

Here, Schäfer et al. investigated whether DNA methylation alterations are involved in aging. Using knockout mice for adapter proteins for site-specific demethylation by TET methylcytosine dioxygenases Gadd45a and Ing1, they show that enhancer methylation can affect aging and imply that C/EBP proteins play an unexpected role in this process.
Keywords: aging, adipogenesis, Cebp, chromatin looping, E2F, progeria
Abstract
Changes in DNA methylation are among the best-documented epigenetic alterations accompanying organismal aging. However, whether and how altered DNA methylation is causally involved in aging have remained elusive. GADD45α (growth arrest and DNA damage protein 45A) and ING1 (inhibitor of growth family member 1) are adapter proteins for site-specific demethylation by TET (ten-eleven translocation) methylcytosine dioxygenases. Here we show that Gadd45a/Ing1 double-knockout mice display segmental progeria and phenocopy impaired energy homeostasis and lipodystrophy characteristic of Cebp (CCAAT/enhancer-binding protein) mutants. Correspondingly, GADD45α occupies C/EBPβ/δ-dependent superenhancers and, cooperatively with ING1, promotes local DNA demethylation via long-range chromatin loops to permit C/EBPβ recruitment. The results indicate that enhancer methylation can affect aging and imply that C/EBP proteins play an unexpected role in this process. Our study suggests a causal nexus between DNA demethylation, metabolism, and organismal aging.
Mammalian DNA methylation is a common epigenetic mark implicated in development and disease (Jones 2012). Alterations of DNA methylation patterns accompanying aging are exceptionally well documented (for reviews, see Issa 2014; Klutstein et al. 2016; Pal and Tyler 2016) to the extent that they are predictive of chronological and biological age (Hannum et al. 2013; Horvath 2013). However, despite this extensive literature, it remains elusive whether DNA methylation changes are causative or bystanders of an underlying aging process. Likewise, it is unknown whether regulators of DNA methylation play a role in aging.
DNA methylation is reversible by enzymatic “active” DNA demethylation, with examples in plants, animal development, adult tissue homeostasis, and disease (Pastor et al. 2013; Wu and Zhang 2017). The currently best-understood mechanism of enzymatic DNA demethylation involves oxidation of methyl groups via the TET (ten-eleven translocation) family of methylcytosine dioxygenases (Kriaucionis and Heintz 2009; Tahiliani et al. 2009; Guo et al. 2011; He et al. 2011). TET enzymes convert 5-methylcytosine (5mC) sequentially to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (He et al. 2011; Ito et al. 2011). DNA repair via thymine DNA glycosylase (TDG) removes 5fC and 5caC to restore unmethylated cytosine (Cortazar et al. 2011; Cortellino et al. 2011; Shen et al. 2013).
An effector of TET/TDG-mediated demethylation is the stress response protein GADD45α (growth arrest and DNA damage protein 45A; a member of a small protein family that also includes GADD45β and GADD45γ), which promotes site-specific demethylation (Barreto et al. 2007; Rai et al. 2008; Ma et al. 2009; Schmitz et al. 2009; Le May et al. 2010; Li et al. 2010; Sen et al. 2010; Matrisciano et al. 2011; Zhang et al. 2011; Gavin et al. 2012; Arab et al. 2014; Sabag et al. 2014). GADD45α directly interacts with TET1 and TDG to enhance turnover of oxidized cytosines (Cortellino et al. 2011; Arab et al. 2014; Kienhöfer et al. 2015; Li et al. 2015). Therefore, GADD45α acts as an adapter that recruits DNA-modifying enzymes to specific sites in the genome and promotes local demethylation. GADD45α relies on bridging factors to be directed to specific loci (Schmitz et al. 2009; Schäfer 2013; Arab et al. 2014; Rajput et al. 2016), including the histone reader ING1 (inhibitor of growth family member 1), whose C terminus mediates direct binding to GADD45α (Schäfer et al. 2013). ING1 contains a plant homedomain (PHD) finger motif, which recognizes trimethylated histone H3 at Lys4 (H3K4me3) (Shi et al. 2006; Pena et al. 2008; Schäfer et al. 2013; Cheng et al. 2014), a promoter-specific mark anti-correlated with DNA methylation (Okitsu and Hsieh 2007; Weber et al. 2007). ING1 is involved in chromatin remodeling and transcriptional regulation, regulating cell growth, apoptosis, senescence, and tumorigenesis (Tallen and Riabowol 2014), and cooperates with GADD45α during in vitro differentiation of C3H10T1/2 cells (Wang et al. 2017). Gadd45a and Ing1 are both stress response genes, and mice deficient for either gene are viable although radiation-sensitive and tumor-prone (Hollander et al. 1999; Kichina et al. 2006; Coles et al. 2007).
Here we characterized Gadd45a/Ing1 homozygous double-knockout mice to reveal synthetic phenotypes, since the function of proteins acting in a complex is often phenotypically buffered (Baryshnikova et al. 2013). Strikingly, Gadd45a/Ing1 double-knockout mice exhibit symptoms of premature aging, including reduced life span, kyphosis, weight reduction, ovarian atrophy, female infertility, bone marrow fattening, and skin senescence. Molecular analysis of mutant cells reveals that GADD45α/ING1 target DNA demethylation of C/EBP (CCAAT/enhancer-binding protein)-dependent superenhancers. Correspondingly, Gadd45a/Ing1-null mice phenocopy C/EBP malfunction, with symptoms that recapitulate lipodystrophy and metabolic defects characteristic of progeroid mice and humans. Our study indicates that GADD45α/ING1 mediate site-specific demethylation to regulate a C/EBP-dependent program, controlling metabolism and aging.
Results
Premature aging in Gadd45a/Ing1 double-knockout mice
We noted that, unlike single mutants (Hollander et al. 1999; Kichina et al. 2006), Gadd45a/Ing1 double-knockout mice showed symptoms of premature aging. Mice had a reduced life span with a median of only 2.5 mo (Fig. 1A), showed prominent spinal kyphosis indicative of osteoporosis or degenerative disc disease (Fig. 1B), had a lower proliferation rate of embryonic fibroblasts (MEFs [mouse embryonic fibroblasts]) (Fig. 1C; Lopez-Otin et al. 2013), and had a thinner dermis containing senescence-associated β-galactosidase (SA-β-Gal)-positive cells (Fig. 1D,E; Supplemental Fig. S1A,B; Dimri et al. 1995; McCullough and Kelly 2006). SA-β-Gal was also mildly elevated in double-knockout MEFs, although the senescence marker p16Ink4a was unaffected (Supplemental Fig. S1C,D). Double-knockout mice also had increased bone marrow fat (Fig. 1F), a hallmark of aging (Moore and Dawson 1990). Phenotypic analyses were carried out in young (4 mo of age), superficially healthy-looking mice to avoid secondary effects.
Figure 1.

Premature aging of Gadd45a/Ing1 double-knockout mice. (A) Kaplan-Meier survival curves of mice of the indicated genotypes. n = 47–65 mice per genotype. (DKO) Gadd45a/Ing1 double knockout. P-values were based on log-rank test. (***) P < 0.001. (B) Lateral view of mice of the indicated genotypes. Kyphosis is apparent in eight of 42 analyzed double-knockout mice. (C) Growth curves of MEF cells. Total cell numbers were counted at the indicated time points. Data are presented as mean values of three independent MEF lines per genotype ±SD. (*) P < 0.05. (D) Histological image (hematoxylin and eosin [H&E] stain) of hindlimb skin. (E) Epidermis; (D) dermis. n = 1–4 animals per genotype. (E) Representative histological images of SA-β-Gal staining of dorsal skin. n = 5 animals per genotype. (F) Representative histological images (H&E stain) of bone marrow within bones from the iindicated regions. n = 3–5 animals per genotype. Lipid vacuole accumulation of varying degrees was observed in four of five Ing1−/− and double-knockout mice, respectively.
Additional premature aging-related symptoms in double knockouts were identified and characterized following the molecular analysis described in detail in the sections below but, in brief, included reduced adipogenic differentiation, elevated cytokine and inflammation-associated gene expression, reduced body weight and lipodystrophy, and female infertility—all symptoms characteristic of aging (Kirkland et al. 2002; Lepperdinger 2011; Tilly and Sinclair 2013). Indeed, Gadd45a/Ing1 double-knockout mice bear a striking resemblance to nucleotide excision repair (NER)-defective progeroid mice (van de Ven et al. 2006), and, like them, double-knockout mice also exhibit hypoglycemia and reduced IGF1 (insulin-like growth factor 1) levels (see below). However, while Gadd45a single-mutant MEFs showed a moderate delay of NER-mediated UV repair as reported (Smith et al. 2000), this delay was not significantly exacerbated in double-knockout MEFs (Supplemental Fig. S1E). Similarly, base excision repair in double-knockout MEFs was normal (Supplemental Fig. S1F).
A number of aging hallmarks (Lopez-Otin et al. 2013) were unaffected in double-knockout MEFs and mice, respectively, including apoptosis (active caspase) (Supplemental Fig. S1G), DNA double-strand breaks (γH2.AX foci) (Supplemental Fig. S1H), telomere length (Supplemental Fig. S1I), reactive oxygen species (ROS) production (Supplemental Fig. S1J,K), WNT signaling (Supplemental Fig. S1L), and the levels of circulating senescence-associated factors (Supplemental Fig. S1M).
We conclude that double-knockout mice manifest premature appearance of some, but not all, of the characteristics (“segments”) observed in natural aging, a condition known as segmental progeria (Kubben and Misteli 2017).
Gadd45a/Ing1 double-knockout MEFs show hypermethylation of C/EBP-dependent superenhancers
To unravel whether DNA methylation changes may underlie the premature aging of double-knockout mice, we analyzed the methylome of MEFs because their embryonic origin avoids accumulation of secondary methylation changes and because they are more homogeneous than adult tissues. For unbiased identification of GADD45α DNA demethylation targets, we performed whole-genome bisulfite sequencing (WGBS-seq) of wild-type and double-knockout MEFs to obtain base-pair-resolution methylomes.
Consistent with other methylomes (Tanaka et al. 1997; Stadler et al. 2011), CpGs in both wild-type and double-knockout MEFs showed an approximately bimodal methylation pattern, with 81.3% highly methylated and 7.3% lowly methylated sites, while 11.4% of CpGs showed intermediate methylation levels (Supplemental Fig. S2A). The overwhelming majority of CpGs had similar methylation levels in wild-type and double-knockout MEFs (R2 = 0.87) (Supplemental Fig. S2B), indicating no global methylation changes. To call differentially methylated CpG regions (DMRs) between wild-type and double-knockout MEFs with high confidence, we used a stringent cutoff of at least 25% methylation difference on both strands of three consecutive CpGs. WGBS-seq does not discriminate 5hmC from 5mC (Booth et al. 2012), but, for simplicity, we refer to these regions as DMRs here, as 5hmC is 10-fold to 100-fold less abundant than 5mC (Globisch et al. 2010). DMRs were broadly distributed on all 19 autosomes except for an enrichment near the Gadd45a and Ing1 knockout loci, situated on chromosomes 6 and 8, respectively (data not shown). These DMRs are likely due to linkage disequilibrium at the knockout loci and therefore were removed from further analysis. The DMR analysis identified 885 hypermethylated and 46 hypomethylated regions in double-knockout MEFs (Supplemental Table S1). The 20-fold bias toward hypermethylation is consistent with impaired site-specific DNA demethylation occurring in double-knockout MEFs.
Twenty-four out of 25 randomly chosen hypermethylated DMRs were confirmed by methylation-specific PCR in double-knockout MEFs. Furthermore, in either Gadd45a or Ing1 single-mutant MEFs, the methylation levels of most of these DMRs were largely unaffected (Supplemental Fig. S2C). The results indicate that GADD45α and ING1 synergize to maintain the hypomethylated state of these DMRs.
Subsequently, we focused on hypermethylated DMRs: Their median size was 78 base pairs (bp) (Supplemental Fig. S2D), the majority overlapped with intronic and intergenic regions (Supplemental Fig. S2E), and they were greatly enriched for lowly methylated regions (LMRs) in wild-type MEFs (Fig. 2A). LMRs are known hot spots for active DNA demethylation, marked by high density of 5hmC (Yu et al. 2012). Indeed, overlay with 5hmC, 5fC, and 5caC data from mouse embryonic stem cells (Shen et al. 2013) revealed that the double-knockout DMRs are enriched for 5hmC and gain 5fC and 5caC upon Tdg knockdown (Fig. 2B), corroborating that these are sites undergoing active TET/TDG-mediated demethylation.
Figure 2.

