

Abstract
Analogies between the damaged tissue and developing organ indicate that a regulatory network that drives embryonic organ development may control aspects of tissue repair. In this regard, there is a growing body of experimental and clinical evidence showing that TH may be critical for recovery after injury. Especially TRα1 has been reported to play an essential role in cell proliferation and differentiation and thus in the process of repair/regeneration in the heart and other tissues. Patients after myocardial infarction, stroke or therapeutic interventions [such as PCI for coronary artery disease (CAD)] with lower TH levels appear to have increased morbidity and mortality. Accordingly, TH treatment in clinical settings of ischemia/reperfusion such as by-pass surgery seems to be cardioprotective against ischemic injury. Furthermore, TH therapy of donors is shown to result in organ preservation and increased numbers of donors and improved post-transplantation graft survival. TH and thyroid analogs may prove novel therapeutic agents for tissue repair.
Keywords: Thyroid hormone (TH), thyroid hormone receptor α1 (TRα1), repair, regeneration, heart failure (HF), myocardial ischemia, brain, skeletal muscle
Introduction
Cardiovascular diseases (CVDs) account for 30% of all deaths worldwide, with 80% of these deaths related to the consequences of ischemic heart disease (IHD). In particular, prognosis of patients with heart failure (HF) is worse than that of several forms of cancer, including breast, colon and ovarian cancer in women and prostate, colon and bladder cancer in men (1). Despite therapeutic advances, the overall 5-year mortality rate remains approximately 50%, while HF is the most common reason for hospitalization, exceeding a million admissions per year both in US and in Europe (2). Among HF causes, the most common is IHD, accounting for around two thirds of all cases. IHD can lead to loss of a given amount of contractile myocardium, with unavoidable consequences on the functional capacity of the heart.
Over the past 30 years, many efficient therapies have been developed to treat IHD, including various reperfusion strategies of occluded coronary arteries, anti-platelet and anticoagulant agents (3). Current therapeutic approaches have reduced acute mortality after acute myocardial infarction (AMI), but have left an increasing number of patients with HF due to a maladaptive healing process resulting in dedifferentiation and fibrosis of the remaining myocardium, dilatation of left ventricle (LV) and eventually failure of myocardial function. Thus, nearly 30% will develop HF despite recent therapeutic advances (4). Current treatments delay the onset of HF or limit the consequences of IHD but do not have the ability to replace the damaged cardiac cells, especially the necrotic and/or apoptotic cardiomyocytes (5), and thus, cannot properly “heal” the injured heart.
A regulatory network that drives embryonic development may control aspects of tissue repair. In this context, tissue repair and regeneration upon injury in principle can utilize two different mechanisms. The first involves the activation of stem cells which, upon injury, start to proliferate and then differentiate. This mechanism seems to be in operation in mammals in a restrictive form particularly after injury (6). Most of cellular therapies aim to augment this regenerative response. The second mechanism for regeneration does not involve any stem cells, but rather involves dedifferentiation of injured cells. In this case, it is the dedifferentiated cells that proliferate and differentiate (6). This regenerative strategy has been extensively studied in newts and mouse myotubes and seems to be of relevance in cardiac regeneration in mammals. Potential mechanisms of this regenerative strategy include transient induction of components of the apoptotic pathway (particularly caspase activation) upon newt limb amputation which leads to myotube fragmentation into mononucleate cells without causing terminal apoptosis, while the subsequent inactivation of these components allows the cells to dedifferentiate fully, proliferate, and ultimately produce new muscle (7). Thus, recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury (8). In accordance with this evidence, zebrafish heart and neonatal mammalian heart regeneration has been shown to occur by cardiomyocyte de-differentiation and proliferation, while stem or progenitor cells are not significantly involved in this process (9,10).
Thyroid hormone (TH) has long been recognized that drives embryonic development due to its actions on cell proliferation and differentiation and thus, TH can control aspects of tissue repair. There is now a great body of preclinical and clinical evidence which reveals the reparative/regenerative action of TH (11,12). TH appears to act on various steps of the regenerative process and be implicated in tissue repair and recovery after injury contributing to complete organ restoration. This approach may change the existing paradigm in the regenerative therapies.
TH regulates gene transcription either by direct binding of nuclear thyroid hormone receptors (TRs) to DNA or indirectly by tethering of TRs to chromatin associated proteins. Furthermore, TH may regulate important intracellular signalling cascades via actions in cytosolic or membrane TRs (13). The predominant mammalian TR isoforms include TRα1, TRβ1, TRβ2, TRβ3, TRβ4 and the TR variants that lack T3-binding capacity TRα2, TRα3 and TRαΔE6. Especially TRα1 and TRβ1 are the best characterized mammalian thyroid receptors. TRα1 is most abundant in cardiac and skeletal muscle, bone, gastrointestinal track and central nervous system. TRβ1 shows high expression in the liver, kidney and inner ear, while TRβ2 is predominant in the pituitary, cochlea and hypothalamus (13,14). Particularly in the heart, TRα1 is present in working myocardium as well as in the peripheral ventricular conduction system, while TRβ1 isoform is confined to the cells that form the peripheral ventricular conduction system (15). In the atria and in the proximal conduction system (sinoatrial node, atrio-ventricular node), TRα1 and TRβ1 isoforms are co-expressed (15). In general, TRs are dynamic proteins, primarily redising in the nucleus but may shuttle rapidly in the cytoplasm. Genomic actions of TRs include interactions with TH response elements in specific genes. In the presence of T3, TRs recruit co-activators and induce transcription of positive regulated genes. In the absence of T3, TRs recruit co-repressors and block transcription. For negative regulated genes, the reverse is the case: liganded TRs act as repressors of transcription while unliganded TRs act as activators (13).
Developmental role of TH and implications for tissue regeneration
Amphibian metamorphosis is the most striking paradigm of adaptation to oxygen rich environment. This biological process is entirely dependent on TH. TH is low during embryonic and early larva development and increases as larva approaches metamorphosis. A similar developmental TH secretion pattern is observed in most species and in humans (16). Furthermore, distinct changes in de-iodinases, and TRs expression occur and thus, a single hormone can coordinate responses among different cell types, and regulate the temporal sequence of remodeling events during amphibian metamorphosis. Several studies on amphibian metamorphosis have been performed on the frog Xenopus laevis. Xenopus TRα1 is expressed early in development (17), before the embryo and larval tadpole have a functional thyroid gland. Just prior to metamorphosis, high levels of TRα1 are detected in the brain, limb buds, skin, and other tissues that are destined to respond to the sharp increase in TH by proliferating and differentiating into adult organs (18,19). In fact, T3 has been found to induce cell proliferation in the ventricular/subventricular neurogenic zones of the tadpole brain predominantly via TRα1. This increase is dependent on T3 until mid-prometamorphosis, after which the phenomenon is self-restricted (20). Furthermore, administration of a TRα1 specific agonist (CO24) in tadpoles resulted in massive hind leg and fore leg development, a noticeably larger body size, and less resorption of larval tissue in the head (21).
