

Abstract

A series of biaryl chromans exhibiting potent and selective agonism for the GPR40 receptor with positive allosteric modulation of endogenous ligands (AgoPAM) were discovered as potential therapeutics for the treatment of type II diabetes. Optimization of physicochemical properties through modification of the pendant aryl rings resulted in the identification of compound AP5, which possesses an improved metabolic profile while demonstrating sustained glucose lowering.
Keywords: GPR40, FFA1, GPCR, diabetes, insulin secretagogue, AgoPAM, chroman
The GPR40 (FFAR1 or FFA1) receptor has recently attracted much interest as a novel target for the treatment of type 2 diabetes due to its ability to stimulate insulin secretion upon activation by fatty acids, in a glucose-dependent fashion.1−3 GPR40 agonists are theorized to reduce the risk of hypoglycemia as compared to other insulin secretagogues.4−7 Thus far, all of the GPR40 agonists that have been assessed in a clinical setting have been pharmacologically classified as partial agonists. However, researchers at Amgen8−10 and Bristol-Myers-Squibb11 have recently disclosed several GPR40 agonists that exhibit superior levels of receptor activation and in vivo efficacy, by operating as full agonists with positive allosteric modulation (AgoPAMs)12,13 of endogenous ligands such as docosahexaenoic acid (DHA). In addition to directly stimulating more insulin secretion via the pancreas, they drive GLP-1 secretion in the gut, potentially accounting for the observed enhancement in preclinical efficacy over partial agonists.14
We recently reported the discovery of a novel class of biaryl chromans (1 and 2) that operate as selective GPR40 AgoPAMs.15 Although these compounds demonstrated superior in vivo glucose lowering efficacy over GPR40 partial agonists such as TAK-875 with no indication of desensitization in rat 2-week and 4-week studies,16 these initial analogs possessed the potential to form phenol metabolites and were potent inhibitors of the bile salt export pump (BSEP).17,18 Herein, we report how these issues were addressed through a systematic exploration of the pendant aryl A and B rings (Figure 1) leading to the discovery of AP5, a metabolically more stable GPR40 AgoPAM.
Figure 1.

GPR40 AgoPAM structural leads.
Our SAR investigation of the aryl B-ring began with a library synthesis based on the model system 4 and culminated in the discovery of key substitutions critical for potency (Table 1). Suzuki coupling of the intermediate 3(19) with the appropriately substituted aryl boronic acids followed by saponification afforded the desired chromans as a mixture of diastereomers (Scheme 1).
Table 1. Exploration of B-Ring SAR.

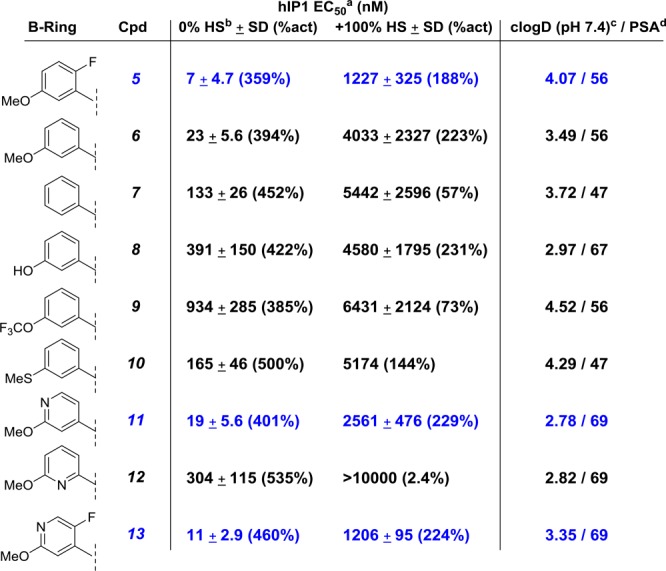
Mean of at least 2 runs.
HS = human serum.
Calculated cLogD at pH 7.4 using ACD Percepta software.
Calculated polar surface area using the TPSA method published in J. Med. Chem. 2000, 43, 3714-3717.
Scheme 1. Library Synthesis for B-Ring Modifications.

Reagents and conditions: (a) R-B(OH)2, 2 M K2CO3, Pd(dtbpf)Cl2, dioxane, 90 °C, 3 h; (b) 1 N NaOH, MeOH, rt, 3 h (8–44% yield over 2 steps).
