

Abstract
Background:
Somatic mutations in the ERBB genes (epidermal growth factor receptor: EGFR, ERBB2, ERBB3, ERBB4) promote oncogenesis and lapatinib resistance in metastatic HER2+ (human epidermal growth factor-like receptor 2) breast cancer in vitro. Our study aimed to determine the frequency of mutations in four genes: EGFR, ERBB2, ERBB3 and ERBB4 and to investigate whether these mutations affect cellular behaviour and therapy response in vitro and outcomes after adjuvant trastuzumab-based therapy in clinical samples.
Methods:
We performed Agena MassArray analysis of 227 HER2+ breast cancer samples to identify the type and frequency of ERBB family mutations. Of these, two mutations, the somatic mutations ERBB4-V721I and ERBB4-S303F, were stably transfected into HCC1954 (PIK3CA mutant), HCC1569 (PIK3CA wildtype) and BT474 (PIK3CA mutant, ER positive) HER2+ breast cancer cell lines for functional in vitro experiments.
Results:
A total of 12 somatic, likely deleterious mutations in the kinase and furin-like domains of the ERBB genes (3 EGFR, 1 ERBB2, 3 ERBB3, 5 ERBB4) were identified in 7% of HER2+ breast cancers, with ERBB4 the most frequently mutated gene. The ERBB4-V721I kinase domain mutation significantly increased 3D-colony formation in 3/3 cell lines, whereas ERBB4-S303F did not increase growth rate or 3D colony formation in vitro. ERBB4-V721I sensitized HCC1569 cells (PIK3CA wildtype) to the pan class I PI3K inhibitor copanlisib but increased resistance to the pan-HER family inhibitor afatinib. The combinations of copanlisib with trastuzumab, lapatinib, or afatinib remained synergistic regardless of ERBB4-V721I or ERBB4-S303F mutation status.
Conclusions:
ERBB gene family mutations, which are present in 7% of our HER2+ breast cancer cohort, may have the potential to alter cellular behaviour and the efficacy of HER- and PI3K-inhibition.
Keywords: breast cancer, HER2+ BC, PI3K, PI3K inhibition, somatic mutations, trastuzumab resistance
Background
Human epidermal growth factor-like receptor 2 (HER2) is overexpressed by ERBB2 gene amplification in approximately 20% of human breast cancers (HER2-positive breast cancers). HER2 and its fellow HER family receptors epidermal growth factor receptor (EGFR), HER3 and HER4 together drive oncogenesis, signalling predominantly through the phosphoinositide 3-kinase (PI3K) and mitogen-activated kinase (MAPK) pathways.1 HER2 overexpression is associated with a worse prognosis2 in breast cancer, and increased risk of metastasis.3
The HER2-targeted therapy, trastuzumab, has established efficacy in the treatment of women with HER2+ breast cancer in the metastatic and adjuvant settings.4,5 Other HER2-targeted therapies such as lapatinib, ado-trastuzumab-emtansine (T-DM1), pertuzumab and afatinib are also currently in use or being trialled for HER2+ breast cancer. However, resistance to HER2-targeted therapies remains a problem.1 Potential mechanisms of trastuzumab resistance include reduced receptor-antibody binding due to HER2 masking, altered signalling through alternative HER family receptor tyrosine kinases (RTKs) or non-HER family receptors, as well as altered intracellular signalling involving loss of phosphatase and tensin homolog (PTEN), reduced p27kip1, or increased PI3K/AKT activity.1 However, it is generally accepted that not all mechanisms that mediate trastuzumab resistance are fully known.1
Emerging evidence suggests that ERBB family gene mutations may play a role in the pathogenesis of HER2+ breast cancer and in response to HER2-targeted therapy. Somatic mutations in ERBB3 are found in 11% of gastric and colon cancers and have demonstrated oncogenic activity in vitro and in vivo.6 Somatic ERBB4 mutations have been seen in breast, gastric, colorectal and non-small cell lung cancers and affect signal transduction in vitro.7 Activating mutations in ERBB2 may increase phosphorylation of EGFR and HER3 in breast cancers which were classed as HER2-negative.8 A previous study identified 12 kinase domain mutants across EGFR (6 mutations), ERBB2 (3 mutations), or ERBB4 (3 mutations) (n = 76) in HER2+ breast cancers.9 Patients whose tumours carried these mutations did not respond to HER2-targeted therapy in the metastatic setting.9 These ERBB2 mutations also conferred a more aggressive phenotype in vitro.9 A T798M somatic mutation in HER2 was identified in an in vitro screen using a randomly mutagenized HER2 library10 and HER2-T798M was shown to confer resistance to lapatinib.11
Our study aimed to determine the frequency of mutations in EGFR, ERBB2, ERBB3, and ERBB4 and to investigate whether these mutations affect cellular behaviour and therapy response in vitro and outcomes after adjuvant trastuzumab-based therapy in clinical samples. In contrast with other known HER family members, increased HER4 protein expression has been associated with a better prognosis12 and increased sensitivity to trastuzumab in metastatic breast cancer.12 However, some studies argue that knockdown of ERBB4 reverses resistance to trastuzumab, and high HER4 expression is associated with a poor outcome in HER2+ breast cancer.13 Given this ambiguous role, the presence of a hotspot mutation, and as it was the most frequently mutated gene in our set, we selected two ERBB4 mutations to functionally interrogate in vitro: ERBB4-S303F and ERBB4-V721I. ERBB4-S303F occurred in three of our samples and as a furin-like domain mutation, offered the potential to affect dimerization; a crucial first step in HER family signalling. ERBB4-V721I was a kinase domain mutation and thus offered the potential to alter HER4 signalling and tyrosine kinase inhibitor (TKI) efficacy.
Methods
Patients
Formalin-fixed, paraffin-embedded (FFPE) patient samples were confirmed to have at least 50% tumour content by the Royal College of Surgeons in Ireland (RCSI) pathology department using haematoxylin and eosin (H&E) staining. Patients were selected for this study if they had received trastuzumab as part of their neoadjuvant or adjuvant treatment for HER2-positive breast cancer. A total of 227 patients were used for Agena analysis. Samples included primary HER2+ breast cancer surgical specimens, initial staging biopsies, and pretreatment biopsies from patients who had received neoadjuvant trastuzumab. Patient characteristics are summarized in Supplementary Table 3. Some patients had to be removed from our survival analysis owing to their receiving trastuzumab for the treatment of metastatic disease. The 133 patients included in our survival analysis were treated between 1994 and 2012 in either St Vincent’s University Hospital or Beaumont Hospital, Ireland. Ethical approval was granted by both institutions for this study. For the relapse free survival analysis 29 events occurred and follow up ranged from 3 to 167 months. For the overall survival analysis 21 events occurred and follow up ranged from 11 to 259 months. In our patient cohort only 11 ERBB family mutations were detected.