Effects of TH in myocardial repair/regeneration after injury
TH and myocardial ischemic injury: the role of TRα1
Although there is accumulating clinical evidence showing that TH may be a novel treatment for cardiac diseases (22,23) its use is limited due to long-held belief that TH may be detrimental for the ischemic myocardium. In practice, TH can increase heart rate and contractile function, which may enhance energy expenditure and thus aggravate ischemia. However, the potential effects of TH on myocardial injury have only recently been explored (24-26). It is now recognized that TH action on the heart depends on its administration to injured or healthy myocardium (11).
In our laboratory, we were the first to observe that TH pretreatment could confer protection against subsequent ischemia-reperfusion injury in a similar pattern as ischemic preconditioning (27,28). Thus, in an experimental model of ischemia–reperfusion using isolated rat hearts, both TH pretreatment and ischemic preconditioning, despite paradoxically exacerbated ischemic contracture, improved post-ischemic recovery. Interestingly, both TH pretreatment and ischemic preconditioning were shown to suppress the I/R-induced activation of the pro-apoptotic p38 mitogen-activated protein kinase (MAPK) (26,27). TH was also shown to upregulate cardio-protective molecules such as heat shock protein 27 (HSP27) and heat shock protein 70 (HSP70), which were also involved in the underlying mechanisms of ischemic preconditioning (24,25).
Similarly, T3 administration at reperfusion (at a dose which had no effect on the normal myocardium) resulted in enhanced post-ischemic recovery of function and less myocardial injury as indicated by apoptosis and tissue necrosis markers (12,29). In this in vitro experimental setting, T4 was shown to have no effect on myocardial injury indicating that this action is likely to require the involvement of TR receptors. Indeed, inactivation of thyroid hormone receptor (TRα1) abolished the T3 effect on limiting myocardial injury (29). The critical role of TRα1 in myocardial ischemia has also been documented in an another study using transgenic animals (30). The effect of T3 on cardiac apoptosis is shown to be mediated, at least in part, by the suppression of the I/R induced activation of the pro-apoptotic p38 (MAPK) (12).
In accordance with this evidence, TH administration in vivo after myocardial infarction in rats resulted in reduced apoptosis and this response involved activation of protein kinase B (Akt) (31) and the miR30a/p53 axis (32). Interestingly, the T3-induced activation of PI3K/Akt/mTOR pathway is found to be regulated by an interaction of the cytosolic TRα1 with the p85α subunit of PI3K (33,34). This molecular footprint induced by T3 upon stress (suppression of p38 MAPK and activation of Akt) was found to provoke cardiomyocyte proliferation and re-differentiation and result in cardiac regeneration. In fact, Engel et al. reported that FGF1 (a well-known Akt activator) combined with a p38 MAPK inhibitor induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction in rats (35).
TH repairs the remaining, viable myocardium after ischemic injury
It is now recognized that after an ischemic event the remaining viable myocardium reactivates an early developmental state. Thus, features of fetal heart metabolism re-emerge and include the preference of glucose metabolism over fatty acids as energy substrates, while isoform switches of many other proteins, including metabolic enzymes and sarcomeric proteins (decrease in α-MHC and increase in β-MHC expression) occur (36-38). Several studies from our group and others during the last decade have revealed the important pathophysiological role of TH signaling in this process. Besides the low T3-syndrome that occurs in about 30% of patients, tissue hypothyroidism that occurs due to increased activity of deiodinase type 3 and alterations in TRs have been found to contribute to molecular and functional changes related to post-ischemic cardiac remodeling (39,40).
More specifically, high expression of TRα1, as this occurs in fetal heart, was found in the remaining viable myocardium (41). Activation of the adrenergic system was found to contribute to this response via the ERK cascade (42). Furthermore, the important causative role of TRα1 in the progression of infarct-related HF has been tested using a TRα1 inhibitor following MI in mice. Inhibition of TRα1 was shown to markedly depress post-ischemic cardiac function, exacerbate myocardial remodeling and further deteriorate calcium handling (43).
Changes in TRα1 have important physiological consequences for the cardiomyocyte. TRα1 is predominantly expressed in the myocardium and regulates genes related to cell growth and differentiation, metabolism, pacemaker activity, conduction and contractile proteins (23,40,44). It has a regulatory role in cardiomyocyte maturation and proliferation depending on its liganded (high T3) or un-liganded state (low T3) acting as a switch. The un-liganded state is prominent during the embryonic stages where TRα1 is highly expressed, while T3 levels are very low. At this stage, the unliganded TRα1 induces the fetal gene program and permits the increase in cardiac mass by continuous cell proliferation. After birth, TRα1 switches to the liganded state due to the rise in T3 levels. This results in cell maturation, physiologic cardiac growth and enhanced myocardial function (42,45). On the basis of these observations, re-expression of high levels of unliganded TRα1 in the viable myocardium after ischemic injury indicate a fetal reprogramming of the heart leading to cardiomyocyte dedifferentiation but could be also interpreted as an opportunity for regeneration. Thus, low T3 levels can retain the myocardium at fetal like phenotype, while high T3 levels can recapitulate the developmental program after birth and restore it to adult phenotype (Figure 1).
Figure 1.
