

Abstract
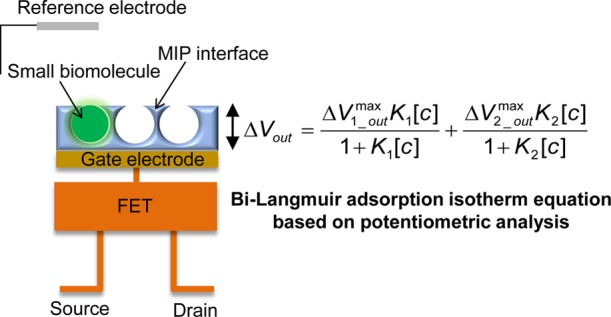
In this paper, we report a direct and quantitative analytical method of small-biomolecule recognition with a molecularly imprinted polymer (MIP) interface, taking advantage of the potentiometric principle of a field-effect transistor (FET) sensor, which enables the direct detection of ionic charges without using labeling materials such as fluorescent dyes. The interaction of low-molecular-weight oligosaccharides such as paromomycin and kanamycin with the MIP interface including phenylboronic acid (PBA) was directly and quantitatively analyzed from the electrical signals of an MIP-coated FET sensor. In particular, the change in the potential response of the FET sensor was derived on the basis of the multi-Langmuir adsorption isotherm equations, considering the change in the molecular charges of PBA caused by the adsorption equilibrium of the analytes with the vinyl PBA-copolymerized MIP membrane. Thus, the potentiometric adsorption isotherm analysis can elucidate the formation of selective binding sites at the MIP interface. The electrochemical analysis of the functional biointerface used in this study supports the design and construction of sensors for small biomarkers.
1. Introduction
A molecularly imprinted polymer (MIP) is an artificially designed polymer for recognition of a specific molecule, where target-selective cavities in a polymeric matrix selectively capture the target molecules.1 In particular, an MIP-based device is important as a bioanalytical material and a tool to capture target biomolecules without requiring specific reactions such as enzymatic reactions and antigen–antibody reactions for its molecular recognition. MIPs can generally be prepared by the following simple steps. First, a target molecule is mixed with functional monomers to form a target–monomer complex via either covalent bonds or noncovalent interactions. Then, the mixture undergoes polymerization in the presence of a high concentration of cross-linkers. After polymerization, the target molecule is extracted from the polymer matrix so that a target-selective binding cavity is formed. This allows the selective rebinding of the target molecules when the target is reintroduced into the polymer (Scheme 1A). MIPs were first developed for applications in molecular separation such as chromatography,2,3 but recent advances in surface modification technologies have made it possible to precisely modify surfaces using polymers, thus opening up the field of MIP-based biosensors. That is, a target biomolecule will selectively adhere to the sensor surface modified with a target-selective MIP, and then the selective adhesion is transduced to a signal in the sensor, hence enhancing selectivity. Owing to the versatility of MIPs, more MIP-based biosensors have recently been developed for the detection of various biocompounds.4−6
Scheme 1. (A) PBA Equilibrium with Sugar in Aqueous Environment.

(B) Schematic illustration of MIP. The illustration is drawn with paromomycin template as an example.
MIPs are most commonly characterized using adsorption isotherm equations because a target–MIP interaction depends on the binding equilibrium between target molecules and target-selective cavities distributed in the polymer matrix.7 In particular, a batch rebinding assay is most commonly employed, by which target compounds are determined by immersing the polymer into a solution with a specific concentration of a target molecule for a certain duration. Then, the results are calibrated using the adsorption isotherm equations. The binding property of MIPs is unique in a way that the binding cavities with different affinities are in most cases heterogeneously distributed throughout the polymer because of the randomness of template–monomer interactions and the process of copolymerization. Therefore, the adsorption isotherm equations for a heterogeneous distribution such as multi-Langmuir adsorption isotherm and Freundlich adsorption isotherm equations are often utilized.8 By using these equations, the binding affinity as well as the homogeneity of the binding-site distribution can be quantified. In general, MIPs, where cavities with high binding affinity are homogeneously distributed, are ideal owing to their highest selectivity for the target molecules.7 Thus, one can design an improved MIP composition on the basis of the information obtained by batch rebinding analysis.
Recently, biosensing devices with an MIP interface have been proposed for the selective detection of biomolecules; however, the novel functionalities of the biointerface of such devices should also be evaluated. A surface plasmon resonance sensor or a quartz crystal microbalance sensor has an attractive detection principle for biomolecular recognition such as antigen–antibody reactions but generally has limitations for use in small-molecule analysis because the signals obtained depend on the molecular weight of the targets to be determined.9−12 On the other hand, fluorescent or chemiluminescent dyes are used as labels to detect biomolecules by fluorescence microscopy and enzyme-linked immunosorbent assay but are generally difficult to use as labels for small molecules.13,14 Therefore, a new analytical method is required to directly and quantitatively evaluate biointerfacial characteristics such as the use of MIPs for small-biomolecule recognition. However, MIP compositions often have to be optimized in a bulk state before MIPs are applied to sensors. Such processes are not only time-consuming but may also lead to differences in adhesion properties between the bulk and the sensor; therefore, a direct analytical methodology for MIPs is highly desirable.