This research was performed in accordance with the Declaration of Helsinki. All clinical samples used in these studies were obtained from Beaumont Hospital and St Vincent’s University Hospital, Ireland with the full approval of each hospital’s ethics committee, who are, respectively, the Beaumont Hospital Ethics Committee (Beaumont Hospital, Beaumont Road, Dublin 9) and the St Vincent’s Healthcare Group Ethics and Medical Research Committee (Education and Research Centre, Elm Park, Dublin 4). Written, informed consent was granted by the patients whose samples were used in this study.
DNA extraction from FFPE HER2+ breast cancer clinical samples
DNA extraction was performed using a QiaAMP DNA FFPE Kit from Qiagen (Hilden, Germany) as per manufacturer’s protocol and quantified using QuBit. We designed an Agena MassARRAY panel to assay for 67 novel ERBB gene family somatic mutations in 227 HER2+ breast cancer patients (Supplementary Table 1). Typically, 10 ng per assay was used for mass spectrometry-based genotyping (Agena MassARRAY, San Diego, CA, USA), which was applied as previously described.14 Reactions where >15% of the resultant mass ran in the mutant site were scored as positive.
Protein extraction and reverse phase protein array analysis of FFPE HER2+ breast cancers
Protein was extracted from 85 FFPE breast cancer samples and reverse phase protein array (RPPA) analysis was carried out as previously described15 (Table 1).
Table 1.
Primary antibodies used in our RPPA experiments.
| Antibody | Manufacturer | Catalogue number | Species | Dilution |
|---|---|---|---|---|
| AKT | Cell Signaling | 9272 | Rabbit | 1:3000 |
| AKT (S473) | Cell Signaling | 9271 | Rabbit | 1:250 |
| AKT (T308) | Cell Signaling | 9275 | Rabbit | 1:500 |
| AKT2 (5B5) | Cell Signaling | 2964 | Rabbit | 1:50 |
| C-Raf | Millipore | 04-739 | Rabbit | 1:250 |
| C-Raf (S338) 56A6) | Cell Signaling | 9427 | Rabbit | 1:200 |
| EGFR | Santa Cruz | SC-03 | Rabbit | 1:100 |
| EGFR (Y1173) | Epitomics | 1124 | Rabbit | 1:50 |
| EGFR (Y992) | Cell Signaling | 2235 | Rabbit | 1:100 |
| EGFY (Y1068) | Cell Signaling | 2234 | Rabbit | 1:100 |
| MAPK – ERK 1/2 | Cell Signaling | 9102 | Rabbit | 1:200 |
| HER2 | Lab Vision | MS-325-P1 | Mouse | 1:1000 |
| HER2 (Y1248) | Upstate | 06-229 | Rabbit | 1:750 |
| HER3 | Santa Cruz | 285 | Rabbit | 1:500 |
| HER3 (Y1289) | Cell Signaling | 4791 | Rabbit | 1:50 |
| HER4 | Cell Signaling | 4795S | Rabbit | 1:50 |
| MAPK (T202/Y204) - ERK 1/2 | Cell Signaling | 4377 | Rabbit | 1:1200 |
| MEK1 | Epitomics | 1235-1 | Rabbit | 1:1200 |
| MEK1/2 (S217/S221) | Cell Signaling | 9121 | Rabbit | 1:1000 |
| mTOR | Cell Signaling | 2972 | Rabbit | 1:400 |
| mTOR (S2448) | Cell Signaling | 2971 | Rabbit | 1:100 |
| mTOR (S2481) | Cell Signaling | 2974 | Rabbit | 1:100 |
| NF-kB-p65 (S536) | Cell Signaling | 3033 | Rabbit | 1:100 |
| p38_MAPK | Cell Signaling | 9212 | Rabbit | 1:300 |
| p38 MAP Kinase (T180/Y182) | Cell Signaling | 9211 | Rabbit | 1:250 |
| p70 S6 Kinase | Epitomics | 1494-1 | Rabbit | 1:250 |
| p70 S6 Kinase (T389) | Cell Signaling | 9205 | Rabbit | 1:250 |
| PDK1 (S241) | Cell Signaling | 3061 | Rabbit | 1:100 |
| PI3K-p110-alpha | Cell Signaling | 4255 | Rabbit | 1:100 |
| PTEN | Cell Signaling | 9552 | Rabbit | 1:1000 |
| S6 Ribosomal Protein (S235/S236) (2F9) | Cell Signaling | 2211 | Rabbit | 1:200 |
| S6 Ribosomal Protein (S240/S244) | Cell Signaling | 2215 | Rabbit | 1:3000 |
| SRC | Upstate | 05-184 | Mouse | 1:200 |
| SRC (Y527) | Cell Signaling | 2105 | Rabbit | 1:400 |
HER, human epidermal growth factor-like receptor; PTEN, phosphatase and tensin homolog; RPPA, reverse phase protein array.
Site-directed mutagenesis
A plasmid encoding wildtype (WT) ERBB4 was obtained from Addgene (29536) and ERBB4-WT DNA isolated using a Qiagen Maxiprep kit and QIAfilter as per the manufacturer’s instructions. This ERBB4 WT DNA was used as a template to generate ERBB4-S303F and ERBB4-V721I DNA with the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent®, Agilent, Santa Clara, California) as per manufacturer’s instructions. Primers (Supplementary Table 2) were designed using Agilent’s QuikChange Primer Design programme at www.agilent.com/genomics/qcpd. MassArray analysis was used to confirm each mutation had been generated.
Transduction of exogenous DNA into HER2+ breast cancer cells
We selected 2 ERBB4 mutations for functional analysis, the potential hotspot mutation S303F (furin-like domain) and V721I (kinase domain). ERBB4-WT, ERBB4-S303F and ERBB4-V721I plasmids, along with the pCDF1-MCS2-EF1-puro empty vector, were stably expressed in HCC1569, BT474 and HCC1954 HER2+ breast cancer cell lines. These cell lines were chosen as transfection hosts as all are WT for all four known ERBB family members. Lentiviral expression constructs were prepared using 20 μl of the pPACKF1 Lentivector Packaging Kit (Systems Biosciences, Palo Alto, California). After 48 h post-transfection, the viral-enriched supernatant was collected from HEK293T cells and filtered through a 0.45 µM syringe filter. Then, 3.5 ml of supernatant was then added to T75 flasks containing host cells. Successfully transfected cells were selected in 2 µg/ml puromycin, beginning 48 h post-transfection, for a minimum of 10 days prior to experiments, and were maintained in this concentration of puromycin thereafter. Cells were removed from puromycin prior to experiments.
Although transfection with ERBB4 WT, ERBB4 V721I and empty vector controls were successfully established across our three cell lines, ERBB4-S303F transfection could be established only in the HCC1569 cell line. We note that, unlike the other two cell lines used in our study, HCC1569 is WT for PIK3CA, and that in our clinical study, ERBB4-S303F did not co-occur with a PIK3CA mutation.
