells and observed that HCT-116 cells highly overexpressed CD44 receptors.50 To evaluate our hypothesis, we pretreated HCT-116 cells with free hyaluronic acid to saturate the CD44 receptors as well as receptors for HA-mediated motility (RHAMM) followed by incubation with HA-FITC-CNPs (Figure 4a). The control cells were treated with only HA-FITC-CNPs without pretreatment with HA. The internalization of green fluorescent HA-FITC-CNPs was observed by high-resolution CLSM. From Figure 4b, it can clearly be observed that HA pretreated cells internalized a highly reduced amount of HA-FITC-CNPs giving considerably less green fluorescence signal in confocal microscopy. Whereas, without any HA pretreatment, HCT-116 cells engulfed a large amount of HA-FITC-CNPs giving rise to high fluorescence intensity in microscopy. Furthermore, the confocal imaging data was validated with flow cytometry analysis. After pretreatment with HA followed by HA-FITC-CNP incubation, the fluorescently labeled HCT-116 cells were quantified by flow cytometry. It was observed from the cytometry analysis that HA pretreated cells were labeled with HA-FITC-CNPs to a remarkably lesser extent. On the other hand, non-HA pretreated cells were highly labeled with green fluorescence from HA-FITC-CNP internalization (Figure 4c). From these fluorescence microscopy and cell sorting analyses, it was established that HA-FITC-CNPs were internalized into lysosomes of HCT-116 cells through HA-CD44 interaction mediated endocytosis.
Figure 4.

(a) Schematic representation of HCT-116 colon cancer cells with or without pretreatment with hyaluronic acid followed by incubation with HA-FITC-CNPs. (b) CLSM images of HCT-116 cells pretreated with or without HA followed by incubation with HA-FITC-CNPs (green fluorescent). Nucleus was stained with DAPI (blue). Scale bar = 35 μm. (c) Flow cytometry analysis of HCT-116 cells pretreated with or without HA followed by HA-FITC-CNP treatment.
2.4. Targeting MAPK-PI3K Signaling Hub
It was hypothesized that after internalization into acidic lysosomes, HA-CNPs will release AZD6244 and PI103 for simultaneous inhibition of MAPK-PI3K signaling hub. To validate our hypothesis, we treated HCT-116 cells with HA-CNPs for 24 h and the expression of MAPK and PI3K signaling proteins was evaluated by western blot analysis. Phosphorylation of extracellular signal regulated kinase (ERK) in MAPK signaling cascade should be inhibited by AZD6244 through perturbation of MEK. From western blot images in Figure 5a, it was observed that expression of p-ERK was highly reduced after treatment with HA-CNPs whereas the amount of total ERK remained unperturbed. Quantification from the protein-expression analysis also validated that HA-CNPs reduced the expression of p-ERK by 2.5-fold compared to that of the non-nanoparticle treated control cells (Figure S16a). On the other hand, PI103 was expected to inhibit PI3K signaling by inhibiting phosphorylation of Akt. To evaluate the effect of the nanoparticles on PI3K signaling, we treated HCT-116 cells with HA-CNPs for 24 h and the expression of p-Akt was determined by western blot analysis. The gel electrophoresis in Figure 5a clearly demonstrates that HA-CNPs reduced the expression of p-Akt while keeping the total amount of Akt protein the same. Further quantification from western blot analysis showed that HA-CNPs reduced the expression of p-Akt by nearly 1.5-fold compared to that of the control cells (Figure S16b). Finally, we evaluated the effect of cisplatin-mediated DNA damage in HCT-116 cells. The poly (ADP-ribose) polymerase (PARP) family of proteins are upregulated as a consequence of DNA damage in cancer cells, making them the markers for DNA damage.67,68 HCT-116 cells were treated with HA-CNPs for 24 h and the expression of PARP was visualized through Western blot analysis. The gel electrophoresis in Figure 5a shows that HA-CNPs increased the expression of PARP through DNA damage. Further quantification from western blot analysis revealed that HA-CNPs increased the expression of PARP by 7.4-fold compared to that of the non-nanoparticle treated cells (Figure S16c). These gel electrophoresis studies evidently confirmed that HA-CNPs inhibited MAPK-PI3K signaling hub simultaneously with DNA damage in colon cancer cells.
Figure 5.

In vitro assays of HA-CNPs in HCT-116 cells at 24 h post-incubation. (a) Expression of p-ERK1/2, p-Akt, and PARP were shown by gel electrophoresis as the marker for MAPK-PI3K inhibition and DNA damage. (b) Cell cycle arrest at the G0/G1 phase induced by HA-CNPs determined by flow cytometry analysis. (c) Induction of early and late apoptosis by HA-CNPs was determined by flow cytometry analysis. (d) Cell viability of HCT-116 cells after treatment with HA-CNPs and free drug cocktail for 24 h was determined by MTT assay.
