

In this study, Liu et al. investigated the molecular mechanisms regulating the proper organization and orderly mitosis of radial glial progenitors (RGPs), which drive the formation of a laminated mammalian cortex in the correct size. They show that RGP behavior and cortical development are controlled by partitioning-defective 3 (PARD3) in concert with dynamic HIPPO and NOTCH signaling regulation, thus providing new insights into mammalian cortical development.
Keywords: Hippo signaling, Notch signaling, megalencephaly with heterotopia, neurogenesis, pard3
Abstract
Proper organization and orderly mitosis of radial glial progenitors (RGPs) drive the formation of a laminated mammalian cortex in the correct size. However, the molecular underpinnings of the intricate process remain largely unclear. Here we show that RGP behavior and cortical development are controlled by temporally distinct actions of partitioning-defective 3 (PARD3) in concert with dynamic HIPPO signaling. RGPs lacking PARD3 exhibit developmental stage-dependent abnormal switches in division mode, resulting in an initial overproduction of RGPs located largely outside the ventricular zone at the expense of deep-layer neurons. Ectopically localized RGPs subsequently undergo accelerated and excessive neurogenesis, leading to the formation of an enlarged cortex with massive heterotopia and increased seizure susceptibility. Simultaneous removal of HIPPO pathway effectors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) suppresses cortical enlargement and heterotopia formation. These results define a dynamic regulatory program of mammalian cortical development and highlight a progenitor origin of megalencephaly with ribbon heterotopia and epilepsy.
The cerebral cortex is responsible for all higher-order brain functions, such as sensory perception, motor control, and cognition. A defining feature of the cortex is its lamination. Individual laminae or layers are segregated largely by cell type and neuronal connections (Kwan et al. 2012; Greig et al. 2013). Therefore, laminar formation is of central importance to both the structural and functional development of the cortex. Cortical lamination relies on the intricate organization and behavior of radial glial cells, the principal progenitor cells responsible for producing neurons as well as glia in the cortex (Malatesta et al. 2000; Miyata et al. 2001; Noctor et al. 2001, 2004; Tamamaki et al. 2001; Anthony et al. 2004; Kriegstein and Alvarez-Buylla 2009; Florio and Huttner 2014; Homem et al. 2015). Radial glial progenitors (RGPs) possess a highly characteristic morphology, with the cell body located in the ventricular zone (VZ), a long radial glial fiber extending to the pia, and a short ventricular endfoot reaching the lateral ventricle (Rakic 2003). Moreover, the cell bodies of RGPs in the VZ are organized in a pseudostratified manner, and the neighboring ventricular endfeet form junctions at the luminal or apical surface of the VZ (i.e., the VZ surface) (Chenn et al. 1998; Bultje et al. 2009).
During cortical development, RGPs actively divide at the VZ surface. At the early stage (e.g., before embryonic days 11–12 [E11–E12] in mice), RGPs largely undergo symmetric proliferative division to amplify the progenitor pool (Kriegstein and Alvarez-Buylla 2009; Florio and Huttner 2014; Homem et al. 2015). After that, RGPs predominantly undergo asymmetric neurogenic division to self-renew and simultaneously produce neurons either directly or indirectly via transit-amplifying progenitors (TAPs), such as outer sub-VZ (SVZ) RGPs (oRGs; also known as basal RGPs or intermediate RGPs) and intermediate progenitors (IPs) that further divide in the SVZ (Haubensak et al. 2004; Noctor et al. 2004; Englund et al. 2005; Fietz et al. 2010; Hansen et al. 2010; Reillo et al. 2011; Wang et al. 2011; Kelava et al. 2012; Betizeau et al. 2013). RGPs also produce short neural precursors located in the VZ (Gal et al. 2006). While IPs are the predominant TAPs in the developing mouse cortex, additional types of TAPs with expanded neurogenic capacity are more abundant in the developing ferret and primate cortices (Fietz et al. 2010; Hansen et al. 2010; Garcia-Moreno et al. 2012; Betizeau et al. 2013; Geschwind and Rakic 2013). Toward the late stage, RGPs mostly undergo terminal neurogenic or differentiation division to exit the cell cycle, whereas a defined fraction remains proliferative to produce glial cells (Anthony et al. 2004; Noctor et al. 2004; Kessaris et al. 2006; Gao et al. 2014). Newborn neurons migrate radially along the mother radial glial fiber to progressively constitute future cortical layers in a birth date-dependent inside-out manner (Angevine and Sidman 1961; Rakic 1971, 1988; Hatten 1999; Noctor et al. 2001; Marin and Rubenstein 2003; Thomsen et al. 2016). Early-born neurons occupy deep layers, whereas late-born neurons occupy superficial layers.