Gadd45a/Ing1 double-knockout MEFs show hypermethylation of C/EBP-dependent adipogenic superenhancers. (A) Overlap of hypermethylated DMRs in Gadd45a/Ing1 double-knockout MEFs with regions in wild-type MEFs that are lowly methylated, fully methylated (FMR), or unmethylated (UMRs). Definitions of FMRs, LMRs, and UMRs are according to Stadler et al. (2011). DMRs in partially methylated regions are not displayed. (B) Heat maps of depicted oxidative DNA modifications centered on the double-knockout hypermethylated DMRs (black triangles). DNA modifications are from DIP-seq (DNA immunoprecipitation combined with sequencing) in embryonic stem cells that were treated with unspecific (shCo) or Tdg-specific shRNA (Shen et al. 2013). Data were sorted based on cumulative normalized signal intensity of individual DNA modifications around DMRs. The red–yellow–blue key indicates high to low DIP-seq signal. (C) Enrichment of hypermethylated DMRs (identified in double-knockout MEFs) of the depicted genomic elements. Enhancers are defined by combined H4K3 monomethylation (H4K3me1) and H3K27 aetylation (H3K27ac) occupancy in MEFs. Promoters are defined as ±2 kb surrounding the transcription start sites (TSSs). The red line indicates no enrichment. (D,E) Localization of hypermethylated and hypomethylated DMRs around enhancer marks H3K4me1 (D) and H3K27ac (E) in MEFs. (F) Gene ontology (GO) enrichments of genes in proximity to enhancer-associated hypermethylated DMRs with corresponding P-values. The full list of GO enrichments is shown in Supplemental Table S1. (G) Transcription factor (TF)-binding motif analysis of hypermethylated DMRs and associated P-values relative to genomic background. (H) Heat maps of the depicted histone marks and TFs centered on the double-knockout hypermethylated DMRs (black triangles). Histone marks are from ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) in MEFs (Yue et al. 2014). TF ChIP-seq data are from 3T3-L1 cells differentiated for 4 h along the adipogenic lineage (Siersbaek et al. 2011, 2014). Data were sorted based on the cumulative normalized signal intensity of analyzed TFs around DMRs. The red–yellow–blue key indicates high to low ChIP-seq signal. (I) ChIP-qPCR (ChIP combined with quantitative PCR [qPCR]) of endogenous C/EBPβ in wild-type, Gadd45a, or Ing1 single-knockout or double-knockout MEFs. qPCR was conducted on nine hypermethylated DMRs, an unrelated C/EBPβ target region (Siersbaek et al. 2014), and two independent negative control regions (Ctrl1–2). Data are presented as mean values of three independent MEF lines per genotype ±SD. Note that these DMRs were also bound by GADD45α in ChIP-seq analysis of Figure 3. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
LMRs are also strongly associated with and predictive for enhancers marked by H3K4 monomethylation (H3K4me1) and occupied by p300 histone acetyltransferase (Stadler et al. 2011). In agreement, comparing DMRs with ENCODE data (Yue et al. 2014) revealed that hypermethylated DMRs were 13-fold enriched at enhancers, while hypomethylated DMRs showed no enrichment (Fig. 2C–E; Supplemental Fig. S2F).
Focusing on the genes in proximity to enhancer-associated hypermethylated DMRs indicated enrichment for functions related to fat cell differentiation, the cell cycle, and cell matrix interaction (Fig. 2F). We searched for transcription factor (TF)-binding motifs that were overrepresented in hypermethylated DMRs using the two independent DNA motif analysis algorithms HOMER (de novo and known motif analysis) (Heinz et al. 2010) and RSAT (data not shown) (Thomas-Chollier et al. 2012). Interestingly, both tools revealed the highest enrichment for C/EBPβ-binding sites, along with other bZip (basic leucine zipper) TFs such as the ATF and AP1 families, which share similar recognition motifs (Fig. 2G). Data mining supported that the above TFs are physiologically bound at these DMRs: Using published ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) profiles from mouse 3T3-L1 fibroblasts undergoing adipogenic differentiation (Siersbaek et al. 2011, 2014), we found that a subset of the enhancer-associated DMRs (22%; n = 156) colocalizes with regions identified previously as adipogenic hot spots (Fig. 2H), which are central constituents in superenhancers (Siersbaek et al. 2014).
Superenhancers are characteristically hypomethylated (Hnisz et al. 2013; Heyn et al. 2016) genomic regions of several kilobases that are enriched for binding of the coactivator Mediator 1 (MED1) and occupied by multiple cooperating TFs (Hnisz et al. 2013; Loven et al. 2013; Whyte et al. 2013). Adipogenic hot spots driving adipogenic reprogramming of gene expression are located mostly within superenhancer regions. They control early induced adipogenic differentiation genes, which are enriched for the gene ontology (GO) terms extracellular matrix–receptor interactions, cell proliferation, and growth factor signaling (Siersbaek et al. 2014). Related GO terms are also enriched for genes associated with hypermethylated enhancer DMRs (Fig. 2F). Besides by MED1, adipogenic superenhancers are occupied by the TFs C/EBPβ, C/EBPδ, cJun, JunB, FOSL2, ATF2, ATF7, KLF4, KLF5, RXRα, VDR, PBX1, and STAT1 (Fig. 2H). A number of these TFs are involved in differentiation along the adipogenic lineage (Sarjeant and Stephens 2012; Siersbaek et al. 2012); notably, C/EBPβ, which acts as a pioneer factor (Tanaka et al. 1997; Tang et al. 2003; Steger et al. 2010; Siersbaek et al. 2011) The C/EBP family of TFs plays important regulatory roles; e.g., in glucose metabolism, cell cycle, hematopoiesis, skeletal development, immune response, and adipocyte differentiation (Tsukada et al. 2011). These bZip proteins bind to common core motifs and display partially overlapping and redundant functions, notably in energy homeostasis and adipocyte differentiation (Tsukada et al. 2011).
DNA methylation can inhibit C/EBPβ binding to cognate sites in vivo (Venza et al. 2012; Wang et al. 2014). Consistently, ChIP-qPCR (ChIP combined with quantitative PCR [qPCR]) of C/EBPβ showed significantly reduced binding to superenhancer DMRs in double-knockout MEFs (Fig. 2I). Once again, in single-mutant MEFs, C/EBPβ binding to DMRs was mostly unaffected, as was binding to an unrelated non-DMR C/EBPβ target site. This indicates a requirement for GADD45α/ING1-dependent DNA demethylation in maintaining adipogenic enhancers accessible to C/EBPβ.
We conclude that in MEFs, (1) GADD45α/ING1 cooperate to demethylate sites near C/EBPβ/δ-binding motifs, which are enriched in adipogenic hot spots and superenhancers, and (2) demethylation is required for efficient C/EBPβ recruitment.
GADD45α binds to C/EBP-dependent enhancers
To analyze whether hypermethylated DMRs in MEFs are direct GADD45α targets, we performed GADD45α ChIP-seq analysis (Supplemental Table S2). The majority of GADD45α occupied sites was hypermethylated in double-knockout MEFs (Fig. 3A), corroborating a direct role for GADD45α in promoting DNA demethylation of these loci.
Figure 3.

GADD45α binds to C/EBP-dependent adipogenic superenhancers. (A) Heat map of mean differential methylation in 1-kb bins around GADD45α ChIP-seq peaks (P; n = 405) identified in HA-Gadd45a transfected MEFs. Data are represented as the DNA methylation difference between Gadd45a/Ing1 double-knockout and wild-type MEFs as determined by WGBS-seq. (B) Enrichment of GADD45α-binding sites in the depicted genomic elements. Enhancers are defined by combined H4K3me1 and H3K27ac occupancy. Promoters are defined as ±2 kb surrounding TSSs. The red line indicates no enrichment. (C) GO enrichment of genes neighboring GADD45α-binding sites, with corresponding P-values. The full list of GO enrichments is shown in Supplemental Table S2. (D) TF-binding motif analysis of GADD45α-binding sites and associated P-values relative to genomic background. (E) ChIP-seq profiles of GADD45α and adipogenic TFs at 4 h of 3T3-L1 adipocyte differentiation (Siersbaek et al. 2011, 2014) and MEF histone marks at an exemplary adipogenic superenhancer. (F) Heat maps of the depicted histone marks and TFs centered on GADD45α-binding sites (black triangles). ChIP-seq data of histone marks are from MEFs (Yue et al. 2014) and for TF binding from 3T3-L1 cells differentiated for 4 h along the adipogenic lineage (Siersbaek et al. 2011, 2014). Shown are three classes of hypermethylated DMRs: cluster I (H3K27ac, H3K4me3, and C/EBP), cluster II (H3K27ac, H3K4me1, and all superenhancer TFs), and cluster III (the same as cluster II except missing enrichment of FOSL2, ATF2, ATF7, and STAT1). The red–yellow–blue key indicates high to low ChIP-seq signal.
Similar to hypermethylated DMRs identified in double-knockout MEFs, GADD45α-binding sites showed a 34-fold enrichment for enhancers and a sevenfold enrichment for promoters (Fig. 3B). Genes neighboring GADD45α peaks showed enrichment for the GO term “fat cell differentiation,” among others (Fig. 3C). Strikingly, as for DMRs, motif analysis of GADD45α peaks revealed by far the highest enrichment for C/EBPβ TF-binding sites (Fig. 3D). Moreover, most enhancer-associated GADD45α peaks lay within adipogenic hot spots/superenhancers (Fig. 3E,F; Siersbaek et al. 2011, 2014).
To corroborate that GADD45α/ING1 act upstream of C/EBPβ recruitment (Fig. 2I), we carried out GADD45α ChIP analysis upon single or double siRNA knockdown of Cebpb or Cebpd. Neither treatment affected GADD45α binding to DMRs (Supplemental Fig. S2G). Collectively, the results show that GADD45α occupies C/EBPβ-dependent adipogenic hot spots and superenhancers and mediates their local demethylation to promote C/EBPβ recruitment.
ING1 binds to promoters occupied by E2F
The results indicate that ING1 and GADD45α mediate DNA demethylation synergistically. To analyze where ING1 binds genome-wide in relation to GADD45α, we performed ChIP-seq analysis of ING1 in MEFs (Supplemental Table S3). Unlike GADD45α, essentially all ING1-binding sites were associated with promoters that were enriched for H3K27 acetylation (H3K27ac) and H3K4me3, depleted for H3K4me1, and in proximity to transcription start sites (TSSs) (Fig. 4A,B). Correlation with the MEF methylome showed no methylation difference between wild-type and double-knockout MEFs around ING1 peaks (Supplemental Fig. S3A) because these sites were located almost exclusively in constitutively unmethylated promoters (data not shown). ING1 sites rarely overlapped with GADD45α peaks (Supplemental Fig. S3B,C). A restriction of ING1 binding to promoters was observed previously (Cheng et al. 2014) and is in line with the protein being a H3K4me3 reader. ING1 bound to only a fraction of all H3K4me3 sites in the genome. ING1 likely has additional binding determinants, as concluded previously (Cheng et al. 2014), which possibly are related to its partial bromodomain or PIP (PCNA-interacting protein) signal. Motif analysis of ING1 peaks showed a very strong enrichment for binding motifs of the E2F TF family (Fig. 4C). Consistently, ING1-binding sites almost completely overlapped with an E2F4 ChIP-seq data set from 3T3-L1 cells (Fig. 4D; MacIsaac et al. 2010). E2F proteins are promoter-binding TFs, which are prominent regulators of cell cycle, apoptosis, and DNA repair (Johnson and Degregori 2006; Putzer and Engelmann 2013) and also adipogenesis (Fajas et al. 2002; Landsberg et al. 2003; Fajas 2013). Accordingly, GO analysis of ING1-bound genes showed enrichment for response to insulin and dexamethasone (Fig. 4E)—two inducers of adipogenesis (Garten et al. 2012)—besides cell cycle terms. We conclude that in MEFs, ING1 binds to promoters occupied by E2F and mostly does not overlap with GADD45α-binding sites.
Figure 4.

ING1 binds to promoters that can loop to GADD45a-bound hypomethylated superenhancers. (A) Heat maps of H3K27ac, H3K4me1, and H3K4me3 histone marks of MEFs (Yue et al. 2014) centered on ING1-binding sites (black triangles). n = 703. Data are from ChIP of HA-Ing1 in transfected MEFs and are sorted based on the cumulative signal intensity of histone marks around ING1-binding sites. The red–yellow–blue key indicates high to low ChIP-seq signal. (B) Distance of ING1-binding sites to adjacent TSSs in percent. (C) TF-binding motif analysis of ING1-binding sites and associated P-values relative to genomic background. (Diamond) Predicted binding of dimers/multimers of the indicated proteins. (D) Overlap between E2F4-binding sites in 3T3-L1 cells (MacIsaac et al. 2010) and ING1-binding sites. (E) GO enrichment of genes neighboring ING1-binding sites, with corresponding P-values. The full list of GO enrichments is shown in Supplemental Table S3. (F) Heat map of ≥1.5-fold down-regulated (green) and up-regulated (red) genes in Gadd45a−/−, Ing1−/−, and double-knockout MEFs at a false discovery rate (FDR) of 10%. The full list of deregulated genes is displayed in Supplemental Table S4. (G) Immunoblot analysis of coimmunoprecipitation (co-IP) experiments using protein lysates of HEK293T cells transiently transfected with GFP, myc-GADD45α, or myc-ING1b with or without Flag-HA-TET1. αTubulin and GFP served as specificity controls. Input shows 2% of lysate was used for immunoprecipitation. (H, top) Model depicting chromatin looping between distant GADD45α - and ING1- bound genomic regions. (Middle) GADD45α and ING1 ChIP-seq peaks, hypermethylated DMRs (identified in Gadd45a/Ing1 double-knockout MEFs), and adipogenic hot spots (based on Siersbaek et al. 2014). (Bottom) NG Capture-C-seq (next-generation Capture-C combined with sequencing) interaction profile of a representative wild-type and double-knockout MEF line in a 450-kb genomic region on chromosome 5. The interaction between the GADD45α-bound viewpoint (orange) and the Tgfbr3 promoter bound by ING1 is highlighted (gray shading). For NG Capture-C interaction profiles of all wild-type, Gadd45a−/−, Ing1−/−, and double-knockout MEFs in triplicates for this region, see Supplemental Figure S3F.