2.5. Cell Cycle Arrest and Apoptosis
Inhibition of MAPK-PI3K signaling hub along with DNA damage leads to cell cycle arrest.69,70 We assessed the potential of HA-CNPs to arrest the cell cycle of colon cancer cells. HCT-116 cells were treated with HA-CNPs for 24 h followed by staining the cellular DNA with propedium iodide (PI), and the cells in different stages of the cell cycle were quantified through flow cytometry analysis. The flow cytometry analysis in Figure 5b reveals that HA-CNP treated cells were in the G0/G1, S, and G2/M phases in 87.86, 6.39, and 4.9%, respectively. Whereas, control cells were found to be in the G0/G1, S, and G2/M phases in 24.26, 11.65, and 65.45%, respectively. From this cytometry analysis, it was confirmed that HA-CNPs stalled the cell cycle in the G0/G1 phase in HCT-116 cells.
Cell cycle arrest into the G0/G1 phase would lead the cancer cells into programmed cell death or apoptosis.64 We further evaluated the induction of apoptosis by HA-CNPs through flow cytometry analysis. HCT-116 cells were treated with HA-CNPs for 24 h and the phosphatidylserine flipped at the outer surface of the apoptotic cells was stained with green fluorescent FITC-labeled Annexin V. The DNA of the late apoptotic and necrotic cells was counter stained with red fluorescent propedium iodide. The cells in different apoptotic and necrotic stages were quantified with fluorescence cell sorting analysis. The flow cytometry analysis in Figure 5c demonstrated that HA-CNPs induced a remarkably higher number of cells into early (60.08%) and late (30.06%) apoptosis. On the other hand, non-nanoparticle treated control cells showed only 7.21 and 6.85% of cells in early and late apoptotic stages, respectively. This cytometry data clearly confirmed that HA-CNP mediated inhibition of MAPK-PI3K signaling hub in combination with DNA damage followed by cell cycle arrest in the G0/G1 phase triggered the colon cancer cells into apoptosis.
2.6. Cell Death
One of the hallmarks of cancer is to resist programmed cell death or apoptosis.71 Hence, induction of apoptosis to the cancer cells would lead to cell death. We evaluated the potential of HA-CNPs to trigger cell death in colon cancer cells. HCT-116 cells were treated with HA-CNPs in a dose-dependent manner over 24 h, and the amount of viable cells was determined by MTT assay. As a control, we treated HCT-116 cells with a cocktail of free inhibitors (AZD6244 and PI103) and cisplatin in the same ratio present in HA-CNPs (AZD6144/PI103/CDDP = 1.5:1.4:1). It was found that HA-CNPs killed HCT-116 cells with an extremely reduced IC50 = 0.91 ± 0.09 μM with only 13.7 ± 1.2% viable cells at 25 μM concentration of AZD6244 (Figure 5d). Whereas a combination of free AZD6244, PI103, and cisplatin showed much a higher IC50 = 5.7 ± 0.8 μM with high cell viability (35.7 ± 5.8%) at 25 μM concentration of AZD6244. To assess whether the HA-CNPs were more effective compared to their dual drug-loaded counterparts; we synthesized HA-AZD-CDDP-NPs, HA-AZD-PI103-NPs, and HA-PI103-CDDP-NPs by using the same method and having the same drug loading. We further treated HCT-116 cells with HA-coated dual drug-loaded nanoparticles in a dose-dependent manner for 24 h and cell viability was quantified by MTT assay. Interestingly, it was observed that HA-AZD-CDDP-NPs, HA-AZD-PI103-NPs, and HA-PI103-CDDP-NPs showed much higher IC50 = 2.96 ± 0.33, 13.0 ± 1.13, and 6.05 ± 0.86 μM, respectively, compared to that of HA-CNPs (Figure S17a–c). Moreover, HA-AZD-CDDP-NPs, HA-AZD-PI013-NPs, and HA-PI103-CDDP-NPs demonstrated much less efficacy in killing HCT-116 cells in the highest concentration (25 μM), as indicated by high cell viability, 24.6 ± 4.4, 47.2 ± 3.1, and 34.9 ± 5.7%.
One of the major challenges of nanoparticle-mediated targeting of MAPK-PI3K signaling hubs in cancer cells specifically is not to induce any off-target toxicity to the healthy cells. To address this issue, we further evaluated the effect of HA-CNPs on L929 fibroblast cells. L929 cells were incubated with HA-CNPs in a dose-dependent manner for 24 h and the cellular viability was measured by MTT assay. Interestingly, it was found that, HA-CNPs showed almost no toxicity to the fibroblast cells even at the highest concentration (cell viability = 107.1 ± 4.5% at 25 μM concentration of AZD6244) (Figure S17d). From these cell viability assays, it was confirmed that HA-CNPs showed remarkable efficacy in HCT-116 colon cancer cells when compared to that of the free drug cocktail as well as the HA-coated dual drug-loaded nanoparticles. Moreover, HA-CNPs showed negligible toxicity toward healthy fibroblast cells, which showed their benign nature toward noncancerous cells.
