

Abstract
Although a range of pharmacologically active compounds has been obtained from the mycelium and fruiting bodies of Ganoderma lucidum, the biological properties of the alkaloids present in this functional food remain unknown. Herein, we report total syntheses of lucidimines B and C, key members of the first family of alkaloids isolated from G. lucidum, and the evaluation of these synthetically derived materials as antioxidants and antiproliferative agents. Lucidimine B proved to be a better antioxidant than congener C. Similarly, lucidimine B exhibited antiproliferative properties toward MCF-7 cells (an EC50 value of 0.27 ± 0.02 μmol/mL), whereas lucidimine C was inactive. The former alkaloid arrested the MCF-7 cell cycle in the S phase by inducing DNA fragmentation, hence reducing the mitochondrial membrane potential. This work thus demonstrates, for the first time, that the alkaloidal constituents derived from G. lucidum are biologically active and may, therefore, contribute to the beneficial health claims made for this nutraceutical.
Introduction
In “new-age” agriculture, functional foods are not just intended to satisfy hunger and provide nutrition. They should also prevent diseases and improve physical and mental well-being.1−4 Such functional foods must contain nutraceuticals, viz., bioactive components that provide the desired functional properties.5 Ideally, nutraceuticals should be pharmaceutical-grade (and standardized) compounds, which are considered to be a natural component of the food, which provides (purportedly, at least) extra health benefits as well, of course, as contributes to the basic nutritional value of the food in question.6−8 Numerous mushrooms have been fundamental components of traditional medicines for centuries. They have been seen as the source of “miracle” cures as well as perceived as having general health-promoting benefits.9−12 Among them, Ganoderma lucidum, popularly known as Lingzhi in China, Reishi in Japan, and Yeongji in South Korea, is recognized as an important medicinal mushroom, even being referred to as the “Mushroom of Immortality”.13 Its pharmacological and nutraceutical applications can be traced back nearly two thousand years.14G. lucidum has been used extensively in Asia to improve well-being and to promote general good health14 as well as employed, alone or in tandem with other treatments, in modern Western medicine to slow the proliferation of certain cancers or to ameliorate the often harsh side effects associated with chemotherapeutic regimes.15−17 Recently, this nutraceutical has seen increasing applications in the prevention and/or treatment of a range of diseases including obesity, bronchitis, insomnia, anorexia, dizziness, hypercholesterolemia, diabetes, allergies, hepatitis, immunological disorders, and cancer.16−20 Because of its various benefits (perceived and real), G. lucidum is cultured on an industrial scale in a number of Asian countries, particularly China, Japan, and South Korea. As a consequence, numerous G. lucidum-derived products have emerged globally in the form of powders, teas, and nutraceuticals.21
Diverse groups of chemical compounds with pharmacological activities have been isolated from the mycelium and fruiting bodies of G. lucidum. These include triterpenoids, polysaccharides, steroids, fatty acids, amino acids, nucleosides, proteins, and alkaloids.22 The triterpenoids and polysaccharides of G. lucidum have attracted considerable attention because of their high content in the fungus, diverse structures, and significant bioactivities.23−30 In general, then, the active triterpenoids inhibit cancer cell proliferation and metastasis, whereas the polysaccharides function as immunomodulators and/or antioxidants.17,31−34 In contrast, the distribution of alkaloids within G. lucidum and their associated biological properties are much less well-documented, even if such compounds obtained from other genera of Ganoderma, such as ganoines I and II from Ganoderma capense,35 sinensines from Ganoderma sinense,36,37 and ganocochlearines C–I from Ganoderma cochlear,38 have been described in detail. To date, only four alkaloids, namely, the polycyclic lucidimine A (1), lucidimine B (2), lucidimine C (3), and lucidimine D (4) (Figure 1), have been isolated from the methanol extracts of the fruiting bodies of G. lucidum,39 and their structures were proposed on the basis of various spectroscopic analyses, particularly those involving 1H and 13C NMR studies. All of the four members possess a cyclopenta[c]pyridine skeleton that is attached to a 1,2,4-trisubstituted phenyl group with variations at the substituents on C-6, 7, 10 as well as the oxygen bridge positions (see Figure 1 for numbering). In a more recent work, lucidimines A (1) and B (2) were also isolated from Ganoderma calidophilum, a rare wild fungi of Ganoderma family, though under different names (ganocalicines A and B).40
Figure 1.
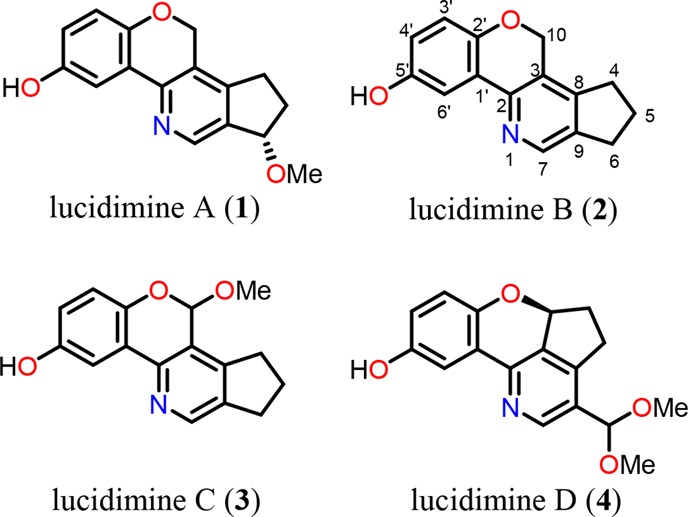
Chemical structures of lucidimines A–D.
Because the number and types of commercially available products derived from G. lucidum are increasing dramatically, consumers are now all the more discerning and seeking proof of the claimed health benefits. As such, the identification and comprehensive biological evaluation of the full complement of chemical constituents of this nutraceutical is assuming greater urgency.21 Furthermore, scientifically acquired information is needed to meet the increasingly stringent regulatory requirements related to health claims surrounding nutraceutical products. As a consequence, the detailed biological evaluation of the lucidimines is becoming an increasingly important matter and one that cannot be readily addressed through the isolation of these materials from the natural source. Indeed, a combination of medium-pressure liquid chromatography, Sephadex LH-20 column chromatography, and multiple prep-HPLC separations was required in the original isolation, which led to miniscule amounts (0.5–3.5 mg) of lucidimines A–D, which precluded any significant evaluation of their biological properties. In principle, chemical synthesis offers a solution to this challenge, which is made all the more appealing because of their novel molecular architectures that embody the rare cyclopenta[c]pyridine substructure. Herein, we report the first total syntheses of lucidimines B and C by a pathway that allows for their preliminary (and revealing) biological evaluation.
Results and Discussion
Total Syntheses of Lucidimines B (2) and C (3)
Scheme 1 summarizes the preparation of lucidimines B (2) and C (3), which started with the condensation of commercially available cyclopentanone 5 with malononitrile under solvent-free conditions,41 to give compound 7 (89%). Treatment of 7 with N,N-dimethylformamide dimethyl acetal (DMF-DMA) in the presence of a substoichiometric amount of acetic anhydride yielded the enamine 8, which was then subjected to Pinner conditions42 to provide the 2-bromonicotinonitrile 9 in 85% yield (over two steps). The resulting compound was then engaged in a Suzuki–Miyaura cross-coupling reaction with (2,5-dimethoxyphenyl)boronic acid 10 to afford diaryl 11 (83%). The nitrile of the latter compound was hydrolyzed to carboxylic acid 12 (90%). In a pivotal step, when acid 12 was treated with three equivalents of BBr3 in dichloromethane followed by quenching with methanol, both of the methyl groups were removed, followed by a spontaneous lactone formation, resulting in compound 13 (58%). On exposure to NaBH4 and BF3·OEt2,43 the lactone 13 was reduced to the target compound 2 (53%); this relatively lower yield could be attributed to the acid/base workup required to remove the impurities. The structure of compound 3 differs from that of compound 2 only at the pyran ring, where it possesses an acetal at C-10, which, we envisaged, could be derived from an aldehyde. However, reduction of nitrile 11 to the corresponding aldehyde was not successful, meaning that the aldehyde functional group needed to be installed in the early stages of the synthesis. An initial diisobutylaluminum hydride (DIBAl-H) reduction of the nitrile 9 yielded an aldehyde 14 (87%), which was then subjected to Suzuki–Miyaura cross-coupling reaction with (2,5-dimethoxyphenyl)boronic acid 10 to afford diaryl 15 (80%). Finally, compound 15 was treated with three equivalents of BBr3 in dichloromethane, to afford a hemiacetal intermediate, which was then submitted to a p-TsOH-catalyzed acetal formation process in methanol, thereby providing the final product 3 (53% in two steps). All spectroscopic data acquired on compounds 2 and 3 matched with those reported39 for lucidimines B and C (see Tables 1 and 2). Using these exact protocols, gram scales of lucidimines B and C were prepared with high purities (97 and 95%, respectively, as indicated by liquid chromatography–mass spectrometry), which allowed further biological evaluations to be performed.