Because biological activities involve ions or molecular charges, potentiometric sensors can be advantageous in the recognition of biological events. In particular, a field-effect transistor (FET) biosensor can directly detect the ionic charges at the gate insulator/electrolyte solution interface on the basis of the field-effect principle (Figure 1).15,16 Therefore, FET biosensors enable the easy detection of not only ions but also small biomolecules, as long as even small biomolecules have charges. Moreover, FET biosensors have the potential to realize low-cost operation/production, portability, and multichannel measurement, which could lead to next-generation medical diagnostics. For these reasons, the applications of FET biosensors in the medical fields have recently been reported, including some biosensors with MIP interfaces incorporated.17,18 In particular, MIP-based FET biosensors showed relatively rapid, concentration-dependent responses and have potential use for building a platform based on molecular sensors for direct characterization.19
Figure 1.
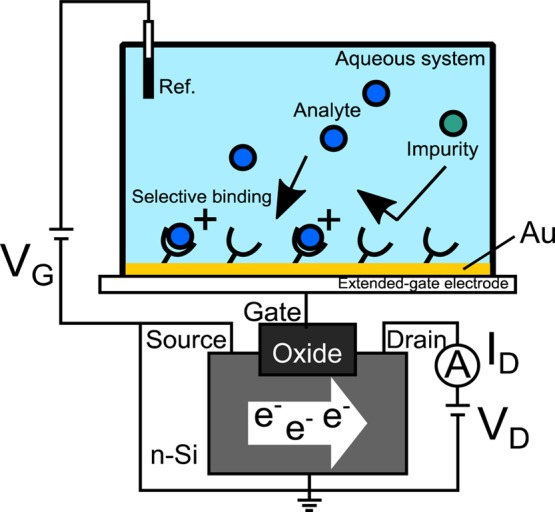
Conceptual design of an FET biosensor. The Au gate electrode was connected to the extended gate of a silicon-based n-channel FET, and gate voltage (VG) was applied through the Ag/AgCl reference electrode. Probe molecules such as MIP, which selectively bind to analytes but not impurities, are tethered on the gate electrode of the FET device. Molecular charges of analytes at the solution/gate electrode interface induce a change in VG at a constant source–drain current (ID) and drain voltage (VD).
Oligosaccharides refer to low-molecular-weight sugar chains, which are involved in a wide range of biological events. For instance, some oligosaccharides are bound to the termini of glycoproteins on the cellular membrane and play important roles in cellular activities such as cellular communication and recognition.20 Because some specific oligosaccharides are associated with various important diseases, they have been increasingly recognized as important biomarkers.21−23 Other oligosaccharides are aminoglycoside antibiotics such as paromomycin and kanamycin, which are widely used as veterinary drugs.24 Because the overuse of such drugs in the food chain has given rise to serious health risks to humans, the development of sensors that target these biomolecules is required.25 Although oligosaccharides have been recognized as important clinical targets, it remains challenging to build a clinical platform owing to the limited availability of lectins and glycan-specific antibodies.26,27 Recently, we have shown that an FET biosensor combined with an MIP interface can be applied to the real-time, quantitative sensing of sialyllactose.19
In this paper, we propose a direct and quantitative analytical method of small-biomolecule recognition with an MIP interface, taking advantage of the potentiometric principle of FET sensors, which enables the direct detection of ionic charges without using labeling materials such as fluorescent dyes. The interaction of low-molecular-weight biomolecules with the MIP interface is directly and quantitatively analyzed from the electrical signals of an MIP-coated FET sensor on the basis of the multi-Langmuir adsorption isotherm equations (Table of Content). In particular, oligosaccharides such as paromomycin and kanamycin (Scheme 2) are utilized as small-biomolecule models to realize a versatile analytical methodology. The electrochemical and analytical method including functional biointerfaces used in this study is expected to support the design and construction of biosensors that directly and quantitatively detect small-molecule biomarker.
Scheme 2. Structural Formulas of Paromomycin (A) and Kanamycin (B).

Paromomycin shares a chemical structure similar to kanamycin.
2. Results and Discussion
2.1. Response of PMIP–FET Biosensors to Various Oligosaccharides
To design a sugar-chain-selective MIP interface on the gate surface, phenylboronic acid (PBA) and methacrylic acid (MAA) were utilized as target-interacting functional monomers. PBA has attracted considerable attention in the field of molecular recognition as it can form stable esters with various biomolecules containing 1,2 or 1,3 cis-diol/polyol groups, such as sugars, in aqueous systems (Scheme 1A).29 Moreover, the esterification of PBA/diol is a reversible reaction, which can be controlled by adjusting the pH of the solution. Thus, the template can be easily extracted from the polymer in the preparation of MIPs by simply adjusting the pH of the solution (Scheme 1B). Moreover, in the esterification, PBA switches from a nonionic form to an anionic form (Scheme 1A). Hence, the change in molecular charge induced by the saccharide/PBA binding can be detected by FET biosensors. Indeed, there have been a few reports on the fabrication of saccharide sensors using PBA-modified FET biosensors.30−32 MAA is the most commonly used functional monomer in the preparation of MIPs because of its versatility in the interaction. MAA was utilized to interact with amino groups in the paromomycin template to further improve the selectivity of the biosensor.
To investigate the detection sensitivity and selectivity of the paromomycin-imprinted polymer (PMIP)–FET for paromomycin and kanamycin, the changes in the surface potential of the PMIP–FET sensor were monitored in real time at various concentrations of each biomolecule, as shown in Figure 2. When paromomycin was introduced into the measurement solution, the surface potential shifted in the negative direction. This is because negatively charged tetrahedral PBA molecules were induced by the paromomycin/PBA complexation on the gate surface/electrolyte interface. However, even kanamycin, which has a chemical structure similar to paromomycin, contributed to the change in the surface potential of the PMIP–FET sensor. Thus, the detection selectivity of the PMIP–FET sensor for paromomycin appeared to be poor. This is because the structural similarity of kanamycin to paromomycin may have enabled kanamycin to enter the cavities of the PMIP membrane, which may have resulted in the strong response. However, because the electrical responses shown in Figure 2 were analyzed at relatively high concentrations of each sample, the detection selectivity of the PMIP–FET sensor should also be evaluated at lower concentrations, as described in the next sections (2.2 and 2.3).