Initially, compounds were tested as diastereomeric mixtures in a cell-based assay, which measures inositol monophosphate (IP1) accumulation in a recombinant hGPR40/HEK293 cell line designed to differentiate the efficacy of full and partial agonists as described in Plummer et al. (2017).15,20 In addition, this assay was performed in the presence of 100% human serum (HS) to assess the impact of plasma protein on potency (EC50).
We began our investigation by determining the impact of the meta-methoxy and ortho-fluoro substituents present in our lead compounds 1 and 2 on in vitro potency. Compound 5(15) is listed in Table 1 for reference. Removing the fluorine substituent in the ortho position as in 6 led to a 3–4-fold loss in potency. Next, in an effort to remove the metabolic liability of the methoxy group, we sought to remove it altogether as in 7 or replace it with other substituents such as a thioether (10) or a trifluoromethyl group (9). These modifications were less tolerated and resulted in a substantial loss in functional potency. The corresponding phenol analog 8 was also shown to be less active. Efforts to further improve physicochemical properties and thereby mitigate potential liabilites for phenol metabolites arising from cleavage of the methoxy group led us to incorporate heteroatoms into the B-ring as in compounds 11–13. Although the introduction of the pyridyl B-ring did increase PSA and reduce cLogD values, it became clear that the placement of the nitrogen atom was critical for potency. The nitrogen in the para-position proved to be optimal as demonstrated by the 16-fold improvement in IP1 potency of 11 over its ortho-regioisomer 12. Interestingly with the pyridyl B-ring, the impact of the ortho-fluorine substituent on potency was less pronouced, resulting in compounds 11 and 13 being essentially equipotent. In particular, compound 13 had a comparable IP1 potency and serum shift profile to the reference compound 5.
With the optimal B-ring substitutions identified, we focused our attention on combining B-ring modifications with A-ring exploration in the more active alpha-methyl series (Table 2, compounds 14–33). As previously reported, installation of a methyl group alpha to the carboxylic acid in the S-configuration (trans-relative configuration) not only afforded a remarkable improvement in potency but also reduced the magnitude of the plasma protein shift on potency.15
Table 2. A and B-Ring SAR in the α-Methyl Series.

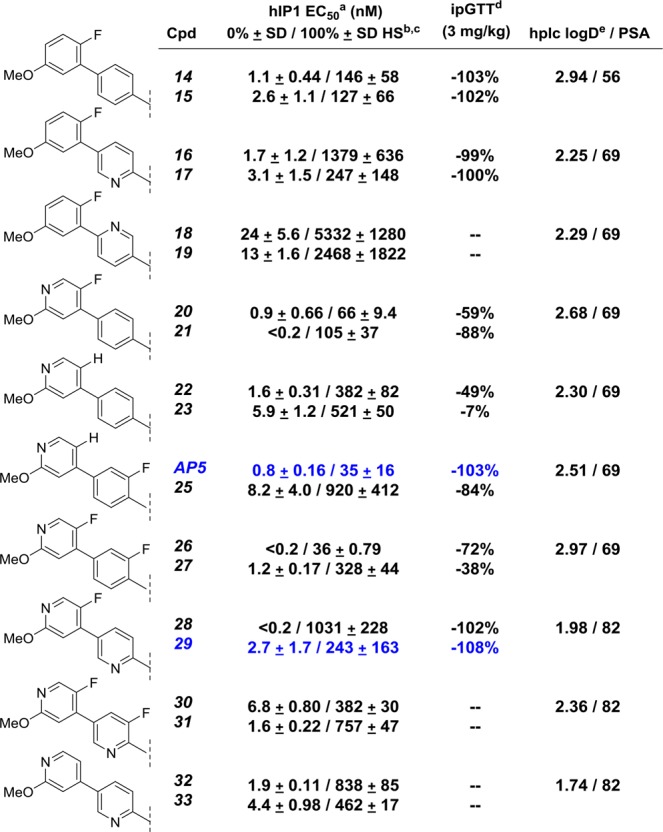
Mean of at least two runs.
HS = human serum.
% Activation >179%22.
% Inhibition of net glucose AUC.
Experimentally determined; see Supporting Information.