In RNA sequencing (RNA-seq) analysis of wild-type and mutant MEFs, more genes were misregulated in Ing1−/− than Gadd45 single-mutant MEFs, suggesting that ING1 is the driver of transcriptional regulation, with GADD45α amplifying the response (Fig. 4F; Supplemental Table S4). There was an overlap of 20% of genes deregulated in Tet1,2 double-knockout MEFs (Wiehle et al. 2015) with genes down-regulated in Ing1−/− double-knockout MEFs (Supplemental Fig. S3D), supporting a common mechanism of regulation. Indeed, not only GADD45α (Kienhöfer et al. 2015) but also ING1 bound to TET1 in coimmunoprecipitation (co-IP) experiments (Fig. 4G). Finally, we intersected DMR-associated genes with genes misregulated in double-knockout MEFs. Gene enrichment analysis showed that the down-regulated but not up-regulated genes were highly enriched for C/EBPδ-binding sites (adjusted P-value 4.1 × 10−11), the top hit of the list (Supplemental Fig. S3E). These results support that hypermethylation of C/EBP-regulated genes in double-knockout MEFs impairs their expression.
Chromatin looping between GADD45α-bound enhancers and ING1-bound promoters
An intriguing finding is that ING1 was bound at promoters yet promoted GADD45α-mediated demethylation at enhancers. This long-distance effect of ING1 mirrors an unsolved conundrum in the demethylation field: TET1 is predominantly bound at promoters, while most 5hmC and 5fC are located at distal elements (for review, see Wu and Zhang 2017). Thus, we hypothesized that ING1 occupies promoters that are engaged in long-range chromatin looping with GADD45α-bound enhancers (Fig. 4H, top).
To explore the possibility of enhancer–promoter looping, we performed NG-Capture-C (next-generation Capture-C) (Davies et al. 2016), a 3C (chromosome conformation capture)-derived method to investigate chromatin conformation between GADD45α and ING1 peaks. As viewpoints, we selected GADD45α-occupied sites in adipogenic superenhancers (Supplemental Table S5). NG Capture-C was carried out with three independent wild-type, single-mutant, and double-mutant MEF lines. An example is a GADD45α/DMR viewpoint in a C/EBPβ/δ superenhancer showing a prominent cis interaction with the ∼300-kb downstream-located Tgfbr3 promoter, which is occupied by ING1 (Fig. 4H, bottom). This cis interaction also occurred in double-knockout MEFs; i.e., independent of the methylation status of the GADD45α-bound site. The peak profiles were highly reproducible among replicates (Supplemental Fig. S3F). For 10 out of 17 tested GADD45α viewpoints in superenhancers, we found long-distance interactions with regions occupied by ING1 located, on average, 107 kb apart and generally independent of the MEF genotype (Supplemental Table S5). We conclude that adipogenic superenhancers occupied by GADD45α can interact over long range with promoters bound by ING1.
GADD45α and ING1 are required for adipocyte differentiation
A TF cascade of C/EBPα, C/EBPβ, and C/EBPδ plays a major role in differentiation of fibroblasts into adipocytes (Fig. 5A; Sarjeant and Stephens 2012; Siersbaek et al. 2012). This raised the possibility that GADD45α/ING1 regulate an adipogenic differentiation program driven by C/EBPβ. We therefore induced adipogenic differentiation in several independent wild-type and Gadd45a/Ing1 mutant MEF lines, scoring adipocytes with Oil Red O staining, and found that differentiation of double-knockout MEFs was greatly impaired (Fig. 5B; Supplemental Fig. S4A). While Gadd45a single-mutant MEFs differentiated normally to adipocytes, a subset of independent Ing1−/− MEF lines already showed some reduced differentiation. RNA-seq analysis at day 6 of adipocyte differentiation confirmed that genes down-regulated in double-knockout cells (n = 411) were strongly enriched for processes such as lipid homeostasis, fatty acid biosynthesis, and fat cell differentiation (Fig. 5C; Supplemental Fig. S4B; Supplemental Table S6). More genes were misregulated in Ing1−/− than in Gadd45 single-mutant MEFs, suggesting that ING1 is the driver of differentiation, with GADD45α amplifying the response (Supplemental Fig. S4B,C). Genes up-regulated in double knockout (n = 183) clustered for cytokine response and inflammation (Fig. 5D; Supplemental Fig. S4C), mirroring the proinflammatory gene signature observed in Cebpg−/− MEFs (Huggins et al. 2013).
Figure 5.

GADD45α and ING1 are required for adipocyte differentiation. (A) Scheme of key TFs induced during adipocyte differentiation and their causal connections. (B) Adipocyte differentiation assays using MEFs. Wild-type, Gadd45a−/−, Ing1−/−, and Gadd45a/Ing1 double-knockout MEFs (n = 4 individual MEF lines per genotype) were differentiated to adipocytes. At the indicated time points of differentiation, cells were stained with Oil Red O (red) to visualize lipid droplets and with hematoxylin (blue) as a counterstain. Representative images are shown. (C,D) GO enrichment of transcriptome changes during adipocyte differentiation. RNA-seq was conducted in wild-type, Gadd45a−/−, Ing1−/−, and double-knockout MEFs differentiated for 6 d along the adipogenic lineage. GO annotations of genes more than twofold down-regulated (C) or more than twofold up-regulated (D) at 10% FDR in double-knockout compared with wild-type cells are shown. The full list of GO enrichments is shown in Supplemental Table S6. (E) qPCR analysis of Gadd45a and Ing1 expression during the time course of MEF-to-adipocyte differentiation. Data are presented as mean values of three independent wild-type MEF lines ±SD. (F) qPCR expression analysis of the key TFs Cebpb, Cebpd, Cebpa, and Pparg during the time course of MEF-to-adipocyte differentiation. Data are presented as mean values of three independent MEF lines per genotype ±SD. (G) Immunoblot analysis of PPARγ and C/EBPβ at day 6 of MEF-to-adipocyte differentiation. Representative blot of n = 3 independent cell lines per genotype. (H) RT-qPCR expression analysis of the indicated adipocyte marker genes at day 6 of MEF-to-adipocyte differentiation in the indicated genotypes and vitamin C treatment (VitC). Data represent mean values of three independent MEF lines per genotype ±SD.
Upon adipocyte differentiation, the adipogenic TFs C/EBPβ and C/EBPδ induce the direct targets Pparg and Cebpa, which mediate terminal adipocyte differentiation (Fig. 5A; Sarjeant and Stephens 2012; Siersbaek et al. 2012). Both Gadd45a and Ing1 are transiently induced during early adipocyte differentiation, coinciding with early adipogenic TFs (Cebpb and Cebpd) (Fig. 5E,F). The following results indicate that a block of the differentiation cascade in double-knockout MEFs occurs at the level of C/EBPβ and C/EBPδ target gene induction. While expression of the early adipogenic TFs Cebpb and Cebpd was unaffected, expression of Pparg and Cebpa and their downstream targets (e.g., Leptin, Adiponectin, and Fabp4) was reduced throughout adipocyte differentiation in double-knockout MEFs as well as in a subset of Ing1−/− MEFs (Fig. 5F; Supplemental Fig. S4D–F). Protein levels of PPARγ were also reduced in differentiating double-knockout cells, confirming an arrest of the adipogenic cascade at this level (Fig. 5G). On the other hand, C/EBPβ, which acts as an adipogenic pioneer factor (Sarjeant and Stephens 2012; Siersbaek et al. 2012), was unaffected in double-knockout MEFs: Cebpb expression (Fig. 5F), protein levels (Fig. 5G), and the characteristic nuclear translocation of C/EBPβ (Supplemental Fig. S4G), which accompanies differentiation (Tang and Lane 1999), occurred normally. Moreover, a transfected C/EBP-luciferase reporter was equally inducible by adipogenic stimuli in wild-type and double-knockout MEFs (Supplemental Fig. S4H). Taken together, these results indicate that C/EBPβ activation occurred normally in double-knockout MEFs, but its ability to bind to hypermethylated adipogenic enhancers was compromised (Fig. 2I).
Reduced expression of adipogenic master regulators in double-knockout cells was rescued by ectopic overexpression of Ing1 but not Gadd45a (Supplemental Fig. S4I), corroborating that ING1 is the main driver of differentiation. In line with the requirement for GADD45α/ING1, adipogenic differentiation is also impaired by TET deficiency, while it is stimulated by vitamin C (Wiehle et al. 2015; Yoo et al. 2017), which activates Fe(II)-dependent and α-ketoglutarate (KG)-dependent dioxygenases, including TETs, by maintaining the ferrous state of iron (Blaschke et al. 2013). Concordantly, vitamin C treatment fully rescued the adipogenic differentiation defects in double-knockout MEFs (Fig. 5H). Since double-knockout MEFs showed normal levels of ROS, superoxide, and oxidative DNA damage intermediates (Supplemental Fig. S1F,J,K,), the differentiation rescue is unlikely to be due to the antioxidative effect of vitamin C.
Taken together, these results demonstrate that (1) Gadd45a and Ing1 are transiently induced by adipogenic stimuli and cooperate in promoting adipocyte differentiation at the level of C/EBPβ, and (2) ING1 acts as a driver for differentiation, and GADD45α amplifies the gene expression program controlled by ING1.
Gadd45a/Ing1 double-knockout mice phenocopy Cebp mutant lipodystrophy
Since a characteristic of Cebp mutant mice is lipodystrophy (Wang et al. 1995; Tanaka et al. 1997), we analyzed adipose tissue in Gadd45a/Ing1 double-knockout mice. There are two types of adipose tissue: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT stores energy as triglycerides and releases free fatty acids in response to energy requirements at other sites, while BAT is involved in thermogenesis (Sarjeant and Stephens 2012). In single-mutant as well as double-knockout mice, WAT and BAT depots were smaller, while weights of other organs were unaffected (Fig. 6A; Supplemental Fig. S5A). Histologically, both WAT and BAT of double-knockout mice contained significantly smaller cells and lipid droplets (Fig. 6B; Supplemental Fig. S5B). Mice lacking Cebpa as well as Cebpb/d also exhibit greatly reduced WAT and BAT depots with smaller lipid droplets, indicative of impaired adipocyte maturation (Wang et al. 1995; Tanaka et al. 1997). Moreover, Cebpb-deficient mice display fat browning (Rahman et al. 2012), and this phenotype was phenocopied in double-knockout mice: Islands within double-knockout WAT displayed a BAT-like morphology (Fig. 6B), and expression microarray analysis of double-knockout WAT indicated that up-regulated genes clustered strongly for functions associated with mitochondria, fatty acid catabolism, and citrate cycle (Fig. 6C,E; Supplemental Fig. S5C; Supplemental Table S7), reflecting the high mitochondrial content and catabolic activity of BAT. These BAT markers were also highly induced in double-knockout BAT (Supplemental Fig. S5D), suggesting a propensity of double-knockout mice for energy dissipation rather than energy storage. Down-regulated genes in double-knockout WAT were enriched for GO terms related to inflammatory response (Fig. 6D). As in MEFs, ING1 was the driver of the phenotype, since many of the ∼800 genes deregulated in double-knockout mutants (Supplemental Fig. S5E) showed a weak subthreshold tendency for deregulation already in Ing1−/−, but not Gadd45a−/−, WAT (Supplemental Fig. S5F), supporting that GADD45α amplifies a gene expression program controlled by ING1. Moreover, in ChIP-qPCR, C/EBPβ binding to a number of the DMRs identified in MEFs was significantly reduced in WAT from double knockouts, while binding in single mutants was mostly unaffected (Fig. 6F).
Figure 6.

Lipodystrophy in Gadd45a/Ing1 double-knockout mice. (A) Weights of control organs (left) and adipose tissue depots (right) of 8-wk-old male mice. (gWAT) Gonadal WAT; (aWAT) axillary WAT; (iWAT) inguinal WAT; (BAT) interscapular BAT; (rWAT) retroperitoneal WAT. n = 5–8 animals per genotype. (DKO) Gadd45a/Ing1 double knockout. (B) Histological analysis of adipocyte abnormalities. (Left) H&E staining of gonadal WAT (gWAT) sections. The panel displaying double-knockout gWAT morphology is divided to display two areas with distinct morphological appearances: classical WAT-like morphology (top left) and beige/brite adipocyte islands (bottom right). (Right) Individual adipocyte cell size in gWAT was quantified by measurement of >200 adipocytes on each section. n = 4–5 mice per genotype. (C,D) GO enrichment and corresponding P-values of genes up-regulated (C) or down-regulated (D) ≥1.5-fold in double-knockout gWAT compared with wild-type gWAT. The full list of GO enrichments is shown in Supplemental Table S7. (E) qPCR confirmation of gWAT gene expression changes in independent animals of the indicated genotypes. n = 5–9 per genotype. (F) ChIP-qPCR of endogenous C/EBPβ in gWAT of wild-type or Gadd45a or Ing1 single-knockout or double-knockout mice. qPCR was conducted on eight DMRs hypermethylated in MEFs (Fig. 2I), an unrelated C/EBPβ target region (Siersbaek et al. 2014), and two independent negative control regions (Ctrl1–2). Data are presented as mean values of paralleled ChIPs using gWAT of independent animals ±SD. n = 5 for wild-type and double-knockout; n = 4 for Gadd45a or Ing1 single knockouts. Data of A, B, E, and F are presented as mean values of the indicated number of samples ±SD. (*) P < 0.05; (**) P < 0.01.
We conclude that GADD45α and ING1 cooperate to promote normal WAT and BAT formation, presumably by ensuring C/EBPβ recruitment, and, furthermore, their combined deficiency phenocopies lipodystrophy of Cebp mutants.