Figure 2.

Real-time measurement of change in surface potential using the PMIP–FET biosensor. The responsivity of the PMIP–FET biosensor to paromomycin and kanamycin was investigated in the range of concentrations from 5 μM to 10 mM.
2.2. Derivation of Adsorption Isotherm Equations for MIP–FET Systems
To understand the chemical basis of interactions between the MIP and the target biomolecules underlying the electrical responses of the PMIP–FET sensor for paromomycin and kanamycin, further quantitative analysis was required. In general, the characteristics of the binding of a target molecule to the MIP are quantified using adsorption isotherm equations, as the binding process involves the reversible adhesion of the target molecule to the target-selective membrane.33 Moreover, the homogeneity and heterogeneity of binding sites distributed in MIPs are critical to the effective enhancement of selectivity. In most cases, the binding sites in MIPs are heterogeneously distributed because of the randomness of copolymerization and the template/functional monomer interaction;34,35 therefore, MIPs include both nonselective and highly selective binding sites at a certain ratio. In this study, Langmuir and bi-Langmuir adsorption isotherm equations, which are often used for MIP characterization,36,37 were utilized as the homogeneous and heterogeneous binding models, respectively, and to derive the corresponding equations for the analysis of the electrical properties of the MIP–FET system. In this way, the potentiometric analyses based on the FET sensor can directly characterize the PMIP interface without the batch rebinding process, which is often required for MIP characterization. According to a previous study,33 the Langmuir adsorption isotherm and bi-Langmuir adsorption isotherm equations for a bulk rebinding system are expressed by
| 1 |
| 2 |
where B refers to a signal observed at equilibrium for the MIP-bound template, [c] to the free concentration of the template at equilibrium, N to the number of available active centers in the MIP per unit volume, and K to the binding constant. Equation 1 assumes homogeneously distributed binding sites with a constant binding constant K. On the other hand, eq 2 assumes two main types of binding sites with different affinities distributed at a ratio of N1/N2 in the polymer, that is, a heterogeneous system.
The operation of a silicon-based FET (Figure 1) in the unsaturated region can generally be described by
| 3 |
where ID is the drain current, μ is the electron mobility in the channel, COX is the gate oxide capacitance, W/L is the channel width-to-length ratio, VD and VG are the applied drain–source and gate–source voltages, respectively, and VT is the threshold voltage, which can be expressed by38
| 4 |
where Eref is the reference electrode potential relative to vacuum; (−ψ0 + χsol) describes the interfacial potential at the electrolyte/Au gate electrode interface (the factor χsol is the surface dipole moment of the solution, which can be considered to be constant); ϕsi/q is the silicon electron work function; Qit, Qf, and QB are the charge of the interface traps, the fixed oxide charge, and the bulk depletion charge per unit area, respectively; and ϕf is the Fermi potential difference between the doped bulk silicon and the intrinsic silicon.
Considering the PMIP membrane on the Au gate electrode of the extended-gate FET (Figure 1), the capacitance and charge in the PMIP membrane should be added to eq 4 and can be expressed by
| 5 |
with
| 6 |
where QMIP is the charge in the PMIP membrane and CCom is the combined capacitance of the FET gate oxide (COX) and the PMIP membrane (CMIP) on the Au gate electrode. In this study, it is assumed that CMIP hardly changed even after the addition of targeted molecules, considering our previous results for a similar hydrogel;28 therefore, CCom was nearly constant regardless of the adsorption of small molecules because COX was also constant. Moreover, the change in interfacial potential (Δψ0) at the electrolyte/Au gate electrode interface should not change because the ionic concentration (i.e., pH) was basically maintained by using the buffer solution. Also, Eref, ϕsi/q, Qit, Qf, QB, and ϕf should be the same before and after the molecular recognition events at the MIP interface. Thus, the signal response obtained using the FET sensor is based on the change in VT (ΔVT); therefore, ΔQMIP should be evaluated in this study, in accordance with eq 5 and the above considerations.
The binding affinity of PBA to a diol is pH-dependent, but it is generally understood that the B(OH)3– complex is much more stable than the B(OH)2 complex, as shown in a previous work.39 For the reversible interaction between paromomycin (P) and PBA in the MIP membrane (Scheme 1B)
| 7 |
the rate of formation of the P·PBA– complex at time t is written as
| 8 |
where ka is the association rate constant and kd is the dissociation rate constant. At time t, [PBA] = [PBA]0 – [P·PBA–], where [PBA]0 is the concentration of PBA at t = 0. This is substituted into eq 8 to give
| 9 |
In this study, the charge QMIP is derived from reaction 7; therefore, it is proportional to the formation of the P·PBA– complex in the PMIP membrane. Additionally, Qmax is proportional to the concentration of PBA in the PMIP membrane ([PBA]0 at t = 0), which indicates the capacity of the immobilized ligand. Therefore, eq 9 is modified to
| 10 |
where 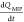 is the rate
of formation of the associated
complex (P·PBA–) in the PMIP membrane (on the
Au gate) and [c] is the concentration of the analyte
(P) in the solutions. Moreover, integrating eq 10 gives
is the rate
of formation of the associated
complex (P·PBA–) in the PMIP membrane (on the
Au gate) and [c] is the concentration of the analyte
(P) in the solutions. Moreover, integrating eq 10 gives
| 11 |
where Ka is the stability constant of P and PBA (ka/kd) in the PMIP membrane. From eq 11, QMIPt=0 = 0. Considering eq 5,
| 12 |
which is estimated after a certain reaction time t. Here, ΔVoutmax is the maximum change in surface potential induced by ΔQmax, which is proportional to the number of binding sites. In this study, ΔVout at the gate was measured at a constant ID using the source follower circuit shown in Figure S1 (Supporting Information). Therefore, the detected ΔVout was regarded as the change in VGS, which was proportional to −ΔVT at a constant ID.
