

Abstract
Background: Overexpression of epidermal growth factor receptor (EGFR) has been reported to be implicated in the pathogenesis of non-small cell lung cancer (NSCLC). Several EGFR inhibitors have been used in clinical treatment of NSCLC, but the emergence of EGFRL858R/T790M resistant mutation has reduced the efficacy of the clinical used EGFR inhibitors. There is an urgent need to develop novel EGFRL858R/T790M inhibitors for better NSCLC treatment.
Methods: By screening a natural product library, we have identified gossypol as a novel potent inhibitor targeting EGFRL858R/T790M. The activity of gossypol on NSCLC cells was evaluated by cell proliferation, cell apoptosis and cell migration assays. Kinase activity inhibition assay and molecular docking were used to study the inhibition mechanism of gossypol to EGFRL858R/T790M. Western blotting was performed to study the molecular mechanism of gossypol inhibiting the downstream pathways of EGFR.
Results: Gossypol inhibited the cell proliferation and cell migration of NSCLC cells, and induced caspase-dependent cell apoptosis of NSCLC cells by upregulating the expression of pro-apoptotic protein BAD. Molecular docking revealed that gossypol could bind to the kinase domain of EGFRL858R/T790M with good binding affinity through hydrogen bonds and hydrophobic interactions. Gossypol inhibited the kinase activity of EGFRL858R/T790M with EC50 of 150.1 nM. Western blotting analysis demonstrated that gossypol inhibited the phosphorylation of EGFR and its downstream signal pathways in a dose-dependent manner.
Conclusion: Gossypol inhibited cell proliferation and induced apoptosis of NSCLC cells by targeting EGFRL858R/T790M. Our findings provided a basis for developing novel EGFRL858R/T790M inhibitors for treatment of NSCLC.
Keywords: gossypol, molecular docking, NSCLC, EGFR, TKI
Introduction
Non-small cell lung cancer (NSCLC) accounts for approximately 85-90% of lung cancers, which has proven to be difficult to be treated due to poorly understood the pathogenesis (Oyewumi et al., 2014; Siegel et al., 2017). Conventional treatment strategies are used for NSCLC including surgical operation, radiotherapy and chemotherapy (Scott et al., 2007; Onishi et al., 2011; Uzel and Abacıoğlu, 2015). In addition, tyrosine kinase-based inhibitors (TKIs) molecular-targeted therapy are also employed to the treatment of NSCLC patients with EGFR mutations. Overexpression of EGFR has been reported and implicated in the pathogenesis of NSCLC, which account for more than 60% of NSCLC (Ohsaki et al., 2000). Therefore, it is increasing in clinic application as molecular targets for NSCLC patients with EGFR mutation.
The role of aberrant activation of the EGFR in NSCLC is well-documented (Sordella et al., 2004; Tracy et al., 2004; Gazdar and Minna, 2005; Sharma and Settleman, 2007; Sharma et al., 2007). The most common activating mutations, including point mutation L858R in exon 21 and deletions within exon 19 (del746-750) (Riely et al., 2006; Sharma et al., 2007), promote EGFR-driven cell proliferation and survival. Both first and second generation EGFR-targeted TKIs (gefitinib and erlotinib) targeting those activating mutants have been demonstrated to have a remarkable clinical response in the treatment of EGFR-mutated NSCLC (Lynch et al., 2004; Paez et al., 2004; Jackman et al., 2009; Rosell et al., 2009; Sequist et al., 2010). Although the early clinical results of first-generation EGFR inhibitors are impressive, unfortunately, most NSCLC patients with activating mutations eventually develop acquired resistance to EGFR inhibitors within several months. The most common mechanism of acquired resistance is the secondary T790M (gatekeeper residue Thr790 to methionine within the EGFR kinase domain) point mutation in exon 20 that occurs with an EGFR mutation (e.g., L858R), which accounts for approximately 60% in these acquired resistances (Balak et al., 2006; Kosaka et al., 2006; Yu et al., 2013). To overcome the acquired resistance to first-generation TKIs, several second- and third-generation EGFR TKIs [such as EKB-569 (Kwak et al., 2005), BIBW2992 (Li et al., 2008) and PF00299804 (Engelman et al., 2007)] have been developed. However, these agents still display limited clinical benefit for NSCLC patients with T790M mutation owing to dose-limiting toxicities (Oxnard et al., 2011; Miller et al., 2012). Recently, third-generation covalent EGFR inhibitor osimertinib (Ward et al., 2013; Cross et al., 2014) has been developed as mutant-selective EGFR inhibitor that specifically targeting EGFRL858R/T790M mutation. However, the effective treatment of patients that harbor the EGFR T790M drug resistance mutation with osimertinib is limited by the emergence of new drug resistances to the tyrosine kinase inhibitor therapy (Thress et al., 2015; Büttner et al., 2017). C797S mutation was reported to be a major mechanism for resistance to third generation EGFR TKIs (Yu et al., 2015). In addition to C797S mutation, other rare tertiary EGFR mutations have also been reported, including novel solvent front mutations (G796S/R), hinge pocket mutations of the leucine residue at position 792 (L792F/H), binding interference at position 798 (L798I), and steric hindrance at position 718 (L718Q) (Bersanelli et al., 2016; Chabon et al., 2016; Chen et al., 2017; Ou Q. et al., 2017; Ou S.-H.I. et al., 2017). With the emergence of resistance mechanisms, there is an urgent need to discover a novel class of EGFR inhibitors that effectively inhibits drug-resistant EGFRL858R/T790M mutation.