Gadd45a/Ing1 double-knockout mice phenocopy increased catabolism and female infertility of Cebp mutants
Collectively, the combination of the aforementioned data from molecular analysis, adipocyte differentiation results, reduced WAT and BAT depots, and WAT browning in double-knockout mice recommends the conclusion that GADD45α/ING1 promote C/EBP-dependent transcriptional responses. Further advancing this notion is the fact that double-knockout mice shared a number of abnormalities in energy expenditure that are also characteristic for Cebp mutants. Cebpa−/− mice die 7–8 h post-partum because of extreme hypoglycemia, defects in glycogen storage, and failure to activate gluconeogenic pathways (Wang et al. 1995). Cebpb−/− mice and conditional Cebpa mutants show reduced expression of phosphoenolpyruvate carboxykinase (Pepck), the rate-limiting enzyme for gluconeogenesis (Wang et al. 1995; Arizmendi et al. 1999). Additionally, Cebpb−/− mice display hypoglycemia and impaired hepatic glucose production after fasting, reduced expression of Igf1, and overall reduced body weight (Liu et al. 1999; Staiger et al. 2009).
Consistently, Gadd45a/Ing1 double-knockout mice had lower body weight (Fig. 7A) and reduced fasting glucose plasma levels (Fig. 7B); in randomly fed mice, glucose levels were unchanged (data not shown). Moreover, double-knockout livers showed reduced expression of Igf1 and Pepck (Fig. 7C), as in Cebpb−/− mice.
Figure 7.
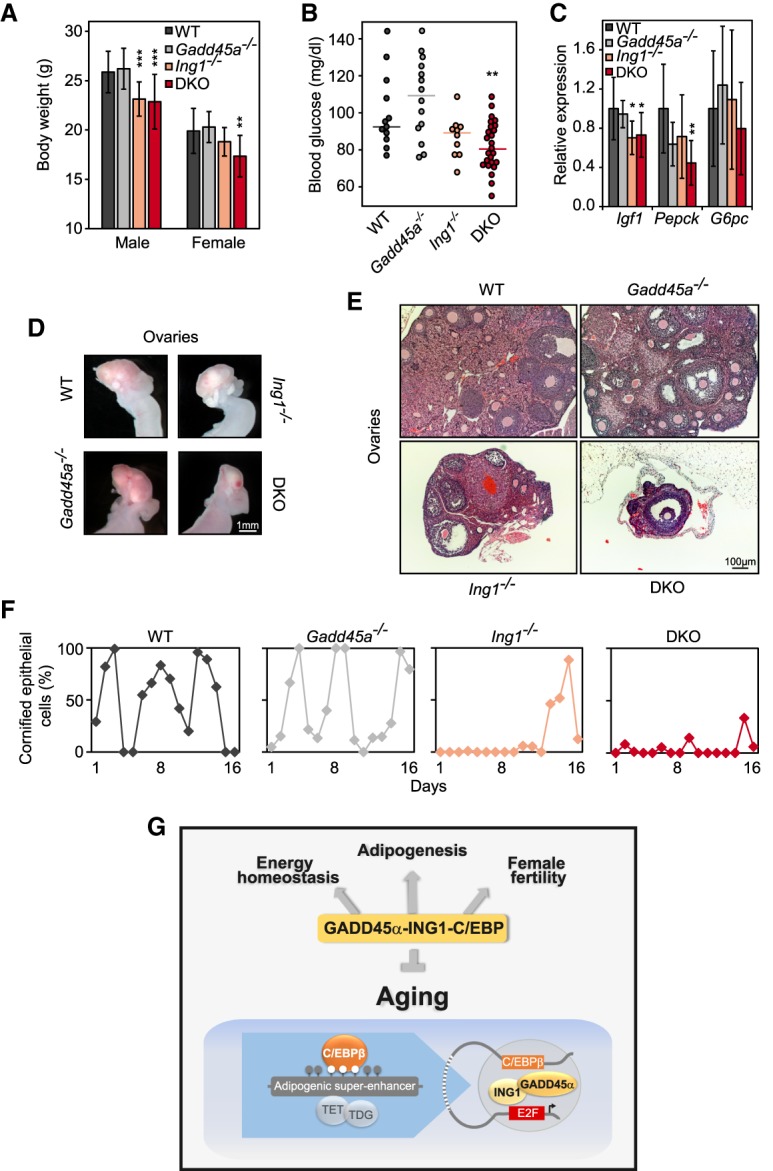
Gadd45a/Ing1 double-knockout mice phenocopy metabolic and ovarian defects of C/EBP mutants. (A) Body weight of male and female 8-wk-old mice from the indicated genotypes. Data are presented as mean values of n = 10–33 animals per sex and genotype ±SD. (**) P < 0.01; (***) P < 0.001. (DKO) Gadd45a/Ing1 double knockout. (B) Blood glucose levels of overnight fasted mice of the indicated genotypes. Each point represents an individual animal, and lines represent median values. n = 8–25 mice per genotype. (**) P < 0.01. (C) qPCR expression analysis of metabolic genes down-regulated in Cebp mutant livers in mice of the indicated genotypes. Data are presented as mean values of n = 8–13 samples per genotype ±SD. (*) P < 0.05; (**) P < 0.01. (D) Representative macroscopic view of mouse ovaries and uteri. n = 11–19 per genotype. Smaller ovaries were observed in 13 of 19 analyzed double-knockout mice. (E) Histological image (H&E stain) of mouse ovaries. n = 5 per genotype. The represented double-knockout phenotype was observed in four of five analyzed samples. (F) Percentage of cornified epithelial cells in vaginal smears, indicating estrus. n = 3–4 animals per genotype. Data from representative animals are shown. (G) A GADD45α–ING1–C/EBP axis regulates energy homeostasis, female fertility, adipogenesis, and organismal aging. GADD45α is an adapter protein for TET and TDG demethylation enzymes, while ING1 is a GADD45α-binding cofactor. In MEFs, GADD45α occupies C/EBP-dependent adipogenic superenhancers and promotes their demethylation via long-range interaction with ING1 bound to promoters to engage demethylating enzymes. Enhancer demethylation permits C/EBP binding and transactivation of target genes.
Female infertility is another hallmark of Cebpb−/− mice (Sterneck et al. 1997), and, indeed, double-knockout females were completely infertile and presented severe ovarian atrophy in 68% of double-knockout mice and milder ovarian atrophy in 11% of Ing1−/− females (Fig. 7D,E). Double-knockout ovaries contained fewer follicles, had more apoptotic cells, and lacked signs of successful ovulation, such as corpora lutea or cornified epithelial cells in vaginal smears (Fig. 7E,F; Supplemental Fig. S6A–C).
Thus, Gadd45a/Ing1 double-knockout mice phenocopy major symptoms of Cebpb and Cebpa mutants: adipogenesis defects, lipodystrophy, increased catabolism, and female infertility. Strikingly, most of these Cebp mutant symptoms are also characteristic for progeroid mice (highlighted in the Venn diagram in Supplemental Fig. S6D; referenced and summarized in Supplemental Table S8). We therefore conclude that the premature aging of double knockouts noticed at the outset of this study (Fig. 1) is, in fact, a manifestation of impaired C/EBP function. This indicates that a GADD45α–ING1–C/EBP axis regulates both energy homeostasis and organismal aging.
Discussion
Organismal aging is associated with a plethora of epigenetic changes, including characteristic signatures of DNA methylation (Issa 2014; Klutstein et al. 2016; Pal and Tyler 2016). Evidence also indicates that epigenetic and metabolic regulators interact to affect aging (Benayoun et al. 2015). However, whether altered DNA methylation is causally involved in aging and whether regulators of DNA methylation play a role in this context have remained elusive. Here we provide evidence that GADD45α and ING1 regulate site-specific demethylation of C/EBP-dependent enhancers and thereby control energy metabolism and organismal aging (Fig. 7G). These results provide the first experimental support for the hypothesis (Vanyushin et al. 1973; Wilson and Jones 1983) that methylation changes cause aging.
Hallmarks of aging are global hypomethylation and local hypermethylation, similar to in cancer cells (Issa 2014; Klutstein et al. 2016; Pal and Tyler 2016). Global hypomethylation occurs at repetitive DNA sequences and accompanies reduced heterochromatin. Loss of repetitive sequence methylation may increase the risk of retrotransposition and genomic instability. In contrast, our study draws attention to local hypermethylation as a proximal cause of aging, since hypermethylation of target superenhancers reduced C/EBPβ binding and function in double knockouts. It was proposed that local hypermethylation results from age-related erosion of TF binding to cognate CpG sites, which allows access to DNA methyltransferases and hence de novo methylation (Jung and Pfeifer 2015). In contrast, our study illustrates that misregulation of DNA demethylation may cause premature aging. Compound Tet1,2,3 and Tdg mutant mice have not been informative regarding aging, since they are embryonic-lethal (Dai et al. 2016). Circumstantial evidence for a role in aging comes from single-Tet mutants showing increased atherosclerosis and cancer (Agathocleous et al. 2017; Cimmino et al. 2017; Fuster et al. 2017). However, TET proteins also have catalytic activity-independent gene repressor functions, which may mask premature aging phenotypes, since, e.g., gene repression actually dominates over activation in mouse embryonic stem cells (Williams et al. 2011).
In MEFs, GADD45α acts on adipogenic C/EBP-dependent superenhancers, which are poised, since binding and demethylation occurred in uninduced naïve cells. Similarly, the demethylation intermediate 5fC occurs preferentially on poised enhancers (Song et al. 2013). Interestingly, age-induced hypermethylation occurs in target genes of Polycomb group proteins (Maegawa et al. 2010; Teschendorff et al. 2010; Beerman et al. 2013), which are known to occupy poised enhancers. Thus, poised/bivalent cis-regulatory elements may be hot spots for hypermethylation during aging (Rakyan et al. 2010).
C/EBPs play an important role in metabolism and nutrient sensing, the deregulation of which is a hallmark of aging (Roesler 2001; Kenyon 2010; Lopez-Otin et al. 2013). If, as the results indicate, C/EBP malfunction underlies the segmental progeria exhibited by Gadd45a/Ing1 double-knockout mice, should there not be earlier indications of C/EBPs functioning in aging? Indeed, previous data mining of genes coexpressed with a seed list of 181 aging-associated genes yielded Cebpa, Cebpb, and Cebpg as top hits (P = 10−30–10−34), with C/EBP-binding motifs enriched in the promoters of aging-associated genes (van Dam et al. 2012). Moreover, levels of C/EBPα and C/EBPδ decrease with aging, thereby reducing fat differentiation (Karagiannides et al. 2001). Additionally, C/EBPδ mediates DNA damage response, and, similar to GADD45α and ING1, C/EBPδ deficiency sensitizes mice to ionizing radiation (Huang et al. 2004; Wu et al. 2011; Pawar et al. 2014). Finally, replacement of the Cebpa gene with Cebpb increases life span by 20% (Chiu et al. 2004). The severity of Cebp single- and compound-mutant phenotypes may have masked premature aging, since, e.g., Cebpa−/− single-mutant and Cebpb/d double-mutant mice die neonatally (Wang et al. 1995; Tanaka et al. 1997).
Gadd45a/Ing1 double knockouts and Cebp mutants resemble progeroid mice deficient for NER effectors, all of which display kyphosis and female infertility as well as reduction in life span, body weight, fat deposits, blood sugar, and IGF1 signaling (de Boer et al. 2002; van de Ven et al. 2006; van der Pluijm et al. 2007). Thus, paradoxically, the starvation phenotype of progeroid mutants resembles the well-established life span-prolonging response of animals under caloric restriction (Guarente and Kenyon 2000). Interestingly, Hoeijmakers and colleagues (Vermeij et al. 2016) demonstrated recently that food restriction in progeroid mutants retards accelerated aging. The investigators therefore suggested that the starvation phenotype in progeroid mice is a compensatory response to reduce steady-state levels of reactive metabolites, thereby preserving genome integrity and delaying aging-related functional decline. On the other hand, C/EBPs directly bind and regulate gene expression of key effectors of energy metabolism such as Cebpa, Ppparg, and Igf (Zuo et al. 2006; Staiger et al. 2009; Siersbaek et al. 2012), which are deregulated in Gadd45a/Ing1 double-knockout mice. Thus, the starvation-like phenotype in double knockouts likely reflects a direct requirement of the GADD45α–ING1–C/EBP axis in regulating metabolic genes rather than an indirect compensatory response to premature aging. It is tempting to speculate that the same is true in DNA repair-defective progeroid mice, since deficiencies in NER can impair DNA demethylation (Barreto et al. 2007; Schmitz et al. 2009; Martinez-Macias et al. 2013; Ho et al. 2017) and thus could lead to hypermethylation of C/EBP sites and a starvation phenotype. In support of this possibility, vitamin C administration rescues median life span of progeric mice deficient in Werner helicase (Massip et al. 2010).
A main conclusion of this study is that GADD45α demethylates C/EBPβ-dependent enhancers. On the one hand, finding DMRs at enhancers is consistent with these elements being preferred sites of active DNA demethylation (Shen et al. 2013; Song and He 2013) and showing lineage-specific regulation of methylation levels (Meissner et al. 2008; Ziller et al. 2013). On the other hand, for GADD45α, this is a wholly surprising finding because previously characterized loci demethylated via GADD45 proteins are localized at promoters (Ma et al. 2009; Schmitz et al. 2009; Zhang et al. 2011; Schäfer et al. 2013; Arab et al. 2014). GADD45α promotes 5mC/5hmC removal by directly binding both TDG and TET1 (Cortellino et al. 2011; Arab et al. 2014; Kienhöfer et al. 2015; Li et al. 2015), and our data support this: (1) Double-knockout DMRs are sites undergoing active TET/TDG-mediated demethylation (Fig. 2B), and (2) the TET activator vitamin C rescues adipogenic MEF differentiation.