Compounds were initially tested in both the human (hGPR40/HEK293) and rat (rGPR40/CHO-K1) cell lines for IP1 accumulation.20 In general, rat IP1 EC50 values were consistent with human IP1 EC50 values and are given for select compounds listed in Table 4.21 Compounds that demonstrated an hIP1 EC50 < 20 nM against the GPR40 receptor (% activation >200%)22 and selectivity over the GPR120 receptor (EC50 > 10 uM) were then further profiled in our key in vivo rodent models, the rat intraperitoneal glucose tolerance test (ipGTT),23 and the Goto Kakizaki rat oral glucose tolerance test (oGTT).24 The rat ipGTT was used to screen compounds for in vivo efficacy at a fixed dose of 3 mg/kg, and the percent inhibition of blood glucose AUC compared to control is reported (Table 2). Further prioritization of active compounds was accomplished by dose titration in the Goto Kakizaki rat oGTT, where glucose lowering was examined across a range of doses (0.1 mg/kg–30 mg/kg). The dose and plasma concentrations at the three hour time point that afforded maximum efficacious glucose lowering (MED) are represented in Table 4.
Table 4. Rat IP1, GK Rat oGTT, BSEP, and Off-Target Activity Comparison of Key Compounds.
| Cmpd | rat IP1a (EC50 nm ± SD) | GK rat oGTT MEDb (pl concc @ 3 h, dose) | Off-target Hitsd # | BSEPe (IC50 μM ± SEM) |
|---|---|---|---|---|
| 14 | 1.2 ± 0.35 | 3.0 μm @ 10 mg/kg | 9 | 1.4 ± 0.1 |
| 16 | 2.3 ± 0.40 | 13.9 μm @ 30 mg/kg | 3 | 0.3 ± 0.04 |
| AP5 | 0.49 ± 0.28 | 4.9 μm @ 10 mg/kg | 4 | 3.5 ± 0.2 |
| 29 | 5.2 ± 1.6 | 32 μm @ 30 mg/kg | 1 | 3.2 ± 0.4 |
Mean of at least two runs, % activation >370%.22
MED = maximum efficacious dose.
pl conc = plasma concentration.
>50% inhibition @ 10 μM; see Supporting Information for specific receptors.
Mean of three measurements.
Utilizing the hetero-Diels–Alder (HDA) methodology described in our early work,15,19 we were able to quickly prepare compounds listed in Table 2, varying substituents and heteroatoms in both the A and B rings in a modular fashion (Scheme 2). In general, electron-rich dienophiles delivered better yields in the HDA reaction. Although the mixture of diastereomers was readily separable by SFC, the absolute configuration of the chroman ring was not determined.
Scheme 2. Achiral Synthesis of 28 and 29 Using HDA.

Reagents and conditions: (a) xylenes, 170 °C, 50 min, 41%; (b) 1 M aqueous LiOH, MeOH/THF, 65 °C, 19 h, 89%; (c) SFC separation: Chiral-pak AD-H (50 × 250 mm; 65% MeOH/CO2, 67%; (d) 1 M aqueous NaOH, MeCN/H2O, 95%.
The diastereomeric pairs, 14/15 and 16/17, described in our earlier work,15 are listed in Table 2 for comparison. Consistent with previous observations, applying the (S)-α methyl phenyl propionic acid headpiece to the analogs (5, 11, and 13) containing the optimal B-ring modifications attenuated serum shift by 100-fold and increased potency in the hIP1 assay (diastereomeric pairs 14/15, 20/21, and 22/23). In general, the chroman stereochemistry had minimal effect on in vitro potency, with both diastereomers being active, and further differentiation was only observed in the magnitude of their serum-shifted IP1 potency. For example, in the case of the diastereomic pair, 28 and 29, the impact of human serum on IP1 potency was far greater for 28 (>5000-fold shift) compared to 29 (170-fold shift).
In an effort to further reduce logD and increase PSA, we replaced the A-ring phenyl with a pyridine ring. As observed with the B-ring SAR, the location of the heteroatom was critical for activity. By moving the nitrogen atom in the ortho- position relative to the chroman ring (16/17), a 10-fold improvement in hIP1 potency compared to the meta-position (18/19) was observed. Further improvement in potency and in vivo efficacy was achieved through the installation of a fluorine atom on either the A or B ring. Diastereomeric pairs AP5/25 and 20/21 demonstrated better potency and glucose lowering than the nonfluorinated compounds 22/23. However, simultaneously installing fluorine in both the A and B ring did not correspond to an additive effect on ipGTT efficacy (26/27 vs AP5/25) despite similar hIP1 potencies.