Scheme 1. Total Syntheses of Lucidimines B (2) and C (3).

Table 1. NMR Spectroscopic Data for Natural Lucidimine B and Synthetic Lucidimine B.
|
13C NMR (δC) |
1H NMR (δH) |
|||
|---|---|---|---|---|
| position numbera | natural lucidimine Bb | synthetic lucidimine Bc | natural lucidimine Bd | synthetic lucidimine Be |
| 2 | 151.4 | 151.3 | ||
| 3 | 124.9 | 124.9 | ||
| 4 | 31.0 | 30.9 | 2.87 (t, J = 7.3 Hz, 2H) | 2.90 (t, J = 7.5 Hz, 2H) |
| 5 | 26.1 | 26.1 | 2.14 (tt, J = 7.3 and 7.3 Hz, 2H) | 2.16 (m, 2H) |
| 6 | 31.1 | 31.1 | 2.95 (t, J = 7.3 Hz, 2H) | 2.99 (t, J = 7.5 Hz, 2H) |
| 7 | 145.0 | 144.9 | 8.28 (s, 1H) | 8.32 (s, 1H) |
| 8 | 147.3 | 147.3 | ||
| 9 | 141.6 | 141.6 | ||
| 10 | 66.5 | 66.5 | 5.09 (s, 2H) | 5.12 (s, 2H) |
| 1′ | 125.1 | 125.0 | ||
| 2′ | 150.8 | 150.8 | ||
| 3′ | 118.8 | 118.7 | 6.76 (d, J = 8.6 Hz, 1H) | 6.80 (d, J = 8.7 Hz, 1H) |
| 4′ | 118.9 | 118.9 | 6.70 (dd, J = 8.6 and 2.8 Hz, 1H) | 6.74 (dd, J = 8.7 and 2.9 Hz, 1H) |
| 5′ | 153.6 | 153.6 | ||
| 6′ | 111.2 | 111.2 | 7.43 (d, J = 2.8 Hz, 1H) | 7.48 (d, J = 2.9 Hz, 1H) |
Table 2. NMR Spectroscopic Data for Natural Lucidimine C and Synthetic Lucidimine C.
|
13C NMR (δC) |
1H NMR (δH) |
|||
|---|---|---|---|---|
| position numbera | natural lucidimine Cb | synthetic lucidimine Cc | natural lucidimine Cd | synthetic lucidimine Ce |
| 2 | 153.5f | 153.4 | ||
| 3 | 123.5g | 123.5 | ||
| 4 | 30.8 | 30.8 | 3.08 (ddd, J = 16.6, 7.6 and 7.6 Hz, 1H) | 3.08 (m, 1H) |
| 2.94 (ddd, J = 16.6, 7.7 and 7.7 Hz, 1H) | 2.94 (m, 1H) | |||
| 5 | 26.1 | 26.1 | 2.18–2.24 (m, 2H) | 2.22 (m, 2H) |
| 6 | 31.2 | 31.2 | 3.03 (t, J = 7.4 Hz, 2H) | 3.03 (t, J = 7.4 Hz, 2H) |
| 7 | 146.7 | 146.7 | 8.47 (s, 1H) | 8.48 (s, 1H) |
| 8 | 145.9 | 145.9 | ||
| 9 | 141.8 | 141.8 | ||
| 10 | 98.1 | 98.1 | 6.10 (s, 1H) | 6.11 (s, 1H) |
| 1′ | 123.6 | 123.6 | ||
| 2′ | 146.7h | 146.7 | ||
| 3′ | 119.6 | 119.6 | 6.95 (d, J = 8.7 Hz, 1H) | 6.97 (d, J = 8.7 Hz, 1H) |
| 4′ | 119.0 | 119.0 | 6.82 (dd, J = 8.7 and 3.0 Hz, 1H) | 6.84 (dd, J = 8.7 and 3.0 Hz, 1H) |
| 5′ | 153.7 | 153.7 | ||
| 6′ | 110.8 | 110.8 | 7.60 (d, J = 3.0 Hz, 1H) | 7.63 (d, J = 3.0 Hz, 1H) |
| OMe | 55.9 | 55.9 | 3.55 (s, 3H) | 3.57 (s, 3H) |
Obtained from ref (39) and recorded in CD3OD at 150 MHz.
Recorded in CD3OD at 150 MHz.
Obtained from ref (39) and recorded in CD3OD at 600 MHz.
Recorded in CD3OD at 400 MHz.
This signal in ref (39) (153.7) is an error, revised by Liu et al.
This signal in the original ref (39) (153.5) is an error, revised by Liu et al.
This signal in ref (39) (153.5) is an error, revised by Liu et al.
Antioxidant Activity
Generally, polysaccharides and triterpenoids identified from G. lucidum were regarded as the main bioactive substances. In recent years, however, the polysaccharides from this nutraceutical have been believed to function as immunomodulators or antioxidants. To reveal the potential pharmacological properties of the recently reported alkaloids, three accepted evaluation methods were applied to explore their antioxidant abilities. Of these, 1,1-diphenyl-2-picrylhydrazyl radical 2,2-diphenyl-1-(2,4,6-trinitrophenyl) hydrazyl (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) radical scavenging assays are quick and sensitive,44 whereas the peroxyl radical scavenging capacity (PSC) method is reliable, rapid, and precise.45 The EC50 values for DPPH radical scavenging activities of lucidimines B and C were 1.24 ± 0.12 and 11.69 ± 1.81 μmol/mL, respectively; meanwhile, the EC50 values obtained from the ABTS assay were 0.19 ± 0.003 and 0.38 ± 0.003 μmol/mL, respectively. The results demonstrated that lucidimine B possessed better antioxidant activity than that observed for lucidimine C. Peroxyl radicals generated by the degradation of 2,2′-azobis-amidinopropane (AAPH) are capable of oxidizing nonfluorescent dichlorofluorescein (DCFH) to fluorescent dichlorofluorescein (DCF). The PSC method incorporates dichlorofluorescein diacetate (DCFH-DA) as a probe to monitor reactions and was applied to further determine the antioxidant potencies of the two alkaloids. The capacities of lucidimines B and C to scavenge peroxyl radicals were summarized on the basis of the variation of DCF fluorescence during the reaction. As shown in Figure 2 A,B, lucidimines B and C decreased the fluorescence as the concentrations increased, indicating that lucidimines B and C had antioxidant activity. The capacity of lucidimines B and C to scavenge peroxyl radicals was summarized on the basis of the variation of DCF fluorescence during the reaction. The PSC values of lucidimines B and C were 175.32 ± 11.74 μmol vitamin C equiv/g and 35.93 ± 13.60 μmol vitamin C equiv/g, respectively. The same tendency was also found in the DPPH and ABTS assays, which clearly indicated that lucidimine B possessed better antioxidant potency than lucidimine C.
Figure 2.
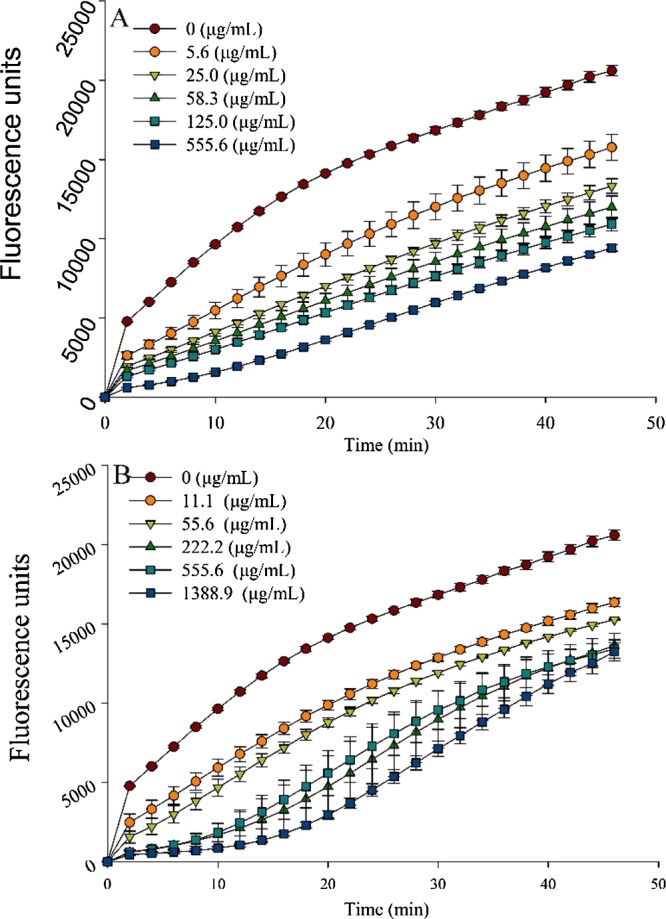
Peroxyl radical-induced oxidation of DCFH to DCF and the inhibition of oxidation by lucidimine B (A) and lucidimine C (B) over time [mean ± standard deviation (SD), n = 3].