3. Conclusions
In conclusion, we have engineered sub-200 nm size hyaluronic acid-coated cholesterol-based chimeric nanoparticles (HA-CNPs) that can simultaneously enclose MAPK-PI3K signaling hub inhibitors (AZD6244 and PI103) along with DNA impairing drug (cisplatin) in a ratiometric manner. These HA-CNPs were internalized into HCT-116 colon cancer cells through a combination of clathrin-mediated endocytosis, macropinocytosis, and CD44 receptor-mediated endocytosis, and they were homed into lysosomes. MAPK-PI3K signaling hub was inhibited in combination with DNA damage by these HA-CNPs. Furthermore, HCT-116 cells were stalled in the G0/G1 phase in the cell cycle, leading to early and late apoptosis by HA-CNPs. Finally, remarkable cell death was induced by HA-CNPs on HCT-116 cells compared to that of the free drug combinations as well as HA-coated dual drug-loaded nanoparticles keeping healthy fibroblast cells unharmed. These HA-CNPs have the potential to be a platform technology for targeting multiple oncogenic signaling hubs toward next-generation targeted therapy for colon cancer.
4. Experimental Section
4.1. Materials
Cisplatin, MTT reagent, DAPI, and propedium iodide were procured from Sigma-Aldrich. PI103 and AZD6244 were purchased from Selleck Chemicals. DMEM media, LysoTracker Red DND-99 were obtained from Life Technologies. HCT-116 cells were obtained from National Centre for Cell Science (NCCS), Pune. Anti-phospho-Akt (Thr308) rabbit monoclonal antibody, anti-ERK1/2 phospho monoclonal antibody, anti-total-AKT antibody, anti-total ERK1/2 antibody, anti-GAPDH antibody, HRP goat anti-mouse IgG antibody, and apoptosis detection kit were purchased from BioLegend. All chemical reactions were carried out under an argon atmosphere using dry solvents unless otherwise stated.
4.2. Synthesis of Cholesterol–Succinic Acid Conjugate (2)
Cholesterol–succinic acid conjugate (2) was synthesized from cholesterol (1) by using the procedure previously described.49 In short, cholesterol (100 mg, 0.26 mmol) was dissolved in 2 mL of a pyridine/dichloromethane (DCM) mixture (1:1 vol/vol) followed by addition of succinic anhydride (130 mg, 1.3 mmol) and a catalytic amount of DMAP. The reaction was stirred at room temperature for 24 h. The progression of the reaction was monitored by thin layer chromatography (TLC). After the starting material was completely consumed, pyridine and DCM were evaporated, and the crude compound was further dissolved in 5 mL of DCM. Crude product in DCM was washed with 1 N HCl (10 mL) and water (10 mL). The organic layer was then separated using a separating funnel and dried over anhydrous Na2SO4. Crude product was filtered and DCM was evaporated under reduced pressure. Pure cholesterol–succinic acid conjugate (2) was obtained in 77% yield after purification through silica gel column chromatography using 1% methanol in DCM as mobile phase.
4.3. Synthesis of Cholesterol–AZD6244 Conjugate (3)
Cholesterol–succinic acid conjugate (2) (5 mg, 0.01 mmol) was dissolved in dry dimethylformamide (DMF) (2 mL) followed by addition of EDCI (5.68 mg, 0.015 mmol) and DMAP (2.5 μL, 0.015 mmol) into the reaction mixture and stirred for 10 min. AZD6244 (6.9 mg, 0.012 mmol) was added into the reaction mixture and stirred for 24 h at room temperature. The progression of the reaction was followed by thin layer chromatography. To quench the reaction, 0.1 N HCl solution was added into the reaction mixture. The organic layer was washed with water (10 mL × 2) and brine (5 mL × 2). The organic layer was dried over anhydrous Na2SO4, filtered, and finally, the solvent was evaporated under reduced pressure. Crude product was purified by silica gel column with dichloromethane/methanol as the eluent to afford a pure cholesterol–AZD6244 conjugate (3) in 78% yield.
4.4. Synthesis of Cholesterol–PI103 Conjugate (4)
Synthesis and characterization of cholesterol–PI103 conjugate were performed by a previously described method.49 In short, cholesterol–succinic acid conjugate (2) (21 mg, 0.04 mmol) was dissolved in dry DMF (2 mL) followed by addition of EDCI (8 mg, 0.04 mmol) and DMAP (5 mg, 0.04 mmol). The reaction was stirred at room temperature for 30 min. PI103 (5 mg, 0.014 mmol) was added into the reaction mixture and stirred for another 24 h at room temperature. After the complete consumption of starting material (monitored by TLC), the reaction was quenched with 1 N HCl (5 mL) and diluted with DCM (10 mL). The organic layer was washed with water (10 mL × 2) and brine (10 mL). The organic layer was then separated and dried over anhydrous Na2SO4 followed by filtration. Organic solvent was further evaporated under vacuum to obtain crude product. Pure cholesterol–PI103 conjugate (4) was obtained in 72% yield after purification through silica gel column chromatography using 1% methanol in DCM as mobile phase.
4.5. Synthesis of Cholesterol–Cisplatin Conjugate (5)
Cholesterol–cisplatin conjugate (5) was synthesized by previously described methods.63 Cholesterol–succinic acid conjugate (2) (5 mg, 0.01 mmol) was dissolved in 1 mL of DMF and aquated cisplatin (600 μL = 3.08 mg, 0.01 mmol) was added into it. The reaction mixture was stirred for 24 h. After 24 h, solvent was evaporated by rotary evaporator and the crude cholesterol–succinic acid–cisplatin conjugate (5) was obtained in 90% yield.