Natural products have been widely regarded as a pivotal source of leading compounds for drug development, recently, several natural products have been identified targeting EGFRL858R/T790M to overcome resistance. (Jung et al., 2015; Xiao et al., 2016). In our previous studies, we have successfully identified several small molecules from natural products library that could inhibit the growth of gefitinib resistant NSCLC via different mechanisms. (Fan et al., 2015; Li et al., 2017). These compounds demonstrated significant anti-proliferative effects on a variety of NSCLC cell lines, including those with T790M and L858R/T790M mutations. In this study, we identified a small molecule gossypol from cottonseed, as a potent inhibitor targeting EGFRL858R/T790M. Gossypol and its derivatives exert antitumor effects on different cancer types in vitro and in vivo, including breast cancer (Xiong et al., 2017), colon cancer (Lan et al., 2015), chronic myeloid leukemia (Goff et al., 2013) and prostate cancer (Volate et al., 2010) by targeting MDM2, VEGFR, Bcl-2 and p53. Herein, the results from our work proved that gossypol could inhibit the proliferation of NSCLC cells by targeting EGFRL858R/T790M. Gossypol also inhibited the phosphorylation of EGFR and suppressed the phosphorylation of extracellular signal–regulated protein kinase (ERK) and AKT. These results indicated that gossypol could be developed as a new potent EGFRL858R/T790M inhibitor and could inhibit the proliferation of NSCLC.
Results and Discussion
Gossypol Inhibits Cell Proliferation in NSCLC Cells
To identify potent small molecule inhibitor of EGFRL858R/T790M, we screened a natural products library with 235 compounds. We evaluated the anti-proliferative effect of each compound on H1975 cell line harboring EGFRL858R/T790M. Gossypol was identified and chosen for further mechanistic investigation due to its significantly anti-proliferative ability. H1975 cells were treated with an increasing concentration of gossypol for 72 h, and then cell viability was determined based on standard MTT assay protocol. As shown in Figure 1, the growth of H1975 cells were obviously inhibited by the treatment of gossypol in a dose-dependent manner, with 50% inhibition concentration (IC50) of 10.89 ± 0.84 μM. In addition, we have tested the cytotoxicity effect of gossypol on human normal lung fibroblast cell line CCD19 (IC50 is 14.89 ± 1.12 μM) and human NSCLC cell line H358 with EGFRWT (IC50 is 35.26 ± 1.09 μM) (the corresponding results can be seen in Supplementary Figure S1). Afatinib was used as positive control (IC50 = 170.4 ± 1.1 nM). The structure and corresponding cytotoxicity of gossypol were showed in Figure 1. We also examined the effect of gossypol on cell colony formation (Figure 2A), in accordance with the cell cytotoxicity, gossypol significantly inhibited the colony formation capacity in a dose-dependent manner in H1975 cell line. Collectively, these results suggested that gossypol could inhibit the proliferation of H1975 cell line.
FIGURE 1.

Cytotoxicity effect of gossypol on EGFR mutant cell line. (A) The structure of gossypol. (B) Evaluation of cell proliferation by gossypol in H1975 cells.
FIGURE 2.