Based on the in vitro IP1 potency, moderate serum shift, and in vivo efficacy demonstrated in the ipGTT, AP5 and 29 were selected for further profiling. A stereoselective route (Scheme 3) featuring a Noyori reduction of the ketone intermediate 36 to establish the chroman stereocenter25 was utilized for the large scale preparation of AP5. Ketone 36 was prepared via a Pd-mediated Heck coupling of the allylic alcohol 34 and the iodophenol 35. Using RuCl[(R,R)-Tsdpen](Mesitylene) catalyst, the key benzylic alcohol intermediate 37 was obtained as a 86:14 mixture of diastereomers enriched in the preferred “S” configuration. Further diastereomeric upgrade to >99% dr was accomplished with chiral SFC separation after a subsequent Mitsunobu reaction to form the chroman intermediate 38. Hydrolysis with aqueous LiOH and conversion to the corresponding sodium salt with aqueous NaOH afforded the final compound AP5. The absolute configuration at the chroman stereocenter of AP5 was further confirmed by electronic circular dichroism (ECD) of its corresponding methyl ester 38.26
Scheme 3. Stereoselective Synthesis of AP5.

Reagents and conditions: (a) t-BuX-Phos Palladacycle, Cy2NMe, toluene, 90 °C, 18 h, 49%; (b) formic acid, NEt3, RuCl[(R,R)-Tsdpen](Mesitylene), EtOAc, 22 h, quant.; (c) tBu3P, DIAD, THF, 1 h, 94%; purity upgrade by SFC, 74%; (d) 1 M aqueous LiOH, MeOH/THF, 55–58 °C, 17 h, 96%; (e) 1 M aqueous NaOH, MeCN/H2O, 95%.
The Wistar-Han rat PK profiles of AP5 and 29 are listed in Table 3 along with 14 and 16 for comparison. Both compounds exhibited acceptable half-lives and exposures projecting to QD dosing in humans and plasma exposures ripe for further evaluation in rodents.
Table 3. PK Comparison of Key Compounds.
| Wistar-Han
Rat PKa |
|||||
|---|---|---|---|---|---|
| Cpd | fu | Cl (mL/min/kg) | Vdss (L/kg) | T1/2 (h) | F (%) |
| 14 | 0.005 | 3.1 | 0.41 | 3.5 | 50 |
| 16 | 0.003 | 1.1 | 0.35 | 6.2 | 50 |
| AP5 | 0.0002 | 2.8 | 0.48 | 3.7 | 81 |
| 29 | 0.001 | 1.2 | 0.41 | 6.4 | 100 |
Administered at a dose of 1 mg/kg iv, 2 mg/kg po.
In the GK rat oGTT, oral administration of AP5 and 29 1 h before an oral dextrose challenge showed that both compounds significantly reduced blood glucose levels compared to the vehicle (Figure 2, Table 4). Compound AP5 was determined to be more efficacious in this model, demonstrating maximally efficacious glucose lowering at a plasma concentration of 4.9 μM at 10 mg/kg. Compounds AP5 and 29 were further evaluated in vitro for inhibition of the BSEP and found to have marginal improvement over our initial analogs, 14 and 16 (Table 4).27 These compounds were also screened against 40 receptors that included a broad panel of GPCR’s, ion channels, transporters, and enzymes at 10 μM (Table 4). The number of off-target hits with greater than 50% inhibition was reduced from nine in 14 to just four in AP5.28 Incorporating nitrogen atoms in both the A and B ring, as in 29, further reduced the number of off-target hits to one, demonstrating an excellent correlation between physical properties and off-target activity.
Figure 2.