Cell culture assays
Human HER2+ breast cancer cell lines were gifted by the National Institute for Cellular Biotechnology, Dublin City University, Ireland. All cell lines were grown in RPMI-1640 media (Sigma Aldrich, St Louis, Missouri) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin and maintained at 37oC with 5% CO2. Cell line identity was confirmed by DNA fingerprinting, performed by Source Biosciences (Nottingham, England). Cell lines were Mycoplasma tested before and after in vitro experiments. Trastuzumab (21 mg/ml), prepared in bacteriostatic water, was obtained from St James University Hospital. Lapatinib (10.8 mM) and Afatinib (20.6 mM) were purchased from Sequoia Chemicals (Pangbourne, United Kingdom) and prepared in dimethylsulfoxide (DMSO). Copanlisib (10 mM) was obtained under materials transfer agreement (MTA) from Bayer Pharmaceuticals (Berlin, Germany) and prepared in DMSO and 5% trifluoracetic acid. The 3D soft agar colony-forming assays were carried out as described previously.16 Proliferation assays over 5 days were used to determine the half maximal inhibitory concentration (IC50) values of copanlisib, afatinib and lapatinib, the growth inhibition at maximum effective dose for trastuzumab, and the combination index (CI) values of the combination of HER2-targeted therapies and copanlisib, as previously described.17 To investigate the effect of our ERBB4 mutations on protein signalling, 4.5 × 105 cells were seeded into each well of six-well plates and left to adhere overnight. Cells were then either treated with 100 ng/μl epidermal growth factor for 5 min or left as untreated controls prior to protein lysis, dilution and RPPA analysis as previously described.17
ERBB4 protein immunoprecipitation and kinase assay
ERBB4 immunoprecipitation was carried out using Dynabeads Protein A (Thermo Fisher Scientific, Waltham, Massachusetts) as per the manufacturer’s instructions, with anti-rabbit ERBB4 antibody (cell signalling, 4795S) at 1:50 dilution. Standard curves were prepared with fixed ratios of adenosine triphosphate (ATP): adenosine diphosphate (ADP). Then, 50 µM of ADP was added to 10 μl sample, 10 μl ERBB4 enzyme and 5 μl DMSO in a 96-well white opaque plate. The plate was covered with tin foil and incubated in the dark for 1 h at room temperature. A standard ATP-to-ADP conversion curve was generated by combining appropriate volumes (µl) of 1 µM ADP and 1 µM ATP stock solutions as follows: ADP:ATP 100:0, 80:20, 60:40, 40:40, 20:80, 10:90, 5:95, 4:96, 3:97, 2:98, 1:99, 0:100. Then, 25 µl ADP-Glo reagent was added to the plate, which was then left at room temperature for 40 min. 50 µl kinase detection reagent was added and the plate incubated in the dark at room temperature for 30 min. Luminescence was read on a PerkinElmer plate reader and the kinase activity calculated by the level of unconverted ATP remaining in the sample.
Statistical analysis
The Kaplan–Meier method was used to estimate the survival curve from the observed survival times. The log rank test was used to compare the survival curves from the input data dichotomized based on the presence or absence of the ERBB mutations. Fisher’s exact test (two variables) or Chi-square test (⩾ three variables) were used to search for significant correlations between mutations and clinical/pathological features. IC50 and CI values at effective dose 50 (ED50) were calculated using CalcuSyn software (BioSoft, Cambridge, UK). A CI value of < 0.9 is considered synergistic, 0.9–1.1 is considered additive and > 1.1 is considered antagonistic. A Student’s t test was used to compare the protein signalling activity of ERBB mutated and WT tumour and cell lines, and to compare the 3D colony formation of the ERBB4-S303F and V721I expressing cells relative to ERBB4-WT cells. Statistical testing was carried out when n ⩾ 3. p < 0.05 was considered statistically significant.
Results
Frequency and function of ERBB mutations in HER2-positive breast cancer
MassArray analysis confirmed the presence of 12 somatic, likely deleterious mutations in the ERBB family genes (Table 2). To our knowledge, these mutations have not been previously reported in HER2+ breast cancer, although the three ERBB3 mutations were reported and functionally tested in gastric and colon cancer.6 Our mutations, which together were present in 7% (16/227) of our samples (Table 2), were predicted as likely deleterious by two independent bioinformatics tools, AVSIFT18 and Mutation Assessor.19 All mutations were confirmed as somatic by sequencing matched normal DNA.
Table 2.
The identity and properties of somatic ERBB family mutations confirmed in our Agena study in 227 HER2-positive breast cancers. A low AVSIFT18 value predicts that a mutation is likely to be deleterious. Mutation Assessor19 is a bioinformatics tool which predicts the likely effect of a mutation. No mutation reported herein was reported in the TCGA HER2-positive breast cancer study as of 1 April 2017. All ERBB family mutations listed here were confirmed somatic by sequencing matched normal DNA.
| Gene/mutation | Domain | AVSIFT value |
Predicted effect (Mutation Assessor) |
COSMIC ID | Accession number |
|---|---|---|---|---|---|
| EGFR V769L | Kinase | 0.24 | T, PD | 6242 | P00533 |
| EGFR A839T | Kinase | 0 | D, PD | 13430 | P00533 |
| EGFR K846R | Kinase | 0.03 | Borderline D/B | 13431 | P00533 |
| HER2 H878Y | Kinase | 0.03 | D, PD | 21985 | P04626.1 |
| HER3 P262H | 1st furin-like | N/A | N/A | N/A | P21860 |
| HER3 Q809R | Kinase | N/A | N/A | N/A | P21860 |
| HER3 E928G | Kinase | 0 | D, PD | 94228, 1363010 | P21860 |
| HER4 S303F | 1st furin-like | 0 | D, PD | N/A | Q15303 |
| HER4 V721I | Kinase | N/A | N/A | N/A | Q15303 |
| HER4 G599W | 2nd furin-like | 0 | D, PD | N/A | Q15303 |
| HER4 D595G | 2nd furin-like | 0.27 | T, PD | 1405173 | Q15303 |
| HER4 K935R | Kinase | 0.1 | T, B | 1405163 | Q15303 |
B, benign; D, deleterious; EGFR, epidermal growth factor receptor; N/A, not available; PD, probably damaging; T, tolerated.
As ERBB4 was the most commonly mutated ERBB family member in our dataset, contained the potential hotspot S303F and due to the ambiguous role of ERBB4 mutations in HER2+ breast cancer, we chose to transfect the ERBB4 V721I (kinase domain) and S303F (furin-like domain) mutations into cell line models of HER2-positive breast cancer, which are either PIK3CA mutant (HCC1954, BT474) or WT (HCC1569).