According to the above considerations, the electrical signal in the entire FET circuit should obey the Langmuir adsorption model. By modifying eqs 1 and 2 in accordance with eq 12, the adsorption isotherm equations for the MIP–FET system are obtained as
| 13 |
| 14 |
where [c] is determined as the concentration of the target biomolecule at equilibrium, which is obtained from the saturated electrical signal in real-time measurement.
2.3. Quantification of the MIP Effect Using Adsorption Isotherm Equations
The data shown in Figure 2 were analyzed using eqs 13 and 14. First, ΔVout was calculated for each concentration of the target biomolecule by subtracting Vout at t = 0 from Vout at each concentration. Then, the resultant data were plotted versus the target concentration, as shown in Figure 3A. ΔVout was the average of 10 data plots taken 5 min after the addition of the target. The best-fit adsorption isotherm equations were determined by optimizing K and ΔVoutmax using the application software to minimize R2 (in Microsoft Excel). Then, ΔVout and the average binding affinity Kavr were expressed by
| 15 |
| 16 |
Figure 3.

(A) Change in surface potential as a function of target concentrations up to 10 mM. Calibration curves were determined by eqs 13 and 14. (Filled circle, open circle, and filled square show the addition of paromomycin into PMIP–FET, paromomycin into NIP–FET, and kanamycin into PMIP–FET, respectively.) (B) Comparison of the addition of paromomycin into PMIP–FET with that of kanamycin at low concentrations of less than 100 μM.
Table 1 shows the optimized values for the paromomycin/PMIP, paromomycin/nonimprinted polymer (NIP), and kanamycin/PMIP interactions in the MIP/NIP–FET measurement systems. The fitting curves are also shown in Figure 3A. From Table 1, the value of R2 shows that the adsorption isotherm equations were successfully applied to the PMIP/NIP–FET measurement systems; thus, the assumptions made in the derivation were valid. K2 and ΔV2_outmax in the case of adding paromomycin to NIP were zero, which indicated that the result fitted the Langmuir adsorption isotherm, indicating in turn that the binding sites were homogeneously distributed in NIP. However, there were no paromomycin-selective binding sites in NIP. Thus, the signal probably originated from nonspecific adsorption on the surface of NIP. Meanwhile, the paromomycin/PMIP interaction fitted the bi-Langmuir adsorption isotherm equation, indicating the heterogeneous distribution of binding sites. That is, from Table 1, two different types of paromomycin-binding sites were found, one with high binding affinity (K1 = 6970 M–1) and the other with low binding affinity (K2 = 73 M–1). Moreover, the number of binding sites (proportional to ΔV1_out) corresponding to K1 was much smaller even for the paromomycin/PMIP interaction, as similarly observed previously in the MIP produced by noncovalent interactions.33 In designing the PMIP for its interaction with paromomycin, PBA was assumed to be a covalently interacting functional monomer and MAA was assumed to be a noncovalently interacting one. Considering the equilibrium reaction shown in Scheme 1B, moreover, some PBAs might be used for the interaction with the template, paromomycin, in the PMIP membrane but not rebind to the target, paromomycin, even upon adding it, resulting in noncovalent interactions in the PMIP membrane. These noncovalently interacting monomers should be heterogeneously distributed and randomly functioned in the PMIP membrane, and then noncovalent, hydrogen bonding may be screened by water molecules in an aqueous solution; thus, the number of well-bound complexes was assumed to be small. In the FET measurement, the largest difference between the PMIP and NIP interfaces that interacted with paromomycin was found in ΔVoutmax, which was proportional to the total number of binding sites. As shown in Figure 3A, the concentration of paromomycin added to the NIP–FET reached a maximum of approximately 5 mM (170 mV), but the PMIP–FET showed a much higher ΔVout (415 mV (according to eq 15) at 850 mM in calculation) upon adding paromomycin. Thus, the difference between MIP and NIP was clearly demonstrated using the potentiometric adsorption isotherm equations derived from the MIP–FET measurement system.