The orderly processes of neurogenesis and neuronal migration supported by RGPs orchestrate the intricate development of the cortex to the correct size and form. In particular, asymmetric division of RGPs represents an elaborate balance of proliferation and differentiation and is responsible for producing virtually all cortical neurons. However, the molecular underpinnings of RGP organization and asymmetric division remain poorly understood. Previous studies in Caenorhabditis elegans and Drosophila have identified key molecular pathways that control asymmetric cell division (Doe et al. 1998; Kemphues 2000; Jan and Jan 2001; Wodarz and Huttner 2003; Knoblich 2008). Among them, the evolutionarily conserved partitioning-defective (PARD) protein complex, consisting of PARD3, PARD6, and atypical protein kinase C (aPKC), is at the top of the genetic regulatory hierarchy (Johnson and Wodarz 2003). Notably, the PARD complex is expressed abundantly in the developing vertebrate nervous system (Manabe et al. 2002). Despite the fact that the PARD complex represents an essential entry point of dissecting the molecular control of vertebrate neural progenitor asymmetric division, its precise function in vertebrate neural progenitors remains debated (Costa et al. 2008; Bultje et al. 2009; Alexandre et al. 2010; Dong et al. 2012). Thus far, a genetic deletion analysis of Pard3 in the mammalian brain has not yet been conducted. Moreover, the key signaling pathway that mediates the action of PARD3 in controlling RGP division behavior and orderly cortical development is largely unknown.
Disruptions in neurogenesis and neuronal migration are well known to cause cortical malformations and human diseases. Early defects in RGP division lead to brain growth dysregulation and changes in cortical size, such as microcephaly (small brain) and megalencephaly (large brain) (Bizzotto and Francis 2015). On the other hand, aberrant neuronal migration results in cortical heterotopia, such as periventricular heterotopia (PH; neurons located along the ventricles) and subcortical band heterotopia (SBH; neurons located in the white matter under the cortex) (Ross and Walsh 2001; Bielas et al. 2004). Interestingly, these abnormalities may coexist in complex forms. For example, there have been individuals reported with megalencephaly and massive subcortical ribbon-like heterotopia, distinct from the classic SBH (Barkovich et al. 2012). The compound abnormalities in volume increase and massive heterotopia represent new types of human cortical malformations that are currently poorly understood.
In this study, we found that loss of PARD3 in mouse cortical RGPs causes severe malformations of the cortex, including increased volume, systematic and drastic changes in neuronal subtype composition, and massive ribbon-like heterotopia. These defects in neuronal production and organization arise progressively from temporally distinct abnormal RGP mitotic behaviors regulated by dynamic HIPPO as well as NOTCH signaling. The mutant mice exhibit elevated seizure susceptibility. Interestingly, simultaneous removal of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), two essential effectors of the HIPPO pathway, suppresses cortical enlargement and giant heterotopia formation caused by PARD3 loss. Together, our findings define a key molecular program controlling RGP behavior and cortical organization and provide insights into unusual cortical malformation characterized by megalencephaly with giant ribbon-like heterotopia and epilepsy.
Results
Pard3 deletion in RGPs leads to an enlarged cortex with giant SBH
To dissect the molecular control of RGP organization and division underlying mammalian cortical development, we generated a conditional Pard3 mutant mouse line, Pard3fl/fl (see the Materials and Methods), and crossed it to the Emx1-Cre mouse line, in which Cre recombinase is selectively expressed in RGPs of the developing cortex starting at approximately E9.5 (Gorski et al. 2002). While PARD3 was abundantly expressed in RGPs, especially at the VZ surface, in the littermate wild-type control cortex, it was depleted in the VZ of the Emx1-Cre;Pard3fl/fl conditional knockout (referred to here as Pard3 cKO) cortex at E12.5 (Supplemental Fig. S1A, arrows and insets). The residual PARD3 expression near the pial surface was likely from migrating interneurons originated from the ventral telencephalon or post-mitotic neurons produced prior to Cre recombination (Supplemental Fig. S1A, asterisks). Western blot assay also showed a drastic loss of PARD3 expression in the Pard3 cKO cortex compared with the control (Supplemental Fig. S1B). Notably, while PARD3 was ablated in RGPs, no obvious defects in junction organization and centrosome localization at the VZ surface were observed at this stage (Supplemental Fig. S1C–E).