ERBB4 mutations affect signalling and kinase activity as well as 3D colony formation in vitro
Transfection of the ERBB4-V721I mutation into HCC1569 cells (Figure 1, p-values in Supplementary Figure 4) resulted in a significant increase in HER4 expression, and HER3 signalling was increased in both HCC1569 ERBB4 mutants. Kinase activity was significantly lower in ERBB4-S303F than ERBB4-WT cells (Figure 2). In HCC1954 cells transfection of the ERBB4-V721I mutation significantly increased HER4 and HER2 expression, and decreased HER3 phosphorylation (Y1289) (Figure 3, p-values in Supplementary Figure 4). BT474 V721I cells showed an increase in phosphorylation of EGFR (Y1173) (Figure 3, p-values in Supplementary Figure 4).
Figure 1.
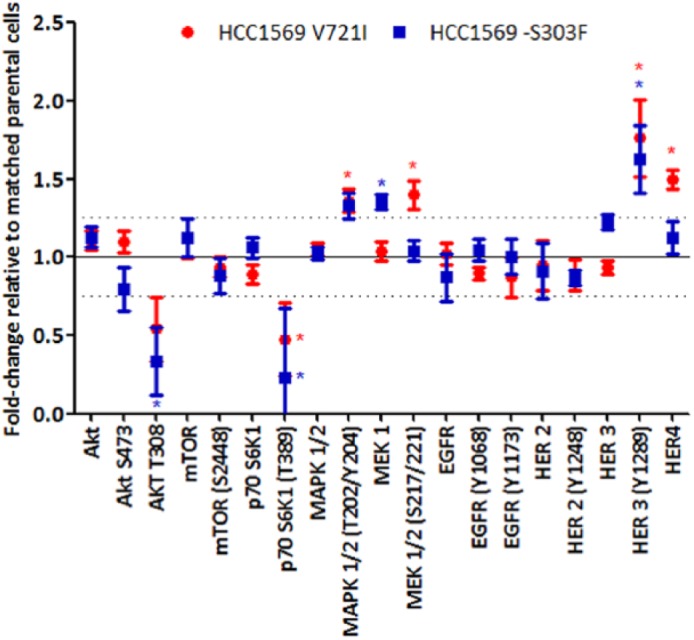
Signalling differences as determined by RPPA in HCC1569 ERBB4 V721I- or ERBB4 S303F- (transfected) HER2-positive cell lines relative to matched HCC1569 cells transfected with WT ERBB4 normalized to 100.
* = p < 0.05 relative to matched ERBB4-WT cells. p-values were calculated using the Student’s t test (two-tailed with equal variance).
HER, human epidermal growth factor-like receptor; RPPA, reverse phase protein array; WT, wildtype.
Figure 2.

The effect of the ERBB4-V721I and ERBB4-S303F mutations on the kinase activity if the HER4 protein in the HER2-positive breast cancer cell lines HCC1569, BT474 and HCC1954. Error bars are representative of independent duplicate experiments.
* = p < 0.05 as calculated by the Student’s t test (two-tailed with equal variance).
HER, human epidermal growth factor-like receptor.
Figure 3.

Signalling differences as determined by RPPA in ERBB4 V721I- (transfected) HCC1569, BT474, and HCC1954 HER2-positive cell lines relative to matched cells transfected with WT ERBB4 normalized to 100.
* = p < 0.05 relative to matched ERBB4-WT cells. p-values were calculated using the Student’s t test (two-tailed with equal variance).
HER, human epidermal growth factor-like receptor; RPPA, reverse phase protein array; WT, wildtype.
Transfection of both the S303F and V721I ERBB4 mutations altered signalling through the ERBB family protein receptors, the PI3K and MAPK pathways (Figure 3, p-values in Supplementary Figure 4).
These results demonstrate that the ERBB4-V721I mutation impacts the expression and phosphorylation of key drivers in both the PI3K/AKT and MAPK/ERK signalling pathways. These impacts are cell line specific and likely dependent on the mutational drivers already present in the cell line models. This however should not diminish the fact that these mutations are activating and will likely have significant impact on cellular function.
To validate these results, we extracted protein from HER2-positive FFPE breast cancer patient tumour samples and analyzed them on the RPPA platform (Figure 4). A limitation of this study is that the ERBB family mutated patients selected for our analysis had mutations in either the EGFR, ERBB2, ERBB3 or ERBB4 genes making it difficult to decipher specific functional impacts of specific ERBB family members. However, we found that ribosomal protein S6 (RPS6), which is activated by the PI3K pathway and involved in translation, has significantly increased phosphorylation (S240/244) (p = 0.0313) in ERBB family mutated versus ERBB family WT breast cancers. SRC kinase phosphorylation was significantly increased at (Y416) and (Y527) in ERBB mutated tumours relative to ERBB family WT tumours (Y416, p < 0.01, Y527, p = 0.03) (Figure 4). Overall, our analysis identifies that ERBB family mutated tumours have elevated signalling in pathways which are PI3K/AKT dependent, a pathway commonly activated in HER2-positive breast cancer; likely indicating that ERBB family mutations will impact response to agents which target the PI3K/AKT pathway.
Figure 4.

RPPA analysis of expression and phosphorylation, levels of (a) HER2 (Y1248), HER3, HER3 (Y1289), (b) AKT, AKT (S473), and AKT (T308), (c) S6 Ribosomal Protein (RPS6) (s235/236), RPS6 (S240/244) and (d) SRC kinase, SRC (Y527), SRC (Y418), (e) MAPK 1/2, MAPK 1/2 (T202/Y204) and (f) MEK 1, MEK 1 (S217/221) in ERBB family WT (n = 75)) versus ERBB family mutant (n = 10) patient tumours. Significant p-values which were calculated using a two-tailed Student’s t test with equal variance are indicated on the figure.
HER, human epidermal growth factor-like receptor; RPPA, reverse phase protein array; WT, wildtype.
When grown in 3D soft agar assays, V721I-expressing cells formed significantly more colonies than the corresponding WT ERBB4 transfected cell lines (HCC1569, 0.74-fold increase, p = 0.02; BT474, 0.68-fold increase, p = 0.02; HCC1954, 1.10-fold increase, p = 0.04) (Figure 5), demonstrating a more aggressive phenotype. In contrast, S303F transfected HCC1569 cells did not show increased colony formation relative to ERBB4 WT-transfected HCC1569 cells (p = 0.17) (Figure 5).
Figure 5.

The effect of the ERBB4-V721I and S303F mutations on 3D colony-forming ability of the HER2- positive breast cancer cell lines HCC1569, BT474 and HCC1954 in 3D soft agar. Error bars are representative of independent triplicate experiments.
* = p < 0.05 as calculated by the Student’s t test (two-tailed with equal variance).
HER, human epidermal growth factor-like receptor.