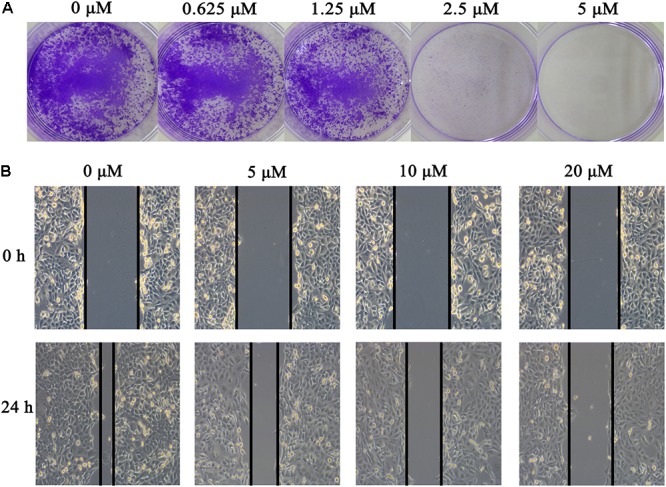
Effect of gossypol on H1975 cell line. (A) Colony formation of H1975 cells was monitored after gossypol (0–5 μM) treatment for 14 days, and photomicrographs of crystal violet stained colonies were depicted. (B) H1975 cells were treated with 0, 5, 10, and 20 μM for 24 h, and were analyzed for wound healing.
Gossypol Induces Cell Apoptosis in NSCLC Cells
To investigate whether the induction of apoptosis also contributed to gossypol-mediated growth inhibition of H1975 cells, Annexin V-FITC/PI staining assay was employed to analyze the number of apoptotic cells after treatment with gossypol using a flow cytometer. As shown in Figures 3A,B, gossypol induced cell apoptosis on H1975 cell line with a concentration-dependent manner.
FIGURE 3.

Apoptosis effect of Gossypol on H1975 cells. Flow cytometric analysis of cell apoptosis with gossypol at different concentrations (0, 5, 10, and 20 μM) for 24 h was determined. (A) Flow cytometry analysis of the apoptosis levels of h1975 cells after treatment with gossypol for 24 h. (B) Data from (A) were statistically analyzed. Mean ± SE. ∗∗P < 0.01. (C) Western blot analysis of apoptotic markers of H1975 cells after treatment of gossypol for 24 h.
Bcl-2 family members play key roles in the regulation of apoptotic progress. To understand how gossypol induced apoptosis, we next examined whether gossypol could alter the expression of apoptotic proteins in H1975 cells. As shown in Figure 3C and Supplementary Figure S4, treatment with gossypol for 24 h remarkably upregulated the expression level of proapoptotic protein Bad in a concentration-dependent manner. Moreover, we also observed that gossypol induced PARP cleavage, a hallmark of caspase-dependent apoptosis, in accordance with the expression level of cleaved caspase-3. Therefore, these results suggested that gossypol induced caspase-dependent apoptotic cell death by upregulating the expression of pro-apoptotic protein Bad in NSCLC cells.
Gossypol Inhibits the Cell Migration of H1975 Cell Line
The effect of gossypol on H1975 cell migration capability was estimated by a wound-healing assay. In the wound-healing assay (see Figure 2B), cells treated with gossypol reduced the rate of wound healing along with the increasing of treatment concentration, which was significantly lower than the untreated cells following incubation. These results demonstrated that gossypol inhibited the migration ability of H1975 cell lines in a dose-dependent manner.
Gossypol Inhibits the Activity of Tyrosine Kinase
To assess the kinase inhibition activities of gossypol, we performed a kinase inhibition profile assay of gossypol against recombinant human EGFRL858R/T790M. The selected compound gossypol exhibited inhibitory activity, which effectively inhibited the enzymatic activity of EGFRL858R/T790M with an EC50 value of 150 ± 30.7 nM (see Supplementary Figure S2). Besides, gossypol also inhibited the enzymatic activity of EGFRWT with an EC50 value of 252.9 ± 26.9 nM, higher than that to EGFRL858R/T790M (the corresponding results can be seen in Supplementary Figure S2). Afatinib was used as positive control (EC50 = 9.6 ± 2.9 nM). The effect of gossypol on cells is very complicated, and it is still difficult to distinguish which part is caused by EGFR targeting. To ensure the consistency of the experimental results, we conducted the entire ELISA enzyme inhibiting assay at the same time. Therefore, EGFRWT could be used as control to compare with EGFRL858R/T790M.