4.6. Synthesis of Cholesterol–Ethylenediamine Conjugate (6)
Cholesterol–ethylenediamine conjugate (6) was synthesized starting from cholesteryl chloroformate (7) and ethylenediamine.63 Briefly, ethylenediamine (1 mL, 15.6 mmol) was dissolved in dry DCM (5 mL) and cooled to 0 °C. Cholesteryl chloroformate (7) (500 mg, 1.1 mmol) was dissolved into 5 mL of dry DCM and added drop wise into cold ethylenediamine solution for 20 min with stirring. The reaction mixture was allowed to come to room temperature and stirred for 24 h. The reaction was quenched with water (10 mL) and washed thoroughly with water (15 mL × 3). The organic layer was further washed with brine (5 mL) followed by drying over anhydrous sodium sulfate, it was then filtered and evaporated under reduced pressure to obtain cholesterol–ethylenediamine conjugate (6) in 76% yield.
4.7. Synthesis of Cholesterol–FITC Conjugate (8)
Conjugate 6 (10 mg, 0.0211 mmol) was dissolved in 3 mL of anhydrous DMF. To this solution, 12.3 mg (0.031 mmol) of fluorescence isothiocyanate (FITC) was added followed by the addition of 3.6 μL (2.2 mmol) of DIPEA. The reaction was stirred in the dark at room temperature for 24 h. Progression of the reaction was followed by TLC. The reaction was quenched with water (5 mL), and 30 mL of dichloromethane was added into it. The organic layer was washed with water (30 mL × 3). The organic layer was separated, dried over anhydrous Na2SO4, and evaporated under reduced pressure. Crude product was purified by silica gel column with dichloromethane/methanol as the eluent to afford pure cholesterol–FITC conjugate (8) in 60% yield.
4.8. Synthesis of Hyaluronic Acid-Coated Chimeric Nanoparticles
Synthesis of chimeric nanoparticles was performed by using the procedure described in ref (49). Briefly, 6 mg of l-α-phosphatidylcholine, 1 mg of each of the cholesterol–drug conjugates (3, 4, and 5), 3 mg of cholesterol–ethylenediamine conjugate (6), and 0.6 mg of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polythylene glycol)2000] (DSPE-PEG2000) were dissolved in 5.0 mL of DCM in a round bottom flask. Organic solvent was slowly evaporated under vacuum to form a thin and uniform lipid layer with constant rotation in the rotary evaporator. Any trace of organic solvent was removed under high vacuum for 30 min. The drug-containing lipid layer was further hydrated with 1 mL of double distilled water for 2 h at 60 °C. The nanoparticles formed after the hydration step were passed through a Sephadex G-25 column to remove unwanted bigger particles. After passing through the Sephadex size exclusion column, the nanoparticles were extruded through (minimum 15 cycles of extrusion) a 200 nm Whatmann polycarbonate membrane at 60 °C to obtain the CNPs. Further, hyaluronic acid was coated on the CNPs to obtain HA-CNPs by the protocol previously described.50 Typically, to the above resultant CNP solution, 0.5 mL of (0.5 mg/mL) hyaluronic acid solution was added drop wise under rigorous stirring. After 10 min, hyaluronic acid-coated CNPs were centrifuged at 45 000g. The HA-CNP pellets were washed with water to remove the excess hyaluronic acid. HA-CNPs were resuspended in water and stored at 4 °C for further use.
4.9. Size, Surface Charge, Shape, and Morphology Determination
Hydrodynamic diameter and surface charge of CNPs and HA-CNPs were determined by dynamic light scattering using a Zetasizer Nano2590 (Malvern, U.K.). CNPs or HA-CNPs (30 μL) were suspended in 1 mL of double distilled water and scattering of light was measured at a 90° angle to obtain the hydrodynamic diameter. The measurements were performed in triplicate.
HA-CNPs (5 μL) in water were drop-casted on either a silicon chip without dopant (for FESEM) or on a mica sheet (for AFM) and dried under reduced pressure for 2 h. For FESEM, the silicon chip was then coated with gold having 30–40 nm layers by a Quorum Q150T-E5. A Carl Zeiss, Ultra plus SEM was used at 4 kV voltage to measure the FESEM images. NanoWizard AFM was used for force microscopy.72
4.10. Quantification of Triple Drug Loading in HA-CNPs
Concentration versus absorbance calibration graphs were determined from UV–vis spectroscopy at λmax = 296, 267, and 706 nm for PI103, AZD6244, and cisplatin, respectively. For quantification of drug loading, HA-CNPs were dissolved in dimethyl sulfoxide (DMSO) in three different concentrations (5, 10, and 15%) and absorbance was measured. Loading of PI103 and AZD6244 was measured from absorbance versus concentration graphs. To evaluate cisplatin loading, 100 μL of HA-CNPs was dissolved in 400 μL of o-phenelenediamine solution in DMSO (1.2 mg/mL). The mixture was heated at 100 °C for 4 h until a bluish-green coloration appeared. Absorbance of the solution was measured at λmax = 706 nm and cisplatin loading was calculated from the calibration curve. Drug loading was quantified in triplicate.49,63
4.11. Drug Release from HA-CNPs
Release of PI103, AZD6244, and cisplatin was determined by a dialysis method in pH = 5.5 and 7.4.49 The amount of PI103, AZD6244, and cisplatin released at predetermined time points was quantified by UV–vis spectroscopy at λmax = 296, 267, and 706 nm, respectively. Released PI103, AZD6244, and cisplatin at each time point were quantified in triplicate.