The ERBB4 mutations tested herein did not appear to affect levels of migration or invasion (Supplementary Figure 1). However, all models were poorly invasive and the BT474 and HCC1569 cells were poorly migratory, making it difficult to confidently draw conclusions from these experiments (Supplementary Figure 1). We noted no change in the morphology of the cells carrying ERBB4-V721I or ERBB4-S303F mutations (Supplementary Figure 2).
ERBB family mutations affect sensitivity to HER2- and PI3K-targeted therapies in vitro
Transfection of ERBB4-V721I resulted in a small but significant increase in resistance to afatinib, relative to cells transfected with ERBB4-WT in 2/3 cell lines, although this (HCC1569 p = 0.02, HCC1954 p = 0.005, Table 3) was not enough to classify the cell line as clinically resistant to afatinib. Again, transfection of ERBB4-S303F resulted in a small but significant increase in the sensitivity of HCC1569 cells to afatinib (p = 0.0006 versus ERBB4-WT, Table 3).
Table 3.
IC50 values of copanlisib, lapatinib and afatinib, and the effect of trastuzumab on percent growth inhibition in a panel of HER2-positive breast cancer cell lines stably expressing ERBB4-WT, ERBB4-V721I, or ERBB4-S303F. Standard deviations are representative of triplicate independent experiments. * = p < 0.05 compared with the matched cell line transfected with ERBB4-WT by Student’s t test. Student’s t tests were two-tailed with equal variance.
| Mutation | Copanlisib IC50 (nM) | Afatinib IC50 (nM) | Lapatinib IC50 (nM) | Trastuzumab % inhibition at 10 µg/ml |
|---|---|---|---|---|
| HCC1569 (PIK3CA WT) | ||||
| ERBB4-WT | 74.74 ± 9.69 | 6.93 ± 0.51 | 514.02 ± 30.50 | 3.59 ± 2.21 |
| ERBB4-V721I | 43.20 ± 9.22* | 12.34 ± 2.38* | 503.67 ± 33.42 | 2.98 ± 2.03 |
| ERBB4-S303F | 47.09 ± 5.09* | 2.98 ± 0.46* | 736.41 ± 51.55* | 0.26 ± 2.89 |
| BT474 (PIK3CA mutated) | ||||
| ERBB4-WT | 4.72 ± 0.65 | 1.49 ± 0.13 | 42.69 ± 4.97 | 43.02 ± 5.63 |
| ERBB4-V721I | 4.27 ± 1.04 | 1.13 ± 0.25 | 44.78 ± 4.12 | 42.05 ± 5.34 |
| HCC1954 (PIK3CA mutated) | ||||
| ERBB4-WT | 5.96 ± 0.29 | 6.54 ± 2.15 | 894.24 ± 42.16 | 3.58 ± 2.76 |
| ERBB4-V721I | 9.25 ± 0.63* | 13.74 ± 0.27* | 789.48 ± 89.44 | 1.41 ± 0.73 |
EV, empty vector; IC50, half maximal inhibitory concentration; WT, wildtype.
ERBB4-V721I transfection did not significantly influence lapatinib sensitivity in any of the cell lines tested (Table 3). Interestingly though, the ERBB4-S303F transfection was associated with resistance to lapatinib in the HCCC1569 model (p = 0.03, Table 3).
Transfection of both V721I and S303F mutations significantly increased sensitivity to the PI3K inhibitor copanlisib in the PIK3CA WT cell line HCC1569 (V721I −0.42-fold change, p = 0.01, S303F −0.37-fold change, p = 0.001). Transfection of V721I increased resistance to copanlisib in the PIK3CA mutated cell line HCC1954 in comparison with the same cell line transfected with WT ERBB4 (p = 0.001), although copanlisib retains low nM IC50s in both cell lines (Table 3).
We have previously shown that co-targeting PI3K and HER2 has the potential to be an improved treatment strategy for patients with HER2+ breast cancer.17 Here, we report that the combination of copanlisib with trastuzumab, lapatinib or afatinib is effective in all cell lines tested, regardless of their ERBB4-S303F or V721I mutation status.
In general, additivity or synergy was noted between afatinib and copanlisib in the HCC1569 and HCC1954 cell models used herein (Figure 6).
Figure 6.

(a) Efficacy of afatinib (-◊-), copanlisib (-□-) and a combination of afatinib and copanlisib (–Δ–) in the HER2-positive cell lines HCC1569, BT474, and HCC1954 stably expressing ERBB4-WT, V721I or S303F and (b) p-values for the difference in efficacy of the combination. Acid phosphatase toxicity assays were used to investigate the effect of a serial dilution of the drugs on these HER2-positive breast cancer cell lines over a 5-day period and the resulting dose-response curves were analyzed with CalcuSyn (Biosoft). Error bars are representative of standard deviations across triplicate experiments.
* = p < 0.05 relative to WT as calculated by the Student’s t test (two-tailed with equal variance). Afatinib:Copanlisib was tested at a 1:1 ratio (top conc 100nM:100nM).
HER, human epidermal growth factor-like receptor; WT, wildtype.
Figure 7.

Impact of somatic ERBB family mutations on RFS (left, n = 133) and OS (right, n = 134) after adjuvant trastuzumab-based therapy in our sample set. p-values were calculated using the log rank (Mantel–Cox) test with Graphpad Prism. ERBB family mutations were identified using Agena’s MassARRAY mass spectrometry-based genotyping platform.
OS, overall survival; RFS, relapse-free survival.
Encouragingly, the minor afatinib resistance induced by the V721I mutation in both HCC1569 and HCC1954 cells was overcome by combining afatinib with copanlisib. No additivity or synergy between afatinib and copanlisib occurred in the BT474 models. However, both drugs were highly potent as single agents in this cell line (Figure 6).
The combination of lapatinib and copanlisib remained synergistic in all of our cell lines regardless of ERBB4 mutational status (Supplementary Figure 3). CIs indicate that the combination of copanlisib and lapatinib is less synergistic in HCC1954 V721I transfected cells relative to ERBB4 WT-transfected HCC1954 cells. However, this could be due to the slightly enhanced sensitivity to lapatinib in the HCC1954-V721I model.
Neither of the ERBB4 mutations we tested affected trastuzumab sensitivity in vitro (Table 3). A log rank test was used to compare the survival curves for both progression-free survival and overall survival (OS) and assign significance (or lack thereof) (Figure 7). While the assumption of the log rank test is that of proportional hazards, small departures from this assumption do not invalidate the test (potentially in the mutant subgroup). Furthermore, the p-values are not even close to significance (relapse-free survival, p = 0.477; OS p = 0.666).
We also found no significant correlation between the presence of ERBB family mutations and clinicopathological features (Supplementary Table 4), although the low numbers in our test groups preclude confidently drawing conclusions.
The combination of trastuzumab and copanlisib inhibited the growth of all cell line models, with no significant difference observed between the transfected models and the WT models. (Supplementary Figure 5).