Antiproliferation in Human Breast Cancer Cells
It is believed that the antiproliferative properties of G. lucidum largely rely on its diversified chemical constituents.46 Numerous studies have demonstrated that the bioactive components from crude extracts of either spores or fruiting bodies of G. lucidum are cytotoxic to a range of cancer cell lines including human hepatoma PLC/PRF/5 and KB,47 human breast cancer MDA-MB-231,48,49 human breast cancer MCF-7,49,50 human prostate cancer PC-3,51 human cervix uteri tumor HeLa,52 and low-grade bladder cancer MTC-11.53 Because the lucidimines were isolated from the methanolic extract of the fruiting bodies of G. lucidum and the same alcoholic extracts were reported48−50 to have antiproliferative activity toward human breast cancer MCF-7 and MDA-MB-231, the cytotoxicity of lucidimines B and C against the two human breast cancer cells was evaluated. MCF-7 and MDA-MB-231 cells are two characteristic feature cells associated with estrogen dependence and estrogen independence, respectively.54 Both lucidimines B and C showed no significant antiproliferative effects toward the estrogen-independent MDA-MB-231 cell line. Nevertheless, lucidimine B (EC50 0.27 ± 0.02 μmol/mL) was more cytotoxic to MCF-7 than lucidimine C (EC50 > 1.86 μmol/mL) in a dose-dependent manner. On the basis of the preliminary results, MCF-7 cells were selected as the target for in-depth exploration of the antiproliferative mechanism of lucidimine B.
Cell Cycle Arrest
Cell cycle plays an important role in cell growth and cell proliferation. During cellular proliferation, the cells cycle through four phases leading to cell division: G1 gap, S phase, G2 gap, and M phase. Blocking of the cell cycle progression of cancer cells is regarded as an effective means of cancer treatment.55 Many phytochemicals act as antiproliferative agents by restraining cell cycle progression or inducing apoptosis in cancer cells.56 In this study, the cell cycle distribution and apoptosis of MCF-7 cells were explored to elucidate the antiproliferation mechanism of lucidimine B. Propidium iodide (PI) is a fluorescent agent and used as a DNA content in cell cycle analysis. The cell cycle distributions of MCF-7 cells after treatment with lucidimine B stained by PI are shown in Figure 3. An obvious increase in the percentage of S phase inhibition of MCF-7 cells was detected in a dose-dependent manner. An increase in DNA concentration was also observed at the G2/M phase in treated cells; however, the proportion of DNA at the G0/G1 phase decreased along with the increases of the S and G2/M phases. The results obtained indicated that lucidimine B could induce DNA fragmentation and impact cell cycle progression at the S phase.
Figure 3.

Effect of different concentrations of lucidimine B [control (A), 10 (B), 50 (C), and 100 μg/mL (D)] on cell cycle in MCF-7 cells, determined by PI; the results of cell cycle in MCF-7 cells (E) (mean ± SD, n = 3). *P < 0.05.
Induction of Apoptosis by Lucidimine B
The apoptosis periods and rates of MCF-7 cells induced by lucidimine B were investigated using flow cytometric analysis. As shown in Figure 4, MCF-7 cells that were treated with lucidimine B (0.04 μmol/mL) displayed significant induction in the rate of early-stage apoptosis (14.2 ± 1.0%) when compared to the control group (0.7 ± 0.1%). In the meantime, a progressive reduction of the rate was obtained for cells treated by 0.21 μmol/mL (5.4 ± 0.6%) and 0.42 μmol/mL (4.3 ± 0.4%) lucidimine B. Moreover, the percentage of late-stage apoptosis in MCF-7 cells showed similar distinct changes. Clearly, these results indicated that lucidimine B induces apoptosis in MCF-7 cells in the early stages of the cell cycle.
Figure 4.
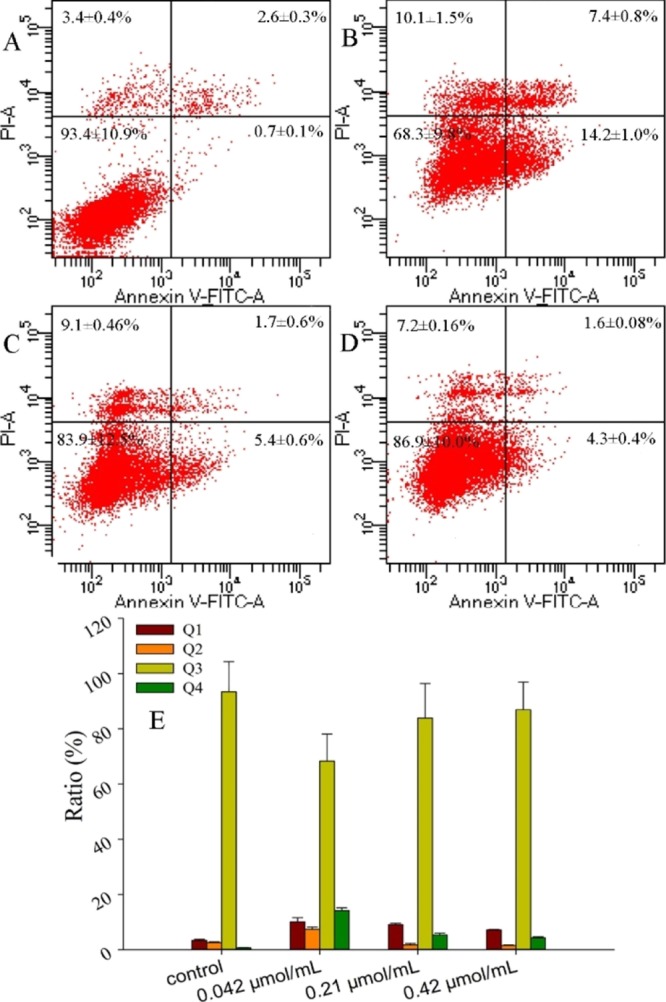
Apoptosis of MCF-7 cells treated with different concentrations of lucidimine B [control (A), 10 (B), 50 (C), and 100 μg/mL (D)], determined by flow cytometry with annexin-V/PI. The upper left quadrant displays necrosis cells (Q1); the upper right quadrant displays late apoptosis cells (Q2); the lower left quadrant displays normal cells (Q3); and the lower right quadrant displays early apoptotic cells (Q4). The ratios of apoptosis in MCF-7 cells induced by lucidimine B (E) (mean ± SD, n = 3). *P < 0.05.
Impact on Mitochondrial Membrane Potential Exerted by Lucidimine B
Mitochondria play an important role in energy production in MCF-7 cells, being regarded as their energy powerhouses.57 Many factors can cause mitochondrial damage, which, in turn, increases mitochondrial membrane permeability and subsequent dysfunction of mitochondrial membrane potential (MMP).58 Normally, when cells were stained with JC-1, red fluorescence was detected in normal cells, whereas green fluorescence was detected in apoptotic cells with impaired mitochondria. The red/green fluorescence intensity quantifies the MMP change; the lower the red/green fluorescence intensity, the stronger impaired the mitochondria. From Figure 5, the cell affected by lucidimine B displayed a decrease in MMP compared with that of the control group, indicating that lucidimine B damaged the mitochondrial function and increased the permeability of the mitochondrial membrane. With increased permeability of the mitochondrial membrane, the release of cytochrome c was promoted, which activates procaspase 9 and thus downstream caspases, which amplify the death process.59 Loss of MMP is considered as a pathway for apoptosis, and increase of MMP loss could lead to the activation of apoptosis-inducing factors from mitochondria, which promotes apoptosis by inactivating inhibitors of apoptosis proteins.60,61
Figure 5.