Molecular Docking Predicts the Potential Binding of Gossypol to EGFR
Molecular docking calculation was performed to gain insight into the binding mode between gossypol and EGFRL858R/T790M. The molecular docking results (see Figure 4 and Supplementary Figure S3) proved that gossypol could be docked into the kinase domain mainly composed of hydrophobic residues of C-helix and A-loop with a docking score of -6.42 ± 0.24 kcal/mol. Five hydrogen bonds were formed between gossypol and the carbonyl group of Q791, amino group of M793, hydroxyl group of T854 and amino group of K875. In addition, the hydrophobic contacts formed between gossypol and surrounded residues, including L718, M790, F723, F858, L792, L844, and M793, which also contributed to the interaction between gossypol and EGFRL858R/T790M. Therefore, the above results suggested that gossypol could bind to EGFRL858R/T790M.
FIGURE 4.

The binding mode between gossypol and EGFRL858R/T790M protein. (A) The 3D structure of EGFRL858R/T790M. (B) Gossypol was docked into the EGFR kinase domain, showing interactions between gossypol and key residues. (C) A two-dimensional interaction map of gossypol and EGFR. (D) The hydrophobic surface of EGFRL858R/T790M.
Gossypol Effectively Suppresses Phosphorylation of EGFR as Well as Its Downstream Signaling Pathway
To determine whether gossypol could inhibit the expression level of EGFR in cells, we investigated the effect of gossypol on the phosphorylation of EGFR in NSCLC cells. H1975 cells were treated with gossypol (0–20 μM) for 24 h. Western blot analysis showed that gossypol inhibited the phosphorylation of EGFR (Tyr 1068) in a concentration dependent manner (see Figure 5). To explore the detailed anti-cancer mechanism of gossypol, we further evaluated the downstream pathways of EGFR, including ERK and AKT signaling pathways. Treatment with gossypol also inhibited the phosphorylation of AKT and ERK in a concentration-dependent manner, consistent with the tendency of phosphorylation level of EGFR. Thus, our results indicated that gossypol could suppress the phosphorylation of EGFR and its downstream AKT and ERK signaling pathways, resulting in induction of apoptosis and proliferation inhibition of H1975 cells.
FIGURE 5.
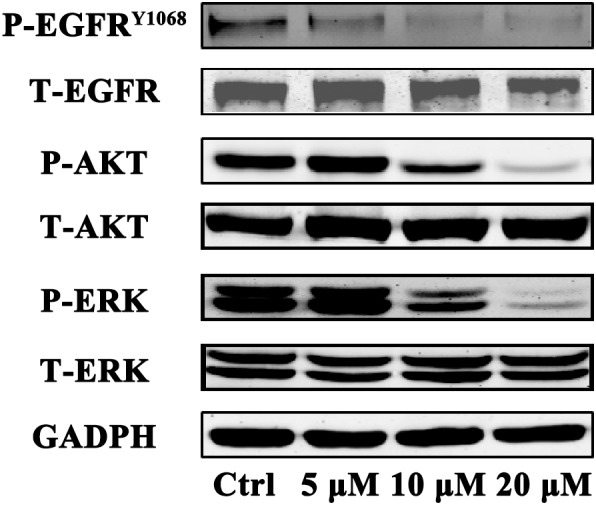
Immunoblot analysis of p-EGFR, EGFR, p-AKT, AKT, p-ERK, and ERK in H1975 cell after treatment with gossypol for 24 h. GAPDH was used as a loading control.
Conclusion
In this study, by screening a natural products library, we have identified that gossypol was a potential anticancer agent targeting EGFRL858R/T790M. Our results proved that gossypol inhibited the proliferation and induced apoptosis of human NSCLC cell line harboring EGFRL858R/T790M. Moreover, gossypol decreased the phosphorylation level of EGFR and its downstream signaling pathways AKT and ERK. Overall, our findings indicate that gossypol is a novel potent EGFRL858R/T790M inhibitor, which may serve as a useful therapeutic agent against NSCLC harboring EGFRL858R/T790M mutation.
Materials and Methods
Reagents
Gossypol was purchased from Selleck Ltd., which was dissolved in dimethyl sulfoxide (DMSO) to form a 20 mM stock solution. Fetal bovine serum (FBS), antibiotics and RPMI medium were purchased from Gibco (Carlsbad, CA, United States). RIPA lysis buffer and antibodies Bad, Bcl-XL, PARP, Cleaved Caspase-3, anti-p-EGFR (1068), anti-p-extracellular signal-regulated kinase 1/2 (Erk1/2) (Thr202/Tyr204), anti-p-Akt (Ser473), anti-Erk1/2, anti-Akt, anti-PERK, and anti-EGFR were purchased from Cell Signaling Technology (Beverly, MA, United States). Anti-GAPDH was purchased from Santa Cruz (Dallas, TX, United States).