4.12. Stability Determination of HA-CNPs
Stability of HA-CNPs was assessed by DLS. 100 μL of HA-CNPs was incubated in DMEM media having 10% FBS at 37 °C for 7 days. Hydrodynamic diameter, polydispersity index, and surface charge were evaluated at different time points and plotted.
4.13. Cellular Internalization by Confocal Microscopy
A total of 50 000 HCT-116 cells were seeded on coverslips in a six-well plate. After 24 h, cells were treated with HA-FITC-CNPs at an FITC concentration of 2 μg/mL at 37 °C for 1, 3, and 6 h. After each time point, the cells were washed with phosphate-buffered saline (PBS) and fixed with paraformaldehyde solution (4%) for 15 min at 4 °C. Lysosomes and nuclei were stained by LysoTracker Red DND-99 and DAPI for 45 min and 5 min, respectively. Cells were washed with PBS and mounted on a glass slide using 5 μL of SlowFade diamond antifade reagent. The cells were visualized by confocal microscopy (Zeis LSM 710). HCT-116 cells were pretreated with different endocytosis inhibitors followed by treatment with HA-FITC-CNPs. The cellular internalization was further visualized by confocal microscopy.50
For hyaluronic acid-mediated cellular uptake, HCT-116 cells in six-well plates were treated with 2 mL of serum-free cell culture media containing 5 mg/mL HA for 2 h followed by treatment with HA-FITC-CNPs for a further 2 h. The cells were then visualized by confocal microscopy.
4.14. Cellular Internalization by Flow Cytometry
A total of 105 HCT-116 cells were treated with various endocytosis inhibitors (amiloride, genistein, and chlorpromazine) at a concentration of 100, 30, and 37 μM, respectively, for 30 min at 37 °C. Control cells were incubated at 37 °C without any treatment of inhibitor. The fluorescently labeled cells were counted by using a BD FACS Calibur.50
For hyaluronic acid-mediated cellular uptake, HCT-116 cells in six-well plates were treated with 2 mL of serum-free cell culture media containing 5 mg/mL HA for 2 h followed by treatment with HA-FITC-CNPs for a further 2 h. The fluorescently labeled cells were counted by using a BD FACS Calibur.
4.15. Western Blot Analysis
A total of 1.5 × 105 HCT-116 cells were treated with HA-CNPs for 24 h followed by cell lysis and suspension in sample buffer. Proteins were visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using primary antibody solution (p-AKT, AKT, p-ERK1/2, and ERK1/2 in 1:2000 dilution, and GAPDH in 1:5000 dilution) and then incubated in HRP conjugated secondary antibody solution (1:10 000 dilution).49,50
4.16. Detection of Apoptosis by Flow Cytometry
A total of 2 × 105 HCT-116 cells were attached in a six-well plate followed by treatment with HA-CNPs for 24 h. After 24 h, cell medium was aspirated and cells were centrifuged at 750 rpm for 2 min and washed with PBS. Cells were resuspended in 100 μL of Annexin V-FITC and propidium iodide solution followed by incubation at room temperature for 5 min in the dark. Further analysis of apoptosis was performed by using a BD FACS Calibur.72
4.17. Cell Cycle Analysis
A total of 2 × 106 HCT-116 cells were attached in a six-well plate followed by treatment with HA-CNPs for 24 h. Cells were then trypsinized, washed with PBS, and centrifuged. Cell palettes were fixed with 70% ice-cold ethanol for 30 min. Fixed cells were further stained with propidium iodide and analyzed with BD FACS Calibur.72
4.18. In Vitro Cytotoxicity Assay
A total of 5000 HCT-116 cells were attached in a 96-well plate and treated with HA-CNPs in a concentration-dependent manner for 24 h. A free drug cocktail was used as control. MTT reagent (20 μL/well) was added and further incubated for 4 h followed by solubilizing formazan crystals. UV–vis spectroscopy was used to measure absorbance at λmax = 550 nm to calculate cell viability. All experiments were performed in triplicate.72
Acknowledgments
S.B. is sincerely thankful to Department of Biotechnology (DBT), Govt. of India (Ramalingaswami Fellowship: BT/RLF/Re-entry/13/2011, BT/PR9918/NNT/28/692/2013, and BT/PR14724/NNT/28/831/2015) for funding. S.P. acknowledges CSIR-UGC for providing fellowship. P.S. is thankful to DST Women Scientist Scheme-A (SR/WOS-A/CS94/2012) for funding, fellowship, and Academy of Scientific & Innovative Research (AcSIR) for PhD registration. We thank IISER Pune Imaging Facility for confocal microscopy. We also recognize the Department of Science and Technology-Nanoscience and DST-FIST for SEM and AFM support, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01315.