Pard3 cKO mice were born at the expected frequency and survived to adulthood. The brains of Pard3 cKO mice appeared similar to those of littermate control mice at postnatal days 21–30 (P21–P30) (Fig. 1A). However, histological examination revealed a striking phenotype in the Pard3 cKO cortex compared with the control (Fig. 1B). A massive ribbon-like tissue mass was found underneath an apparently layered but thinner cortex. The heterotopic cortical tissue (“HC”) (Fig. 1B, asterisks) exceeded the seemingly normotopic cortex (“NC”) in thickness in the mediodorsal region, where the expression of Cre recombinase initiates (Gorski et al. 2002; Li et al. 2003), and the overall cortical region was thicker in the Pard3 cKO brain than in the control (Fig. 1B, right). Given that the “NC” and “HC” in the Pard3 cKO brain harbored the neural progeny of cortical RGPs marked by Emx1-Cre, they were both considered as the cortical tissue. Of note, selective deletion of Pard3 in post-mitotic neurons using the Nex-Cre mice (Schwab et al. 2000) did not cause any obvious defects in cortical development (Supplemental Fig. S1F–I). Together, these results suggest that selective removal of PARD3 in RGPs, but not in post-mitotic neurons, leads to a thicker cortex with massive ribbon-like heterotopia.
Figure 1.
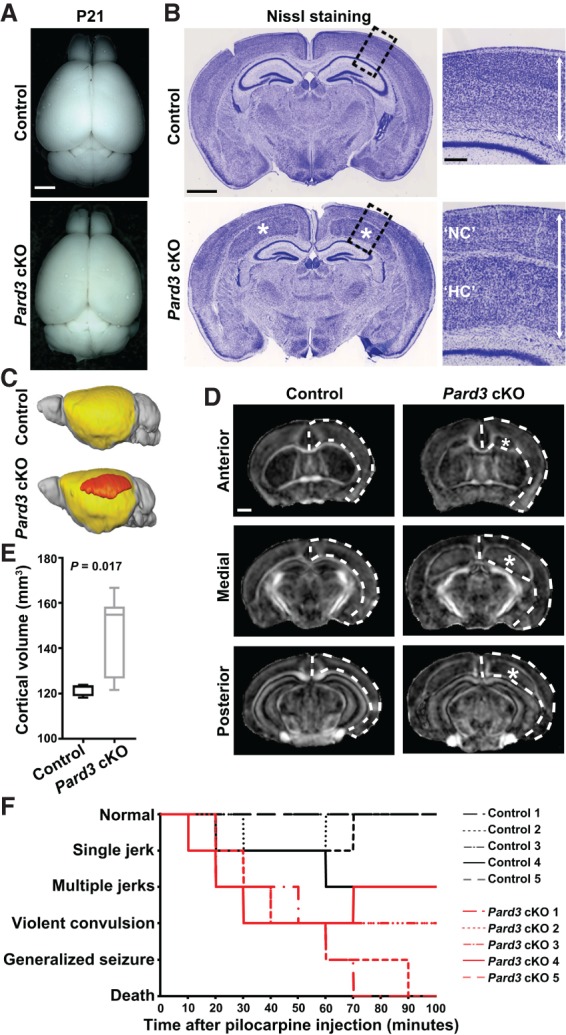
Pard3 deletion in RGPs leads to an enlarged cortex with giant SBH. (A) Representative whole-mount images of control and Pard3 cKO brains at P21. Bar, 2.5 mm. (B) Representative Nissl staining images of the control and Pard3 cKO brain coronal sections at P21. (Asterisks) Giant heterotopia. High-magnification images of the cortex (dashed lines) are shown at the right. (Double-headed arrows) Overall thickness of the cortex. Bars: left, 1.25 mm; right, 25 µm. (C) Three-dimensional (3D) rendering images of the control and Pard3 cKO brains at P21 based on magnetic resonance imaging (MRI) data. The NC is highlighted in yellow, and the heterotopia is highlighted in orange. (D) Representative fractional anisotropy (FA) maps of the control and Pard3 cKO brains at P21 along the rostrocaudal axis. Dashed lines indicate the overall cortical tissue, and asterisks indicate the giant heterotopia. Bar, 1 mm. (E) Quantification of the whole cortical volume. n = 7 brains per genotype; unpaired two-tailed t-test with Welch's correction. In the box and whisker plot, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum. (F) Behavioral responses of the control (black lines; n = 5) and Pard3 cKO (red lines; n = 5) mice at 3–4 mo of age upon pilocarpine administration.