Cell Culture
The human NSCLC cell line H1975 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, United States). Cells were cultured in RPMI1640 medium supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. All the cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Cell Proliferation Assay
Cell viability was evaluated by using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide MTT assay. Briefly, 3 × 103 cells per well were plated in 96-well plates and cultured overnight for cell adhesion. The cells were treated with DMSO or various concentrations of gossypol for 72 h. Subsequently, 10 μL MTT was added into each well and incubated for 4 h, and then the dark blue crystals were dissolved with 100 μl of the resolved solution (99% DMSO). Finally, the absorbance at 570 nm was measured by microplate reader (Tecan, Morrisville, NC, United States). The cell viability was calculated relative to controls, with results based on at least three independent experiments. Cells treated with the vehicle (DMSO) alone served as a control.
Colony Formation Assay
Briefly, H1975 cells were seeded in 6-well plates (1000 cells/well), after attachment overnight, cells were exposed to various concentration of gossypol with medium changes every 3 days until visible colonies formed. The colonies were washed with cold PBS, then fixed in 4% paraformaldehyde (PFA) for 15 min, and then stained with 0.5% crystal violet (1% PFA, 0.5% crystal violet, and 20% methanol in ddH2O) for 20 min. The colonies were photographed.
Apoptosis Analysis Assay
NSCLC cells were plated on 6-well plate with cell density of 2 × 105 cells per well and cultured overnight for adhesion. Subsequently, the cells were treated with different concentrations of gossypol for 24 h. After treatment, the cells were harvested by trypsin digestion and washed twice with ice-cold PBS, and resuspended in 100 μl 1 × binding buffer. Next, 4 μl of propidium iodide (PI, 1 mg/ml) and 1 μl Annexin-V fluorescein dye were added to the solution and mixed well at room temperature in the dark for 15 min. After that, the cells were resuspended in 300 μl of 1 × binding buffer from BD Biosciences (San Jose, CA, United States). The percentage of apoptotic cells was quantitatively measured using a BD FACSAria III flow cytometer from BD Bioscience (San Jose, CA, United States).
Enzyme-Linked Immunosorbent Assay (ELISA)
The kinase activity was evaluated with ELISA assay based on the kinase domain of dual-mutant EGFR (EGFRL858R/T790M) recombinant human protein (Peng et al., 2014). Briefly, 20 μg/mL Poly (Glu, Tyr) 4:1 (Sigma, St. Louis, MO, United States) was precoated in 96-well plates as substrate. Active kinases were added and incubated with indicated gossypol in 1 × reaction buffer containing 5 μmol/L ATP at 37°C for 1 h. Then, the wells were washed with PBS and then incubated with an anti-phosphotyrosine (PY99) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, United States) followed by a horseradish peroxidase (HRP)-conjugated secondary antibody. The wells were read with a multiwell spectrophotometer (VERSAmaxTM, Molecular Devices, Sunnyvale, CA, United States) at 492 nm. The inhibitory rate (%) was calculated with the following formula: [1–(A492 treated/A492 control)] × 100%, and responding EC50 values were calculated from the fitting inhibitory curves.
Molecular Docking
The X-ray structure of EGFRL858R/T790M with a resolution of 2.5 Å complexed with diaminopyrimidine derivative was retrieved from the Protein Data Bank [PDB ID code 4RJ8 (Hanan et al., 2014)] for docking with gossypol. Molecular structures were prepared using the standard procedure from the Protein Preparation Wizard module in Schrödinger 2015. The docking grid box was defined using the Receptor Grid Generation tool in Glide by centering on native ligand in the EGFRL858R/T790M structure. The structure of gossypol was derived from the PubChem database1, which was imported to the LigPrep module (Version 2.3, Schrödinger, LLC, New York, NY, United States) based on OPLS-2005 force field (Kaminski et al., 2001). The ionized state was assigned by using Epik (Version 2.0, Schrödinger, LLC, New York, NY, United States) at a pH value of 7.0 ± 2.0. Gossypol was docked into the kinase domain of the EGFRL858R/T790M using the Glide (Version 5.5, Schrödinger, LLC, New York, NY, United States) with the extra precision (XP) scoring mode. In the process of molecular docking, 5000 poses were generated during the initial phase of the docking calculation. The best binding pose for Gossypol was conserved for the further analysis.