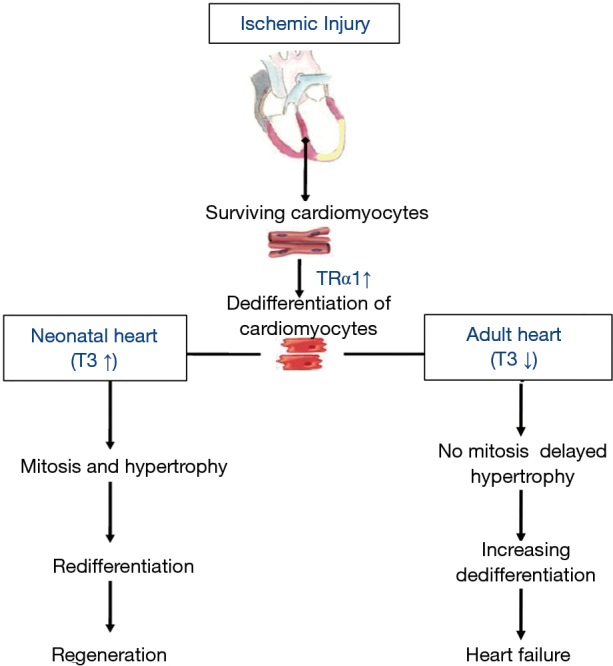
Changes in thyroid hormone signaling determine the response of the heart after ischemic injury. The initial response of the adult heart to injury involves enhanced expression of thyroid hormone receptor α1 (TRα1) contributing to dedifferentiation. In this context, low T3 blocks the regenerative response and leads to increasing dedifferentiation, dysfunction and heart failure. However, high T3 levels combined with increased expression of TRα1 (neonatal heart) lead to increased cardiac mass (proliferation and hypertrophy), redifferentiation and regeneration. ↑, increase; ↓, decrease.
The potential of TH to serve as a novel treatment has been extensively investigated in animal models of infarct-related HF resulting in consistent improvement in left ventricular function (31,32,46-52). More specifically, TH treatment following MI has been shown to result in increased viable cardiac mass with a physiological adult phenotype via favorable changes in the expression of myosin heavy chain (MHC), calcium cycling proteins and enhanced pro-survival signaling such as Akt. TH improved cardiac function even in the presence of co-morbidities, such as diabetes (48).
Potential role of TH in the formation of new cardiomyocytes
Naqvi et al. showed that a surge in TH during the first postnatal days in mice causes a brief burst of proliferative activity via IGF1/Akt pathway and results in an increase of 500,000 cardiomyocytes in the third week of life (53). Increased postnatal wall stress seems to act synergistically with TH to produce this proliferative burst (53). It is interesting that new cardiomyocytes in this process derived from larger, elongated, binuclear cardiomyocytes that completed cytokinesis. Furthermore, TH is reported to elongate neonatal cardiomyocytes via transient activation of extracellular signal-regulated kinases (ERK) (54). Thus, an increase in cardiac mass by restricted cell proliferation is achieved via an endocrine-mechanical interplay. There is now preliminary evidence that such a mechanism may also be operable in the injured myocardium. Cardiac stretch increases during the early phase of ischemia before the development of cardiac hypertrophy. This is a mechanism to preserve mechanical performance according to the Starling’s Law. TH treatment at this early stage was shown to result in an increase in cardiac mass (50). In contrast, TH treatment, starting at the time at which wall stretch was normalized by the development of compensatory hypertrophy, failed to increase cardiac mass (55). This novel mechanism of TH-mechanical interplay induced endogenous regeneration is now under investigation (Figure 1).
Tremendous research efforts are currently invested to achieve tissue regeneration via transplantation of stem cells. Recent evidence suggests that triiodothyronine promotes the proliferation of epicardial progenitor cells through the ERK pathway in a dose and time-dependent manner (56). Interestingly, TH can also drive maturation of human induced pluripotent stem cell-derived cardiomyocytes (hi-PSC-CM) with important implications in tissue regeneration (57,58). Furthermore, treatment of hi-PSC-CM with combined triiodothyronine and dexamethasone promotes the cardiac differentiation process via T-tubule formation, synchronizing intracellular Ca release and inducing ventricular-like excitation-contraction coupling (59).
TH and tissue repair beyond the heart
Pathophysiological changes in TH signaling after cerebral ischemia seem to be in accordance with myocardial ischemia. In a model of middle cerebral artery occlusion in rats, we found decreased TH levels in serum and increased expression of TRα1 in microglial cells within the infarct zone and in neural cells around the infarct (60).
The effect of TH on limiting the extent of injury has been demonstrated in models of ischemia/reperfusion in brain. TH administration suppressed the pro-apoptotic p38 MAPK and significantly reduced cerebral infarction in a model of I/R in rat brain (61). Interestingly, TH treatment resulted in suppressed activation of microglia and increased the expression of neurotrophic factors (BDNF, GDNF) (61). Similarly, T3 treatment resulted in significant reduction of tissue infarct and decrease in edema in in vivo models of ischemia/reperfusion in brain, both pre- and post-stroke. T3 also suppressed the aquaporin 4 which could be a probable mechanism of its anti-edema effect (62). In a recent study, Li et al. showed that TH treatment after traumatic brain injury not only reduces brain edema but also promotes important neurogenesis markers such as Sox2, Dcx and Gap43 (63).
TH is shown to play an essential role in myogenesis, the key process in skeletal muscle development and regeneration. More, specifically, T3 increases myoblast differentiation via TRα1 in cultures. Genetic approaches also confirmed that TRα1 plays an important role in normal myoblast proliferation and differentiation and acts through the Wnt/β-catenin signaling pathway. Myoblasts that do not express TRα or express a mutant TRα unable to bind T3 (RTH-TRα PV mice), show reduced proliferation and myogenic differentiation. Moreover, skeletal muscle from the TRα1PV mutant mouse has impaired in vivo regeneration after injury (45). In fact, in a recent study, TRα was found to have an important role in the activation, migration and proliferation of the satellite cells which are major players in the processes of skeletal muscle maintenance and regeneration after injury (64).
TRα1 has been reported to play a critical role in expansion of the pancreatic β-cell mass during postnatal development. Further studies have shown that T3 treatment in pancreatic acinar cells after induced overexpression of TRα1 results in reprogramming of these into insulin-producing cells via activation of the PI3K/Akt pathway (65). In vivo, treatment with an adenovirus vector that expresses TRα1 in immunodeficient mice with streptozotocin-induced diabetes also results in the reprogramming of pancreatic acinar cells to insulin-producing cells (65). Furthermore, we have found that TH treatment in streptozotocin-induced diabetic rats with myocardial infarction, not only improves left ventricular function but also increases insulin levels and improves high blood glucose (48). Interestingly, in another study, both treatment with palmitate in cultures and a high fat diet in vivo (endoplasmic reticulum stress) were shown to result in reduced survival of TRα-deficient pancreatic beta-cells and reduced insulin production (66).