Discussion
To date, little work has been undertaken to determine the frequency and implications of ERBB family mutations in HER2+breast cancer. Herein we report somatic ERBB family mutations in 7% of our HER2+breast cancer cohort. To the best of our knowledge, these have not previously been reported in HER2-positive breast cancer. Within the full the cancer genome atlas (TCGA) breast cancer study (but not the TCGA HER2-positive study), ERBB3-E928G and ERBB4-S303F were reported at a similar frequency as to our dataset. The frequencies we report are in line with previous reports of mutations in 5% (4/78) of HER2-positive breast tumours9 (ERBB2), but lower than the 11% reported in gastric and colon cancers6 (ERBB3). We argue that a 7% frequency, while low, may be clinically relevant, as low frequency mutations can be targeted in cancer. For example, ALK mutations, although occurring in approximately 5% of lung cancers, guide therapy in that disease.20 Low frequency mutations in 8% or less were found to be an independent predictor of poor treatment-free survival in chronic lymphocytic leukemia (CLL) and monoclonal B-cell lymphocytosis,21 and low frequency KRAS mutations (<10%) have been shown to predict a worse response to anti-EGFR therapies in metastatic colorectal cancers.22
The three ERBB3 mutations reported herein have been previously reported in colon and gastric cancer by Jaiswal and colleagues who found that ERBB3 mutations could transform cells in culture and in vivo, promote anchorage-independent growth of breast and colonic epithelial cells, and promote cell survival.6 Intriguingly they found that ERBB3 mutants’ oncogenic effects were dependent on the expression of kinase-active ERBB2.6
Our ERBB mutations clustered in either the furin-like domain or the kinase domain. Kinase domain mutations can affect both cell signalling and drug sensitivity, with EGFR kinase domain mutations known to sensitize lung cancers to therapies such as gefitinib and erlotinib. The furin-like domain mediates the formation of homo- and heterodimers, which is a crucial first step in ERBB family signalling.1 Further, crystallography studies suggest these mutations may lie at the base of the dimerization arm of ERBB4 and be involved in the dimerization contacts by the receptor.23 An analysis of data from over 5000 tumours across 22 cancer types from TCGA found that mutations cluster in domains involved in tyrosine kinase receptor signalling, including the kinase and furin-like domain, and identified ERBB4-S303F as a mutation warranting further study.24
ERBB4-S303F, which shows an effect only in some of our functional studies, is analogous to ERBB2-S310F, which has been observed at low frequencies in lung, breast and ovarian cancers, and in a bladder cancer cell line.25 This mutation-induced anchorage-independent growth, and cells bearing it were more sensitive to anti-ERBB2 small molecule inhibitors than corresponding ERBB4-WT cells.25 Although not directly analogous, ERBB4-V721I is just four residues away from the ERBB4 residue analogous to EGFR-G719S. EGFR-G719S has been shown to activate the kinase by activating the p-loop, and to alter nucleotide binding.26
HER4 overexpression may be associated with a better outcome clinically12,27 and an antiproliferative response in vitro.28 Conversely, ERBB4 has been shown in vitro to promote the proliferation of breast cancer cells,29 while there is evidence to suggest HER4 has a role in resistance to therapy in breast and other cancers.30 In fact, a recent study demonstrated that in both HER2-positive and triple negative breast cancer, HER4 expression was significantly associated with a favourable prognosis in a univariate analysis, however the results were not significant after multivariate analysis.31 In part, due to the conflicting studies described above, and because it was the most frequently mutated gene in our study, and contained the unpublished hotspot S303F, we selected the ERBB4 gene as a candidate for functional analysis in vitro.
Neither ERBB4-V721I nor ERBB4-S303F co-occurred with PIK3CA mutations in our clinical samples. In vitro we could only establish the ERBB4-S303F mutation in the HCC1569 cell line (PIK3CA-WT), possibly indicating that the combination of ERBB4-S303F with a PIK3CA mutation is lethal. 3D colony formation is a surrogate for anchorage-independent growth, one of the hallmarks of cancer.32 All ERBB4-V721I mutated cells formed significantly more 3D colonies than ERBB4-WT cells. This finding was in contrast with the HCC1569-S303F cell line which did not form greater numbers of colonies than the HCC1569 ERBB4-WT cells, thus suggesting ERBB4-V721I mutations may be oncogenic, whereas the S303F mutation may be less so. Transfection of an ERBB4-mutation (both V721I and S303F) resulted in altered activation and expression of EGFR, ERBB2 and ERBB3; however, these effects were cell line dependent.
Interestingly we found that in the HCC1569 cells, p-HER3 (Y1289) signalling was increased while p70S6 kinase phosphorylation (T389) was decreased in response to transfection with either ERBB4-V721I or ERBB4-S303F. We also observed an increase in either expression or phosphorylation of MAPK (Y202/T204) and MEK (S221/222) in both cell lines. These findings support the idea that in the HCC1569 cells (PIK3CA WT), transfection of either ERBB4-V721I or ERBB4-S303F increases MAPK/ERK signalling.
However, in the BT474 and HCC1954 cell lines which are PIK3CA mutated, transfection of ERBB4-V721I resulted in an increase in AKT phosphorylation AKT (S473 and T308) in HCC1954 cells and an increase in mTOR (S2448) in BT474 cells. These results indicate that in PIK3CA-Mutant cells, transfection of these ERBB4 mutations will likely promote an increase in PI3K/AKT signalling.
ERBB4 can signal through ectodomain cleavage and translocation of the remaining intracellular half of the protein into the nucleus, a process which requires ADAM 17 protease.33 We used the cleavable isoform of ERBB4 in our in vitro studies,34 and although a full study of ERBB4 cleavage was beyond the scope of our work, it may present an interesting future study (although the unclear physiological relevance of ERBB4 cleavage in breast cancer and the difficulties in detecting the cleaved intracellular domain will present difficulties).33
As mentioned previously, our FFPE experiments required the pooling together of all ERBB family mutations, due to the limited number of mutations in our dataset. However, our RPPA analysis of FFPE-preserved HER2+breast cancer tumours demonstrates that ERBB family mutant tumours show increased phosphorylation of AKT (T308), and ribosomal protein S6 (RPS6) (S240/244) relative to the ERBB-WT tumours. Phosphorylation of AKT (T308) is essential for activation of mTORC1, which activates protein synthesis and can further activate S6 Kinase, which subsequently activates RPS6. Inhibition of RPS6 has been shown to correlate with trastuzumab-mediated growth inhibition in vitro35 indicating that ERBB-mutant cells may proliferate at a faster rate.
We also found that phosphorylation of SRC kinase (Y527 and Y416) was significantly increased in ERBB-mutant cell lines. This finding suggests that future studies should examine the targeting of ERBB-mutant cancers with SRC kinase inhibitors such as dasatinib, which have recently been reported to overcome trastuzumab resistance in HER2-amplified gastric and biliary tract cancer cell lines.36
Pan-HER family inhibitors such as neratinib have and afatinib have recently entered clinical trials for the treatment of cancer patients who have a mutated or altered HER family gene or protein. We chose to use trastuzumab, lapatinib and afatinib in this study as trastuzumab and lapatinib are currently the standard of care for HER2+ advanced breast cancer, and afatinib it is currently being tested clinically in patients with altered HER family members.