Western Blot Analysis
Preparation of whole-cell protein lysates for western blot analysis was conducted as follows. After treatment, cells were lysed in RIPA lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris–HCl, pH 8.0,1% Triton X-100, 0.1% SDS, and 1% deoxycholate) containing protease inhibitor cocktail from Roche (Basel, Lewes, United Kingdom) for 15 min on ice and then boiled for 10 min. The concentration of total protein was determined with a Bio-Rad DCTM Protein Assay Kit (Bio-Rad, Hercules, CA, United States). Equal amounts of total protein (30 μg) protein lysate were loaded and separated by 10% SDS–polyacrylamide gel electrophoresis and then transferred to a nitrocellulose (NC) membrane from Millipore (Billerica, MA, United States). The membranes were blocked with 5% milk without fat in 1 × TBST for 2 h at room temperature, and then incubated with various primary antibodies, including phospho-AKT, phospho-ERK, t-AKT, t-ERK, phospho-EGFR (Tyr1068), t-EGFR at 1:1000 dilutions and anti-GADPH antibody at a 1:800 dilution overnight at 4°C. After washing the membranes in TBST three times (5 min per time), secondary fluorescent antibodies, either anti-rabbit or anti-mouse secondary antibodies depending on the source of the primary anti-bodies, were added to the membrane at 1:10,000 dilutions at room temperature for 2 h. GAPDH was used as the loading control and for normalization. The signal intensity of the membranes was detected using an LI-COR Odessy scanner (Belfast, ME, United States).
Statistical Analysis
The results were expressed as mean values ± standard error (mean ± SE). Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s post-tests. Significance was accepted at P < 0.05.
Author Contributions
EL, LL, and XY conceived this research, led the project, and revised the manuscript. YW, HL, XF, FD, ZJ, QW, and LL carried out the experiments and analyzed the data. YW and XY wrote the manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by Macau Science and Technology Development Fund (Project Nos. 082/2013/A3, 086/2015/A3, 082/2015/A3, 005/2014/AMJ, and 046/2016/A2).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00728/full#supplementary-material
References
- Balak M. N., Gong Y., Riely G. J., Somwar R., Li A. R., Zakowski M. F., et al. (2006). Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor–mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 12:6494. 10.1158/1078-0432.CCR-06-1570 [DOI] [PubMed] [Google Scholar]
- Bersanelli M., Minari R., Bordi P., Gnetti L., Bozzetti C., Squadrilli A., et al. (2016). L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC. J. Thoracic Oncol. 11 e121–e123. 10.1016/j.jtho.2016.05.019 [DOI] [PubMed] [Google Scholar]
- Büttner R., Wolf J., Thomas R. K., Sos M. L. (2017). Resistance mechanisms to AZD9291 and rociletinib—response. Clin. Cancer Res. 23 3967–3968. 10.1158/1078-0432.CCR-17-0948 [DOI] [PubMed] [Google Scholar]
- Chabon J. J., Simmons A. D., Lovejoy A. F., Esfahani M. S., Newman A. M., Haringsma H. J., et al. (2016). Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 7:11815. 10.1038/ncomms11815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Zhou F., Shen W., Jiang T., Wu X., Tong X., et al. (2017). Novel mutations on EGFR Leu792 potentially correlate to acquired resistance to osimertinib in advanced NSCLC. J. Thoracic Oncol. 12 e65–e68. 10.1016/j.jtho.2016.12.024 [DOI] [PubMed] [Google Scholar]
- Cross D. A. E., Ashton S. E., Ghiorghiu S., Eberlein C., Nebhan C. A., Spitzler P. J., et al. (2014). AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4 1046–1061. 10.1158/2159-8290.CD-14-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A., Zejnullahu K., Gale C.-M., Lifshits E., Gonzales A. J., Shimamura T., et al. (2007). PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 67 11924–11932. 10.1158/0008-5472.CAN-07-1885 [DOI] [PubMed] [Google Scholar]