Table 1. Summary of the Optimized Values of K and ΔV Based on the Bi-Langmuir Adsorption Isotherm Equation.
| K1 (M–1) | ΔV1_outmax (mV) | K2 (M–1) | ΔV2_outmax (mV) | Kavr (M–1) | ΔVoutmax (mV) | R2 | |
|---|---|---|---|---|---|---|---|
| P/PMIP | 6970 | 95 | 73 | 320 | 1070 | 415 | 0.998 |
| P/NIP | 2060 | 170 | 0 | 0 | 2060 | 170 | 0.994 |
| K/PMIP | 2800 | 240 | 0 | 0 | 2800 | 240 | 0.998 |
A similar trend was observed by comparing the addition of paromomycin with that of kanamycin to the PMIP–FET devices. Although the binding affinity of kanamycin was higher on average (Kavr = 2800 M–1 according to eq 16), the selectivity of PMIP for paromomycin was better than expected at concentrations of less than 100 μM, as shown in Figure 3B. Initially, the addition of kanamycin to the PMIP–FET system fitted the Langmuir adsorption isotherm equation, similar to the addition of paromomycin to the NIP–FET system. This indicated that the detection of kanamycin using the PMIP–FET system was also based on the nonspecific adsorption of kanamycin to the binding sites with low affinity (K2 and ΔV2_outmax in the case of adding kanamycin to PMIP were zero). On the other hand, the binding sites with higher affinity were crucial at lower concentrations of target molecules. Figure 3B shows the change in surface potential at the lower concentrations of less than 100 μM of paromomycin and kanamycin using the PMIP–FET sensors. From this result, paromomycin was detected more sensitively than kanamycin at the lower concentrations. That is, a target molecule at a lower concentration will first bind to high-affinity binding sites. Thus, the effect of K1 on the selectivity of PMIP for paromomycin was very important at the low concentrations of paromomycin until the signal reached ΔV1_out. Additionally, the limit of detection for paromomycin using the PMIP–FET sensor in this study was predicted to be 2.3 μM from the semilogarithmic plots in the range of 100 μM to 5.8 mM shown in Figure 3A, obtained from the Kaiser limit theory.40 This means that the higher selectivity of PMIP for paromomycin than kanamycin at concentrations of less than 100 μM should be ensured down to 2.3 μM. Therefore, the potentiometric adsorption isotherm analysis using the MIP–FET device can elucidate the formation of selective binding sites at the MIP interface. The electrochemical analysis of the functional biointerface used in this study is expected to support the design and construction of sensors for small biomarkers.
Additionally, the heterogeneous distribution of binding sites in PMIP was also analyzed using the potentiometric Freundlich adsorption isotherm in the same way as in the Langmuir analysis, which is expressed by
| 17 |
where a includes the number of available active centers N in the MIP per unit volume and the adsorption constant KF and m indicates the heterogeneity of the MIP interface (0 < m < 1). A change in the amount of adsorbate (paromomycin) induces a change in the molecular charge (ΔQ) on the basis of the equilibrium reaction 7, resulting in a change in the output voltage of ΔVout. In fact, the paromomycin/PMIP interaction, which fitted the bi-Langmuir adsorption isotherm equation, also fitted the Freundlich adsorption isotherm equation, giving a = 1.35 and m = 0.38. These calculated values are similar to those obtained for the adsorption of hydrocarbons on a hydrophobic adsorbent such as activated carbon.41 However, the Freundlich equation is based on an empirical rule and may not be valid for evaluating the detection selectivity of an MIP interface for target molecules at low concentrations by comparison with the Langmuir and bi-Langmuir adsorption isotherms. The Henry isotherm may be used to show the correlation between ΔVout and [c] in a dilute solution, although the potentiometric bi-Langmuir adsorption isotherm analysis clarified the detection selectivity at low concentrations of less than 100 μM, as mentioned in this paper.
3. Conclusions
In this paper, we introduced a direct and quantitative analytical method of small-biomolecule recognition with an MIP interface, taking advantage of the potentiometric principle of FET sensors. To realize the direct and quantitative analysis of the interaction of target molecules with the MIP or NIP interface, Langmuir and bi-Langmuir isotherm equations were derived depending on the changes in surface potential, which were caused by molecular charges of the P·PBA– complexes in the PMIP membrane, at the sensor surface. Then, the derived equations were applied to the measured change in the surface potential for the addition of paromomycin and kanamycin to the PMIP– and PNIP–FET systems. Although a better signal was observed upon the addition of kanamycin to PMIP–FET than that of paromomycin on average, and hence the detection selectivity of PMIP–FET to paromomycin seemed to be poor, the isotherm analysis revealed the differences in adsorption characteristics between paromomycin and kanamycin and the high selectivity of paromomycin to the PMIP interface at lower concentrations. In conclusion, a platform based on the potentiometric adsorption isotherm analysis is suitable for the elucidation of the formation of selective binding sites at the MIP interface. In particular, the advantage of FET sensors for the evaluation of MIP interfaces is that they enable the recognition of small biomolecules because ionic charges can be easily detected using FET sensors, even for small biomolecules. Such an electrochemical analysis contributes to the design and construction of sensors for small biomarkers.
Finally, we comment on the magnitude of the electrical responses. The PMIP–FET sensor produced larger signals than the other sensors even upon adding kanamycin. This may be because the PMIP–FET sensor had template-specific cavities for paromomycin in PMIP; therefore, paromomycin was captured by these cavities, making it more difficult for it to pass through the MIP interface to the Au substrate, while kanamycin more easily passed through the MIP interface to the Au substrate because such template-specific cavities became vacancies for kanamycin. Hence, the PMIP–FET sensor may have readily responded to kanamycin at the Au electrode. This is also true for the NIP–FET sensor because both cases fitted the Langmuir adsorption isotherm equation rather than the bi-Langmuir adsorption isotherm equation. Therefore, the effect of the material used as a substrate on nonspecific signals should be investigated in the future.