ERBB4-V721I HCC1569 and HCC1954 cells had significantly higher levels of HER4 expression than the corresponding ERBB-WT cells. However, it is important to note that both HCC1569 and HCC1954 ERBB4-V721I mutant cell lines retain their sensitivity to afatinib. HCC1569-S303F cells demonstrated increased resistance to lapatinib, but not afatinib. Although lapatinib and afatinib are both anti-HER2 TKIs, their mechanism differs in that lapatinib is a reversible inhibitor of HER2 and EGFR, whereas afatinib is an irreversible, pan-HER inhibitor, which has been shown to bind strongly to ERBB4.12 These different drug mechanisms may explain the difference in sensitivity.
Both HCC1569 S303F and HCC1569 V721I have increased sensitivity to the small molecule PI3K inhibitor copanlisib, previously found by us to have single-agent efficacy and, when used in combination with the HER2-targeted therapies trastuzumab, lapatinib and afatinib, to restore sensitivity to these HER2-targeted therapies in cell lines with acquired resistance.17 Therefore we tested combinations of copanlisib and HER2-targeted therapies in our WT and S303F/V721I-expressing cell lines. We found these combinations largely remain synergistic regardless of ERBB4 mutation status. In the case of the S303F hotspot mutation, synergy between afatinib and copanlisib may be enhanced.
We did not find any significant association between ERBB family mutations and clinical/histological features at surgery or diagnosis (Supplementary table 4), or survival after adjuvant trastuzumab therapy (Figure 7). This analysis though is limited by the low frequency of ERBB family mutations in our dataset which required us to group all mutations. This grouping also limits the opportunity to evaluate individual ERBB gene mutations and their impact on clinical response.
Conclusions
We found ERBB family members to be mutated in 7% of our sample set (n = 227). ERBB4 was the most frequently mutated gene of all four ERBB family members in our sample set, carrying five mutations, including the ERBB4 S303F hotspot. ERBB4 V721I and S303F affect the biology and a preclinical study suggests they may affect therapy responsiveness of HER2-positive breast cancer. This work suggests that ERBB family mutations have potential to alter the efficacy of PI3K-inhibition and may be potential biomarkers, if not to trastuzumab, then to emerging anti-HER TKIs such as afatinib.
Supplemental Material
Supplemental material, Supplementary_material for Frequency, impact and a preclinical study of novel ERBB gene family mutations in HER2-positive breast cancer by Naomi Elster, Sinead Toomey, Yue Fan, Mattia Cremona, Clare Morgan, Karolina Weiner Gorzel, Una Bhreathnach, Malgorzata Milewska, Madeline Murphy, Stephen Madden, Jarushka Naidoo, Joanna Fay, Elaine Kay, Aoife Carr, Sean Kennedy, Simon Furney, Janusz Mezynski, Oscar Breathhnach, Patrick Morris, Liam Grogan, Arnold Hill, Susan Kennedy, John Crown, William Gallagher, Bryan Hennessy and Alex Eustace in Therapeutic Advances in Medical Oncology
Acknowledgments
We thank Dr Scott Wilhelm and Bayer Pharmaceuticals for providing us with copanlisib. We thank Padraig Doolan for his helpful comments on the manuscript.
We also acknowledge the following for their contributions to this manuscript: NE performed Chi-square and Fisher’s exact tests on the clinical data, performed the site-directed mutagenesis and performed all in vitro experiments with the stably transfected cell lines excluding the kinase assay, analyzed those results, and drafted this manuscript. ST designed the Agena Panel and performed the Agena analysis. YF performed bioinformatics analysis. MC and CM completed the RPPA experiments (subsequent RPPA analysis by NE and AJE, with advice from MC). KW-M, UB and MM (Murphy) stably transfected the construct cell lines used in this study. MM (Milewska) assisted in the design of the creation of the construct cell lines, and extracted protein from FFPE samples for use in the RPPA experiment. SM and SJF provided statistical expertise and guidance to the clinical study, including the generation of KM curves. JF and EWK performed H&E staining and quality control on all clinical samples used in this study. EWK also contributed to the revision of this manuscript. AC extracted DNA from FFPE clinical samples for use in Agena experiments. SK (RCSI) performed the kinase assays. JN, JM, OSB, PGM, LG, ADH, SK (SVUH) and JC facilitated this study through the provision of clinical samples and data, and assistance in the analysis of the data. PGM also contributed to the revision of this manuscript. WMG provided bioinformatics and NGS supervision and expertise to the project. BTH and AJE conceived of the need for this study, designed and supervised all stages of the project, and were major contributors to the revision and preparation of the final manuscript. All authors read and approved the final manuscript
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Funding: This work was supported by the Irish Cancer Society Collaborative Cancer Research Centre, BREAST-PREDICT (CCRC13GAL) (http://www.breastpredict.com), an Irish Cancer Society Research Scholarship (CRS11ELS), the Health Research Board (HRA/POR2012/054), NECRET, the North-Eastern Cancer Research and Education Trust, and a Breast Cancer Now Catalyst Grant (2016NovPCC002).
ORCID iDs: Elster N  https://orcid.org/0000-0003-3865-5662
https://orcid.org/0000-0003-3865-5662
Eustace AJ
https://orcid.org/0000-0002-4092-1360
Supplementary material: Supplementary material for this article is available online.
Contributor Information
Naomi Elster, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland.
Sinead Toomey, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Yue Fan, Conway Institute, University College Dublin, Ireland.
Mattia Cremona, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Clare Morgan, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Karolina Weiner Gorzel, Conway Institute, University College Dublin, Ireland.
Una Bhreathnach, Conway Institute, University College Dublin, Ireland.
Malgorzata Milewska, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Madeline Murphy, Conway Institute, University College Dublin, Ireland.
Stephen Madden, Data Science Centre, Royal College of Surgeons in Ireland, Dublin, Ireland.
Jarushka Naidoo, Memorial Sloan-Kettering Cancer Centre, New York, USA.
Joanna Fay, Department of Pathology, Royal College of Surgeons in Ireland, Dublin, Ireland.
Elaine Kay, Department of Pathology, Royal College of Surgeons in Ireland, Dublin, Ireland.
Aoife Carr, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Sean Kennedy, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland; St Vincent’s University Hospital, Dublin, Ireland.
Simon Furney, Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland.
Janusz Mezynski, Beaumont Hospital, Dublin, Ireland.
Oscar Breathhnach, Beaumont Hospital, Dublin, Ireland.