- Fan X. X., Yao X. J., Xu S. W., Wong V. K., He J. X., Ding J., et al. (2015). (Z)3,4,5,4’-trans-tetramethoxystilbene, a new analogue of resveratrol, inhibits gefitinb-resistant non-small cell lung cancer via selectively elevating intracellular calcium level. Sci Rep 5 16348. 10.1038/srep16348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A. F., Minna J. D. (2005). Inhibition of EGFR signaling: all mutations are not created equal. PLoS Med 2:e377. 10.1371/journal.pmed.0020377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff D. J., Court Recart A., Sadarangani A., Chun H.-J., Barrett C. L., Krajewska M., et al. (2013). A Pan-BCL2 Inhibitor Renders Bone-Marrow-Resident Human Leukemia Stem Cells Sensitive to Tyrosine Kinase Inhibition. Cell Stem Cell 12 316–328. 10.1016/j.stem.2012.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanan E. J., Eigenbrot C., Bryan M. C., Burdick D. J., Chan B. K., Chen Y., et al. (2014). Discovery of Selective and Noncovalent Diaminopyrimidine-Based Inhibitors of Epidermal Growth Factor Receptor Containing the T790M Resistance Mutation. J. Med. Chem. 57 10176–10191. 10.1021/jm501578n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman D. M., Miller V. A., Cioffredi L. A., Yeap B. Y., Janne P. A., Riely G. J., et al. (2009). Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: results of an online tumor registry of clinical trials. Clin. Cancer Res. 15 5267–5273. 10.1158/1078-0432.CCR-09-0888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S. K., Lee M.-H., Lim D. Y., Lee S. Y., Jeong C.-H., Kim J. E., et al. (2015). Butein, a novel dual inhibitor of MET and EGFR, overcomes gefitinib-resistant lung cancer growth. Mol. Carcinog. 54 322–331. 10.1002/mc.22191 [DOI] [PubMed] [Google Scholar]
- Kaminski G. A., Friesner R. A., Tirado-Rives J., Jorgensen W. L. (2001). Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105 6474–6487. 10.1021/jp003919d [DOI] [Google Scholar]
- Kosaka T., Yatabe Y., Endoh H., Yoshida K., Hida T., Tsuboi M., et al. (2006). Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res 12 5764–5769. 10.1158/1078-0432.CCR-06-0714 [DOI] [PubMed] [Google Scholar]
- Kwak E. L., Sordella R., Bell D. W., Godin-Heymann N., Okimoto R. A., Brannigan B. W., et al. (2005). Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. U.S.A. 102 7665–7670. 10.1073/pnas.0502860102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L., Appelman C., Smith A. R., Yu J., Larsen S., Marquez R. T., et al. (2015). Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 9 1406–1420. 10.1016/j.molonc.2015.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Ambrogio L., Shimamura T., Kubo S., Takahashi M., Chirieac L. R., et al. (2008). BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27 4702–4711. 10.1038/onc.2008.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Fan X. X., Jiang Z. B., Loo W. T., Yao X. J., Leung E. L., et al. (2017). Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 115 45–55. 10.1016/j.phrs.2016.11.011 [DOI] [PubMed] [Google Scholar]
- Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 350 2129–2139. 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- Miller V. A., Hirsh V., Cadranel J., Chen Y.-M., Park K., Kim S.-W., et al. (2012). Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 13 528–538. 10.1016/S1470-2045(12)70087-6 [DOI] [PubMed] [Google Scholar]
- Ohsaki Y., Tanno S., Fujita Y., Toyoshima E., Fujiuchi S., Nishigaki Y., et al. (2000). Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol. Rep. 7 603–610. 10.3892/or.7.3.603 [DOI] [PubMed] [Google Scholar]
- Onishi H., Shirato H., Nagata Y., Hiraoka M., Fujino M., Gomi K., et al. (2011). Stereotactic body radiotherapy (SBRT) for operable stage I non–small-cell lung cancer: can SBRT be comparable to surgery? Int. J. Radiat. Oncol. Biol. Phys. 81 1352–1358. 10.1016/j.ijrobp.2009.07.1751 [DOI] [PubMed] [Google Scholar]
- Ou Q., Wu X., Bao H., Tong X., Wang X., Zhang X., et al. (2017). Investigating novel resistance mechanisms to third generation EGFR TKI osimertinib in non-small cell lung cancer patients using next generation sequencing. J. Clin. Oncol. 35 2572–2572. [Google Scholar]
- Ou S.-H. I., Cui J., Schrock A. B., Goldberg M. E., Zhu V. W., Albacker L., et al. (2017). Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 108 228–231. 10.1016/j.lungcan.2017.04.003 [DOI] [PubMed] [Google Scholar]