4. Experimental Section
4.1. Chemicals
The following chemicals used in the experiments in this study were purchased. Paromomycin sulfate, kanamycin monosulfate, 4-vinylphenylboronic acid (VPBA), and N,N,N′,N′-tetramethylethylenediamine (TEMED) were purchased from Tokyo Chemical Industry Co., Ltd. N-3-(Dimethylamino)propylmethacrylamide (DMAPMAAm), dimethyl sulfoxide (DMSO), MAA, N,N′-methylenebisacrylamide (MBAAm), 2-hydroxyethyl methacrylate (HEMA), ammonium peroxodisulfate (APS), sulfuric acid (H2SO4), hydrogen peroxide (H2O2), 40 mM phosphate buffer, phosphate-buffered saline (PBS), 1 M hydrochloric acid (HCl), saturated KCl, solid KCl, agarose, nitric acid (HNO3), methanol, and ethanol were purchased from Wako Pure Chemical Industries Ltd.
4.2. Designing a Sugar-Chain-Imprinted Polymer Interface
In the design of the paromomycin-selective MIP, VPBA and MAA were utilized as functional monomers. MAA was targeted to interact with the amino groups of paromomycin. As shown in Scheme 1A, VPBA was used to interact with diols on the sugar rings. Moreover, VPBA can induce a change in surface charges through the ionization derived by the complexation reaction. The sensing surface was functionalized by MIPs through free-radical copolymerization. Because paromomycin can only be dissolved in an aqueous solution, MBAAm was chosen as a hydrophobic cross-linker. DMAPMAAm was used to control the pH, and HEMA was used to improve the hydrophilicity of the polymer. TEMED was used as an initiator. Controlling the pH was important because the PBA–sugar binding is pH-dependent. The fundamental properties of the polymers, such as thickness, morphology, and swelling properties, were characterized in a previous work,28 where the chemical structure of the characterized hydrogel was similar to that of the polymers used in this study.
4.3. Fabrication of the MIP-Modified Extended Au Gate Electrode
An approximately 100 nm thick Au thin film over an approximately 15 nm thick Cr layer was sputtered on a transparent glass slide (Matsunami Glass). A polycarbonate ring (18 mm inner diameter/20 mm outer diameter) was encapsulated on the Au substrate using an epoxy resin (Pelnox ZC-203T) excluding the sensing surface. The Au substrate was immersed in a piranha solution (1/3 vol % mixture of H2O2 and H2SO4) for 10 min and then thoroughly rinsed with distilled water. The Au sensing surface was then kept in a UV ozone cleaner (Meiwafosis) to remove and prevent the adhesion of organic compounds before the copolymerization of the hydrogel.
PMIP was prepared on the Au electrode by free-radical polymerization. A prepolymer solution (1 mL) was prepared by dissolving the following chemicals in a mixture of 3/2 vol % H2O/DMSO in a 1.5 mL Eppendorf tube: paromomycin (30 mM), VPBA (60 mM), MAA (300 mM), MBAAm (200 mM), HEMA (400 mM), and DMAPMAAm (300 mM). TEMED (2 μL) was added to the mixture, and the mixture was deoxygenated with N2 gas for 20 min. Then, 100 μL of the solution was transferred to a 500 μL tube, and 5 μL of a stock solution of 50 mg/mL APS was added. The 5 μL mixture was placed on the Au sensing surface, which was then covered with a thin fluorine-coated poly(ethylene terephthalate) PET film, and the Au surface was allowed to undergo polymerization for 24 h at room temperature (rt) in a N2 environment. After polymerization, the PET film was carefully removed, and the resulting polymer-coated Au electrode was immersed in 0.1 M HCl in 1/1 vol % methanol/water for 24 h to extract the template molecule, paromomycin. For the comparative experiment, a NIP was prepared using the same procedure but without the addition of paromomycin to the prepolymer solution.
4.4. FET Real-Time Measurement
The MIP/NIP Au electrode was connected to the extended gate of a silicon-based n-channel junction-type FET (K246-Y9A, Toshiba), and gate voltage was applied through the Ag/AgCl reference electrode. Gate surface potential was measured in real time using an FET real-time monitoring system (Optogenesys). In this study, constant-charge mode operation was used for all the measurements, where VG, VD, and IDS were set to constant values and ΔVout at the gate was measured using the source follower circuit shown in Figure S1 (Supporting Information). Thus, Vout is a measure of changes in the surface potential or threshold voltage.
In the measurement, the sensing surface was covered with 1.5 mL of PBS (pH 7.4), and the source–drain current flow was controlled to 700 μA with a gate voltage of 0 V. After the stabilization of the surface potential, analytes of various concentrations were added to the solution. The concentration was controlled by exchanging 15 μL of the buffer and the analyte solution to give a 100-fold dilution. A stock solution of analyte solution was prepared beforehand and stored at 4 °C. The solution was allowed to warm to rt 1 h before the measurement to avoid the effect of a temperature change.
Acknowledgments
We would like to thank Dr. T. Kajisa of PROVIGATE Inc. for his help and useful discussion.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00627.