Clinical relevance
On the basis of the existing experimental evidence, TH appears to be essential for the adaptive response after injury. These observations are in accordance with several epidemiological studies in patients with ischemic heart disease (IHD). Accordingly, proof of concept clinical studies show that TH treatment is beneficial also in clinical settings of myocardial ischemia-reperfusion, such as by-pass surgery and cardiac transplantation.
Epidemiological evidence: TH determines prognosis in patients with IHD
It has long been recognized that several neurohormonal changes occur after acute tissue injury but the physiological relevance of this response remains largely unknown. In this context, a condition described as non-thyroidal illness often accompanies acute and chronic diseases (67). There is a growing body of clinical evidence showing a strong association between TH levels and adverse clinical outcomes even within TH normal range. Thus, in patients with AMI subjected to primary PCI, T3 levels at 48h and six months were positively associated with cardiac function. Furthermore, T3 levels at 6months appear to be an independent determinant of recovery of cardiac function as this was assessed by left ventricular ejection fraction (68). Similarly, in a series of 1,047 AMI patients undergoing mechanical revascularization, low fT3 levels were shown to be associated with high in- hospital mortality and higher PCI failure. As shown in Table 1, long term follow up median [31.2 (12–44.9) months] revealed that patients aged less than 75 years with lower fT3 levels had higher mortality as compared to those with fT3 in the euthyroid range (69).
Table 1. Observational studies showing an association of thyroid hormone changes with clinical outcome in clinical settings of ischemic heart disease treated with reperfusion.
| Clinical study | Patients (N) | Setting | Outcome |
|---|---|---|---|
| Lymvaios et al. 2011 (68) | 47 | STEMI patients with primary PCI | T3 levels at 6 months appear to be an independent determinant of recovery of cardiac function |
| Lazzeri et al. 2012 (69) | 1,047 | STEMI patients with primary PCI | Patients aged less than 75 years with lower fT3 levels had higher mortality |
| Ndrepepa et al. 2015 (70) | 8,010 | Elective PCI in CAD | Patients at the upper limit of TSH had higher 30-day and 3-year mortality compared to groups with lower TSH levels |
| Zhang et al. 2016 (71) | 2,430 | Elective PCI in CAD | Association of hypothyroidism (clinical and subclinical) to major cardiovascular and cerebral events (MACCE) |
| Lee et al. 2018 (72) | 936 | Patients with elective or primary PCI | Subclinical hypothyroidism negatively impacted repeat revascularization and cardiac death following PCIs |
CI, Cardiac Index; CO, cardiac output; SV, stroke volume; AE, adverse events; CBP, cardiopulmonary bypass; ACC, aortic cross-clamp.
A potential link of low TH state to adverse clinical outcomes was also shown in two studies after elective PCI in patients with chronic coronary artery disease (CAD). The first study investigated 8,010 patients with CAD treated with PCI. All patients had TSH levels at physiological range (0.3–4.0 mU/L). However, those at the upper range (1.67–4.0) had higher 30-day and 3-year mortality compared to the groups with lower TSH levels. Furthermore, the incidence of cardiogenic shock and peri-PCI bleeding was increased in patients in upper normal range (70).
In support to this notion, an association of hypothyroidism (clinical and subclinical) with major cardiovascular and cerebral events (MACCE) was also documented in a second study which included 2,430 CAD patients undergoing PCI. Interestingly, this study further showed that adequately treated hypothyroid patients had a lower risk of MACCE (71).
These observations were also confirmed in a very recent study that included 936 CAD patients with primary or elective PCI. This study showed that subclinical hypothyroidism negatively impacted repeat revascularization and cardiac death following PCI in these patients with a hazard ratio of 1.52 after adjustment for several known cardiovascular risk factors (72). In fact, subclinical hypothyroidism was not associated with repeat PCI for de novo stenotic lesions but for in-stent restenotic lesions (72). A summary of observational studies showing an association of TH changes with clinical outcome in clinical scenarios of CAD with reperfusion is shown in Table 1.
Low T3 syndrome has been recorded in 32–62% of patients following an acute cerebrovascular event. More specifically, there is a trend towards a decrease in T4 and free T4 during the first seven days after admission post-stroke. T3 values are shown to remain low until day 5 and recover on days 7 and 9 after stroke (73). There is significant evidence to suggest low T3 as strong predictor of worse stroke outcome. In a large study, patients in the first or lower free T3 tertile reported greater neurological impairment and greater 1 year mortality than those in 3rd or higher free T3 tertile (74). In addition, low T3 patients were shown to present with acute stroke in greater numbers and low T3 levels predicted greater mortality at 1 year (75).
Taken together, this ongoing clinical evidence suggests that TH may be essential for the recovery after injury.
TH treatment in CABG and cardiac transplantation
TH was first used as postoperative treatment after coronary artery bypass grafting (CABG) to support hemodynamics. The efficacy of TH on cardiac hemodynamics and its side effects have been tested in several trials. Meta-analysis of data provided by a great number of studies showed that TH may have a beneficial effect on cardiac hemodynamics without severe side effects (76). Both high- and low-dose intravenous (iv) T3 treatment resulted in increased cardiac index after CABG. Mortality was not found to be significantly altered in high dose iv T3 (76). The potential effect of T3 therapy on reperfusion injury after CABG was also explored in a randomized double-blind placebo-control trial that included 440 patients undergoing first time isolated on pump CABG (77). In this study, the effect of postoperative T3 administration was compared to placebo and to glucose-insulin-potassium (GIK) treatment. Serial hemodynamic measurements were taken up to 12 h, and troponin I levels were assayed to 72 h. T3 significantly increased cardiac index, which reached the peak at 12 h. T3 significantly lowered the troponin release, and this effect was greater versus placebo and GIK. Furthermore, T3 significantly reduced the mean norepinephrine use in first 6 h after removal of the aortic cross-clamp. In contrast, GIK significantly increased the requirement for norepinephrine use (77). Here it should be noted that norepinephrine aggravates ischemia/reperfusion injury and its use to support hemodynamics in the ischemic myocardium is now questionable (78,79). In accordance with these findings, T3 pretreatment (125 µg/day for 7 days) in patients with CABG and impaired left ventricular function improved post-ischemic recovery of function and significantly lowered the mean inotropic requirements (80) (Table 2).