- Oxnard G. R., Arcila M. E., Chmielecki J., Ladanyi M., Miller V. A., Pao W. (2011). New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 17 5530–5537. 10.1158/1078-0432.CCR-10-2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyewumi M. O., Alazizi A., Wehrung D., Manochakian R., Safadi F. F. (2014). Emerging lung cancer therapeutic targets based on the pathogenesis of bone metastases. Int. J. Cell Biol. 2014:236246. 10.1155/2014/236246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez J. G., Jänne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to Gefitinib therapy. Science 304 1497–500. 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- Peng T., Wu J.-R., Tong L.-J., Li M.-Y., Chen F., Leng Y.-X., et al. (2014). Identification of DW532 as a novel anti-tumor agent targeting both kinases and tubulin. Acta Pharmacol. Sin. 35 916–928. 10.1038/aps.2014.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riely G. J., Politi K. A., Miller V. A., Pao W. (2006). Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin. Cancer Res. 12 7232. 10.1158/1078-0432.CCR-06-0658 [DOI] [PubMed] [Google Scholar]
- Rosell R., Moran T., Queralt C., Porta R., Cardenal F., Camps C., et al. (2009). Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 361 958–967. 10.1056/NEJMoa0904554 [DOI] [PubMed] [Google Scholar]
- Scott W. J., Howington J., Feigenberg S., Movsas B., Pisters K. (2007). Treatment of non-small cell lung cancer stage I and stage II: ACCP evidence-based clinical practice guidelines (2nd Edition). Chest 132 234S–242S. 10.1378/chest.07-1378 [DOI] [PubMed] [Google Scholar]
- Sequist L. V., Besse B., Lynch T. J., Miller V. A., Wong K. K., Gitlitz B., et al. (2010). Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 28 3076–3083. 10.1200/JCO.2009.27.9414 [DOI] [PubMed] [Google Scholar]
- Sharma S. V., Bell D. W., Settleman J., Haber D. A. (2007). Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7 169–181. 10.1038/nrc2088 [DOI] [PubMed] [Google Scholar]
- Sharma S. V., Settleman J. (2007). Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 21 3214–3231. 10.1101/gad.1609907 [DOI] [PubMed] [Google Scholar]
- Siegel R. L., Miller K. D., Jemal A. (2017). Cancer statistics, 2017. CA Cancer J. Clin. 67 7–30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- Sordella R., Bell D. W., Haber D. A., Settleman J. (2004). Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 305 1163–1167. 10.1126/science.1101637 [DOI] [PubMed] [Google Scholar]
- Thress K. S., Paweletz C. P., Felip E., Cho B. C., Stetson D., Dougherty B., et al. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21 560–562. 10.1038/nm.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S., Mukohara T., Hansen M., Meyerson M., Johnson B. E., Jänne P. A. (2004). Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 64 7241–7244. 10.1158/0008-5472.CAN-04-1905 [DOI] [PubMed] [Google Scholar]
- Uzel E. K., Abacıoğlu U. (2015). Treatment of early stage non-small cell lung cancer: surgery or stereotactic ablative radiotherapy? Balkan Med. J. 32 8–16. 10.5152/balkanmedj.2015.15553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volate S. R., Kawasaki B. T., Hurt E. M., Milner J. A., Kim Y. S., White J., et al. (2010). Gossypol induces apoptosis by activating p53 in prostate cancer cells and prostate tumor-initiating cells. Mol. Cancer Ther. 9 461–470. 10.1158/1535-7163.MCT-09-0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R. A., Anderton M. J., Ashton S., Bethel P. A., Box M., Butterworth S., et al. (2013). Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem. 56 7025–7048. 10.1021/jm400822z [DOI] [PubMed] [Google Scholar]
- Xiao X., He Z., Cao W., Cai F., Zhang L., Huang Q., et al. (2016). Oridonin inhibits gefitinib-resistant lung cancer cells by suppressing EGFR/ERK/MMP-12 and CIP2A/Akt signaling pathways. Int. J. Oncol. 48 2608–2618. 10.3892/ijo.2016.3488 [DOI] [PubMed] [Google Scholar]
- Xiong J., Li J., Yang Q., Wang J., Su T., Zhou S. (2017). Gossypol has anti-cancer effects by dual-targeting MDM2 and VEGF in human breast cancer. Breast Cancer Res. 19:27. 10.1186/s13058-017-0818-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. A., Arcila M. E., Rekhtman N., Sima C. S., Zakowski M. F., Pao W., et al. (2013). Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19 2240–22407. 10.1158/1078-0432.CCR-12-2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. A., Tian S. K., Drilon A. E., Borsu L., Riely G. J., Arcila M. E., et al. (2015). Acquired resistance of egfr-mutant lung cancer to a t790m-specific egfr inhibitor: emergence of a third mutation (c797s) in the egfr tyrosine kinase domain. JAMA Oncology 1 982–984. 10.1001/jamaoncol.2015.1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.