Schematic of the follower circuit for the real-time FET measurement system (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wulff G. Enzyme-like Catalysis by Molecularly Imprinted Polymers. Chem. Rev. 2002, 102, 1–28. 10.1021/cr980039a. [DOI] [PubMed] [Google Scholar]
- Piletsky S. A.; Panasyuk T. L.; Piletskaya E. V.; Nicholls I. A.; Ulbricht M. Receptor and Transport Properties of Imprinted Polymer Membranes – A Review. J. Membr. Sci. 1999, 157, 263–278. 10.1016/s0376-7388(99)00007-1. [DOI] [Google Scholar]
- Turiel E.; Martín-Esteban A. Molecularly Imprinted Polymers for Sample Preparation: A Review. Anal. Chim. Acta 2010, 668, 87–99. 10.1016/j.aca.2010.04.019. [DOI] [PubMed] [Google Scholar]
- Pichon V.; Chapuis-Hugon F. Role of Molecularly Imprinted Polymers for Selective Determination of Environmental Pollutants – A Review. Anal. Chim. Acta 2008, 622, 48–61. 10.1016/j.aca.2008.05.057. [DOI] [PubMed] [Google Scholar]
- Haupt K.; Mosbach K. Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors. Chem. Rev. 2000, 100, 2495–2504. 10.1021/cr990099w. [DOI] [PubMed] [Google Scholar]
- Whitcombe M. J.; Chianella T.; Larcombe L.; Piletsky S. A.; Noble J.; Porter R.; Horgan A. The Rational Development of Molecularly Imprinted Polymer-Based Sensors for Protein Detection. Chem. Soc. Rev. 2011, 40, 1547–1571. 10.1039/c0cs00049c. [DOI] [PubMed] [Google Scholar]
- Spivak D. Optimization, Evaluation, and Characterization of Molecularly Imprinted Polymers. Adv. Drug Delivery Rev. 2005, 57, 1779–1794. 10.1016/j.addr.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Umpleby R.; Baxter S.; Rampey A.; Rushton G.; Chen Y.; Shimizu K. Characterization of the Heterogeneous Binding Site Affinity Distributions in Molecularly Imprinted Polymers. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2004, 804, 141–149. 10.1016/j.jchromb.2004.01.064. [DOI] [PubMed] [Google Scholar]
- Gillis E. H.; Gosling J. P.; Sreenan J. M.; Kane M. Development and Validation of a Biosensor-Based Immunoassay for Progesterone in Bovine Milk. J. Immunol. Methods 2002, 267, 131–138. 10.1016/s0022-1759(02)00166-7. [DOI] [PubMed] [Google Scholar]
- Mitchell J. S.; Wu Y.; Cook C. J.; Main L. Sensitivity Enhancement of Surface Plasmon Resonance Biosensing of Small Molecules. Anal. Biochem. 2005, 343, 125–135. 10.1016/j.ab.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Mitchell J. Small Molecule Immunosensing Using Surface Plasmon Resonance. Sensors 2010, 10, 7323–7346. 10.3390/s100807323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata T.; Fukuda R. Simultaneous Biosensing with Quartz Crystal Microbalance with Dissipation Coupled-Gate Semiconductor Device. Anal. Chem. 2013, 85, 5796–5800. 10.1021/ac400468m. [DOI] [PubMed] [Google Scholar]
- Pradelles P.; Grassi J.; Creminon C.; Boutten B.; Mamas S. Immunometric Assay of Low Molecular Weight Haptens Containing Primary Amino Groups. Anal. Chem. 1994, 66, 16–22. 10.1021/ac00073a005. [DOI] [PubMed] [Google Scholar]
- Giraudi G.; Anfossi L.; Rosso I.; Baggiani C.; Giovannoli C.; Tozzi C. A General Method to Perform a Noncompetitive Immunoassay for Small Molecules. Anal. Chem. 1999, 71, 4697–4700. 10.1021/ac981282c. [DOI] [PubMed] [Google Scholar]
- Bergveld P. Development of an Ion-Sensitive Solid-State Device for Neurophysiological Measurements. IEEE Trans. Biomed. Eng. 1970, BME-17, 70–71. 10.1109/tbme.1970.4502688. [DOI] [PubMed] [Google Scholar]
- Bergveld P. Thirty Years of ISFETOLOGY: What Happened in the Past 30 Years and What may Happen in the Next 30 Years. Sens. Actuators, B 2003, 88, 1–20. 10.1016/s0925-4005(02)00301-5. [DOI] [Google Scholar]
- Iskierko Z.; Checinska A.; Sharma P. S.; Golebiewska K.; Noworyta K.; Borowicz P.; Fronc K.; Bandi V.; D’Souza F.; Kutner W. Molecularly Imprinted Polymer Based Extended-Gate Field-Effect Transistor Chemosensors for Phenylalanine Enantioselective Sensing. J. Mater. Chem. C 2017, 5, 969–977. 10.1039/c6tc03812c. [DOI] [Google Scholar]
- Rayanasukha Y.; Pratontep S.; Porntheeraphat S.; Bunjongpru W.; Nukeaw J. Non-Enzymatic Urea Sensor Using Molecularly Imprinted Polymers Surface Modified Based-on Ion-Sensitive Field Effect Transistor (ISFET). Surf. Coat. Technol. 2016, 306, 147–150. 10.1016/j.surfcoat.2016.05.060. [DOI] [Google Scholar]
- Nishitani S.; Kajisa T.; Sakata T. Development of Molecularly Imprinted Polymer-Based Field Effect Transistor for Sugar Chain Sensing. Jpn. J. Appl. Phys. 2017, 56, 04CM02. [Google Scholar]
- Munro S. Essentials of Glycobiology. Trends Cell Biol. 2000, 10, 552–553. 10.1016/s0962-8924(00)01855-9. [DOI] [Google Scholar]