To determine whether the heterotopia in the Pard3 cKO cortex represents PH or SBH, we stained brain sections with antibodies against SMI-312 (Supplemental Fig. S1J, green), a pan-axonal neurofilament protein, and isolectin B4 (Supplemental Fig. S1J, red), which labels blood vessels (Supplemental Fig. S1J). We found that the heterotopia was surrounded by the axonal fibers and contained blood vessels, indicative of SBH in nature. No obvious change in the number of blood vessels was observed (Supplemental Fig. S1J), consistent with the fact that Cre is not expressed in blood vessels in Emx1-Cre. In line with the increase in cortical thickness, the whole-brain magnetic resonance imaging (MRI) analysis showed that the cortical volume in the Pard3 cKO brain, including the giant heterotopia (Fig. 1C–E, orange, asterisks), was substantially larger than that of the control (Fig. 1C–E). These results demonstrate that PARD3 removal in RGPs results in an enlarged cortex with giant SBH.
Pard3 cKO mice display increased seizure susceptibility
Cortical heterotopia is often associated with epilepsy in humans (Watrin et al. 2015). Mice with cortical heterotopia show different seizure susceptibilities, depending on the genetic mutations (Croquelois et al. 2009; Gabel et al. 2013). To examine the seizure susceptibility of adult (3- to 4-mo-old) Pard3 cKO and littermate control mice, we administered pilocarpine, a chemoconvulsant commonly used for seizure induction (Kandratavicius et al. 2014), to animals and continuously monitored their behavioral response for 100 min (Fig. 1F). Remarkably, all five Pard3 cKO mice began to seize as soon as 10 min after injection and progressed to severe seizure behaviors ranging from violent convulsions to generalized seizures that lasted for 30–60 sec, and two of them died from the seizures toward the end of the observation period. In contrast, none of the littermate controls exhibited severe seizure symptoms similar to that of Pard3 cKO mice. While three out of five control mice developed mild clonic episodes such as single or multiple short jerks for 1–2 sec, the other two mice did not show any obvious seizure-like activity throughout the entire period. These results suggest that Pard3 cKO mice exhibit increased seizure susceptibility compared with the control.
Systematic alterations in neuronal composition in the Pard3 cKO cortex
The enlarged cortex in the Pard3 cKO brain, including the NC and HC, indicates abnormalities in neuronal production. To determine this, we stained P21 brain sections with antibodies against FOXP2 (a layer VI neuronal marker) (Fig. 2A) or SATB2 (a pan-neuronal marker with enrichment in superficial layers) (Fig. 2C; Greig et al. 2013). As expected, FOXP2+ neurons were located predominantly in the deep layer VI in the control cortex (Fig. 2A, left). Notably, a much thinner band of FOXP2+ neurons in the NC, in addition to a few scattered ones in the HC, was found in the Pard3 cKO brain (Fig. 2A, right). The density as well as the total number of FOXP2+ neurons were significantly decreased in the Pard3 cKO cortex compared with the control (Fig. 2A,B; Supplemental Fig. S2A,B). In contrast, we observed a substantial increase in the density and total number of SATB2+ neurons in the Pard3 cKO cortex compared with the control (Fig. 2C,D; Supplemental Fig. S2C,D). Together, these results suggest that PARD3 removal in RGPs leads to a reduced production of deep layer neurons and a concomitant overproduction of superficial layer neurons.
Figure 2.