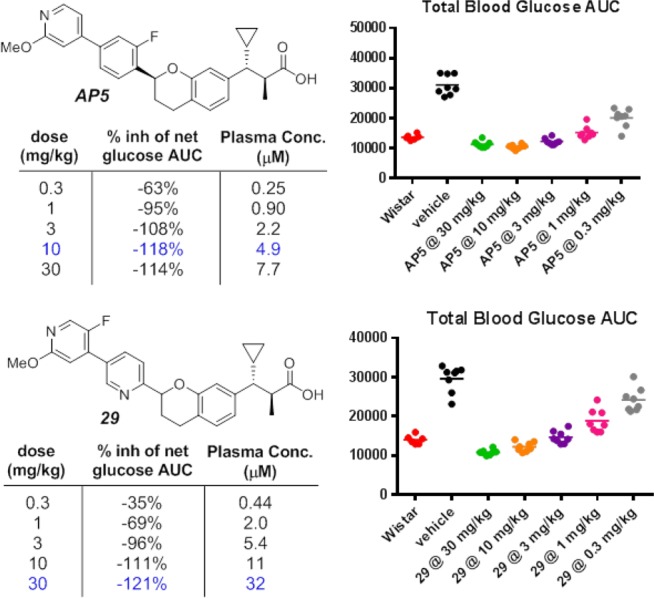
GK rat oGTT titrations for AP5 and 29.
In addition, AP5 showed enhanced GLP-1 secretion that was dose-dependent (Figure 3), consistent with the observed GPR40 AgoPAM mechanism of action.16 In contrast, measurable GLP-1 release is not observed with partial agonists such as TAK-875.
Figure 3.
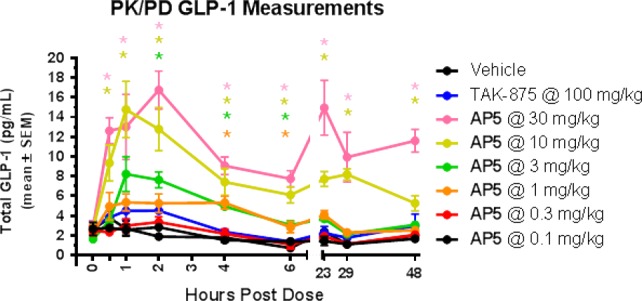
Dose-dependent GLP-1 secretion31 with AP5.
Furthermore, AP5’s acceptable rat PK profile translated into higher species including dog and monkey with a projected human oral bioavailability of 70% (Table 5). Using allometric scaling29 from the rat, dog (beagle), and rhesus, AP5 is projected to have a human dose of 7–15 mg along with a human half-life of 10–16 h making the compound suitable for QD dosing. In addition, in vitro metabolism studies in human hepatocytes showed that formation of phenol metabolites was not observed in AP5 through the introduction of the B-ring heteroatom, with the major metabolite being formation of the acyl-gluoronide (75%).30 In contrast, the starting analog, 14, showed that the B-ring phenol and its sulfonated derivative accounted for 67% of metabolites formed in vitro in human hepatocytes.30
Table 5. PK Profile of AP5 in Preclinical Speciesa.
| Species | fu | Clint, hep (mL/min/kg) | Clp (mL/min/kg) | Vdss (L/kg) | T1/2, eff (h) | F (%) |
|---|---|---|---|---|---|---|
| Rat | 0.00021 | 241 | 2.8 | 0.48 | 3.7 | 81 |
| Dog | 0.015 | 126 | 1.6 | 0.45 | 6.9 | 79 |
| Rhesus | 0.00071 | 102 | 0.72 | 0.48 | 10.0 | 64 |
| Human | 0.00044 | 111 | 0.58–0.98 | 0.45 | 5.3–9.0 | 70 |
Administered at a dose of 1 mg/kg iv, 2 mg/kg po.
Human pK prediction based on allometry method.29
In view of compound AP5’s significant glucose lowering effect in the GK rat oGTT, demonstrated increase in GLP-1 secretion, and improved off-target and PK profile over 14, AP5 was selected to undergo further assessment in a subchronic 4-day high dose safety and tolerability study in Wistar rats dosed at 200 and 750 mg/kg/day. Compound AP5’s plasma concentration levels reached 2309 μM·h and 7354 μM·h, respectively. At these exposures, no mortalities, serum biochemistry changes (including cardiac troponin I), liver weight increases, gross findings, or changes in the functional observational battery were observed. The only observed effect was a decrease in body weight gain reported in Table 6.