Unlike GADD45α, ING1 expectedly bound to promoters, and the binding motifs were highly enriched for TFs of the E2F family, which aligns with the described promoter preference of ING1 (Shi et al. 2006; Pena et al. 2008; Schäfer et al. 2013; Cheng et al. 2014). ING1 acted as the driver for MEF and WAT differentiation, while GADD45α amplified the gene expression program controlled by ING1. An intriguing finding is that ING1 bound at promoters was required to prevent DMR hypermethylation at enhancer sites. This requirement could reflect some indirect effect of ING1 on demethylation. However, the fact that (1) ING1 GADD45α-bound elements interact via promoter–enhancer loops (pre-existing, not dependent on the two proteins), (2) ING1 via its C terminus directly binds GADD45α (Schäfer et al. 2013), and (3) both GADD45α and ING1 directly bind TET1 (Kienhöfer et al. 2015; this work) supports a more direct mode of ING1 action. These results suggest that ING1 and GADD45α acting long and short range, respectively, cooperate to promote the assembly of a local TET/TDG complex, demethylating C/EBP-dependent enhancers. Since both proteins can bind TET1, they may compensate for each other in single mutants, but, upon combined deficiency, TET/TDG targeting becomes limiting, leading to demethylation defects. Final proof of the proposed GADD45α–ING1–TET1 ternary complex awaits detection of the relevant endogenous interactions.
Materials and methods
Cell culture and electroporation
MEFs were isolated as described (Nagy et al. 2003) from embryonic day 15.5 embryos. MEFs were cultured at 37°C, 95% humidity, 5% CO2, and 5% O2 in Dulbecco's modified Eagle's medium (DMEM), 10% fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin for up to four passages unless indicated otherwise.
Electroporation of MEFs was conducted using a Neon transfection system (Invitrogen) according to the manufacturer's instructions. For each confluent 10-cm dish of MEFs, 20 µg of plasmid was electroporated with one pulse of 1250 V for 20 msec in a 100-µL Neon tip followed by 36 h of incubation. eGFP was used as control plasmid for electroporation of the control samples for ChIP-seq. The expression constructs used in this study were pCS2-myc-hIng1b (Schäfer et al. 2013), pCS2-myc-hGadd45a (Kienhöfer et al. 2015), pCS2-HA-hIng1b, pHA-hGadd45a (Barreto et al. 2007), pCS2-eGFP, p95/47T81 C/EBP-Luciferase (Sterneck et al. 1997), and pRL (Promega). pCS2-HA-hIng1b was generated by inserting the hIng1b sequence from pCS2-Myc-Ing1b (Barreto et al. 2007; Schäfer et al. 2013) into pCS2-Nterm-HA (M Gierl, unpubl.) via XbaI and BamHI restriction sites. MEFs were transfected with 40 nM Cebpb- or Cebpd-specific or control Dharmacon siRNA Smart pools using Lipofectamine 2000 (Invitrogen). For Figure 2I, MEF cells were incubated for 24 h with siRNA prior to plasmid DNA electroporation. Cebpb and Cebpd knockdown was controlled by qPCR and was >80% or 50%, respectively (data not shown).
For growth curve analyses, 1.9 × 104 MEFs were seeded to wells of 12-well dishes in technical duplicates. At intervals of 1, 3, 5, and 7 d after cell seeding, MEFs were detached and counted.
Adipocyte differentiation and Oil Red O staining
MEFs were cultured in DMEM containing 10% fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin until 2 d after confluence. Adipocyte differentiation was induced in post-confluent MEFs using culture medium supplemented with 0.5 mM 3-isobutyl-1-methylxanthine, 1 µM dexamethasone, 2 µM rosiglitazone, and 10 µg/mL insulin. After 48 h, differentiation was continued with culture medium supplemented with 10 mg/mL insulin. Adipocyte differentiation medium was exchanged every 2 d. For vitamin C treatment, 2-phospho-L-ascorbic acid was added along with the differentiation cocktail at 10 µg/mL final concentration.
For Oil Red O staining, cells grown on coverslips were fixed for 30 min in 4% paraformaldehyde, equilibrated for 5 min in 60% isopropanol, and stained for 8 min in freshly prepared and filtered 3 g/L Oil Red O in 60% isopropanol. After removing excess staining with 60% isopropanol, coverslips were washed in water, counterstained for 5–8 min with Harris’ hematoxylin, and imaged with a Leica DM2500 light microscope. Quantification of Oil Red O staining was performed on nine randomly chosen micrographs per condition in biological triplicates.
RNA isolation, cDNA synthesis, and qPCR
Adipose tissue was collected and snap-frozen in liquid nitrogen. For RNA isolation, samples were immersed in 1 mL of Qiazol and homogenized with an Ultra Turrax homogenizer. After 5 min of incubation at room temperature, 200 µL of chloroform was added, and samples were shaken vigorously for 5 min. Following centrifugation at 12,000 rpm for 15 min at 4°C, the upper aqueous phase was transferred to a fresh tube. All other tissues were excised and stored in RNAlater (Sigma) at −80°C. For RNA isolation, tissues were transferred to RLT buffer (Qiagen), including 1% β-mercaptoethanol, and homogenized with an Ultra Turrax homogenizer. Cells grown in tissue culture were washed in PBS and resuspended in RLT buffer, including 1% β-mercaptoethanol. Subsequent steps of RNA isolation for all tissues and cells were performed using a Qiagen RNeasy Mini or Qiagen RNeasy 96-well kit according to the manufacturer's instructions, including on-column DNaseI digest. cDNA synthesis was conducted with SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. For each batch of cDNA synthesis, negative control samples lacking either RNA or reverse transcriptase were included. qPCR reactions were performed in technical duplicates using the Roche Universal probe library (UPL) system with a Roche LightCycler 480 in 384-well format. UPL-compatible primers were designed using the Roche assay design center (http://lifescience.roche.com/shop/en/mx/overviews/brand/universal-probe-library). Primer sequences are listed in the Supplemental Material. Roche LightCycler analysis software was used for quantification. Expression levels were normalized to two housekeeping genes (Gapdh and Tbp) as well as to respective wild-type expression levels within the experiment.
RNA expression microarrays
RNA isolation from gonadal WAT from 59- to 67-d-old male mice of all four genotypes was performed as described above. RNA integrity (RIN) was determined with a RNA 6000 Nano kit on a Bioanalyzer (Agilent) and yielded RIN values ranging from 8.3 to 9.6, indicating suitable RNA quality without significant RNA degradation. Total RNA was pooled from three individual mice per genotype, and three pools per genotype were used for RNA expression microarray analysis. Samples were processed and hybridized to SurePrint G3 Mouse GE 8X60K microarrays (Agilent) according to the manufacturer's recommendations and scanned on Agilent Technologies scanner G2505C with version 10.7.3.1 feature extraction software at 3-µm resolution. The raw array data were processed using the Bioconductor package limma version 3.26.9 (https://bioconductor.org/packages/release/bioc/html/limma.html) following the standard workflow for single-channel Agilent arrays (chapter 17.4 of the user's guide). Following normexp background correction and quantile intersample normalization, the control and low-signal probes were filtered out, the replicate probes were averaged, and the knockout samples were compared against the wild-type samples for differential gene expression (lmFit, eBayes). Differential expression cutoff was set at 10% false discovery rate (FDR) and 1.5-fold change compared with the wild-type samples. Heat map of double-knockout-deregulated genes was produced with MeV (http://www.tm4.org/mev.html) using hierarchical clustering with Manhattan distance and average linkage. Gene ontology (GO) analysis was performed using GOrilla (http://cbl-gorilla.cs.technion.ac.il) and focused on biological process GO terms with up-regulated or down-regulated genes as a target set and background list of detected genes.
RNA-seq
Wild-type, Gadd45a−/−, Ing1−/−, and Gadd45a−/−/Ing1−/− (double-knockout) MEFs were differentiated to adipocytes for 6 d in biological triplicates. Total RNA from MEFs at days 0 and 6 was isolated using the Qiagen RNeasy kit according to the manufacturer's recommendations, including on-column DNaseI digest. RNA integrity was verified using an RNA 6000 Nano kit on a 2100 Bioanalyzer (Agilent), which yielded RIN values consistently >9.5.
RNA-seq libraries were prepared from 280 ng of RNA using the TruSeq RNA sample preparation kit version 2 (Illumina) according to manufacturer's recommendations and amplified in 12 PCR cycles. Libraries were profiled in a high-sensitivity DNA chip on a 2100 Bioanalyzer and quantified using the Qubit dsDNA HS assay kit in a Qubit 2.0 fluorometer (Life technologies). The 24 libraries were pooled in equimolar ratio and sequenced on a HiSeq 2000 Illumina sequencer, single-read mode, for 51 cycles plus seven cycles for the index read.
Each sample yielded, on average, 50 million reads of 51-bp length. Raw reads were mapped to the NCBI37/mm9 build of the mouse genome using TopHat version 2.0.9 (https://ccb.jhu.edu/software/tophat), and HT-Seq count version 0.5.4 (http://www-huber.embl.de/HTSeq) was used for summarizing the reads over exons. Using the DESeq (https://www.bioconductor.org/packages/devel/bioc/html/DESeq.html) package in R, differentially expressed genes were identified at a FDR of 10% and 1.5-fold (untreated) or twofold (day 6 of adipocyte differentiation) change compared with wild-type samples. GO analysis was performed using GOrilla (http://cbl-gorilla.cs.technion.ac.il) and focused on biological process GO terms, with gene symbols of up-regulated or down-regulated genes as a target set and background list of detected genes.
ChIP
ChIP in MEFs (Schäfer et al. 2013) and abdominal WAT (Haim et al. 2013) was carried out essentially as described. Briefly, MEFs were cross-linked with 0.75% formaldehyde for 10 min at room temperature and quenched with 125 mM glycine for 5 min. After harvest of cells by scraping in PBS, cells were lysed for 10 min at 4°C in cell lysis buffer (5 mM PIPES at pH 8.0, 85 mM KCl, 0.5% NP-40, complete protease inhibitor cocktail [PI; Roche]). WAT was rinsed in PBS, including PI; minced into 1- to 3-mm3 pieces; and fixed using 1% formaldehyde for 12 min at room temperature, and cross-linking was stopped by 125 mM glycine addition for 5 min. After two washes with PBS, including PI, WAT was dissolved in adipocyte lysis buffer (500 mM PIPES, 80 mM KCl, 1% igepal with PI), homogenized using a Dounce homogenizer (40 strokes; Sigma), and incubated for 15 min at 4°C while vortexing every 3 min. Nuclei of MEFs and WAT were pelleted and lysed for 10 min (MEFs) or overnight (WAT) at 4°C in nuclei lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl at pH 8.1 with PI). Chromatin was sonicated to 150- to 500-bp fragments using a Bioruptor Plus (Diagenode), cleared by centrifugation for 10 min at 21.000g, and diluted 1:10 in immunoprecipitation dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl at pH 8.1, 167 mM NaCl including PI). After preclearing for 20 min at 4°C with 3.125 µL/mL Protein A/G Dynabeads (Invitrogen) preblocked with BSA and herring sperm DNA, chromatin was incubated with 4 µg/mL antibody overnight at 4°C. Antibodies were rabbit a-HA ChIP-grade (Abcam, ab9110) and mouse a-C/EBPb ChIP-grade (Abcam, 1H7, ab15050). Antibody–chromatin complexes were captured with 25 µL/mL blocked Protein A/G Dynabeads (Invitrogen) for 90 min at 4°C followed by five washes in ChIP wash buffer (150 mM NaCl, 20 mM Tris at pH 8.1, 2 mM EDTA, 10% glycerol, 1% Triton X-100, including PI). Captured fragments were eluted in elution buffer (1% SDS, 50 mM Tris at pH 8.1, 10 mM EDTA). After addition of NaCl to a final concentration of 375 mM, samples were decross-linked overnight at 65°C, digested for 2 h with 40 µg of Proteinase K at 56°C, and purified with QIAquick PCR purification columns (Qiagen). Resulting samples were used for qPCR or further processed for high-throughput sequencing.
ChIP-seq
ChIP-seq library preparation was performed using NuGEN's Ovation Ultralow system version 2 1-16 (2014) following the manufacturer's recommendations. Libraries were prepared from 1-ng or 48-ng ChIP-DNA and amplified in 14 or nine PCR cycles for HA-ING1 or HA-GADD45α, respectively. For ING1 ChIP-seq, size selection (300- to 1550-bp fragments) was performed using a XT DNA 750 chip (Perkin Elmer). For GADD45α ChIP-seq, double-bead size selection with ratios of 0.65:1 and 0.85:1 (beads:DNA ratio) was performed to enrich the library for 230- to 730-bp fragments using Agencourt AMPure XP (Beckman Coulter). Libraries were profiled in a high-sensitivity DNA chip on a 2100 Bioanalyzer (Agilent Technologies) and quantified using the Qubit dsDNA HS assay kit in a Qubit 2.0 fluorometer (Life technologies). ING1 libraries were pooled in equimolar ratio, as were GADD45α libraries, and each pool was sequenced on two HiSeq 2500 lanes in rapid mode and single-read for 68 cycles for read 1 plus seven cycles for the index read.