Pard3 deletion leads to systematic alterations in neuronal composition in the cortex. (A) Representative confocal images of P21 control and Pard3 cKO cortices stained for layer VI neuronal marker FOXP2 (green) and counterstained with DAPI (blue). Bar, 70 µm. (B) Quantification of the number of FOXP2+ cells per 300-µm radial column in control and Pard3 cKO mice at P21. n = 4 brains per genotype; unpaired two-tailed t-test with Welch's correction. (C) Representative confocal images of P21 control and Pard3 cKO cortices stained for layer II/III/V neuronal marker SATB2 (red) and counterstained with DAPI (blue). Bars, 70 µm. (D) Quantification of the number of SATB2+ cells per 300-µm radial column in control and Pard3 cKO mice at P21. n = 4 brains per genotype; unpaired two-tailed t-test with Welch's correction. (E) Representative images of P21 control and Pard3 cKO cortices stained for layer V/VI neuronal marker CTIP2 (green) and layer II–IV neuronal marker CUX1 (red) and counterstained with DAPI (blue). Bars, 70 µm. (F) Quantification of the number of CTIP2+ (left) and CUX1+ (right) cells per 300-µm radial column in control and Pard3 cKO mice at P21. n = 4 brains per genotype; unpaired two-tailed t-test with Welch's correction. (G) Quantification of the relative ratio of the respective neuronal (FOXP2, CTIP2, SATB2, and CUX1) numbers in Pard3 cKO versus control cortices. n = 4 brains per genotype; χ2 test for linear trend. Note that there is a progressive change in neuronal composition. (H) The percentage of FOXP2+ (blue), CTIP2+ (red), and CUX1+ (green) cells in control and the “NC” and “HC” of Pard3 cKO cortices at P21. n = 4 brains per genotype; χ2 test. Data are presented as mean ± SEM (standard error of the mean). For all box and whisker plots, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum.
To further characterize neuronal changes, we stained sections with antibodies against CTIP2 (a layer V/VI neuronal marker (Fig. 2E, green) and CUX1 (a layer II/III/IV neuronal marker) (Fig. 2E, red; Greig et al. 2013). Compared with the control, we observed similarly positioned but thinner layers of CUX1+ and CTIP2+ neurons in the NC of the Pard3 cKO brain (Fig. 2E). On the other hand, neurons in the HC were predominantly CUX1+ neurons with a few scattered CTIP2+ neurons. Consequently, while the total density of CTIP2+ neurons was comparable between the control and Pard3 cKO cortices (Fig. 2F, left), the total density of CUX1+ neurons in the Pard3 cKO cortex was nearly double that of the control cortex (Fig. 2F, right). These results further demonstrate that PARD3 removal in RGPs leads to an excessive production of superficial layer neurons that predominantly occupy the HC in addition to the NC.
Interestingly, in comparison with the control, the changes in neuronal composition in the Pard3 cKO cortex were systematic and progressive, depending on laminar identity (Fig. 2G). While the deep layer VI FOXP2+ neurons were significantly reduced, the superficial layers II–IV CUX1+ as well as SATB2+ neurons were concurrently and robustly increased. As a result, the total number of neurons in the cortex and the cortical volume were substantially increased. In addition, while the NC in the Pard3 cKO brain contained a proportion of deep and superficial layer neurons similar to that of the control brain, the HC harbored predominantly superficial layer neurons (Fig. 2H). Coinciding with an overall increase in neuronal production, we also observed an increase in the number of glial cells in the Pard3 cKO cortex (Supplemental Fig. S2E–G). Together, these results suggest that PARD3 removal in RGPs leads to a gradual but dramatic shift toward the production of late-born neurons at the expense of early-born neurons, resulting in the formation of an enlarged cortex with two distinct parts: the NC with a relatively normal lamination but reduced in size and the HC with no lamination and excessive superficial layer neurons.
Pard3 deletion causes abnormal RGP dynamics and organization
To understand the origins of altered neurogenesis and lamination in the Pard3 cKO cortex, we next examined RGP behavior and organization at the embryonic stage. We systematically stained brain sections across the entire period of embryonic cortical development (i.e., E11.5–E17.5) with an antibody against PAX6, a transcription factor highly expressed in cortical RGPs (Gotz et al. 1998; Englund et al. 2005). While there was no obvious change in the number or localization of PAX6+ RGPs at E11.5, we observed a significant increase in the number of PAX6+ RGPs in the VZ as well as outside of the VZ (i.e., extra-VZ) (Fig. 3A, asterisks) at E12.5 in the Pard3 cKO cortex compared with the control (Fig. 3A). Moreover, as development proceeded, RGPs in the Pard3 cKO cortex exhibited distinct dynamic