Table 6. Subchronic 4-Day High Dose Safety and Tolerability Study with AP5 in Wistar Ratsa.
| Dose (mg/kg/day) | AUC0–24 h (μM·h) | Cmax (μM) | Cmin (μM) | Tmax (h) | Body Weight Gainb |
|---|---|---|---|---|---|
| 200 | 2309 ± 804 | 223 ± 34 | 70 | 1–2 | 72% decrease |
| 750 | 7354 ± 2321 | 545 ± 81 | 328 | 1–2 | 90% decrease |
Vehicle = 0.5% (w/v) methylcellulose in deionized water.
Compared to concurrent controls.
In conclusion, through a systematic exploration of A and B ring SAR, AP5 was identified as a potent and selective GPR40 AgoPAM that demonstrates excellent in vivo efficacy with a projected human dose of 7–15 mg. By introducing key modifications, such as nitrogen atom incorporation into the B ring and fluorine substitution in the A-ring, phenol metabolite formation was mitigated, off-target activity was reduced, and marginal lowering was seen in BSEP inhibition, all of which led to a clean safety and tolerability profile of AP5 in a sub chronic 4-day high dose study.
Acknowledgments
The authors would like to thank Leo Joyce, Ed Sherer, and Joe Shpungin for the structural determination of compound 38 by ECD analysis, Scott Borges for SFC Separations, Qi Gao for NMR analysis, and Rong-Sheng Yang for HRMS support.
Glossary
Abbreviations
- GPR40
g-protein-coupled receptor 40
- GPCR
g-protein coupled receptors
- FFAR1
free fatty acid receptor 1
- AgoPAM
agonist-allosteric modulator
- DHA
docosahexaenoic acid
- GLP-1
glucagon-like peptide 1
- BSEP
bile salt export pump
- SAR
structure–activity relationship
- h1P1
human inositol monophosphate
- HS
human serum
- act.
activation
- SD
standard deviation
- SEM
standard error of mean
- PSA
polar surface area
- oGTT
oral glucose tolerance test
- ipGTT
intraperitoneal glucose tolerance test
- MED
maximum efficacious dose
- PK
pharmacokinetics
- HAD
Hetero-Diels–Alder
- HEK293
human embryonic kidney (cell line)
- CHO-K1
Chinese Hamster Ovary-K1 (cell line)
- SFC
supercritical fluid chromatography
- THF
tetrahydrofuran
- ACN
acetonitrile
- HPLC
high performance liquid chromatography
- AUC
area under curve
- dr
diastereomeric ratio
- dtbpf
di-(tert-butylphosphino)ferrocene
- DIAD
diisopropylazodicarboxylate
- t-BuXphos Palladacycle
[2-(di-tert-butylphosphino)-2′,4′,6′-triisopropyl-1,1′-biphenyl][2-(2-aminoethyl)phenyl]palladium(II) chloride
- Cy2NMe
N,N-dicyclohexylmethylamine.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00149.
Experimental procedures, analytical data, assay protocols, and metabolism data. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Defronzo R. A. From the triumvirate to the ominous octet: a new paradigm for the treatment of type II diabetes. Diabetes 2009, 58, 773–795. 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Kawamata Y.; Harada M.; Kobayashi M.; Fujii R.; Fukusumi S.; Ogi K.; Hosoya M.; Tanaka Y.; Uejima H.; Tanaka H.; Maruyama M.; Satah R.; Okubo S.; Kizawa H.; Komatsu H.; Matsumura F.; Moguchi Y.; Shinohara T.; Hinuma S.; Fujisawa Y.; Fujino M. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Stoddart L. A.; Smith N. J.; Milligan G. International union of pharmacology. LXXI. Free fatty acid receptors FFA1, −2, and −3: pharmacology and pathophysiological functions. Pharmacol. Rev. 2008, 60, 405–417. 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- Negoro N.; Sasaki S.; Mikami S.; Ito M.; Suzuki M.; Tsujihata Y.; Ito R.; Harada A.; Takeuchi K.; Suzuki N.; Miyazaki J.; Santou T.; Odani T.; Kanzaki N.; Funami M.; Tanaka T.; Kogame A.; Matsunaga S.; Yasuma T.; Momose Y. Discovery of TAK-875: A potent, selective, and orally bioavailable GPR40 agonist. ACS Med. Chem. Lett. 2010, 1, 290–294. 