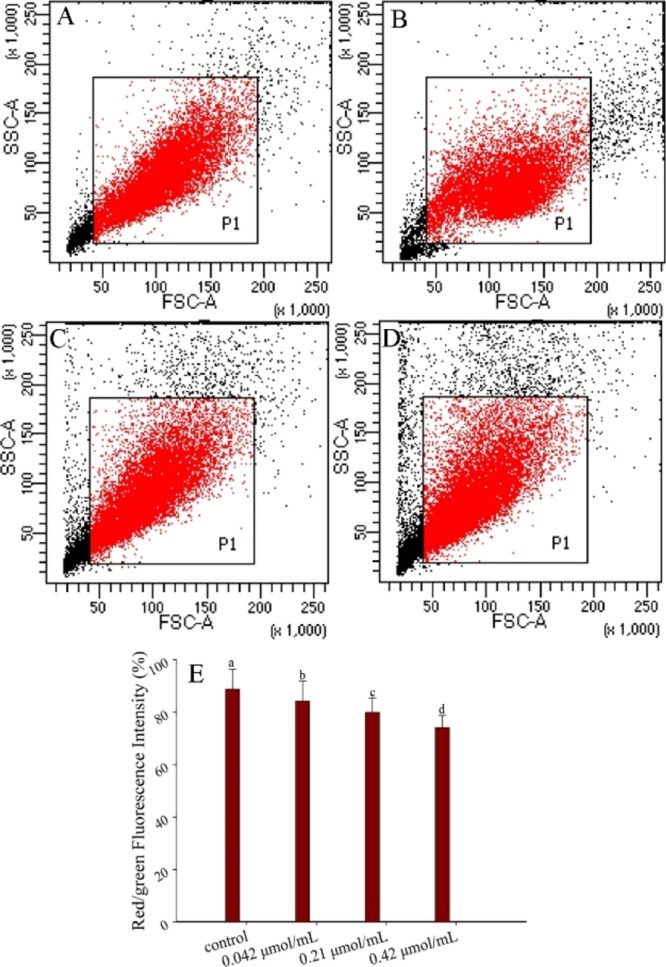
Effect of different concentrations of lucidimine B [control (A), 10 (B), 50 (C), and 100 μg/mL (D)] on MMP in MCF-7 cells, determined by flow cytometry with JC-1; P1 represents red fluorescence (aggregated by JC-1). Red/green fluorescence intensity of variation in MMP affected by lucidimine B (E) (mean ± SD, n = 3). *P < 0.05.
Conclusions
The work detailed above, when considered in conjunction with various studies conducted on other components from G. lucidum, has provided valuable scientific evidence to support the health claims with respect to G. lucidum as nutraceuticals. Our work has proved that alkaloid components such as lucidimine B from G. lucidum display valuable yet underestimated antioxidant and antiproliferative potencies. Further mechanistic studies revealed that lucidimine B could induce MCF-7 cell cycle arrest in the S phase, inducing DNA fragmentation and blocking cell cycle progression of MCF-7 cells. The proliferation of MCF-7 cells being treated with lucidimine B was suppressed in early-stage apoptosis, and they displayed decreased MMP. The synthetic protocols used to obtain the title natural products and certain analogues are robust ones and provide the capacity to generate a significant number of variants of the title compounds.
Experimental Section
Materials
Cyclopentanone, malononitrile, morpholine, methanesulfonic acid, acetic anhydride, DMF-DMA, acetic acid (AcOH), hydrobromic acid (HBr), (2,5-dimethoxyphenyl)boronic acid, palladium acetate [Pd(OAc)2], 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (S-Phos), potassium phosphate (K3PO4), boron tribromide (BBr3), boron trifluoride diethyl etherate (BF3·OEt2), sodium borohydride (NaBH4), DIBAl-H, p-toluenesulfonic acid, DPPH, ABTS, DCFH-DA, and AAPH were all purchased from Sigma-Aldrich (St. Louis, MO, USA). Aluminum-backed 0.2 mm thick silica gel 60 F254 plates for analytical thin-layer chromatography as well as silica gel 60 (40–63 μm) for flash chromatographic separations were supplied by Merck (Darmstadt, Germany). Cell culture media were purchased from Gibco U.S. Biotechnology Co. Fetal bovine serum (FBS) was bought from Tianhang Biotech Co. Inc. (Zhejiang, China). 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1), PI, and annexin V-FITC/PI kits were obtained from Becton, Dickinson and Company (Franklin Lakes, NJ, USA).
General Synthetic Protocols
Proton (1H) and carbon (13C) NMR spectra were recorded at 25 °C on a Bruker spectrometer operated at 400 MHz for proton and 100 MHz for carbon nuclei. For 1H NMR spectra, signals arising from the residual protio-forms of the solvent were used as the internal standards. 1H NMR data were recorded as follows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz), relative integral], where multiplicity is defined as s = singlet; d = doublet; t = triplet; q = quartet; and m = multiplet or combinations of the above. Low-resolution electrospray ionization mass spectra were recorded on a single quadrupole liquid chromatograph–mass spectrometer, whereas high-resolution measurements were conducted on a time-of-flight instrument. Low- and high-resolution electron ionization (EI) mass spectra were recorded on a magnetic-sector machine. Melting points were measured on an OptiMelt automated melting point system and are uncorrected. Infrared spectra (νmax) were recorded on a PerkinElmer 1800 Series FTIR spectrometer. Samples were analyzed as thin films on KBr plates. Eluted plates were visualized using a 254 nm UV lamp. Where necessary, reactions were performed under a nitrogen atmosphere.
Specific Chemical Transformations
2-Cyclopentylidenemalononitrile 7
The procedure for the preparation of 7 was adapted from the literature.41 Thus, a magnetically stirred mixture of cyclopentanone (5) (10.0 g, 118.9 mmol), malononitrile (6) (9.4 g, 142.7 mmol), morpholine (0.5 g, 5.7 mmol), and methanesulfonic acid (0.5 g, 5.2 mmol) was heated at 120 °C for 1 h before being cooled to 18 °C. The mixture thus obtained was subjected to flash chromatography (silica, hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.8), malononitrile 7 (14.0 g, 89%) as clear colorless oil. 1H NMR (400 MHz, CDCl3): δ 2.81–2.77 (complex m, 4H), 1.94–1.90 (complex m, 4H). 13C NMR (100 MHz, CDCl3): δ 192.4, 111.7 (×2), 81.2, 36.1 (×2), 25.9 (×2). IR (KBr) νmax: 2974, 2954, 2888, 2231, 1612, 1454, 1408, 1316, 1231, 1140, 1024, 939, 853, 673, 630 cm–1. MS (EI, +70 eV) m/z: 132 (M+•, 75%), 104 (32), 41 (100). HRMS M+• calcd for C8H8N2: 132.0687; found, 132.0690.
3-Bromo-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile 9
Step (i): Following a protocol reported by McQuade et al.,42 a magnetically stirred mixture of malononitrile 7 (14.0 g, 105.9 mmol) in toluene (150 mL) was treated with Ac2O (2.0 mL, 21.2 mmol) and DMF-DMA (16.9 mL, 127.1 mmol), and the resulting mixture was stirred at 18 °C for 12 h. Hexane (200 mL) was then added to the reaction mixture, which was cooled to 0 °C, and the solid thus formed was collected by vacuum filtration. The resulting crude product, which contains enamine 8, was dried under reduced pressure and then used directly in the next step. Step (ii): A magnetically stirred mixture of the abovementioned enamine 8 in AcOH (60 mL) was treated dropwise with HBr (110 mL of a 33 wt % solution in AcOH, 628 mmol) and then heated at 55 °C for 1 h before being cooled to 0 °C. Water (100 mL) was then slowly added, and the precipitate was collected by vacuum filtration and rinsed with water (100 mL). The off-white solid was dried in an oven, then dissolved in a minimum amount of dichloromethane, and subjected to flash chromatography (silica, dichloromethane elution) to afford, after concentration of the relevant fractions (Rf = 0.7), compound 9 (20.1 g, 85%) as an amorphous white powder. 1H NMR (400 MHz, CDCl3): δ 8.33 (s, 1H), 3.13 (t, J = 7.6 Hz, 2H), 2.98 (t, J = 7.6 Hz, 2H), 2.23 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.8, 147.9, 140.3, 140.2, 114.9, 110.6, 33.0, 29.9, 24.7. IR (KBr) νmax: 2975, 2954, 2229, 1559, 1445, 1428, 1359, 1305, 1235, 1183, 984, 861, 734, 636 cm–1. MS (EI, +70 eV) m/z: 223 and 225 ([M + H]+•, both 100%). HRMS M+• calcd for C9H8N279Br, 222.9871; found, 222.9874. Calcd for C9H8N281Br, 224.9850; found, 224.9846.