Table 2. Clinical studies with T3 administration in settings of myocardial ischemia-reperfusion.
| Clinical study | Patients | Setting | Dose of i.v. T3 | Outcome | Safety |
|---|---|---|---|---|---|
| Mullis-Jansson et al. (81) | 170 | CABG | 0.4 ìg/kg bolus and 0.1 ìg/kg for 6 hours | Increased cardiac index at 4 to 6 h in pooled comparisons at meta-analysis, no change in hospital mortality (76) | No AEs |
| Klemperer et al. (82) | 142 | CABG with depressed EF% | 0.8 ìg/kg bolus and 0.113 ìg/kg/h for 6 hours | No AEs | |
| Güden et al. (83) | 60 | CABG | 0.8 ìg/kg bolus and 0.12 ìg/kg/h for 6 hours | No AEs | |
| Bennett-Guerrero et al. (84) | 211 | CABG | 0.8 ìg/kg bolus and 0.12 ìg/kg/h for 6 hours | No AEs | |
| Ranasinghe et al. (77) | 440 | CABG | 0.8 ìg/kg bolus and 0.113 ìg/kg/h for 6 hours | Increased CI, lowered troponin release, reduced mean norepinephrine use | No AEs |
| Sirlak et al. (80) | 80 | CABG with EF% <30% | 125 ìg/day per os for 7 d before and for 5 d after CABG | Increased CI | No AEs |
| Hamilton et al. (85) | 23 | Advanced heart failure | 0.15–2.7 ìg/kg for 6–12 h | Increased CO and reduction of SVR | No AEs |
| Pingitore et al. (86) | 20 | Heart failure | 35.6 ìg in the first 24 h and 15 ìg/day till 72 h | Increased SV and lower HR, decrease of NT-proBNP, noradrenaline and aldosterone | No AEs |
| Portman et al. (87) | 193 children <2 years old | Heart surgery with CPB | 0.4 ìg/kg bolus before CPB, 0.4 ìg/kg on the release of ACC, 0.2 ìg/kg at 3, 6, and 9 h | Reduction in inotropic agent use and improved cardiac function | No AEs |
| Novitzky et al. (88) | 63,593 | Brain-dead organ donors | 4 ìg bolus and 4 ìg/h guidelines of UNOS | Enhanced procurement of hearts and improved graft survival | – |
UNOS, US United Network fo Organ Sharing; CI, Cardiac Index; CO, cardiac output; SV, stroke volume; SVR, systemic vascular resistance; AE, adverse events; CPB, cardiopulmonary bypass; ACC, aortic cross-clamp.
TH therapy for supporting donor heart hemodynamics has been also extensively used in cardiac transplantation. A pronounced effect of TH on unstable donors (due to myocardial ischemia) was observed indicating that TH may protect the donor heart against ischemic injury as previously was shown in patients undergoing CABG (89). This unique effect was documented in a series of 66,629 organ donors. Interestingly, T3/T4 treatment of cardiac donors was associated with procurement of significantly greater numbers of hearts. Furthermore, the effect of TH treatment was independent of other factors and associated with improved post-transplantation graft survival (88,90). A summary of clinical studies with T3 administration in settings of myocardial ischemia-reperfusion is shown in Table 2.
Concluding remarks
There is a growing body of experimental and clinical evidence showing that TH may be critical for recovery after injury. Especially TRα1 has been reported to play an essential role in cell proliferation and differentiation and thus in the process of repair/regeneration in the heart and other tissues. Patients after myocardial infarction, stroke or therapeutic interventions (such as PCI for CAD) with lower TH levels appear to have worse prognosis. Accordingly, TH treatment in clinical settings of ischemia/reperfusion such as by-pass surgery seems to be cardioprotective against ischemic injury. Furthermore, TH therapy of donors is shown to result in organ preservation and increased numbers of donors and improved post-transplantation graft survival. TH and/or thyroid analogs may prove novel therapeutic agents for tissue repair.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Stewart S, MacIntyre K, Hole DJ, et al. More 'malignant' than cancer? Five-year survival following a first admission for heart failure. Eur J Heart Fail 2001;3:315-22. 10.1016/S1388-9842(00)00141-0 [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 2015;131:e29-322. 10.1161/CIR.0000000000000152 [DOI] [PubMed] [Google Scholar]
- 3.McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012;33:1787-847. 10.1093/eurheartj/ehs104 [DOI] [PubMed] [Google Scholar]
- 4.Ambrosy AP, Fonarow GC, Butler J, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 2014;63:1123-33. 10.1016/j.jacc.2013.11.053 [DOI] [PubMed] [Google Scholar]
- 5.Ausoni S, Sartore S. The cardiovascular unit as a dynamic player in disease and regeneration. Trends Mol Med 2009;15:543-52. 10.1016/j.molmed.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 6.Yutzey KE. Cardiomyocyte Proliferation: Teaching an Old Dogma New Tricks. Circ Res 2017;120:627-9. 10.1161/CIRCRESAHA.116.310058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frasch M. Dedifferentiation, Redifferentiation, and Transdifferentiation of Striated Muscles During Regeneration and Development. Curr Top Dev Biol 2016;116:331-55. 10.1016/bs.ctdb.2015.12.005 [DOI] [PubMed] [Google Scholar]
- 8.Witman N, Murtuza B, Davis B, et al. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol 2011;354:67-76. 10.1016/j.ydbio.2011.03.021 [DOI] [PubMed] [Google Scholar]
- 9.Jopling C, Sleep E, Raya M, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010;464:606-9. 10.1038/nature08899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senyo SE, Steinhauser ML, Pizzimenti CL, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013;493:433-6. 10.1038/nature11682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pantos C, Mourouzis I, Cokkinos DV. New insights into the role of thyroid hormone in cardiac remodeling: time to reconsider? Heart Fail Rev 2011;16:79-96. 10.1007/s10741-010-9185-3 [DOI] [PubMed] [Google Scholar]
- 12.Pantos C, Mourouzis I, Saranteas T, et al. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol 2009;104:69-77. 10.1007/s00395-008-0758-4 [DOI] [PubMed] [Google Scholar]