More than 17 million and 40 million 68-bp reads per sample were obtained for ING1 and GADD45α ChIP-seq, respectively. The reads were aligned to the NCBI37/mm9 genome with Bowtie version 0.12.9 (http://bowtie.cbcb.umd.edu) with parameters “-m 1 --best --strata.” Peak calling analysis of GADD45α ChIP versus sontrol ChIP was performed for each replicate with MACS version 1.4.2 (http://liulab.dfci.harvard.edu/MACS/index.html). The common peaks from the replicate experiments were determined with BedTools version 2.26.
WGBS-seq
Genomic DNA from a wild-type and a Gadd45a−/−/Ing1−/− (double-knockout) MEF line was purified with a DNeasy blood and tissue kit (Qiagen) according to the manufacturer's instructions. Genomic DNA was fragmented with a Covaris S2 Ultrasonicator. For end repair of DNA fragments, the paired-end DNA sample preparation kit (Illumina) was used. Adapter ligation was carried out with an early access methylation adapter oligo kit (Illumina) and the paired-end DNA sample preparation kit (Illumina). After size selection of fragments using the E-Gel electrophoresis system (Invitrogen), DNA was bisulfite-converted with a EZ DNA methylation kit (Zymo Research). PCR-amplified and purified products were subjected to 100-bp paired-end sequencing on an Illumina HiSeq 2000 high-throughput sequencer with 10-fold mean coverage for each strand.
The WGBS data sets contained 1.1 billion and 0.9 billion reads for wild-type and double-knockout MEF samples, respectively. Raw reads were aligned over the reference mouse genome (NCBI37/mm9) using Bismark version 0.9.0 (http://www.bioinformatics.babraham.ac.uk/projects/bismark). Besides the default parameters for paired-end reads, “-n 3 -l 60 -e 200” was used for a unique mapping rate of >75%. Methylation calling for individual cytosines was performed using the Bismark tool bismark_methylation_extractor with parameters “-p --no_overlap --merge_non_CpG --ignore_r2 3 --bedGraph --counts” and bedGraph2cytosine with default parameters. CpG methylation was compared between wild-type and double-knockout samples using custom-made scripts. DMRs between double-knockout and wild-type MEFs were identified by clustering the differentially methylated CpGs with custom-made scripts according to the following parameters: (1) minimum read coverage: 10× per strand, (2) minimum methylation difference: 25%, and (3) minimum number of consecutive CpGs affected: three on both strands.
NG Capture-C
NG Capture-C was carried out essentially as described (Davies et al. 2016) with some modifications. Three independent wild-type, Gadd45a−/−, Ing1−/−, and Gadd45a–Ing1 double-knockout MEF cell lines were grown to 80% confluency. Cells were fixed in 2% formaldehyde for 10 min at room temperature in batches of 2 × 107 cells, and the reaction was quenched with 125 mM glycine (final concentration). After two PBS washes, cells were lysed in 5 mL of ice-cold lysis buffer (10 mM Tris at pH 8, 10 mM NaCl, 0.2 % Igepal CA-630, PI [Sigma]). After snap-freezing in liquid nitrogen, cell pellets were resuspended in a total of 1.4 mL of 1× DpnII buffer (New England Biolabs) and homogenized on ice with 55 strokes of a 5-mL Dounce homogenizer. Cells were pelleted and resuspended in 600 µL of 1× DpnII buffer. Cells were shaken horizontally at 1400 rpm for 1 h at 37°C after dilution with 2.5 vol of 1× DpnII buffer containing 0.4% SDS. Triton X-100 was added to a final concentration of 1.67%, and lysates were shaken for 1 h at 37°C. For restriction digest, 1.500 U of DpnII was added three times within 24 h (total of 4.500 U) while lysates were shaking horizontally at 1400 rpm at 37°C. After heat inactivation for 20 min at 65°C, 1 vol of 2× T4 DNA HC ligase buffer containing 30 µL of T4 DNA HC ligase (Life Technologies) was added and incubated overnight with shaking at 1400 rpm at 16°C. After decross-linking at 65°C in the presence of 65 µg/mL Proteinase K (Thermo Scientific) overnight and digestion using 3.45 µg/mL RNase A (Roche) for 30 min at 37°C, DNA was phenol-chloroform-isoamylalcohol-extracted and precipitated using sodium acetate. As a control of efficient digest and ligation, aliquots were taken during the procedure without enzyme addition and controlled via agarose gel electrophoresis (data not shown). In addition, digest efficiency was confirmed to be >80% using DpnII-sensitive qPCR (data not shown). Fifteen micrograms of the 3C library of each sample in 120 µL was sonified in a Covaris microtube (duty cycle 10%, intensity 5, 200 cycles per burst for 6 min in frequency sweeping mode) to yield an average fragment size of ∼180 bp. After sonication, the DNA was purified using Ampure XP SPRI bead cleanup. Illumina sequencing adaptors were added to 1.5 µg of the 3C library using NEBNext DNA library preparation reagent set (New England Biolabs) according to manufacturer's instruction with two exceptions: First, 15 µL instead of 10 µL of adaptor was used, and, second, Herculase II fusion polymerase (Agilent) was used for PCR-based addition of indices instead of New England Biolabs polymerase. For the first capture, 2 µg of the 12 indexed libraries was mixed in an exact stoichiometric ratio. Capture was performed using the Nimblegen SeqCap EZ kits (Roche) based on the Nimblegen SeqCap SR user guide instructions. Note that Nimblegene HE-Oligo kit A was used, and the New England Biolabs multiplex primers during indexing were chosen accordingly from NEBNext primer sets 1 and 2. Sixty-three different 120-bp oligonucleotides covering 37 regions were designed using the Hughes laborartory Web tool (http://apps.molbiol.ox.ac.uk/CaptureC/cgi-bin/CapSequm.cgi) and ordered from Sigma, carrying a 5′biotin. Oligonucleotides were dissolved individually at 2.9 µM final concentration and mixed in a 1:1 ratio. For the hybridization, 4.5 µL of this oligonucleotide pool was used per sample (54 µL for 12 samples). Capture was performed based on the manufacturer's instructions of the Nimblegen SeqCap EZ kit in four paralleled reactions to preserve complexity. For amplification, 12 PCR cycles using Kapa HiFi polymerase were performed, and hybridization was conducted for 72 h at 47°C. The second capture was performed exactly the same as the first capture with the exception that only 500 ng of the captured library was used. qPCR served as quality control and confirmed a >5 × 106-fold enrichment of the control locus Hba1-2 over unspecific control regions after the second capture (data not shown). The pooled library was diluted to 4 nM (based on QX200 digital droplet PCR measurement [Bio-Rad]) using the ddPCR library quantification kit for Illumina TruSeq (Bio-Rad). The library was denatured and further diluted according to Illumina's recommendations for the NextSeq500 flow cell. Of a 1.2 pM dilution of the denatured pool, 1.3 mL (with 2% spiked-in ΦX) was loaded and sequenced in High-Output mode, paired end, for 2 × 150 bp, resulting in >30 million reads per sample. Sample demultiplexing and FastQ file generation were performed using Illumina conversion software bcl2fastq version 2.18.
The raw data were processed following the standard workflow described in Davies et al. (2016) (https://github.com/telenius/captureC/releases) in the following steps. After read quality assessment with FastQC version 0.11.5, adaptor sequences were trimmed using Trim Galore version 0.4.1 (https://www.bioinformatics.babraham.ac.uk/projects) with parameters “--stringency 3 --paired.” The paired-end reads were merged into fragments using Flash version 1.2.11 (https://sourceforge.net/projects/flashpage) with parameters “--interleaved-output -m 10 -M 150 -x 0.1 -t 1 -z.” In silico DpnII restriction digest of the merged reads was done with the custom script dpnII2E.pl (https://github.com/telenius/captureC/releases). Alignment of the digested reads to the NCBI37/mm9 mouse genome was carried out using Bowtie version 1.1.2 (http://bowtie-bio.sourceforge.net/index.shtml) with parameters “-p 1 -m 2 -y --best --strata --chunkmbs 1024.” Capture-C-tailored filtering, visualization, and interaction quantitation of the mapped data were performed using the custom script CCanalyser3.pl (https://github.com/telenius/captureC/releases) with default parameters.
Next-generation sequencing (NGS) data integration and visualization
Genome-wide methylation profiling was conducted using methylKit (https://github.com/al2na/methylKit) in R. Genome partitioning into unmethylated regions (UMRs), LMRs, and fully methylated regions (FMRs) was performed with MethylSeekR (https://www.bioconductor.org/packages/devel/bioc/html/MethylSeekR.html). The heat map for the 5hmC, 5fC, 5caC levels (normalized read density) in control and Tdg knockdown mouse embryonic stem cells (Shen et al. 2013) was generated using deepTools version 2.5.1 (Ramirez et al. 2014), taking reference points from the centre of hypermethylated DMRs. Positional correlations of DMRs across MEF histone marks (Yue et al. 2014) was conducted with Genome Inspector (Genomatix https://www.genomatix.de). GO analysis for the enhancer-associated DMRs was carried out using PANTHER (http://pantherdb.org). GADD45α and ING1 ChIP-seq peaks were correlated with CpG methylation levels from WGBS using self-created scripts, and the results were plotted with GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E).
For annotation analyses and heat maps, promoters and enhancers were defined using available MEF H3K4me1, H3K4me3, and H3K27ac ChIP-seq data sets (Yue et al. 2014). Gene regulatory region information was extracted from the University of California at Santa Cruz (UCSC) table browser (https://genome.ucsc.edu/cgi-bin/hgTables). Enrichment scores were estimated by taking the ratio between proportions of the feature-associated DMRs in the total number of DMRs and proportions of the features in the whole-mouse genome. Heat maps of TF binding were generated using custom-made scripts and published ChIP-seq data sets (Siersbaek et al. 2011, 2014). Ing1 peaks were correlated with E2F4 ChIP-seq data sets from 3T3-L1 cells (MacIsaac et al. 2010) using closestBed (BedTools version 2.26) with the parameter “-d 1000”.
GREAT version 0.3.0 (http://bejerano.stanford.edu/great) was used to associate ChIP-seq peaks with adjacent genes and for TF motif analysis of ING1 ChIP-seq peaks. De novo and known motif analysis of DMRs and GADD45α ChIP-seq was performed with HOMER (http://homer.salk.edu/homer/motif). GO analysis for DMRs and peaks from the ChIP-seq analysis was conducted with GOrilla using nearby genes as a target set and all mm9 genes as a background set. Heat maps of differentially expressed genes were produced with gplots (https://cran.r-project.org/web/packages/gplots) in R, and Venn diagrams were generated with Venny (http://bioinfogp.cnb.csic.es/tools/venny) or BioVenn (http://www.cmbi.ru.nl/cdd/biovenn/index.php; semiproportional). UCSC genome browser (https://genome.ucsc.edu) was used for NGS data visualization.
Methylation-sensitive PCR
Methylation-sensitive PCR takes advantage of the different sensitivities of restriction endonucleases to DNA methylation. While MspI, cutting at the CCGG sequences, is insensitive to DNA methylation, its isoschizomere, HpaII, cuts only unmethylated DNA. HhaI, another methylation-sensitive restriction enzyme, cuts GCGC sequences only in their unmethylated state. PvuII was used to sterically relax genomic DNA by cutting in nonrelevant sequence contexts.
Genomic DNA was purified with a Qiagen blood and tissue kit according to the manufacturer's recommendations. One-hundred-fifty nanograms of genomic DNA was digested overnight with 10 U of PvuII and 10 U of either HpaII, HhaI, or MspI (all Promega) or no enzyme. Five nanograms of digested DNA was used for qPCR analysis in technical duplicates. Obtained values were normalized to PvuII-only control digests to estimate DNA methylation at examined CpGs.
Animal experiments
Gadd45a-deficient mice (Hollander et al. 1999) were a kind gift from M.C. Hollander. Ing1-deficient mice (Kichina et al. 2006) were obtained from Jackson Laboratories. Both strains were backcrossed several generations into the C57BL/6N background and interbred to generate wild-type, Gadd45a−/−, Ing1−/−, and Gadd45a−/−/Ing1−/− (double-knockout) mice from homogenous genetic backgrounds. Mice were housed in accordance with national and international guidelines with ad libitum access to water and chow diet and a 12-h/12-h light–dark rhythm. Mice were sacrificed by CO2 asphyxiation followed by cervical dislocation before experimental manipulation. All procedures were performed with the approval of the ethical committees on animal care and use of the federal states of Baden-Württemberg and Rheinland-Pfalz, Germany.
Hematoxylin and eosin (H&E) staining
Mouse tissues were dissected and fixed for 48 h at 4°C in freshly prepared 4% paraformaldehyde in PBS. Tissues were embedded in paraffin, sectioned to 5–10 µm, and stained with H&E according to standard procedures. Images were taken on a Leica DM2500 light microscope.