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen E.; Due-Hansen M. E.; Urban C.; Merten N.; Pfleiderer M.; Karlsen K. K.; Rasmussen S. S.; Steensgaard M.; Hamacher A.; Schmidt J.; Drewke C.; Petersen R. K.; Kristiansen K.; Ullrich S.; Kostenis E.; Kassack M. U.; Ulven T. Structure activity study of dihydrocinnamic acids and the discovery of the potent FFA1 (GPR40) agonist TUG-469. ACS Med. Chem. Lett. 2010, 1, 345–349. 10.1021/ml100106c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houze J. B.; Zhu L.; Akerman M.; Qiu W.; Zhang A. J.; Sharma R.; Schmidt M.; Wang Y.; Liu J.; Medina J. C.; Regan J. D.; Luo J.; Tonn G.; Zhang J.; Lu J. Y-L.; Chen M.; Lopez E.; Nguyen K.; Yang L.; Tang L.; Tian H.; Shuttleworth S. J.; Lin D. C-H. AMG-837: A potent, orally bioavailable GPR40 agonist. Bioorg. Med. Chem. Lett. 2012, 22, 1267–1270. 10.1016/j.bmcl.2011.10.118. [DOI] [PubMed] [Google Scholar]
- Ge M.; Yang L.; Zhou C.; Lin S.; Tang H.; Cline E. D.; Malkani S.. Preparation of bicyclic compounds as antidiabetics. (2006) WO 2006083781 A1.
- Brown S. P.; Dransfield P. J.; Vimolratana M.; Jiao X.-Y.; Zhu L.; Pattaropong V.; Sun Y.; Liu J.; Luo J.; Zhang J.; Wong S.; Zhuang R.; Guo Q.; Li F.; Medina J. C.; Swaminath G.; Lin D. C-H.; Houze J. B. Discovery of AM-1638: A Potent and Orally Bioavailable GPR40/FFA1 Full Agonist. ACS Med. Chem. Lett. 2012, 3, 726–730. 10.1021/ml300133f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu J.; Dransfield P. J.; Zhu L.; Wang Z.; Du X.; Jiao X.; Su Y.; Li A.-R.; Brown S. P.; Kasparian A.; Vimolratana M.; Yu M.; Pattaropong V.; Houze J. B.; Swaminath G.; Tran T.; Nguyen K.; Guo Q.; Zhang J.; Zhuang R.; Li F.; Miao L.; Bartberger M. D.; Correll T. L.; Chow D.; Wong S.; Luo J.; Lin D. C-H.; Medina J. C. Discovery and optimization of potent GPR40 full agonists containing tricyclic spirocycles. ACS Med. Chem. Lett. 2013, 4, 551–555. 10.1021/ml300427u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X.; Dransfield P. J.; Lin D. C-H.; Wong S.; Wang Y.; Wang Z.; Kohn T.; Yu M.; Brown S. P.; Vimolratana M.; Zhu L.; Li A.-R.; Su Y.; Jiao X.; Liu J.; Swaminath G.; Tran T.; Luo J.; Zhuang R.; Zhang J.; Guo Q.; Li F.; Connors R.; Medina J. C.; Houze J. B. Improving the pharmacokinetics of GPR40/FFA1 full agonists. ACS Med. Chem. Lett. 2014, 5, 384–389. 10.1021/ml4005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth B. A.; Shi J.; Jurica E. A.; Nielsen L. L.; Wu X.; Hernandez A. H.; Wang Z.; Gu Z.; Williams K. N.; Chen B.; Cherney E. C.; Ye X.; Wang Y.; Zhou M.; Cao G.; Xie C.; Wilkes J. J.; Liu H.; Kunselman L. K.; Gupta A. K.; Jayarama R.; Ramar T.; Rao J. P.; Zinker B. A.; Sun Q.; Dierks E. A.; Foster K. A.; Wang T.; Cvijic M. E.; Whaley J. M.; Robl J. A.; Ewing W. R.. Discovery of BMS-986118, a dual MOA GPR40 agonist that produces glucose-dependent insulin and GLP-1 secretion. 248th ACS National Meeting, San Francisco, CA, United States,, August 10–14, 2014. [Google Scholar]