3-(2,5-Dimethoxyphenyl)-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile 11
A magnetically stirred mixture of nitrile 9 (5.0 g, 22.4 mmol) and (2,5-dimethoxyphenyl)boronic acid 10 (4.9 g, 26.9 mmol) in dioxane/water (400 mL of a 3:1 v/v mixture) was purged with argon for 0.33 h before being treated with Pd(OAc)2 (50 mg, 0.22 mmol), S-Phos (184 mg, 0.44 mmol), and K3PO4 (4.9 g, 23.1 mmol). The ensuing solution was heated at 80 °C for 16 h while being maintained under a nitrogen atmosphere. The cooled solution was poured into NH4Cl solution (150 mL of a saturated aqueous solution) and extracted by EtOAc (3 × 150 mL). The organic layer was separated and then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, 1:3 v/v ethyl acetate/hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.6), compound 11 (5.2 g, 83%) as an amorphous white powder. 1H NMR (400 MHz, CDCl3): δ 8.65 (s, 1H), 6.96 (m, 3H), 3.81 (s, 3H), 3.78 (s, 3H), 3.15 (t, J = 7.6 Hz, 2H), 3.03 (t, J = 7.6 Hz, 2H), 2.22 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 158.4, 156.8, 153.5, 150.8, 147.7, 138.7, 127.6, 116.5, 116.2, 115.8, 112.4, 107.4, 55.8, 55.7, 32.7, 30.3, 24.4. IR (KBr) νmax: 2947, 2835, 2227, 1584, 1557, 1499, 1487, 1259, 1216, 1178, 1042, 808, 760, 735 cm–1. MS (EI, +70 eV) m/z: 303.1 ([M + Na]+•, 100%), 281.3 ([M + H]+•, 50). HRMS M+• calcd for C17H17N2O2, 281.1290; found, 281.1288.
3-(2,5-Dimethoxyphenyl)-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carboxylic Acid 12
A magnetically stirred mixture of compound 11 (5.0 g, 17.8 mmol) in MeOH (300 mL) was treated with 8 M NaOH aqueous solution (100 mL). The ensuing solution was heated at 90 °C for 12 h before being concentrated under reduced pressure. The residue thus formed was then dissolved in water, the pH was adjusted to 2 by concentrated HCl, and the mixture was extracted by EtOAc (3 × 150 mL). The organic layer was separated and then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, 1:10 v/v MeOH/CH2Cl2 elution) to afford, after concentration of the relevant fractions (Rf = 0.5), compound 12 (4.8 g, 90%) as an amorphous white powder. 1H NMR (400 MHz, CD3OD): δ 8.66 (s, 1H), 7.15 (m, 3H), 3.79 (s, 6H), 3.32 (m, 2H), 3.23 (t, J = 7.6 Hz, 2H), 2.32 (m, 2H). 13C NMR (100 MHz, CD3OD): δ 167.9, 165.2, 155.0, 152.4, 147.3, 145.5, 137.8, 134.5, 120.67, 119.7, 117.3, 113.9, 56.6, 56.4, 34.4, 31.5, 25.9. IR (KBr) νmax: 3284, 3118, 2990, 2828, 1670, 1614, 1499, 1425, 1272, 1224, 1042, 1024, 896, 828, 752, 715, 669 cm–1. MS (EI, +70 eV) m/z: 299.1 (M+•, 15%), 298.1 ([M – H]+•, 70), 267.1 (90), 254.1 (100). HRMS M+• calcd for C17H16NO4, 298.1079; found, 298.1085.
8-Hydroxy-2,3-dihydrochromeno[4,3-b]cyclopenta[d]pyridin-4(1H)-one 13
A magnetically stirred mixture of acid 12 (4.5 g, 15.0 mmol) in dry CH2Cl2 (300 mL) at 0 °C was treated with BBr3 (4.3 mL, 45.0 mmol). The ensuing solution was then warmed to 18 °C and stirred for 16 h before being quenched by MeOH (10 mL). The solution was then poured into brine (200 mL) and was extracted by CH2Cl2 (3 × 100 mL). The organic layer was separated and then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, EtOAc elution) to afford, after concentration of the relevant fractions (Rf = 0.8), lactone 13 (2.2 g, 58%) as an amorphous white powder. 1H NMR (400 MHz, DMSO-d6): δ 9.74 (broad s, 1H), 8.85 (s, 1H), 7.81 (d, J = 3.0 Hz, 1H), 7.25 (d, J = 8.8 Hz, 1H), 7.01 (dd, J = 8.8 and 3.0 Hz, 1H), 3.37 (m, 2H), 3.01 (t, J = 7.6 Hz, 2H), 2.14 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 160.0, 157.5, 154.1, 150.9, 149.4, 145.1, 141.6, 119.8, 119.4, 117.7, 114.0, 108.7, 33.7, 29.6, 24.0. IR (KBr) νmax: 3222, 2990, 2828, 1679, 1554, 1462, 1440, 1345, 1254, 1223, 1180, 1126, 979, 888, 821, 714, 697 cm–1. MS (EI, +70 eV) m/z: 276.2 ([M + Na]+•, 60%), 254.2 ([M + H]+•, 10), 84.9 (100). HRMS M+• calcd for C15H12NO3, 254.0812; found, 254.0814.
Lucidimine B 2
Following a similar procedure reported by Xu et al.,43 a solution of lactone 13 (500 mg, 1.98 mmol) in dry THF (20 mL) and BF3·OEt2 (5 mL) was added over 15 min to a suspension of NaBH4 (250 mg, 6.60 mmol) in THF (5 mL) under nitrogen while maintaining the reaction temperature below 10 °C. The ensuing solution was then heated to 70 °C and stirred for 4 h before being cooled to 0 °C. Cold HCl (8 mL of a 2 N aqueous solution) was then cautiously added, and the solution was warmed up to increase to 25 °C. Water (40 mL) was added, and the reaction mixture was extracted with CH2Cl2 (3 × 50 mL). The combined extracts were evaporated, and the ensuing residue was heated at 80 °C with NaOH (80 mL of 2 N aqueous solution) for 30 min. The resulting mixture was cooled and extracted with EtOAc (4 × 50 mL). The organic extracts were combined, then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, 1:3 v/v ethyl acetate/hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.4), lucidimine B 2 (250 mg, 53%), as an amorphous white powder, which was recrystallized in methanol to give a crystalline solid mp 147–150 °C. 1H NMR (400 MHz, CD3OD): δ see Table 1. 13C NMR (100 MHz, CD3OD): δ Table 1. IR (KBr) νmax: 2952, 2839, 1504, 1555, 1491, 1470, 1385, 1288, 1265, 1183, 1041, 963, 919, 885, 824, 805, 761, 613 cm–1. MS (EI, +70 eV) m/z: 262.1 ([M + Na]+•, 100%), 240.2 ([M + H]+•, 60). HRMS M+• calcd for C15H14NO2, 240.1019; found, 240.1021.
3-Bromo-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbaldehyde 14
A magnetically stirred mixture of nitrile 9 (2.3 g, 10.0 mmol) in dry toluene (60 mL) at −78 °C was treated with DIBAl-H (12 mL of a 1 M solution in toluene, 12.0 mmol), and the mixture was warmed to 18 °C over a period of 12 h. The reaction was then quenched by 2 M sulfuric acid (35.0 mL), the resulting mixture was stirred for another 1 h at 18 °C before being diluted with water (300 mL), the organic layer was separated, and the aqueous layer was further extracted by EtOAc (2 × 100 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, 1:9 v/v ethyl acetate/hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.4), aldehyde 14 (2.0 g, 87%), as an amorphous white powder. 1H NMR (400 MHz, CDCl3): δ 10.43 (s, 1H), 8.32 (s, 1H), 3.28 (t, J = 7.6 Hz, 2H), 2.91 (t, J = 7.6 Hz, 2H), 2.16 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 193.2, 159.4, 148.7, 142.9, 142.2, 126.6, 33.2, 29.1, 25.1. IR (KBr) νmax: 2956, 2860, 1684, 1559, 1553, 1442, 1429, 1338, 1232, 1183, 1140, 1001, 859, 729 cm–1. MS (EI, +70 eV) m/z: 227.1 and 225.1 (M+•, both 100%). HRMS M+• calcd for C9H8NO79Br, 224.9789; found, 224.9791. Calcd for C9H8NO81Br, 226.9769; found, 226.9765.