- 13.Anyetei-Anum CS, Roggero VR, Allison LA. Thyroid hormone receptor localization in target tissues. J Endocrinol 2018;237:R19-R34. 10.1530/JOE-17-0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skah S, Uchuya-Castillo J, Sirakov M, et al. The thyroid hormone nuclear receptors and the Wnt/β-catenin pathway: An intriguing liaison. Dev Biol 2017;422:71-82. 10.1016/j.ydbio.2017.01.003 [DOI] [PubMed] [Google Scholar]
- 15.Stoykov I, Zandieh-Doulabi B, Moorman AF, et al. Expression pattern and ontogenesis of thyroid hormone receptor isoforms in the mouse heart. J Endocrinol 2006;189:231-45. 10.1677/joe.1.06282 [DOI] [PubMed] [Google Scholar]
- 16.Johnson CK, Voss SR. Salamander paedomorphosis: linking thyroid hormone to life history and life cycle evolution. Curr Top Dev Biol 2013;103:229-58. 10.1016/B978-0-12-385979-2.00008-3 [DOI] [PubMed] [Google Scholar]
- 17.Banker DE, Bigler J, Eisenman RN. The thyroid hormone receptor gene (c-erbA alpha) is expressed in advance of thyroid gland maturation during the early embryonic development of Xenopus laevis. Mol Cell Biol 1991;11:5079-89. 10.1128/MCB.11.10.5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry DL, Rose CS, Remo BF, et al. The expression pattern of thyroid hormone response genes in remodeling tadpole tissues defines distinct growth and resorption gene expression programs. Dev Biol 1998;203:24-35. 10.1006/dbio.1998.8975 [DOI] [PubMed] [Google Scholar]
- 19.Kawahara A, Baker B, Tata J. Developmental and regional expression of thyroid hormone receptor genes during Xenopus metamorphosis. Development 1991;112:933-43. [DOI] [PubMed] [Google Scholar]
- 20.Denver RJ, Hu F, Scanlan TS, et al. Thyroid hormone receptor subtype specificity for hormone-dependent neurogenesis in Xenopus laevis. Dev Biol 2009;326:155-68. 10.1016/j.ydbio.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 21.Ocasio CA, Scanlan TS. Characterization of thyroid hormone receptor alpha (TRalpha)-specific analogs with varying inner- and outer-ring substituents. Bioorg Med Chem 2008;16:762-70. 10.1016/j.bmc.2007.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pantos C, Mourouzis I, Cokkinos DV. Thyroid hormone as a therapeutic option for treating ischaemic heart disease: from early reperfusion to late remodelling. Vascul Pharmacol 2010;52:157-65. 10.1016/j.vph.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 23.Pantos C, Mourouzis I, Xinaris C, et al. Thyroid hormone and "cardiac metamorphosis": potential therapeutic implications. Pharmacol Ther 2008;118:277-94. 10.1016/j.pharmthera.2008.02.011 [DOI] [PubMed] [Google Scholar]
- 24.Pantos C, Malliopoulou V, Mourouzis I, et al. Thyroxine pretreatment increases basal myocardial heat-shock protein 27 expression and accelerates translocation and phosphorylation of this protein upon ischaemia. Eur J Pharmacol 2003;478:53-60. 10.1016/j.ejphar.2003.08.030 [DOI] [PubMed] [Google Scholar]
- 25.Pantos C, Malliopoulou V, Mourouzis I, et al. Hyperthyroid hearts display a phenotype of cardioprotection against ischemic stress: a possible involvement of heat shock protein 70. Horm Metab Res 2006;38:308-13. 10.1055/s-2006-925404 [DOI] [PubMed] [Google Scholar]
- 26.Pantos C, Malliopoulou V, Paizis I, et al. Thyroid hormone and cardioprotection: study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Mol Cell Biochem 2003;242:173-80. 10.1023/A:1021162417490 [DOI] [PubMed] [Google Scholar]
- 27.Pantos CI, Malliopoulou VA, Mourouzis IS, et al. Long-term thyroxine administration protects the heart in a pattern similar to ischemic preconditioning. Thyroid 2002;12:325-9. 10.1089/10507250252949469 [DOI] [PubMed] [Google Scholar]
- 28.Pantos CI, Malliopoulou VA, Mourouzis IS, et al. Long-term thyroxine administration increases heat stress protein-70 mRNA expression and attenuates p38 MAP kinase activity in response to ischaemia. J Endocrinol 2001;170:207-15. 10.1677/joe.0.1700207 [DOI] [PubMed] [Google Scholar]
- 29.Pantos C, Mourouzis I, Saranteas T, et al. Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRalpha1 receptor. Mol Cell Biochem 2011;353:235-41. 10.1007/s11010-011-0791-8 [DOI] [PubMed] [Google Scholar]
- 30.Suarez J, Wang H, Scott BT, et al. In vivo selective expression of thyroid hormone receptor alpha1 in endothelial cells attenuates myocardial injury in experimental myocardial infarction in mice. Am J Physiol Regul Integr Comp Physiol 2014;307:R340-6. 10.1152/ajpregu.00449.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen YF, Kobayashi S, Chen J, et al. Short term triiodo-l-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol 2008;44:180-7. 10.1016/j.yjmcc.2007.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forini F, Kusmic C, Nicolini G, et al. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014;155:4581-90. 10.1210/en.2014-1106 [DOI] [PubMed] [Google Scholar]
- 33.Hiroi Y, Kim HH, Ying H, et al. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A 2006;103:14104-9. 10.1073/pnas.0601600103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenessey A, Ojamaa K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem 2006;281:20666-72. 10.1074/jbc.M512671200 [DOI] [PubMed] [Google Scholar]
- 35.Engel FB, Hsieh PC, Lee RT, et al. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci U S A 2006;103:15546-51. 10.1073/pnas.0607382103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajabi M, Kassiotis C, Razeghi P, et al. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 2007;12:331-43. 10.1007/s10741-007-9034-1 [DOI] [PubMed] [Google Scholar]
- 37.Swynghedauw B. Molecular mechanisms of myocardial remodeling Physiol Rev 1999;79:215-62. 10.1152/physrev.1999.79.1.215 [DOI] [PubMed] [Google Scholar]