1H, 13C, and 19F NMR spectroscopy data and spectra, DLS, ζ potential, drug release, protein-expression quantification, MTT assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Colorectal Cancer Facts and Figures 2011–2013; American Cancer Society, 2011–2013. https://www.cancer.org/research/cancer-facts-statistics/colorectal-cancer-facts-figures.html. [Google Scholar]
- Ferlay J.; Soerjomataram I.; Dikshit R.; Eser S.; Mathers C.; Rebelo M.; Parkin D. M.; Forman D.; Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Rubbia-Brandt L.; Audard V.; Sartoretti P.; Roth A. D.; Brezault C.; Le Charpentier M.; Dousset B.; Morel P.; Soubrane O.; Chaussade S.; Mentha G.; Terris B. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann. Oncol. 2004, 15, 460–466. 10.1093/annonc/mdh095. [DOI] [PubMed] [Google Scholar]
- André T.; Boni C.; Mounedji-Boudiaf L.; Navarro M.; Tabernero J.; Hickish T.; Topham C.; Zaninelli M.; Clingan P.; Bridgewater J.; Tabah-Fisch I.; de Gramont A. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- Klotz H. P.; Weder W.; Largiadèr F. Local and systemic toxicity of intra-hepatic-arterial 5-FU and high-dose or low-dose leucovorin for liver metastases of colorectal cancer. Surg. Oncol. 1994, 3, 11–16. 10.1016/0960-7404(94)90019-1. [DOI] [PubMed] [Google Scholar]
- DeVita V. T. Jr.; Chu E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- Sawyers C. Targeted cancer therapy. Nature 2004, 432, 294–297. 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Sharma S. V.; Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- Aggarwal S. Targeted cancer therapies. Nat. Rev. Drug Discov. 2010, 9, 427–428. 10.1038/nrd3186. [DOI] [PubMed] [Google Scholar]
- Yap T. A.; Carden C. P.; Kaye S. B. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- Shaw R. J.; Cantley L. C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Bader A. G.; Kang S.; Zhao L.; Vogt P. K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- Gschwind A.; Fischer O. M.; Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold J. S.; Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Hennessy B. T.; Smith D. L.; Ram P. T.; Lu Y.; Mills G. B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Courtney K. D.; Corcoran R. B.; Engelman J. A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Wilson T. R.; Fridlyand J.; Yan Y.; Penuel E.; Burton L.; Chan E.; Peng J.; Lin E.; Wang Y.; Sosman J.; Ribas A.; Li J.; Moffat J.; Sutherlin D. P.; Koeppen H.; Merchant M.; Neve R.; Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard P. L.; Hansen A. R.; Ratain M. J.; Siu L. L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila M. R.; de Sauvage F. J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- Sos M. L.; Fischer S.; Ullrich R.; Peifer M.; Heuckmann J. M.; Koker M.; Heynck S.; Stückrath I.; Weiss J.; Fischer F.; Michel K.; Goel A.; Regales L.; Politi K. A.; Perera S.; Getlik M.; Heukamp L. C.; Ansén S.; Zander T.; Beroukhim R.; Kashkar H.; Shokat K. M.; Sellers W. R.; Rauh D.; Orr C.; Hoeflich K. P.; Friedman L.; Wong K. K.; Pao W.; Thomas R. K. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18351–18356. 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel B.; Blair J. A.; Gonzalez B.; Nazif T. M.; Feldman M. E.; Aizenstein B.; Hoffman R.; Williams R. L.; Shokat K. M.; Knight Z. A. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Chen L.; Tan X.; Crosby K.; Guimaraes A. R.; Upadhyay R.; Maira M.; McNamara K.; Perera S. A.; Song Y.; Chirieac L. R.; Kaur R.; Lightbown A.; Simendinger J.; Li T.; Padera R. F.; García-Echeverría C.; Weissleder R.; Mahmood U.; Cantley L. C.; Wong K. K. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T.; Tolcher A. W.; Papadopoulos K. P.; Beeram M.; Rasco D. W.; Smith L. S.; Gunn S.; Smetzer L.; Mays T. A.; Kaiser B.; Wick M. J.; Alvarez C.; Cavazos A.; Mangold G. L.; Patnaik A. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- Posch C.; Moslehi H.; Feeney L.; Green G. A.; Ebaee A.; Feichtenschlager V.; Chong K.; Peng L.; Dimon M. T.; Phillips T.; Daud A. I.; McCalmont T. H.; LeBoit P. E.; Ortiz-Urda S. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 4015–4020. 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès G.; Guérin S.; Grall D.; Bonino F.; Smith A.; Anjuere F.; Auberger P.; Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Giroux S.; Tremblay M.; Bernard D.; Cardin-Girard J. F.; Aubry S.; Larouche L.; Rousseau S.; Huot J.; Landry J.; Jeannotte L.; Charron J. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 1999, 9, 369–376. 10.1016/S0960-9822(99)80164-X. [DOI] [PubMed] [Google Scholar]