3-(2,5-Dimethoxyphenyl)-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbaldehyde 15
A magnetically stirred mixture of nitrile 14 (1.8 g, 8.0 mmol) and (2,5-dimethoxyphenyl)boronic acid 10 (1.7 g, 9.6 mmol) in dioxane/water (200 mL of a 3:1 v/v mixture) was purged with argon for 0.33 h before being treated with Pd(OAc)2 (18 mg, 0.08 mmol), S-Phos (67 mg, 0.16 mmol), and K3PO4 (1.8 g, 8.0 mmol). The ensuing solution was heated at 80 °C for 16 h while being maintained under a nitrogen atmosphere. The cooled solution was poured into NH4Cl solution (100 mL of a saturated aqueous solution) and extracted by EtOAc (3 × 100 mL). The organic layer was separated and then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was subjected to flash chromatography (silica, 1:3 v/v ethyl acetate/hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.6), compound 15 (1.8 g, 80%) as an amorphous white powder. 1H NMR (400 MHz, CDCl3): δ 9.83 (s, 1H), 8.68 (s, 1H), 7.08 (d, J = 3.1 Hz, 1H), 6.98 (dd, J = 9.0 and 3.1 Hz, 1H), 6.90 (d, J = 3.1 Hz, 1H), 3.82 (s, 3H), 3.69 (s, 3H), 3.34 (t, J = 7.6 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.19 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 193.4, 157.2, 155.0, 153.9, 150.2, 148.5, 140.8, 127.7, 126.5, 116.5, 116.3, 112.0, 55.9, 55.8, 33.3, 29.6, 24.9. IR (KBr) νmax: 2951, 2835, 1690, 1583, 1555, 1497, 1460, 1269, 1217, 1177, 1043, 808, 760, 717 cm–1. MS (EI, +70 eV) m/z: 283.3 (M+•, 50%), 254.3 (100). HRMS M+• calcd for C17H17NO3, 283.1208; found, 283.1206.
Lucidimine C 3
A magnetically stirred mixture of aldehyde 15 (0.8 g, 2.82 mmol) in dry CH2Cl2 (100 mL) at 0 °C was treated with BBr3 (0.8 mL, 8.48 mmol). The ensuing solution was then warmed to 18 °C and stirred for 16 h before being quenched by MeOH (1 mL). The solution was then poured into brine (100 mL) and was extracted by CH2Cl2 (3 × 100 mL). The organic layer was separated and then dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue thus obtained was dissolved in MeOH (10 mL) and was treated with p-toluenesulfonic acid (1.0 g, 5.64 mmol). The solution was stirred at 18 °C for 1 h before being concentrated, and the residue was subjected to flash chromatography (silica, 1:3 v/v ethyl acetate/hexane elution) to afford, after concentration of the relevant fractions (Rf = 0.4), lucidimine C 3 (0.4 g, 53%) as an amorphous white powder, which was recrystallized in methanol to give a crystalline solid mp 167–169 °C. 1H NMR (400 MHz, CD3OD): δ see Table 2. 13C NMR (100 MHz, CD3OD): δ see Table 2. IR (KBr) νmax: 2910, 2839, 1505, 1570, 1465, 1353, 1257, 1179, 1090, 1028, 1000, 992, 964, 854, 828, 739, 613 cm–1. MS (EI, +70 eV) m/z: 269.1 (M+•, 30%), 238.1 (100). HRMS M+• calcd for C16H15NO3, 269.1052; found, 269.1053.
DPPH Radical Scavenging Assay
A DPPH radical scavenging assay was developed following the published method and with minor modification.44 Briefly, 2 mL of DPPH (0.2 mM, in methanol) was mixed with 1 mL of the sample at different concentrations, and methanol was used as a control. Then, the various solutions were completely mixed and allowed to stand in dark at room temperature for 30 min before being tested for absorbance at 517 nm using a microplate reader (Tecan M200 PRO, Switzerland). The DPPH radical scavenging activity of the sample was calculated using the following formula:
The percentages of DPPH radical scavenging activities were plotted against the sample concentrations to obtain the corresponding EC50 values, which were defined as the concentration of sample necessary to cause a 50% scavenging effect.
ABTS Radical Scavenging Assay
An ABTS radical scavenging assay was employed following a published protocol.44 The ABTS radical cation working solution with an absorbance of 0.70 ± 0.02 at 734 nm was prepared by mixing ABTS and potassium persulfate. A sample solution (0.15 mL) mixed with the ABTS working solution (2.85 mL) was left at room temperature for 10 min before being tested for absorbance at 734 nm using a microplate reader (Tecan M200 PRO, Switzerland). The control containing the solvent instead of the sample solution was also prepared. The ABTS radical scavenging activity of the sample was calculated by the following formula:
Again, the percentages of ABTS radical scavenging activities were plotted against the sample concentration to obtain the EC50 values, which were defined as the concentration of sample necessary to cause a 50% scavenging effect.
PSC Assay
Peroxyl radical scavenging assay was performed using a reported method with modification.45 Vitamin C was selected as a standard, and phosphate buffer was used as the reaction medium. Fluorescence was monitored with a Fluoroskan Ascent fluorescence spectrophotometer (Tecan M200 PRO, Switzerland) for 23 cycles. Results were shown as μmol vitamin C equiv/g.
Cell Lines and Cell Culture
Human breast cancer cell lines (MDA-MB-231 and MCF-7) were maintained in DMEM (high glucose) supplemented with streptomycin (50 units/mL), penicillin (100 units/mL), and 10% FBS at 37 °C in a humidified incubator with a 5% CO2 atmosphere.
Antiproliferative Ability of Lucidimine B and Lucidimine C
An antiproliferation test was carried out using a methylene blue assay.62 Cells were seeded at a density of 1 × 104 cells/well on a clear 96-well microplate in 100 μL of growth medium/well. After 6 h at 37 °C, the growth medium was removed and the cells were washed with phosphate-buffered saline (PBS) solution. The free FBS medium with different concentrations of samples was then added to the well, whereas the same medium without sample served as the control. After an additional 48 h of incubation, the wells were stained with methylene blue solution. The absorbance was measured at 570 nm in a microplate reader.
Determination of Cell Cycle and Apoptosis through Flow Cytometry
Samples were prepared with free FBS medium under different concentrations. The cell cycle distribution of MCF-7 cells after having been treated for 24 h were washed with PBS and harvested with trypsinization. According to the PI detection kit, the harvested cells were treated with 70% ethanol solution at −20 °C for a period of 12 h and then stained by PI-containing RNAse at 37 °C for 15 min. For each sample, 10 000 events were recorded by flow cytometry. The results were determined by the amount of PI intercalated to DNA and analyzed by CellQuest research software (FACSCanto, Becton, Dickinson and Company, USA). Proportion of apoptotic MCF-7 cells was determined through an annexin V-FITC/PI apoptosis detection kit. The samples were prepared with free FBS media at different concentrations. The MCF-7 cells treated with various concentrations of lucidimine B for 24 h were washed with PBS and harvested with trypsinization. The harvested cells were stained with annexin V-FITC/PI at 37 °C for 15 min. For each sample, 10 000 events were recorded by flow cytometry. The data were collected and analyzed by CellQuest research software.
Determination of MMP
The MMPs of MCF-7 cells were measured and analyzed with a JC-1 detection kit. Samples were prepared with free FBS media at various concentrations. MCF-7 cells were treated with different concentrations of lucidimine B for 24 h, washed with PBS, and harvested with trypsinization. The harvested cells were suspended and dyed using JC-1 at room temperature for 15 min. Then, the dyed cells were centrifuged, washed once with PBS, and resuspended with 200 μL of PBS. The excitation wavelength for flow cytometric analysis was 488 nm, and the emission wavelengths were 525 nm for green fluorescence analysis and 590 nm for red fluorescence. For each sample, 10 000 events were recorded by flow cytometry.
Statistical Analysis
Each bioactivity evaluation experiment was performed three times. The results were presented as mean values ± SD. Statistical analysis was performed using the SPSS software (version 19.0) to determine the significant differences. All values were assessed by one-way analysis of variance in conjunction with Duncan’s new multiple-range test. A difference was considered significant when P < 0.05.
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities, China, and the China Postdoctoral Science Foundation (no. 2017M622910).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00295.