- Adamczyk B.; Tharmalingam T.; Rudd P. M. Glycans as Cancer Biomarkers. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1347–1353. 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Schauer R. Achievements and Challenges of Sialic Acid Research. Glycoconjugate J. 2000, 17, 485–499. 10.1023/a:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster M. M.; Esko J. D. The Sweet and Sour of Cancer: Glycans as Novel Therapeutic Targets. Nat. Rev. Cancer 2005, 5, 526–542. 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- Megoulas N. C.; Koupparis M. A. Direct Determination of Kanamycin in Raw Materials, Veterinary Formulation and Culture Media Using a Novel Liquid Chromatography-Evaporative Light Scattering method. Anal. Chim. Acta 2005, 547, 64–72. 10.1016/j.aca.2004.11.031. [DOI] [Google Scholar]
- Song K.-M.; Cho M.; Jo H.; Min K.; Jeon S. H.; Kim T.; Han M. S.; Ku J. K.; Ban C. Gold Nanoparticle-Based Colorimetric Detection of Kanamycin Using a DNA Aptamer. Anal. Biochem. 2011, 415, 175–181. 10.1016/j.ab.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Fujitani N.; Furukawa J.-i.; Araki K.; Fujioka T.; Takegawa Y.; Piao J.; Nishioka T.; Tamura T.; Nikaido T.; Ito M.; Nakamura Y.; Shinohara Y. Total Cellular Glycomics Allows Characterizing Cells and Streamlining the Discovery Process for Cellular Biomarkers. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 2105–2110. 10.1073/pnas.1214233110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings R. D.; Etzler M. E.. Chapter 28 R-Type Lectins. Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press, 2009; pp 633–648. [PubMed] [Google Scholar]
- Kajisa T.; Sakata T. Glucose Responsive Hydrogel Electrode for Biocompatible Glucose Transistor. Sci. Technol. Adv. Mater. 2017, 18, 26–33. 10.1080/14686996.2016.1257344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Gao X.; Wang B. Boronic Acid Compounds as Potential Pharmaceutical Agents. Med. Res. Rev. 2003, 23, 346–368. 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- Matsumoto A.; Sato N.; Kataoka K.; Miyahara Y. Noninvasive Sialic Acid Detection at Cell Membrane by Using Phenylboronic Acid Modified Self-Assembled Monolayer Gold Electrode. J. Am. Chem. Soc. 2009, 131, 12022–12023. 10.1021/ja902964m. [DOI] [PubMed] [Google Scholar]
- Kajisa T.; Sakata T. Fundamental Property of Phenylboronic-Acid-Coated Gate Field-Effect Transistor for Saccharide Sensing. ChemElectroChem 2014, 1, 1647–1655. 10.1002/celc.201402193. [DOI] [Google Scholar]
- Lin C.-H.; Hsiao C.-Y.; Hung C.-H.; Lo Y.-R.; Lee C.-C.; Su C.-J.; Lin H.-C.; Ko F.-H.; Huang T.-Y.; Yang Y.-S. Ultrasensitive Detection of Dopamine Using a Polysilicon Nanowire Field-Effect Transistor. Chem. Commun. 2008, 5749–5751. 10.1039/b812968a. [DOI] [PubMed] [Google Scholar]
- García-Calzón J. A.; Díaz-García M. E. Characterization of Binding Sites in Molecularly Imprinted Polymers. Sens. Actuators, B 2007, 123, 1180–1194. 10.1016/j.snb.2006.10.068. [DOI] [Google Scholar]
- Stanley B. J.; Szabelski P.; Chen Y.-B.; Sellergren B.; Guiochon G. Affinity Distributions of a Molecularly Imprinted Polymer Calculated Numerically by the Expectation-Maximization Method. Langmuir 2003, 19, 772–778. 10.1021/la020747y. [DOI] [Google Scholar]
- Lanza F.; Rüther M.; Hall A. J.; Dauwe C.; Sellergren B. Studies on the Process of Formation, Nature and Stability of Binding Sites in Molecularly Imprinted Polymer. Mater. Res. Soc. Symp. Proc. 2002, 723, 93–103. 10.1557/proc-723-m5.6. [DOI] [Google Scholar]
- Umpleby R. J.; Baxter S. C.; Chen Y.; Shah R. N.; Shimizu K. D. Characterization of Molecularly Imprinted Polymers with the Langmuir–Freundlich Isotherm. Anal. Chem. 2001, 73, 4584–4591. 10.1021/ac0105686. [DOI] [PubMed] [Google Scholar]
- Sajonz P.; Kele M.; Zhong G.; Sellergren B.; Guiochon G. Study of the Thermodynamics and Mass Transfer Kinetics of Two Enantiomers on a Polymeric Imprinted Stationary Phase. J. Chromatogr. A 1998, 810, 1–17. 10.1016/s0021-9673(98)00247-7. [DOI] [Google Scholar]
- Bergveld P. The Impact of MOSFET-Based Sensors. Sens. Actuators 1985, 8, 109–127. 10.1016/0250-6874(85)87009-8. [DOI] [Google Scholar]
- Furikado Y.; Nagahata T.; Okamoto T.; Sugaya T.; Iwatsuki S.; Inamo M.; Takagi H. D.; Odani A.; Ishihara K. Universal Reaction Mechanism of Boronic Acids with Diols in Aqueous Solution: Kinetics and the Basic Concept of a Conditional Formation Constant. Chem.—Eur. J. 2014, 20, 13194–13202. 10.1002/chem.201403719. [DOI] [PubMed] [Google Scholar]
- Kaiser H. Die Berechnung der Nachweisempfindlichkeit. Spectrochim. Acta 1947, 3, 40–67. 10.1016/0371-1951(47)80005-0. [DOI] [Google Scholar]
- Abe I.; Hayashi K.; Hirashima T.; Kitagawa M. Relationship between the Freundlich adsorption constants K and 1/N hydrophobic adsorption. J. Am. Chem. Soc. 1982, 104, 6452–6453. 10.1021/ja00387a052. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.