SA-β-Gal staining
SA-β-Gal activity in MEFs was determined with a 96-well cellular senescence assay kit (Cell Biolabs) according to the manufacturer's instructions with the following modifications: Cell lysis was conducted for 10 min, and clarification of the lysate was performed in the original multiwell plate. Fluorescence was normalized to total protein concentrations to give relative SA-β-Gal activity. For determination of SA-β-Gal activity in mouse skin, dissected tissues were fixed overnight in freshly prepared 4% paraformaldehyde in PBS. Following several washes in PBS, tissues were infiltrated overnight in 15% sucrose and, subsequently, 30% sucrose in PBS. Tissues were immersed in OCT TissueTek embedding medium and stored at −80°C. After sectioning tissues at 5–10 µm, SA-β-Gal staining was performed essentially as described (Dimri et al. 1995; Debacq-Chainiaux et al. 2009). Briefly, tissue sections were rinsed twice for 10 min in PBS at 4°C to remove the embedding medium and stained overnight at 37°C in freshly prepared SA-β-Gal staining solution {40 mM citric acid, 40 mM dibasic sodium phosphate, 5 mM K4[Fe(CN)6], 5mM K3[Fe(CN)6], 150 mM NaCl, 2 mM MgCl, 1 mg/mL X-Gal [5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside]} adjusted to pH 6. Samples were washed in PBS, mounted with aqueous mounting medium, and imaged on a Leica DM2500 light microscope. SA-β-Gal staining was quantified in Photoshop PS5 as percent of total area stained using iteratively increased image contrast and the histogram tool.
Quantification of DNA modifications
The alkaline elution technique in combination with repair enzymes as probes was used to quantify various types of DNA modifications, as described previously (Epe and Hegler 1994; Pflaum et al. 1997). The assay makes use of the fact that the elution rate of chromosomal DNA from a membrane filter (2-µm pores) depends on the length of the DNA molecules and therefore the number of strand breaks. Briefly, the sum of single-strand breaks (SSBs) and DNA modifications sensitive to a repair enzyme (here Fpg protein, which recognizes oxidized purines and AP sites, or T4-endonuclease V, which recognizes cyclobutane pyrimidine dimers [CPDs] and AP sites) was obtained from experiments in which the cellular DNA was incubated with the repair enzyme immediately after cell lysis. To quantify SSBs, the incubation was carried out without repair enzyme. The numbers of enzyme-sensitive modifications were obtained by subtracting the number of SSBs. For the analysis of the repair kinetics of CPDs, cells were exposed to 2.9 J/m2 UVB, and the number of T4 endonuclease V-sensitive lesions was determined at various time points after damage induction. In this case, the numbers of DNA modifications observed in untreated control cells (background levels) were subtracted. The slope of an elution curve obtained with γ-irradiated cells was used for calibration (6 Gy = 1 SSB/106 bp).
Quantification of ROS
ROS were measured in MEFs using the total ROS/superoxide detection kit (Enzo Life Sciences) according to the manufacturer's instructions. ROS and superoxide anions were detected on a LSR-Fortessa Sorp flow cytometer. Cell debris and doublets were gated out, and unstained N-acetylcysteine-treated negative control as well as pyocyanin-treated positive control cells were used for channel compensation corrections and population gating. Quantification of results was performed with FlowJo data analysis software (FlowJo LLC).
Quantification of telomere length
To determine telomere length, genomic DNA from mouse livers was purified with a Qiagen blood and tissue kit according to the manufacturer's recommendations. Genomic DNA (1.35 µg per sample) was analyzed by Southern blot using the Telo TAAGG telomere length assay kit (Roche) according to the manufacturer's instructions. Signals were detected with a ChemiDoc imager (Bio-Rad) and analyzed using the ImageLab software (Bio-Rad).
Quantification of serum cytokines
Mouse blood was collected by cardiac puncture after CO2 asphyxiation. Blood was allowed to clot for 30 min at room temperature and centrifuged at 2000g for 10 min at 4°C. The serum supernatant was stored at −20°C. For cytokine detection, 50 µL of serum each from three animals per genotype was pooled and processed using a mouse cytokine antibody array (C series, array 3) from RayBiotech according to the manufacturer's recommendations. Both the serum and antibody cocktail incubation steps were conducted overnight at 4°C. Signals were detected with a ChemiDoc imager (Bio-Rad). Background signals were subtracted and technical duplicate values were averaged.
Estrus cycle analysis
Estrus cycle phases were determined as described (Caligioni 2009) and quantified based on the relative amount of cornified epithelial cells, which mark the estrus phase and are strongly reduced or absent in other phases of the estrus cycle.
Blood glucose measurement
Plasma blood glucose levels were assessed in 6- to 8-wk-old mice in both randomly fed and overnight fast conditions. Blood samples were drawn by tail clipping, and glycemia was assessed using an Accu-Chek glucometer (Roche).
Luciferase reporter assays
For luciferase assays, 10-cm dishes of confluent MEFs were electroporated with 19 µg of p95/47T81 C/EBP-Luciferase (Sterneck et al. 1997) and 1 µL of pRL (Promega) and, 24 h later, differentiated along the adipogenic lineage. Dual-luciferase reporter assays (Promega) were performed according to the manufacturer's instructions and measured on an Infinite200 Pro microplate reader (Tecan).
Co-IP and immunoblotting
Co-IP was performed as described previously (Kienhöfer et al. 2015). For immunoblot analysis, cells were lysed in RIPA buffer (150 mM NaCl, 1% Triton-X100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-HCl at pH 8.0) supplemented with PI (Roche). Cell disruption was achieved by four cycles of freezing in liquid nitrogen and thawing at 37°C followed by sonication for 10 cycles (15 sec on/off at high setting) in a Bioruptor (Diagenode). Lysates were cleared by centrifugation, and protein concentration was estimated by bicinchoninic acid (BCA) assay. Absorption at 562 nm was measured with an Infinite 200 Pro microplate reader (Tecan) and compared with a standard curve of bovine serum albumin samples with known protein concentrations that was run in parallel to samples of interest. Lysates were boiled in 4× stock Lämmli sample buffer (60 mM Tris-Cl at pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue), separated on polyacrylamide gels, and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in 5% skimmed milk in Tris-buffered saline with 0.1% Tween-20 (TBS-T), membranes were incubated first overnight at 4°C with primary antibodies diluted in blocking buffer and subsequently for 1 h at room temperature with peroxidase-coupled secondary antibodies diluted in blocking buffer. Signals were developed with SuperSignal West Pico or Femto chemiluminescent substrate (Thermo Scientific) and analyzed using ChemiDoc (Bio-Rad) with ImageLab software.
Immunostaining
MEFs were cultured on coverslips and fixed for 30 min with 4% paraformaldehyde in PBS. Neutralization of residual paraformaldehyde and permeabilization were performed by a 30-min incubation in 200 mM glycine and 0.1% Triton X-100 in PBS. After blocking for 1 h in PBGNT (PBS, 0.5% BSA, 0.2% cold water fish gelatin, 0.5 M NaCl, 0,1% Triton-X100), coverslips were incubated overnight at 4°C with primary antibody diluted in PBGNT and subsequently incubated for 1 h at room temperature with Alexa fluor 546-coupled secondary antibody and 0.1 µg/mL DAPI in PBGNT. Samples were analyzed with a Leica SP5 confocal microscope and Leica LAS image software.
Antibodies
Primary antibodies used in this study were rabbit anti-HA (Abcam, ab9110), rabbit anti-C/EBPβ (Santa Cruz Biotechnology, sc-150X), mouse anti-PPARγ (Abcam, ab41928), rabbit anti-GADD45a (Santa Cruz Biotechnology, sc-797), goat anti-ING1 (Santa Cruz Biotechnology, sc-7566), mouse anti-GFP epitope tag (Dianova, DLN-07227), rabbit anti-Caspase 3 (Cell Signaling, 9662), rabbit anti-p16 (Santa Cruz Biotechnology, sc-1207), mouse anti-phosho-Histone H2A.X (Millipore, 05-636), rabbit anti-Histone H3 (Abcam, ab1791), and mouse anti-α-Tubulin (Sigma, T5186). Secondary antibodies were HRP-coupled goat anti-mouse IgG (Dianova, 115-035-146), HRP-coupled goat anti-rabbit IgG (Dianova, 111-035-144), and Alexa fluor 546-coupled goat anti-rabbit IgG (Invitrogen, A11035).
Statistical analysis
Data throughout this report are displayed as arithmetic mean ± SD. Statistical significance was determined with an unpaired two-tailed Student's t-test in Excel 2013 (Microsoft) and is displayed as P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). Statistical significance of mouse survival was determined by log-rank test (https://www.evanmiller.org/ab-testing/survival-curves.html).
Data availability
All NGS and microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO) under superseries accession number GSE99607. Primer sequences are listed in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank H.-J. Gröne for expert histopathological analysis, C. Hollander for providing Gadd45a mutant mice, and A. Leutz and I. Grummt for reagents. Technical support by the Institute of Molecular Biology Core Facilities for Genomics (especially Maria Méndez-Lago), Microscopy, Histology, and Cytometrie and the Animal Facility is gratefully acknowledged. We further acknowledge the contribution of the German Cancer Research Center (DKFZ) Core Facility for Genomics and the DKFZ Central Animal Laboratory. We are indebted to J. Hughes for help with NG Capture-C. We thank M. Pradhan for critical reading. This work was supported by a European Research Council senior investigator grant to C.N. (“DNAdemethylase”).
Author contributions: C.N. and A.S. conceived the study. A.S., B.M., V.V., D.S., C.v.d.L., and B.E. performed the invesitgations. M.M. and E.K. performed the formal analysis. C.N. and B.M. wrote the original draft of the manuscript. C.N. and A.S. supervised the study. C.N. acquired the funding.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311969.118.
References
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. 2017. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549: 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab K, Park YJ, Lindroth AM, Schafer A, Oakes C, Weichenhan D, Lukanova A, Lundin E, Risch A, Meister M, et al. 2014. Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol Cell 55: 604–614. [DOI] [PubMed] [Google Scholar]
- Arizmendi C, Liu S, Croniger C, Poli V, Friedman JE. 1999. The transcription factor CCAAT/enhancer-binding protein β regulates gluconeogenesis and phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J Biol Chem 274: 13033–13040. [DOI] [PubMed] [Google Scholar]
- Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, Döderlein G, Maltry N, Wu W, Lyko F, et al. 2007. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445: 671–675. [DOI] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Myers CL, Andrews B, Boone C. 2013. Genetic interaction networks: toward an understanding of heritability. Annu Rev Genomics Hum Genet 14: 111–133. [DOI] [PubMed] [Google Scholar]
- Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, Rossi DJ. 2013. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Brunet A. 2015. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16: 593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, et al. 2013. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500: 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. 2012. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336: 934–937. [DOI] [PubMed] [Google Scholar]
- Caligioni CS. 2009. Assessing reproductive status/stages in mice. Curr Protoc Neurosci 48: A.4I.1–A.4I.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Blum R, Bowman C, Hu D, Shilatifard A, Shen S, Dynlacht BD. 2014. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell 53: 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CH, Lin WD, Huang SY, Lee YH. 2004. Effect of a C/EBP gene replacement on mitochondrial biogenesis in fat cells. Genes Dev 18: 1970–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. 2017. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170: 1079–1095.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles AH, Liang H, Zhu Z, Marfella CG, Kang J, Imbalzano AN, Jones SN. 2007. Deletion of p37Ing1 in mice reveals a p53-independent role for Ing1 in the suppression of cell proliferation, apoptosis, and tumorigenesis. Cancer Res 67: 2054–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, et al. 2011. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470: 419–423. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. 2011. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai HQ, Wang BA, Yang L, Chen JJ, Zhu GC, Sun ML, Ge H, Wang R, Chapman DL, Tang F, et al. 2016. TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 538: 528–532. [DOI] [PubMed] [Google Scholar]
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, Hughes JR. 2016. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat Methods 13: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. 2009. Protocols to detect senescence-associated β-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4: 1798–1806. [DOI] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, et al. 2002. Premature aging in mice deficient in DNA repair and transcription. Science 296: 1276–1279. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci 92: 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epe B, Hegler J. 1994. Oxidative DNA damage: endonuclease fingerprinting. Methods Enzymol 234: 122–131. [DOI] [PubMed] [Google Scholar]
- Fajas L. 2013. Re-thinking cell cycle regulators: the cross-talk with metabolism. Front Oncol 3: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. 2002. E2Fs regulate adipocyte differentiation. Dev Cell 3: 39–49. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. 2017. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355: 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Schuster S, Kiess W. 2012. The insulin-like growth factors in adipogenesis and obesity. Endocrinol Metab Clin North Am 41: 283–295. [DOI] [PubMed] [Google Scholar]
- Gavin DP, Sharma RP, Chase KA, Matrisciano F, Dong E, Guidotti A. 2012. Growth arrest and DNA-damage-inducible, β (GADD45b)-mediated DNA demethylation in major psychosis. Neuropsychopharmacology 37: 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. 2010. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5: e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Kenyon C. 2000. Genetic pathways that regulate ageing in model organisms. Nature 408: 255–262. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. 2011. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim Y, Tarnovscki T, Bashari D, Rudich A. 2013. A chromatin immunoprecipitation (ChIP) protocol for use in whole human adipose tissue. Am J Physiol Endocrinol Metab 305: E1172–E1177. [DOI] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49: 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, et al. 2016. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 17: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. 2013. Super-enhancers in the control of cell identity and disease. Cell 155: 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JJ, Cattoglio C, McSwiggen DT, Tjian R, Fong YW. 2017. Regulation of DNA demethylation by the XPC DNA repair complex in somatic and pluripotent stem cells. Genes Dev 31: 830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, et al. 1999. Genomic instability in Gadd45a-deficient mice. Nat Genet 23: 176–184. [DOI] [PubMed] [Google Scholar]