1H and 13C NMR spectra of compounds 2, 3, 7, 9, and 11–15 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tang N.; Skibsted L. H. Zinc bioavailability from phytate-rich foods and zinc supplements. Modeling the effects of food components with oxygen, nitrogen, and sulfur donor ligands. J. Agric. Food Chem. 2017, 65, 8727–8743. 10.1021/acs.jafc.7b02998. [DOI] [PubMed] [Google Scholar]
- Ferreira I. C. F. R.; Vaz J. A.; Vasconcelos M. H.; Martins A. Compounds from wild mushrooms with antitumor potential. Anti-Cancer Agents Med. Chem. 2010, 10, 424–436. 10.2174/1871520611009050424. [DOI] [PubMed] [Google Scholar]
- Giménez-Bastida J. A.; Zieliński H. Buckwheat as a functional food and its effects on health. J. Agric. Food Chem. 2015, 63, 7896–7913. 10.1021/acs.jafc.5b02498. [DOI] [PubMed] [Google Scholar]
- Cakebread J. A.; Humphrey R.; Hodgkinson A. J. Immunoglobulin A in bovine milk: a potential functional food?. J. Agric. Food Chem. 2015, 63, 7311–7316. 10.1021/acs.jafc.5b01836. [DOI] [PubMed] [Google Scholar]
- Brower V. Nutraceuticals: poised for a healthy slice of the healthcare market?. Nat. Biotechnol. 1998, 16, 728–731. 10.1038/nbt0898-728. [DOI] [PubMed] [Google Scholar]
- Ting Y.; Zhao Q.; Xia C.; Huang Q. Using in vitro and in vivo models to evaluate the oral bioavailability of nutraceuticals. J. Agric. Food Chem. 2015, 63, 1332–1338. 10.1021/jf5047464. [DOI] [PubMed] [Google Scholar]
- Chung X.; Rojanasasithara T.; Mutilangi W.; McClements D. J. Stabilization of natural colors and nutraceuticals: inhibition of anthocyanin degradation in model beverages using polyphenols. Food Chem. 2016, 212, 596–603. 10.1016/j.foodchem.2016.06.025. [DOI] [PubMed] [Google Scholar]
- Santini A.; Novellino E.; Armini V.; Ritieni A. State of the art of ready-to-use therapeutic food: a tool for nutraceuticals addition to foodstuff. Food Chem. 2013, 140, 843–849. 10.1016/j.foodchem.2012.10.098. [DOI] [PubMed] [Google Scholar]
- Donadio S.; Monciardini P.; Alduina R.; Mazza P.; Chiocchini C.; Cavaletti L.; Sosio M.; Puglia A. M. Microbial technologies for the discovery of novel bioactive metabolites. J. Biotechnol. 2002, 99, 187–198. 10.1016/s0168-1656(02)00209-2. [DOI] [PubMed] [Google Scholar]
- Mizuno T. Bioactive biomolecules of mushrooms: Food function and medicinal effect of mushroom fungi. Food Rev. Int. 1995, 11, 5–21. 10.1080/87559129509541017. [DOI] [Google Scholar]
- Wasser S. P.; Weis A. L. Medicinal properties of substances occurring in higher Basidiomycetes mushrooms: current perspectives. Int. J. Med. Mushrooms 1999, 1, 31–62. 10.1615/intjmedmushrooms.v1.i1.30. [DOI] [PubMed] [Google Scholar]
- Wasser S. Medicinal mushrooms as a source of antitumor and immunomodulating polysaccharides. Appl. Microbiol. Biotechnol. 2002, 60, 258–274. 10.1007/s00253-002-1076-7. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhang J.; Chen H.; Chen X.; Lan J.; Liu C. Complete mitochondrial genome of the medicinal mushroom Ganoderma lucidum. PLoS One 2013, 8, e72038. 10.1371/journal.pone.0072038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanodiya B.; Thakur G.; Baghel R.; Prasad G.; Bisen P. Gonoderma lucidum: a potent pharmacological macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. 10.2174/138920109789978757. [DOI] [PubMed] [Google Scholar]
- Wang C.-Z.; Basila D.; Aung H. H.; Mehendale S. R.; Chang W.-T.; McEntee E.; Guan X.; Yuan C.-S. Effects of Ganoderma lucidum extract on chemotherapy-induced nausea and vomiting in a rat model. Am. J. Chin. Med. 2005, 33, 807–815. 10.1142/s0192415x05003429. [DOI] [PubMed] [Google Scholar]
- Sliva D. Ganoderma lucidum(Reishi) in cancer treatment. Integr. Cancer Ther. 2003, 2, 358–364. 10.1177/1534735403259066. [DOI] [PubMed] [Google Scholar]
- Sliva D. Cellular and physiological effects of Ganoderma lucidum (Reishi). Mini-Rev. Med. Chem. 2004, 4, 873–879. 10.2174/1389557043403323. [DOI] [PubMed] [Google Scholar]
- Cheng S.; Sliva D. Ganoderma lucidum for cancer treatment: we are close but still not there. Integr. Cancer Ther. 2015, 14, 249–257. 10.1177/1534735414568721. [DOI] [PubMed] [Google Scholar]
- Paterson R. R. M. Ganoderma—a therapeutic fungal biofactory. Phytochemistry 2006, 67, 1985–2001. 10.1016/j.phytochem.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Sanodiya B.; Thakur G.; Baghel R.; Prasad G.; Bisen P. Ganoderma lucidum: a potent pharmacological macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. 10.2174/138920109789978757. [DOI] [PubMed] [Google Scholar]
- Bishop K. S.; Kao C. H. J.; Xu Y.; Glucina M. P.; Paterson R. R. M.; Ferguson L. R. From 2000 years of Ganoderma lucidum to recent developments in nutraceuticals. Phytochemistry 2015, 114, 56–65. 10.1016/j.phytochem.2015.02.015. [DOI] [PubMed] [Google Scholar]
- Boh B.; Berovic M.; Zhang J.; Lin Z.-B. Ganoderma lucidum and its pharmaceutically active compounds. Biotechnol. Annu. Rev. 2007, 13, 265–301. 10.1016/s1387-2656(07)13010-6. [DOI] [PubMed] [Google Scholar]
- Zhao X.-R.; Huo X.-K.; Dong P.-P.; Wang C.; Huang S.-S.; Zhang B.-J.; Zhang H.-L.; Deng S.; Liu K.-X.; Ma X.-C. Inhibitory effects of highly oxygenated lanostane derivatives from the fungus Ganoderma lucidum on P-glycoprotein and α-glucosidase. J. Nat. Prod. 2015, 78, 1868–1876. 10.1021/acs.jnatprod.5b00132. [DOI] [PubMed] [Google Scholar]
- Radwan F. F. Y.; Perez J. M.; Haque A. Apoptotic and immune restoration effects of ganoderic acids define a new prospective for complementary treatment of cancer. J. Clin. Cell. Immunol. 2011, 01, S3–S4. 10.4172/2155-9899.s3-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.-T.; Deng Y.; Chen L.-X.; Zhao J.; Bzhelyansky A.; Li S.-P. Evaluation on quality consistency of Ganoderma lucidum dietary supplements collected in the United States. Sci. Rep. 2017, 7, 7792. 10.1038/s41598-017-06336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taofiq O.; Heleno S. A.; Calhelha R. C.; Alves M. J.; Barros L.; González-Paramás A. M.; Barreiro M. F.; Ferreira I. C. F. R. The potential of Ganoderma lucidum extracts as bioactive ingredients in topical formulations, beyond its nutritional benefits. Food Chem. Toxicol. 2017, 108, 139–147. 10.1016/j.fct.2017.07.051. [DOI] [PubMed] [Google Scholar]
- Wang C.-H.; Hsieh S.-C.; Wang H.-J.; Chen M.-L.; Lin B.-F.; Chiang B.-H.; Lu T.-J. Concentration variation and molecular characteristics of soluble (1,3;1,6)-β-D-glucans in submerged cultivation products of Ganoderma lucidum mycelium. J. Agric. Food Chem. 2014, 62, 634–641. 10.1021/jf404533b. [DOI] [PubMed] [Google Scholar]
- Wu G.-S.; Guo J.-J.; Bao J.-L.; Li X.-W.; Chen X.-P.; Lu J.-J.; Wang Y.-T. Anti-cancer properties of triterpenoids isolated from Ganoderma lucidum—a review. Expert Opin. Invest. Drugs 2013, 22, 981–992. 10.1517/13543784.2013.805202. [DOI] [PubMed] [Google Scholar]
- Yan Y.-M.; Wang X.-L.; Luo Q.; Jiang L.-P.; Yang C.-P.; Hou B.; Zuo Z.-L.; Chen Y.-B.; Cheng Y.-X. Metabolites from the mushroom Ganoderma lingzhi as stimulators of neural stem cell proliferation. Phytochemistry 2015, 114, 155–162. 10.1016/j.phytochem.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Luo Q.; Wang X.-L.; Di L.; Yan Y.-M.; Lu Q.; Yang X.-H.; Hu D.-B.; Cheng Y.-X. Isolation and identification of renoprotective substances from the mushroom Ganoderma lucidum. Tetrahedron 2015, 71, 840–845. 10.1016/j.tet.2014.12.052. [DOI] [Google Scholar]
- Xu Z.; Chen X.; Zhong Z.; Chen L.; Wang Y. Ganoderma lucidum polysaccharides: immunomodulation and potential anti-tumor activities. Am. J. Chin. Med. 2011, 39, 15–27. 10.1142/s0192415x11008610. [DOI] [PubMed] [Google Scholar]