Patrick Morris, Beaumont Hospital, Dublin, Ireland; Department of Medicine, Royal College of surgeons in Ireland, Dublin, Ireland.
Liam Grogan, Beaumont Hospital, Dublin, Ireland.
Arnold Hill, Beaumont Hospital, Dublin, Ireland.
John Crown, St Vincent’s University Hospital, Dublin, Ireland.
William Gallagher, Conway Institute, University College Dublin, Ireland.
Alex Eustace, Medical Oncology Group, Department of Molecular Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
References
- 1. Elster N, Collins DM, Toomey S, et al. HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res Treat 2015; 149: 5–15. [DOI] [PubMed] [Google Scholar]
- 2. Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235: 177–182. [DOI] [PubMed] [Google Scholar]
- 3. Gonzalez-Angulo AM, Litton JK, Broglio KR, et al. High risk of recurrence for patients with breast cancer who have human epidermal growth factor receptor 2-positive, node-negative tumors 1 cm or smaller. J Clin Oncol 2009; 27: 5700–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 2005; 353: 1659–1672. [DOI] [PubMed] [Google Scholar]
- 5. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792. [DOI] [PubMed] [Google Scholar]
- 6. Jaiswal BS, Kljavin NM, Stawiski EW, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 2013; 23: 603–617. [DOI] [PubMed] [Google Scholar]
- 7. Tvorogov D, Sundvall M, Kurppa K, et al. Somatic mutations of ErbB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. J Biol Chem 2009; 284: 5582–5591. [DOI] [PubMed] [Google Scholar]
- 8. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 2013; 3: 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boulbes DR, Arold ST, Chauhan GB, et al. HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol Oncol 2015; 9: 586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trowe T, Boukouvala S, Calkins K, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res 2008; 14: 2465–2475. [DOI] [PubMed] [Google Scholar]
- 11. Rexer BN, Ghosh R, Narasanna A, et al. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin Cancer Res 2013; 19: 5390–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sassen A, Diermeier-Daucher S, Sieben M, et al. Presence of HER4 associates with increased sensitivity to Herceptin in patients with metastatic breast cancer. Breast Cancer Res 2009; 11: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohd Nafi SN, Generali D, Kramer-Marek G, et al. Nuclear HER4 mediates acquired resistance to trastuzumab and is associated with poor outcome in HER2 positive breast cancer. Oncotarget 2014; 5: 5934–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toomey S, Eustace AJ, Fay J, et al. Impact of somatic PI3K pathway and ERBB family mutations on pathological complete response (pCR) in HER2-positive breast cancer patients who received neoadjuvant HER2-targeted therapies. Breast Cancer Res 2017; 19: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hennessy BT, Lu Y, Gonzalez-Angulo AM, et al. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin Proteomics 2010; 6: 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Brien NA, Browne BC, Chow L, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther 2010; 9: 1489–1502. [DOI] [PubMed] [Google Scholar]
- 17. Elster N, Cremona M, Morgan C, et al. A preclinical evaluation of the PI3K alpha/delta dominant inhibitor BAY 80–6946 in HER2-positive breast cancer models with acquired resistance to the HER2-targeted therapies trastuzumab and lapatinib. Breast Cancer Res Treat 2015; 149: 373–383. [DOI] [PubMed] [Google Scholar]
- 18. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015; 24: 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q, Sun T, Kang P, et al. Combined analysis of rearrangement of ALK, ROS1, somatic mutation of EGFR, KRAS, BRAF, PIK3CA, and mRNA expression of ERCC1, TYMS, RRM1, TUBB3, EGFR in patients with non-small cell lung cancer and their clinical significance. Cancer Chemother Pharmacol 2016; 77: 583–593. [DOI] [PubMed] [Google Scholar]
- 21. Winkelmann N, Rose-Zerilli M, Forster J, et al. Low frequency mutations independently predict poor treatment-free survival in early stage chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis. Haematologica 2015; 100: e237–e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tougeron D, Lecomte T, Pages JC, et al. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol 2013; 24: 1267–1273. [DOI] [PubMed] [Google Scholar]
- 23. Bouyain S, Longo PA, Li S, et al. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci USA 2005; 102: 15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller ML, Reznik E, Gauthier NP, et al. Pan-cancer analysis of mutation hotspots in protein domains. Cell Syst 2015; 1: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greulich H, Kaplan B, Mertins P, et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci USA 2012; 109: 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007; 11: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujiwara S, Ibusuki M, Yamamoto S, et al. Association of ErbB1–4 expression in invasive breast cancer with clinicopathological characteristics and prognosis. Breast Cancer 2014; 21: 472–481. [DOI] [PubMed] [Google Scholar]
- 28. Das PM, Thor AD, Edgerton SM, et al. Reactivation of epigenetically silenced HER4/ERBB4 results in apoptosis of breast tumor cells. Oncogene 2010; 29: 5214–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sundvall M, Peri L, Maatta JA, et al. Differential nuclear localization and kinase activity of alternative ErbB4 intracellular domains. Oncogene 2007; 26: 6905–6914. [DOI] [PubMed] [Google Scholar]
- 30. Canfield K, Li J, Wilkins OM, et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 2015; 14: 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Machleidt A, Buchholz S, Diermeier-Daucher S, et al. The prognostic value of Her4 receptor isoform expression in triple-negative and Her2 positive breast cancer patients. BMC Cancer 2013; 13: 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Borowicz S, Van Scoyk M, Avasarala S, et al. The soft agar colony formation assay. J Vis Exp 2014: e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blobel CP, Carpenter G, Freeman M. The role of protease activity in ErbB biology. Exp Cell Res 2009; 315: 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carpenter G, Liao HJ. Trafficking of receptor tyrosine kinases to the nucleus. Exp Cell Res 2009; 315: 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang-Kolodji G, Mumenthaler SM, Mehta A, et al. Phosphorylated ribosomal S6 (p-rpS6) as a post-treatment indicator of HER2 signalling targeted drug resistance. Biomarkers 2015; 20: 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin MH, Nam AR, Park JE, et al. Resistance mechanism against trastuzumab in HER2-positive cancer cells and its negation by SRC inhibition. Mol Cancer Ther 2017; 16: 1145–1154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_material for Frequency, impact and a preclinical study of novel ERBB gene family mutations in HER2-positive breast cancer by Naomi Elster, Sinead Toomey, Yue Fan, Mattia Cremona, Clare Morgan, Karolina Weiner Gorzel, Una Bhreathnach, Malgorzata Milewska, Madeline Murphy, Stephen Madden, Jarushka Naidoo, Joanna Fay, Elaine Kay, Aoife Carr, Sean Kennedy, Simon Furney, Janusz Mezynski, Oscar Breathhnach, Patrick Morris, Liam Grogan, Arnold Hill, Susan Kennedy, John Crown, William Gallagher, Bryan Hennessy and Alex Eustace in Therapeutic Advances in Medical Oncology