- 38.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart Ann N Y Acad Sci 2010;1188:191-8. 10.1111/j.1749-6632.2009.05100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen R, Muller A, Simonides WS. Cardiac Thyroid Hormone Metabolism and Heart Failure. Eur Thyroid J 2017;6:130-7. 10.1159/000469708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pantos C, Mourouzis I. The emerging role of TRalpha1 in cardiac repair: potential therapeutic implications. Oxid Med Cell Longev 2014;2014:481482. 10.1155/2014/481482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pantos C, Mourouzis I, Galanopoulos G, et al. Thyroid hormone receptor alpha1 downregulation in postischemic heart failure progression: the potential role of tissue hypothyroidism. Horm Metab Res 2010;42:718-24. 10.1055/s-0030-1255035 [DOI] [PubMed] [Google Scholar]
- 42.Pantos C, Xinaris C, Mourouzis I, et al. Thyroid hormone receptor alpha 1: a switch to cardiac cell "metamorphosis"? J Physiol Pharmacol 2008;59:253-69. [PubMed] [Google Scholar]
- 43.Mourouzis I, Kostakou E, Galanopoulos G, et al. Inhibition of thyroid hormone receptor alpha1 impairs post-ischemic cardiac performance after myocardial infarction in mice. Mol Cell Biochem 2013;379:97-105. 10.1007/s11010-013-1631-9 [DOI] [PubMed] [Google Scholar]
- 44.Pantos C, Mourouzis I, Cokkinos DV. Thyroid hormone and cardiac repair/regeneration: from Prometheus myth to reality? Can J Physiol Pharmacol 2012;90:977-87. 10.1139/y2012-031 [DOI] [PubMed] [Google Scholar]
- 45.Milanesi A, Lee JW, Kim NH, et al. Thyroid Hormone Receptor alpha Plays an Essential Role in Male Skeletal Muscle Myoblast Proliferation, Differentiation, and Response to Injury. Endocrinology 2016;157:4-15. 10.1210/en.2015-1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forini F, Lionetti V, Ardehali H, et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodeling in rats. J Cell Mol Med 2011;15:514-24. 10.1111/j.1582-4934.2010.01014.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson KK, Danzi S, Paul JT, et al. Physiological replacement of T3 improves left ventricular function in an animal model of myocardial infarction-induced congestive heart failure. Circ Heart Fail 2009;2:243-52. 10.1161/CIRCHEARTFAILURE.108.810747 [DOI] [PubMed] [Google Scholar]
- 48.Mourouzis I, Giagourta I, Galanopoulos G, et al. Thyroid hormone improves the mechanical performance of the post-infarcted diabetic myocardium: a response associated with up-regulation of Akt/mTOR and AMPK activation. Metabolism 2013;62:1387-93. 10.1016/j.metabol.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 49.Mourouzis I, Mantzouratou P, Galanopoulos G, et al. Dose dependent effects of thyroid hormone on post-ischaemic cardiac performance: potential involvement of Akt and ERK signaling. Mol Cell Biochem 2012;363:235-43. 10.1007/s11010-011-1175-9 [DOI] [PubMed] [Google Scholar]
- 50.Pantos C, Mourouzis I, Markakis K, et al. Thyroid hormone attenuates cardiac remodeling and improves hemodynamics early after acute myocardial infarction in rats. Eur J Cardiothorac Surg 2007;32:333-9. 10.1016/j.ejcts.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 51.Pantos C, Mourouzis I, Markakis K, et al. Long-term thyroid hormone administration reshapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic Res Cardiol 2008;103:308-18. 10.1007/s00395-008-0697-0 [DOI] [PubMed] [Google Scholar]
- 52.Rajagopalan V, Zhang Y, Pol C, et al. Modified Low-Dose Triiodo-L-thyronine Therapy Safely Improves Function Following Myocardial Ischemia-Reperfusion Injury. Front Physiol 2017;8:225. 10.3389/fphys.2017.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naqvi N, Li M, Calvert JW, et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 2014;157:795-807. 10.1016/j.cell.2014.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pantos C, Xinaris C, Mourouzis I, et al. Thyroid hormone changes cardiomyocyte shape and geometry via ERK signaling pathway: potential therapeutic implications in reversing cardiac remodeling? Mol Cell Biochem 2007;297:65-72. 10.1007/s11010-006-9323-3 [DOI] [PubMed] [Google Scholar]
- 55.Pantos C, Mourouzis I, Tsagoulis N, et al. Thyroid hormone at supra-physiological dose optimizes cardiac geometry and improves cardiac function in rats with old myocardial infarction. J Physiol Pharmacol 2009;60:49-56. [PubMed] [Google Scholar]
- 56.Deng SB, Jing XD, Wei XM, et al. Triiodothyronine promotes the proliferation of epicardial progenitor cells through the MAPK/ERK pathway. Biochem Biophys Res Commun 2017;486:372-7. 10.1016/j.bbrc.2017.03.048 [DOI] [PubMed] [Google Scholar]
- 57.Hellen N, Wheeler J, Pinto Riccardo C, et al. Effect of T3 on human induced pluripotent stem cell-derived cardiomyocyte maturation. Cardiovascular Research 2014;103:S62 10.1093/cvr/cvu091.29 [DOI] [Google Scholar]
- 58.Ivashchenko CY, Pipes GC, Lozinskaya IM, et al. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol Heart Circ Physiol 2013;305:H913-22. 10.1152/ajpheart.00819.2012 [DOI] [PubMed] [Google Scholar]
- 59.Parikh SS, Blackwell DJ, Gomez-Hurtado N, et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res 2017;121:1323-30. 10.1161/CIRCRESAHA.117.311920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lourbopoulos A, Mourouzis I, Karapanayiotides T, et al. Changes in thyroid hormone receptors after permanent cerebral ischemia in male rats. J Mol Neurosci 2014;54:78-91. 10.1007/s12031-014-0253-3 [DOI] [PubMed] [Google Scholar]
- 61.Genovese T, Impellizzeri D, Ahmad A, et al. Post-ischaemic thyroid hormone treatment in a rat model of acute stroke. Brain Res 2013;1513:92-102. 10.1016/j.brainres.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 62.Sadana P, Coughlin L, Burke J, et al. Anti-edema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J Neurol Sci 2015;354:37-45. 10.1016/j.jns.2015.04.042 [DOI] [PubMed] [Google Scholar]
- 63.Li J, Donangelo I, Abe K, et al. Thyroid hormone treatment activates protective pathways in both in vivo and in vitro models of neuronal injury. Mol Cell Endocrinol 2017;452:120-30. 10.1016/j.mce.2017.05.023 [DOI] [PubMed] [Google Scholar]