- Xiang Q.-D.; Yu Q.; Wang H.; Zhao M.-M.; Liu S.-Y.; Nie S.-P.; Xie M.-Y. Immunomodulatory activity of Ganoderma atrum polysaccharide on purified T lymphocytes through Ca2+/CaN and mitogen-activated protein kinase pathway based on RNA sequencing. J. Agric. Food Chem. 2017, 65, 5306–5315. 10.1021/acs.jafc.7b01763. [DOI] [PubMed] [Google Scholar]
- Li W.-J.; Tang X.-F.; Shuai X.-X.; Jiang C.-J.; Liu X.; Wang L.-F.; Yao Y.-F.; Nie S.-P.; Xie M.-Y. Mannose receptor mediates the immune response to Ganoderma atrum polysaccharides in macrophages. J. Agric. Food Chem. 2017, 65, 348–357. 10.1021/acs.jafc.6b04888. [DOI] [PubMed] [Google Scholar]
- Zhu K.-X.; Nie S.-P.; Tan L.-H.; Li C.; Gong D.-M.; Xie M.-Y. A polysaccharide from Ganoderma atrum improves liver function in type 2 diabetic rats via antioxidant action and short-chain fatty acids excretion. J. Agric. Food Chem. 2016, 64, 1938–1944. 10.1021/acs.jafc.5b06103. [DOI] [PubMed] [Google Scholar]
- Yu J. G.; Chen R. Y.; Yao Z. X.; Zhai Y. F.; Yang S. L.; Ma J. L. Constituents of Ganoderma capense IV. The chemical structures of ganoine, ganodine and ganoderpurine. Acta Pharmacol. Sin. 1990, 25, 612–616. [PubMed] [Google Scholar]
- Liu C.; Zhao F.; Chen R. A novel alkaloid from the fruiting bodies of Ganoderma sinense. Chin. Chem. Lett. 2010, 21, 197–199. 10.1016/j.cclet.2009.07.023. [DOI] [Google Scholar]
- Liu J.-Q.; Wang C.-F.; Peng X.-R.; Qiu M.-H. New alkaloids from the fruiting bodies of Ganoderma sinense. Nat. Prod. Bioprospect. 2011, 1, 93–96. 10.1007/s13659-011-0026-4. [DOI] [Google Scholar]
- Wang X.-L.; Dou M.; Luo Q.; Cheng L.-Z.; Yan Y.-M.; Li R.-T.; Cheng Y.-X. Racemic alkaloids from the fungus Ganoderma cochlear. Fitoterapia 2017, 116, 93–98. 10.1016/j.fitote.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Zhao Z.-Z.; Chen H.-P.; Feng T.; Li Z.-H.; Dong Z.-J.; Liu J.-K. Lucidimine A-D, four new alkaloids from the fruiting bodies of Ganoderma lucidum. J. Asian Nat. Prod. Res. 2015, 17, 1160–1165. 10.1080/10286020.2015.1119128. [DOI] [PubMed] [Google Scholar]
- Huang S. Z.; Cheng B. H.; Ma Q. Y.; Wang Q.; Kong F. D.; Dai H. F.; Qiu S. Q.; Zheng P. Y.; Liu Z. Q.; Zhao Y.-X. Anti-allergic prenylated hydroquinones and alkaloids from the fruiting body of Ganoderma calidophilum. RSC Adv. 2016, 6, 21139–21147. 10.1039/c6ra01466f. [DOI] [Google Scholar]
- Góra M.; Kozik B.; Jamroży K.; Łuczyński M. K.; Brzuzan P.; Woźny M. Solvent-free condensations of ketones with malononitrile catalyzed by methanesulfonic acid/morpholine system. Green Chem. 2009, 11, 863–867. 10.1039/b820901d. [DOI] [Google Scholar]
- Longstreet A. R.; Campbell B. S.; Gupton B. F.; McQuade D. T. Improved synthesis of mono- and disubstituted 2-halonicotinonitriles from alkylidene malononitriles. Org. Lett. 2013, 15, 5298–5301. 10.1021/ol4025265. [DOI] [PubMed] [Google Scholar]
- Dai J.-J.; Xu W.-T.; Wu Y.-D.; Zhang W.-M.; Gong Y.; He X.-P.; Zhang X.-Q.; Xu H.-J. Silver-catalyzed C(sp2)–H functionalization/C–O cyclization reaction at room temperature. J. Org. Chem. 2015, 80, 911–919. 10.1021/jo5024238. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhang R.; Liu C.; Zheng X.; Liu B. Enhancing antioxidant activity and antiproliferation of wheat bran through steam flash explosion. J. Food Sci. Technol. 2016, 53, 3028–3034. 10.1007/s13197-016-2274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Chen G.; Fu X.; Liu R.-H. Phytochemical profiles and antioxidant activity of different varieties of Adinandra tea (Adinandra Jack). J. Agric. Food Chem. 2015, 63, 169–176. 10.1021/jf503700v. [DOI] [PubMed] [Google Scholar]
- Yuen J. W. M.; Gohel M. D. Anticancer effects of Ganoderma lucidum: a review of scientific evidence. Nutr. Cancer 2005, 53, 11–17. 10.1207/s15327914nc5301_2. [DOI] [PubMed] [Google Scholar]
- Lin C.-N.; Tome W.-P.; Won S.-J. Novel cytotoxic principles of Formosan Ganoderma lucidum. J. Nat. Prod. 1991, 54, 998–1002. 10.1021/np50076a012. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Slivova V.; Harvey K.; Valachovicova T.; Sliva D. Ganoderma lucidum suppresses growth of breast cancer cells through the inhibition of Akt/NF-κB signaling. Nutr. Cancer 2004, 49, 209–216. 10.1207/s15327914nc4902_13. [DOI] [PubMed] [Google Scholar]
- Liu Y.-W.; Gao J.-L.; Guan J.; Qian Z.-M.; Feng K.; Li S.-P. Evaluation of antiproliferative activities and action mechanisms of extracts from two species of Ganoderma on tumor cell lines. J. Agric. Food Chem. 2009, 57, 3087–3093. 10.1021/jf900011f. [DOI] [PubMed] [Google Scholar]
- Hu H.; Ahn N.-S.; Yang X.; Lee Y.-S.; Kang K.-S. Ganoderma lucidum extract induces cell cycle arrest and apoptosis in MCF-7 human breast cancer cell. Int. J. Cancer 2002, 102, 250–253. 10.1002/ijc.10707. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Slivova V.; Valachovicova T.; Harvey K.; Sliva D. Ganoderma lucidum inhibits proliferation and induces apoptosis in human prostate cancer cells PC-3. Int. J. Oncol. 2004, 24, 1093–1099. 10.3892/ijo.24.5.1093. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yuan J.-P.; Chung C.-K.; Chen X.-J. Antitumor activity of the sporoderm-broken germinating spores of Ganoderma lucidum. Cancer Lett. 2002, 182, 155–161. 10.1016/s0304-3835(02)00080-0. [DOI] [PubMed] [Google Scholar]
- Lu Q.-Y.; Jin Y.-S.; Zhang Q.; Zhang Z.; Heber D.; Go V. L. W.; Li F. P.; Rao J. Y. Ganoderma lucidum extracts inhibit growth and induce actin polymerization in bladder cancer cells in vitro. Cancer Lett. 2004, 216, 9–20. 10.1016/j.canlet.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Rutqvist L. E. The significance of hormone receptors to predict the endocrine responsiveness of human breast cancer. Acta Oncol. 1990, 29, 371–377. 10.3109/02841869009090017. [DOI] [PubMed] [Google Scholar]
- Stewart Z. A.; Westfall M. D.; Pietenpol J. A. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. 10.1016/s0165-6147(03)00026-9. [DOI] [PubMed] [Google Scholar]
- Tait S. W. G.; Green D. R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S.; Ren Y.; Zheng H.; Zhao M.; Zhang X.; Zhu Y.; Yang J.; Zheng S. Salvianolic acid B inhibits intermittent high glucose-induced INS-1 cell apoptosis through regulation of Bcl-2 proteins and mitochondrial membrane potential. Eur. J. Pharmacol. 2017, 814, 56–62. 10.1016/j.ejphar.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Denk G. U.; Kleiss C. P.; Wimmer R.; Vennegeerts T.; Reiter F. P.; Schulz S.; Zischka H.; Rust C. Tauro-β-muricholic acid restricts bile acid-induced hepatocellular apoptosis by preserving the mitochondrial membrane potential. Biochem. Biophys. Res. Commun. 2012, 424, 758–764. 10.1016/j.bbrc.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Petronilli V.; Penzo D.; Scorrano L.; Bernardi P.; Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death—correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. 10.1074/jbc.m010604200. [DOI] [PubMed] [Google Scholar]
- Green D. R.; Reed J. C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Smiley S. T.; Reers M.; Mottola-Hartshorn C.; Lin M.; Chen A.; Smith T. W.; Steele G. D.; Chen L. B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 3671–3675. 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Wang G.; Wang H.; Cheng C.; Zang G.; Guo X.; Liu R. H. Phytochemical profiles and antioxidant activities in six species of ramie leaves. PLoS One 2014, 9, e108140. 10.1371/journal.pone.0108140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.