- Bendell J. C.; Rodon J.; Burris H. A.; de Jonge M.; Verweij J.; Birle D.; Demanse D.; De Buck S. S.; Ru Q. C.; Peters M.; Goldbrunner M.; Baselga J. Phase I, Dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- Liao L.; Liu J.; Dreaden E. C.; Morton S. W.; Shopsowitz K. E.; Hammond P. T.; Johnson J. A. A convergent synthetic platform for single-nanoparticle combination cancer therapy: ratiometric loading and controlled release of cisplatin, doxorubicin, and camptothecin. J. Am. Chem. Soc. 2014, 136, 5896–5899. 10.1021/ja502011g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolishetti N.; Dhar S.; Valencia P. M.; Lin L. Q.; Karnik R.; Lippard S. J.; Langer R.; Farokhzad O. C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17939–17944. 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Xia J.; Zhao Q.; Liu L.; Zhang Z. Functional graphene oxide as a nanocarrier for controlled loading and targeted delivery of mixed anticancer drugs. Small 2010, 6, 537–544. 10.1002/smll.200901680. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Mo R.; Bellotti A.; Zhou J.; Gu Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv. Funct. Mater. 2014, 24, 2295–2304. 10.1002/adfm.201303222. [DOI] [Google Scholar]
- Sengupta S.; Eavarone D.; Capila I.; Zhao G.; Watson N.; Kiziltepe T.; Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature 2005, 436, 568–572. 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; Farokhzad O. C.; Margalit R.; Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Delivery Rev. 2013, 65, 71–79. 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E.; Bhatia S. N.; Sailor M. J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narain R.; Gonzales M.; Hoffman A. S.; Stayton P. S.; Krishnan K. M. Synthesis of monodisperse biotinylated p(NIPAAm)-coated iron oxide magnetic nanoparticles and their bioconjugation to streptavidin. Langmuir 2007, 23, 6299–6304. 10.1021/la700268g. [DOI] [PubMed] [Google Scholar]
- Kurosaki T.; Morishita T.; Kodama Y.; Sato K.; Nakagawa H.; Higuchi N.; Nakamura T.; Hamamoto T.; Sasaki H.; Kitahara T. Nanoparticles electrostatically coated with folic acid for effective gene therapy. Mol. Pharmaceutics 2011, 8, 913–919. 10.1021/mp2001268. [DOI] [PubMed] [Google Scholar]
- Farokhzad O. C.; Cheng J.; Teply B. A.; Sherifi I.; Jon S.; Kantoff P. W.; Richie J. P.; Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6315–6320. 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. W.; Hong K.; Kirpotin D. B.; Colbern G.; Shalaby R.; Baselga J.; Shao Y.; Nielsen U. B.; Marks J. D.; Moore D.; Papahadjopoulos D.; Benz C. C. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 2002, 8, 1172–1181. [PubMed] [Google Scholar]
- Peer D.; Margalit R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia 2004, 6, 343–353. 10.1593/neo.03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Lee H.; Kim Y. B.; Kim J.; Hyeon T.; Park H.; Messersmith P. B.; Park T. G. Bioinspired surface immobilization of hyaluronic acid on monodisperse magnetite nanocrystals for targeted cancer imaging. Adv. Mater. 2008, 20, 4154–4157. 10.1002/adma.200800756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat M.; El-Boubbou K.; Zhu D. C.; Lansdell T.; Lu X.; Li W.; Huang X. Hyaluronic acid immobilized magnetic nanoparticles for active targeting and imaging of macrophages. Bioconjugate Chem. 2010, 21, 2128–2135. 10.1021/bc100354m. [DOI] [PubMed] [Google Scholar]
- Choi K. Y.; Jeon E. J.; Yoon H. Y.; Lee B. S.; Na J. H.; Min K. H.; Kim S. Y.; Myung S. J.; Lee S.; Chen X.; Kwon I. C.; Choi K.; Jeong S. Y.; Kim K.; Park J. H. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials 2012, 33, 6186–6193. 10.1016/j.biomaterials.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palvai S.; More P.; Mapara N.; Basu S. Chimeric Nanoparticle: A platform for simultaneous targeting of phosphatidylinositol-3-kinase signaling and damaging DNA in cancer cells. ACS Appl. Mater. Interfaces 2015, 7, 18327–35. 10.1021/acsami.5b04015. [DOI] [PubMed] [Google Scholar]
- Palvai S.; Kuman M. M.; Basu S. Hyaluronic acid cloaked oleic acid nanoparticles inhibit MAPK signaling with sub-cellular DNA damage in colon cancer cells. J. Mater. Chem. B 2017, 5, 3658–3666. 10.1039/C7TB00656J. [DOI] [PubMed] [Google Scholar]