- 64.Milanesi A, Lee JW, Yang A, et al. Thyroid Hormone Receptor Alpha is Essential to Maintain the Satellite Cell Niche During Skeletal Muscle Injury and Sarcopenia of Aging. Thyroid 2017;27:1316-22. 10.1089/thy.2017.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furuya F, Shimura H, Yamashita S, et al. Liganded thyroid hormone receptor-alpha enhances proliferation of pancreatic beta-cells. J Biol Chem 2010;285:24477-86. 10.1074/jbc.M109.100222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi K, Furuya F, Shimura H, et al. Impaired oxidative endoplasmic reticulum stress response caused by deficiency of thyroid hormone receptor α. J Biol Chem 2014. 289:12485-93. 10.1074/jbc.M113.544122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome J Clin Endocrinol Metab 1999;84:151-64. 10.1210/jcem.84.1.5364 [DOI] [PubMed] [Google Scholar]
- 68.Lymvaios I, Mourouzis I, Cokkinos DV, et al. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: A strong association? Eur J Endocrinol 2011;165:107-14. 10.1530/EJE-11-0062 [DOI] [PubMed] [Google Scholar]
- 69.Lazzeri C, Sori A, Picariello C, et al. Nonthyroidal illness syndrome in ST-elevation myocardial infarction treated with mechanical revascularization. Int J Cardiol 2012;158:103-4. 10.1016/j.ijcard.2012.03.100 [DOI] [PubMed] [Google Scholar]
- 70.Ndrepepa G, Braun S, Mayer K, et al. Prognostic value of thyroid-stimulating hormone within reference range in patients with coronary artery disease. Metabolism 2015;64:1308-15. 10.1016/j.metabol.2015.07.009 [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Sara JD, Matsuzawa Y, et al. Clinical outcomes of patients with hypothyroidism undergoing percutaneous coronary intervention. Eur Heart J 2016;37:2055-65. 10.1093/eurheartj/ehv737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee Y, Lim YH, Shin JH, et al. Impact of subclinical hypothyroidism on clinical outcomes following percutaneous coronary intervention. Int J Cardiol 2018;253:155-60. 10.1016/j.ijcard.2017.09.192 [DOI] [PubMed] [Google Scholar]
- 73.Schwarz S, Schwab S, Klinga K, et al. Neuroendocrine changes in patients with acute space occupying ischaemic stroke. J Neurol Neurosurg Psychiatry 2003;74:725-7. 10.1136/jnnp.74.6.725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ambrosius W, Kazmierski R, Gupta V, et al. Low free triiodothyronine levels are related to poor prognosis in acute ischemic stroke. Exp Clin Endocrinol Diabetes 2011;119:139-43. 10.1055/s-0030-1267918 [DOI] [PubMed] [Google Scholar]
- 75.Alevizaki M, Synetou M, Xynos K, et al. Low triiodothyronine: a strong predictor of outcome in acute stroke patients. Eur J Clin Invest 2007;37:651-7. 10.1111/j.1365-2362.2007.01839.x [DOI] [PubMed] [Google Scholar]
- 76.Kaptein EM, Sanchez A, Beale E, et al. Clinical review: Thyroid hormone therapy for postoperative nonthyroidal illnesses: a systematic review and synthesis. J Clin Endocrinol Metab 2010;95:4526-34. 10.1210/jc.2010-1052 [DOI] [PubMed] [Google Scholar]
- 77.Ranasinghe AM, Quinn DW, Pagano D, et al. Glucose-insulin-potassium and tri-iodothyronine individually improve hemodynamic performance and are associated with reduced troponin I release after on-pump coronary artery bypass grafting. Circulation 2006;114:I245-50. 10.1161/CIRCULATIONAHA.105.000786 [DOI] [PubMed] [Google Scholar]
- 78.Kapoor MC. Phenylephrine in cardiac surgery: will it have a place? Ann Card Anaesth 2014;17:209-10. [PubMed] [Google Scholar]
- 79.Mourouzis I, Saranteas T, Ligeret H, et al. Phenylephrine postconditioning increases myocardial injury: are alpha-1 sympathomimetic agonist cardioprotective? Ann Card Anaesth 2014;17:200-9. 10.4103/0971-9784.135850 [DOI] [PubMed] [Google Scholar]
- 80.Sirlak M, Yazicioglu L, Inan MB, et al. Oral thyroid hormone pretreatment in left ventricular dysfunction. Eur J Cardiothorac Surg 2004;26:720-5. 10.1016/j.ejcts.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 81.Mullis-Jansson SL, Argenziano M, Corwin S, et al. A randomized double-blind study of the effect of triiodothyronine on cardiac function and morbidity after coronary bypass surgery. J Thorac Cardiovasc Surg 1999;117:1128-34. 10.1016/S0022-5223(99)70249-7 [DOI] [PubMed] [Google Scholar]
- 82.Klemperer JD, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery. N Engl J Med 1995;333:1522-7. 10.1056/NEJM199512073332302 [DOI] [PubMed] [Google Scholar]
- 83.Güden M, Akpinar B, Saggbas E, et al. Effects of intravenous triiodothyronine during coronary artery bypass surgery. Asian Cardiovasc Thorac Ann 2002;10:219-22. 10.1177/021849230201000306 [DOI] [PubMed] [Google Scholar]
- 84.Bennett-Guerrero E, Jimenez JL, White WD, et al. Cardiovascular effects of intravenous triiodothyronine in patients undergoing coronary artery bypass graft surgery. A randomized, double-blind, placebo- controlled trial. Duke T3 study group. JAMA 1996;275:687-92. 10.1001/jama.1996.03530330031025 [DOI] [PubMed] [Google Scholar]
- 85.Hamilton MA, Stevenson LW, Fonarow GC, et al. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am J Cardiol 1998;81:443-7. 10.1016/S0002-9149(97)00950-8 [DOI] [PubMed] [Google Scholar]
- 86.Pingitore A, Galli E, Barison A, et al. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized, placebo-controlled study. J Clin Endocrinol Metab 2008;93:1351-8. 10.1210/jc.2007-2210 [DOI] [PubMed] [Google Scholar]
- 87.Portman MA, Slee A, Olson AK, et al. Triiodothyronine Supplementation in Infants and Children Undergoing Cardiopulmonary Bypass (TRICC): a multicenter placebo-controlled randomized trial: age analysis. Circulation 2010;122:S224-33. 10.1161/CIRCULATIONAHA.109.926394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Novitzky D, Mi Z, Sun Q, et al. Thyroid hormone therapy in the management of 63,593 brain-dead organ donors: a retrospective review. Transplantation 2014;98:1119-27. 10.1097/TP.0000000000000187 [DOI] [PubMed] [Google Scholar]
- 89.Jeevanandam V. Triiodothyronine: spectrum of use in heart transplantation. Thyroid 1997;7:139-45. 10.1089/thy.1997.7.139 [DOI] [PubMed] [Google Scholar]
- 90.Novitzky D, Cooper DK. Thyroid hormones and the stunned myocardium. J Endocrinol 2014;223:R1-8. 10.1530/JOE-14-0389 [DOI] [PubMed] [Google Scholar]