- For a review of GPCR allostery, see:Wootten D.; Christopoulos A.; Sexton P. M. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discovery 2013, 12 (8), 630–644. 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- For precedent of agoPAMs applied to other GPCRs, see:Noetzel M. J.; Rook J. M.; Vinson P. N.; Cho H. P.; Days E.; Zhou Y.; Rodriguez A. L.; Lavreysen H.; Stauffer S. R.; Niswender C. M.; Xiang Z.; Daniels J. S.; Jones C. K.; Lindsley C. W.; Weaver C. D.; Conn P. J. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol. Pharmacol. 2012, 81 (2), 120–133. 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfalk S.; Steneberg P.; Edlund H. GPR40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer C. W.; Clements M. J.; Chen H.; Rajagopalan M.; Josien H.; Hagmann W. K.; Miller M.; Trujillo J. E.; Kirkland M.; Kosinski D.; Mane J.; Pachanski M.; Cheewatrakoolpong B.; Nolting A. F.; Orr R.; Christensen M.; Campeau L. C.; Wright M. J.; Bugianesi R.; Souza S.; Zhang X.; Di Salvo J.; Weinglass A. B.; Tschirret-Guth R.; Nargund R.; Howard A. D.; Colletti S. L. Design and Synthesis of Novel, Selective GPR40 AgoPAMs. ACS Med. Chem. Lett. 2017, 8, 221–226. 10.1021/acsmedchemlett.6b00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo M. E.; Pachanski M. J.; Kirkland M. E.; Kosinski D. T.; Mane J.; Cheewatrakoolpong B.; Xue J.; Szeto D.; Forrest G.; Miller C.; Bunzel M.; Plummer C. W.; Chobanian H. R.; Miller M. W.; Souza S.; Thomas-Fowlkes B. S.; Ogawa A. M.; Weinglass A. B.; Di Salvo J.; Li X.; Feng Y.; Tatosian D. A.; Howard A. D.; Colletti S. L. GPR40 partial agonists and AgoPAMs: Differentiating effects on glucose and hormonal secretions in the rodent. PLoS One 2017, 12 (10), e0186033 10.1371/journal.pone.0186033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleo M. D.; Luo Y.; Swiss R.; Bonin P. D.; Potter D. M.; Will Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60 (3), 1015–1022. 10.1002/hep.27206. [DOI] [PubMed] [Google Scholar]
- Dawson S.; Stahl S.; Paul N.; Barber J.; Kenna J. G. In Vitro Inhibition of the Bile Salt Export Pump Correlates with Risk of Cholestatic Drug-Induced Liver Injury in Humans. Drug Metab. Dispos. 2012, 40 (1), 130–138. 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- Brockunier L. L.; Chen H.; Chobanian H. R.; Clements M. J.; Crespo A., Demong D. E.; Guo Y.; Hagmann W. K.; Marcantonio K. M.; Miller M.; Pio B.; Plummer C. W.; Xiao D.. Antidiabetic bicyclic compounds (2014), WO2014130608 A1.
- See Supporting Information for in vitro inositol phosphate turnover (hIP1 and rIP1) study protocols.
- See Supporting Information for rat IP1 EC50s of compounds in Table 2 with in vivo ipGTT data.
- Comparable level of receptor activation to AM-1638 as discussed in footnote 16.
- See Supporting Information for in vivo ipGTT study protocol.
- See Supporting Information for in vivo GK rat oGTT study protocol.
- Orr R. K.; Campeau L. C.; Chobanian H. R.; McCabe Dunn J. M.; Pio B.; Plummer C. W.; Nolting A.; Ruck R. T. A Modular Synthesis of 2-Alkyl- and 2-Arylchromans via a Three-Step Sequence. Synthesis 2017, 49 (3), 657–666. 10.1055/s-0036-1588075. [DOI] [Google Scholar]
- See Supporting Information for ECD determination.
- See Supporting Information for in vitro BSEP study protocol.
- See Supporting Information for specific receptors.
- Tang H.; Hussain A.; Leal M.; Mayersohn M.; Fluhler E. Interspecies Prediction of Human Drug Clearance Based on Scaling Data from One or Two Animal Species. Drug Metab. Dispos. 2007, 35 (10), 1886–1893. 10.1124/dmd.107.016188. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for metabolites of AP5 and 14.
- See Supporting Information for in vivo GLP-1 study protocol.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.