- Horvath S. 2013. DNA methylation age of human tissues and cell types. Genome Biol 14: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AM, Montagna C, Sharan S, Ni Y, Ried T, Sterneck E. 2004. Loss of CCAAT/enhancer binding protein δ promotes chromosomal instability. Oncogene 23: 1549–1557. [DOI] [PubMed] [Google Scholar]
- Huggins CJ, Malik R, Lee S, Salotti J, Thomas S, Martin N, Quinones OA, Alvord WG, Olanich ME, Keller JR, et al. 2013. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol Cell Biol 33: 3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP. 2014. Aging and epigenetic drift: a vicious cycle. J Clin Invest 124: 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Degregori J. 2006. Putting the oncogenic and tumor suppressive activities of E2F into context. Curr Mol Med 6: 731–738. [DOI] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Jung M, Pfeifer GP. 2015. Aging and DNA methylation. BMC Biol 13: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannides I, Tchkonia T, Dobson DE, Steppan CM, Cummins P, Chan G, Salvatori K, Hadzopoulou-Cladaras M, Kirkland JL. 2001. Altered expression of C/EBP family members results in decreased adipogenesis with aging. Am J Physiol Regul Integr Comp Physiol 280: R1772–R1780. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. 2010. The genetics of ageing. Nature 464: 504–512. [DOI] [PubMed] [Google Scholar]
- Kichina JV, Zeremski M, Aris L, Gurova KV, Walker E, Franks R, Nikitin AY, Kiyokawa H, Gudkov AV. 2006. Targeted disruption of the mouse ing1 locus results in reduced body size, hypersensitivity to radiation and elevated incidence of lymphomas. Oncogene 25: 857–866. [DOI] [PubMed] [Google Scholar]
- Kienhöfer S, Musheev MU, Stapf U, Helm M, Schomacher L, Niehrs C, Schäfer A. 2015. GADD45a physically and functionally interacts with TET1. Differentiation 90: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T, Pirtskhalava T, Han J, Karagiannides I. 2002. Adipogenesis and aging: does aging make fat go MAD? Exp Gerontol 37: 757–767. [DOI] [PubMed] [Google Scholar]
- Klutstein M, Nejman D, Greenfield R, Cedar H. 2016. DNA methylation in cancer and aging. Cancer Res 76: 3446–3450. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. 2009. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324: 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Misteli T. 2017. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol 18: 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg RL, Sero JE, Danielian PS, Yuan TL, Lee EY, Lees JA. 2003. The role of E2F4 in adipogenesis is independent of its cell cycle regulatory activity. Proc Natl Acad Sci 100: 2456–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May N, Mota-Fernandes D, Velez-Cruz R, Iltis I, Biard D, Egly JM. 2010. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell 38: 54–66. [DOI] [PubMed] [Google Scholar]
- Lepperdinger G. 2011. Inflammation and mesenchymal stem cell aging. Curr Opin Immunol 23: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhao M, Yin H, Gao F, Wu X, Luo Y, Zhao S, Zhang X, Su Y, Hu N, et al. 2010. Overexpression of the growth arrest and DNA damage-induced 45α gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum 62: 1438–1447. [DOI] [PubMed] [Google Scholar]
- Li Z, Gu TP, Weber AR, Shen JZ, Li BZ, Xie ZG, Yin R, Guo F, Liu X, Tang F, et al. 2015. Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res 43: 3986–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Croniger C, Arizmendi C, Harada-Shiba M, Ren J, Poli V, Hanson RW, Friedman JE. 1999. Hypoglycemia and impaired hepatic glucose production in mice with a deletion of the C/EBPβ gene. J Clin Invest 103: 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153: 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153: 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. 2009. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323: 1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Lo KA, Gordon W, Motola S, Mazor T, Fraenkel E. 2010. A quantitative model of transcriptional regulation reveals the influence of binding location on expression. PLoS Comput Biol 6: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. 2010. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20: 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Macias MI, Cordoba-Canero D, Ariza RR, Roldan-Arjona T. 2013. The DNA repair protein XRCC1 functions in the plant DNA demethylation pathway by stimulating cytosine methylation (5-meC) excision, gap tailoring, and DNA ligation. J Biol Chem 288: 5496–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massip L, Garand C, Paquet ER, Cogger VC, O'Reilly JN, Tworek L, Hatherell A, Taylor CG, Thorin E, Zahradka P, et al. 2010. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J 24: 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F, Dong E, Gavin DP, Nicoletti F, Guidotti A. 2011. Activation of group II metabotropic glutamate receptors promotes DNA demethylation in the mouse brain. Mol Pharmacol 80: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough JL, Kelly KM. 2006. Prevention and treatment of skin aging. Ann N Y Acad Sci 1067: 323–331. [DOI] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SG, Dawson KL. 1990. Red and yellow marrow in the femur: age-related changes in appearance at MR imaging. Radiology 175: 219–223. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. 2003. Manipulating the mouse embryo: a laboratory manual . Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Okitsu CY, Hsieh CL. 2007. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol 27: 2746–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Tyler JK. 2016. Epigenetics and aging. Sci Adv 2: e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. 2013. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14: 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar SA, Shao LJ, Chang JH, Wang WZ, Pathak R, Zhu XY, Wang JR, Hendrickson H, Boerma M, Sterneck E, et al. 2014. C/EBPδ deficiency sensitizes mice to ionizing radiation-induced hematopoietic and intestinal injury. PLoS One 9: e94967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RP, Subach OM, Champagne KS, Zhao R, Verkhusha VV, et al. 2008. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J Mol Biol 380: 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflaum M, Will O, Epe B. 1997. Determination of steady-state levels of oxidative DNA base modifications in mammalian cells by means of repair endonucleases. Carcinogenesis 18: 2225–2231. [DOI] [PubMed] [Google Scholar]
- Putzer BM, Engelmann D. 2013. E2F1 apoptosis counterattacked: evil strikes back. Trends Mol Med 19: 89–98. [DOI] [PubMed] [Google Scholar]
- Rahman SM, Janssen RC, Choudhury M, Baquero KC, Aikens RM, de la Houssaye BA, Friedman JE. 2012. CCAAT/enhancer-binding protein β (C/EBPβ) expression regulates dietary-induced inflammation in macrophages and adipose tissue in mice. J Biol Chem 287: 34349–34360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. 2008. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135: 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. 2010. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 20: 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesler WJ. 2001. The role of C/EBP in nutrient and hormonal regulation of gene expression. Annu Rev Nutr 21: 141–165. [DOI] [PubMed] [Google Scholar]
- Sabag O, Zamir A, Keshet I, Hecht M, Ludwig G, Tabib A, Moss J, Cedar H. 2014. Establishment of methylation patterns in ES cells. Nat Struct Mol Biol 21: 110–112. [DOI] [PubMed] [Google Scholar]
- Sarjeant K, Stephens JM. 2012. Adipogenesis. Cold Spring Harb Perspect Biol 4: a008417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A. 2013. Gadd45 proteins: key players of repair-mediated DNA demethylation. Adv Exp Med Biol 793: 35–50. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Karaulanov E, Stapf U, Döderlein G, Niehrs C. 2013. Ing1 functions in DNA demethylation by directing Gadd45a to H3K4me3. Genes Dev 27: 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schäfer A, Grummt I, Mayer C. 2009. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell 33: 344–353. [DOI] [PubMed] [Google Scholar]
- Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. 2010. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 463: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Wu H, Diep D, Yamaguchi S, D'Alessio AC, Fung HL, Zhang K, Zhang Y. 2013. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153: 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. 2006. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442: 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersbaek R, Nielsen R, John S, Sung MH, Baek S, Loft A, Hager GL, Mandrup S. 2011. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J 30: 1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersbaek R, Nielsen R, Mandrup S. 2012. Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol Metab 23: 56–64. [DOI] [PubMed] [Google Scholar]
- Siersbaek R, Rabiee A, Nielsen R, Sidoli S, Traynor S, Loft A, La Cour Poulsen L, Rogowska-Wrzesinska A, Jensen ON, Mandrup S. 2014. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep 7: 1443–1455. [DOI] [PubMed] [Google Scholar]
- Smith ML, Ford JM, Hollander MC, Bortnick RA, Amundson SA, Seo YR, Deng CX, Hanawalt PC, Fornace AJ Jr. 2000. p53-mediated DNA repair responses to UV radiation. studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol 20: 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, He C. 2013. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci 38: 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, et al. 2013. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495. [DOI] [PubMed] [Google Scholar]
- Staiger J, Lueben MJ, Berrigan D, Malik R, Perkins SN, Hursting SD, Johnson PF. 2009. C/EBPβ regulates body composition, energy balance-related hormones and tumor growth. Carcinogenesis 30: 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Grant GR, Schupp M, Tomaru T, Lefterova MI, Schug J, Manduchi E, Stoeckert CJ, Lazar MA. 2010. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev 24: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneck E, Tessarollo L, Johnson PF. 1997. An essential role for C/EBPβ in female reproduction. Genes Dev 11: 2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallen G, Riabowol K. 2014. Keep-ING balance: tumor suppression by epigenetic regulation. FEBS Lett 588: 2728–2742. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S. 1997. Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO J 16: 7432–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Lane MD. 1999. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev 13: 2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. 2003. CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci 100: 850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. 2010. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20: 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, Herrmann C, Defrance M, Sand O, Thieffry D, van Helden J. 2012. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res 40: e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly JL, Sinclair DA. 2013. Germline energetics, aging, and female infertility. Cell Metab 17: 838–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada J, Yoshida Y, Kominato Y, Auron PE. 2011. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 54: 6–19. [DOI] [PubMed] [Google Scholar]
- van Dam S, Cordeiro R, Craig T, van Dam J, Wood SH, de Magalhaes JP. 2012. GeneFriends: an online co-expression analysis tool to identify novel gene targets for aging and complex diseases. BMC Genomics 13: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pluijm I, Garinis GA, Brandt RM, Gorgels TG, Wijnhoven SW, Diderich KE, de Wit J, Mitchell JR, van Oostrom C, Beems R, et al. 2007. Impaired genome maintenance suppresses the growth hormone–insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol 5: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ven M, Andressoo JO, Holcomb VB, von Lindern M, Jong WM, de Zeeuw CI, Suh Y, Hasty P, Hoeijmakers JH, van der Horst GT, et al. 2006. Adaptive stress response in segmental progeria resembles long-lived dwarfism and calorie restriction in mice. PLoS Genet 2: e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanyushin BF, Nemirovsky LE, Klimenko VV, Vasiliev VK, Belozersky AN. 1973. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia 19: 138–152. [PubMed] [Google Scholar]
- Venza I, Visalli M, Fortunato C, Ruggeri M, Ratone S, Caffo M, Caruso G, Alafaci C, Tomasello F, Teti D, et al. 2012. PGE2 induces interleukin-8 derepression in human astrocytoma through coordinated DNA demethylation and histone hyperacetylation. Epigenetics 7: 1315–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij WP, Dolle ME, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, Wu H, Roks AJ, Botter SM, van der Eerden BC, et al. 2016. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537: 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. 1995. Impaired energy homeostasis in C/EBPα knockout mice. Science 269: 1108–1112. [DOI] [PubMed] [Google Scholar]
- Wang F, Demura M, Cheng Y, Zhu A, Karashima S, Yoneda T, Demura Y, Maeda Y, Namiki M, Ono K, et al. 2014. Dynamic CCAAT/enhancer binding protein-associated changes of DNA methylation in the angiotensinogen gene. Hypertension 63: 281–288. [DOI] [PubMed] [Google Scholar]
- Wang B, Fu X, Zhu MJ, Du M. 2017. Retinoic acid inhibits white adipogenesis by disrupting GADD45A-mediated Zfp423 DNA demethylation. J Mol Cell Biol 9: 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehle L, Raddatz G, Musch T, Dawlaty MM, Jaenisch R, Lyko F, Breiling A. 2015. Tet1 and Tet2 protect DNA methylation canyons against hypermethylation. Mol Cell Biol 36: 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. 2011. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473: 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson VL, Jones PA. 1983. DNA methylation decreases in aging but not in immortal cells. Science 220: 1055–1057. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang Y. 2017. TET-mediated active DNA demethylation. mechanism, function and beyond. Nat Rev Genet 18: 517–534. [DOI] [PubMed] [Google Scholar]
- Wu SR, Li CF, Hung LY, Huang AM, Tseng JT, Tsou JH, Wang JM. 2011. CCAAT/enhancer-binding protein δ mediates tumor necrosis factor α-induced Aurora kinase C transcription and promotes genomic instability. J Biol Chem 286: 28662–28670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo Y, Park JH, Weigel C, Liesenfeld DB, Weichenhan D, Plass C, Seo DG, Lindroth AM, Park YJ. 2017. TET-mediated hydroxymethylcytosine at the Pparγ locus is required for initiation of adipogenic differentiation. Int J Obes 41: 652–659. [DOI] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RP, Shao JZ, Xiang LX. 2011. GADD45A protein plays an essential role in active DNA demethylation during terminal osteogenic differentiation of adipose-derived mesenchymal stem cells. J Biol Chem 286: 41083–41094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT-Y, Kohlbacher O, de Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. 2013. Charting a dynamic DNA methylation landscape of the human genome. Nature 500: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Qiang L, Farmer SR. 2006. Activation of CCAAT/enhancer-binding protein (C/EBP) α expression by C/EBPβ during adipogenesis requires a peroxisome proliferator-activated receptor-γ-associated repression of HDAC1 at the C/ebpα gene promoter. J Biol Chem 281: 7960–7967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All NGS and microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO) under superseries accession number GSE99607. Primer sequences are listed in the Supplemental Material.