- Dreaden E. C.; Kong Y. W.; Morton S. W.; Correa S.; Choi K. Y.; Shopsowitz K. E.; Renggli K.; Drapkin R.; Yaffe M. B.; Hammond P. T. Tumor-targeted synergistic blockade of MAPK and PI3K from a layer-by-layer nanoparticle. Clin. Cancer Res. 2015, 21, 4410–4419. 10.1158/1078-0432.CCR-15-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A. A.; Roy B.; Rao P. S.; Wyant G. A.; Mahmoud A.; Ramachandran M.; Sengupta P.; Goldman A.; Kotamraju V. R.; Basu S.; Mashelkar R. A.; Ruoslahti E.; Dinulescu D. M.; Sengupta S. Supramolecular nanoparticles that target phosphoinositide-3-kinase overcome insulin resistance and exert pronounced antitumor efficacy. Cancer Res. 2013, 73, 6987–6997. 10.1158/0008-5472.CAN-12-4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalanotti F.; Solit D. B.; Pulitzer M. P.; Berger M. F.; Scott S. N.; Iyriboz T.; Lacouture M. E.; Panageas K. S.; Wolchok J. D.; Carvajal R. D.; Schwartz G. K.; Rosen N.; Chapman P. B. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in iatients with BRAFV600E/K-mutated melanoma. Clin. Cancer Res. 2013, 19, 2257–2264. 10.1158/1078-0432.CCR-12-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B. R.; Logie A.; McKay J. S.; Martin P.; Steele S.; Jenkins R.; Cockerill M.; Cartlidge S.; Smith P. D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 2007, 6, 2209–2219. 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- Workman P.; Clarke P. A.; Raynaud F. I.; van Montfort R. L. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010, 70, 2146–2157. 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W.; Knight Z. A.; Goldenberg D. D.; Yu W.; Mostov K. E.; Stokoe D.; Shokat K. M.; Weiss W. A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklein D.; Graab U.; Naumann I.; Yan T.; Ridzewski R.; Nitzki F.; Rosenberger A.; Dittmann K.; Wienands J.; Wojnowski L.; Fulda S.; Hahn H. PI3K inhibition enhances doxorubicin-induced apoptosis in sarcoma cells. PLoS One 2012, 7, e52898 10.1371/journal.pone.0052898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opel D.; Westhoff M. A.; Bender A.; Braun V.; Debatin K. M.; Fulda S. Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res. 2008, 68, 6271–6280. 10.1158/0008-5472.CAN-07-6769. [DOI] [PubMed] [Google Scholar]
- Yang S. Y.; Miah A.; Sales K. M.; Fuller B.; Seifalian A. M.; Winslet M. Inhibition of the p38 MAPK pathway sensitises human colon cancer cells to 5-fluorouracil treatment. Int. J. Oncol. 2011, 38, 1695–1702. 10.3892/ijo.2011.982. [DOI] [PubMed] [Google Scholar]
- Richly H.; Henning B. F.; Kupsch P.; Passarge K.; Grubert M.; Hilger R. A.; Christensen O.; Brendel E.; Schwartz B.; Ludwig M.; Flashar C.; Voigtmann R.; Scheulen M. E.; Seeber S.; Strumberg D. Results of a phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann. Oncol. 2006, 17, 866–873. 10.1093/annonc/mdl017. [DOI] [PubMed] [Google Scholar]
- Shao L.; Zhang R.; Lu J.; Zhao C.; Deng X.; Wu Y. Mesoporous silica coated polydopamine functionalized reduced graphene oxide for synergistic targeted chemo-photothermal therapy. ACS Appl. Mater. Interfaces 2017, 9, 1226–1236. 10.1021/acsami.6b11209. [DOI] [PubMed] [Google Scholar]
- DJung H. S.; Kong W. H.; Sung D. K.; Lee M. Y.; Beack S. E.; Keum D. H.; Kim K. S.; Yun S. H.; Hahn S. K. Nanographene oxide-hyaluronic acid conjugate for photothermal ablation therapy of skin cancer. ACS Nano 2014, 8, 260–268. 10.1021/nn405383a. [DOI] [PubMed] [Google Scholar]
- Sengupta P.; Basu S.; Soni S.; Pandey A.; Roy B.; Oh M. S.; Chin K. T.; Paraskar A. S.; Sarangi S.; Connor Y.; Sabbisetti V. S.; Kopparam J.; Kulkarni A.; Muto K.; Amarasiriwardena C.; Jayawardene I.; Lupoli N.; Dinulescu D. M.; Bonventre J. V.; Mashelkar R. A.; Sengupta S. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11294–11299. 10.1073/pnas.1203129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen-Lacey M.; Carregal-Romero S.; Liz-Marzán L. M. Current challenges toward in vitro cellular validation of inorganic nanoparticles. Bioconjugate Chem. 2017, 28, 212–221. 10.1021/acs.bioconjchem.6b00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliu N.; Docter D.; Heine M.; Del Pino P.; Ashraf S.; Kolosnjaj-Tabi J.; Macchiarini P.; Nielsen P.; Alloyeau D.; Gazeau F.; Stauber R. H.; Parak W. In vivo degeneration and the fate of inorganic nanoparticles. Chem. Soc. Rev. 2016, 45, 2440–2457. 10.1039/C5CS00699F. [DOI] [PubMed] [Google Scholar]
- Mayor S.; Pagano R. E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. 10.1038/nrm2216. [DOI] [PubMed] [Google Scholar]
- Ciccia A.; Elledge S. J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin N. J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- Meloche S.; Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- Roy S. K.; Srivastava R. K.; Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal. 2010, 5, 10–22. 10.1186/1750-2187-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. The hallmarks of cancer. Cell 2000, 100, 57–70. 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Mallick A.; More P.; Ghosh S.; Chippalkatti R.; Chopade B. A.; Lahiri M.; Basu S. Dual drug conjugated nanoparticle for simultaneous targeting of mitochondria and nucleus in cancer cells. ACS Appl. Mater. Interfaces 2015, 7, 7584–98. 10.1021/am5090226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.