changes in number and localization (Fig. 3A,B). While the density of RGPs in the control cortex remained largely comparable between E13.5 and E17.5, it exhibited a time-dependent biphasic change in the Pard3 cKO cortex (Fig. 3B, left). It increased and peaked at E15.5 and then rapidly decreased after that. The changes in the VZ and extra-VZ appeared to be different (Fig. 3B, middle and right). Compared with the control, the density of RGPs in the VZ briefly increased at E12.5 and then gradually decreased after E13.5 (Fig. 3B, middle), whereas the density in the extra-VZ gradually increased until E15.5 and then decreased (Fig. 3B, right). Consistent with their RGP identity, PAX6+ cells in both the VZ and extra-VZ expressed brain lipid-binding protein (BLBP; a bona fide RGP marker) (Supplemental Fig. S3A; Hartfuss et al. 2001; Anthony et al. 2004) but not HOPX (Supplemental Fig. S4A), PTPRZ1 (Supplemental Fig. S4B), or TNC (Supplemental Fig. S4C)—previously suggested oRG markers (Pollen et al. 2015; Thomsen et al. 2016). While the VZ PAX6+ cells possessed a typical bipolar morphology with a long radial glial fiber reaching the pia (Supplemental Fig. S3B), the ectopic PAX6+ cells in the extra-VZ possessed mostly a multipolar morphology with several short processes but no obvious single long basal process (Supplemental Fig. S3C,D), indicating that they are not typical oRGs previously characterized in the developing mouse cortex (Wang et al. 2011). Based on their expression of PAX6 and BLBP as well as their mitotic behavior (e.g., symmetric proliferation) (see Figs. 4, 5), we considered them RGPs or RG-like progenitors. Together, these results show that PARD3 removal leads to abnormal dynamic changes in RGP number and organization, but not oRG generation, in the embryonic cortex. Consistent with the giant SBH formation, PARD3 removal also disrupted neuronal migration in the developing cortex (Supplemental Fig. S3E).
Figure 3.

Pard3 deletion causes abnormal RGP dynamics and organization in the embryonic cortex. (A) Representative confocal images of control and Pard3 cKO cortices stained for RGP marker PAX6 (green) and counterstained with DAPI (blue) from E11.5 to E17.5. Double-headed arrows indicate the VZ, and asterisks indicate ectopic RGPs or RG-like progenitors. Bars, 30 µm. (B) Quantification of the number of PAX6+ cells per 200-µm radial column in the entire cortex (left), the VZ (middle), and outside the VZ (i.e., extra-VZ; right) in control (blue) and Pard3 cKO (red) mice. E11.5: n = 4 control; n = 6 Pard3 cKO. E12.5: n = 6 control; n = 4 Pard3 cKO. E13.5: n = 4 control; n = 4 Pard3 cKO. E14.5: n = 6 control; n = 5 Pard3 cKO. E15.5: n = 5 control; n = 4 Pard3 cKO. E16.5: n = 4 control; n = 10 Pard3 cKO. E17.5: n = 4 control; n = 4 Pard3 cKO. Data are presented as mean ± SEM. Two-tailed Mann-Whitney test was used to assess statistical significance.
The changes in RGP number and localization in the Pard3 cKO cortex occurred at E12.5 and E13.5, prior to any obvious defect in the junction and apical domain organization at the VZ surface (Supplemental Fig. S1C–E,K), suggesting that the original defects in the Pard3 cKO cortex arise from abnormal behavior of RGPs but not junction or VZ disorganization. Consistent with this, we observed a significant increase in the number of dividing cells at the VZ surface in the Pard3 cKO cortex at E11.5 (Supplemental Fig. S5A,B). Notably, this increase in mitosis appeared before any significant change in the overall density of RGPs (Fig. 3). Together, these results suggest that PARD3 removal changes the mitotic behavior of RGPs. The increase in mitotic cells at the VZ surface persisted at E12.5 (Supplemental Fig. S5B, top). Interestingly, we observed a robust and progressive increase in the number of mitotic cells away from the VZ surface between E11.5 and E15.5 in the Pard3 cKO cortex (Supplemental Fig. S5B, middle). As a result, the total number of mitotic cells increased until E15.5 (Supplemental Fig. S5B, bottom). Consistent with the biphasic changes in RGP number and localization, we also observed a rapid and significant decrease in the number of mitotic cells at the VZ surface as well as away from the VZ surface after E15.5 (Supplemental Fig. S5B).
During cortical neurogenesis, RGPs divide at the VZ surface to produce neurons as well as IPs that continue to divide in the SVZ (Haubensak et al. 2004; Noctor et al. 2004; Englund et al. 2005). The increase in extra-VZ surface division in the Pard3 cKO cortex may be attributable to IPs. To test this, we examined the production and organization of IPs (Supplemental Fig. S6). We stained brain sections with an antibody against TBR2 (Supplemental Fig. S6, red), a T-box transcription factor highly expressed in IPs (Englund et al. 2005). As expected, TBR2+ IPs were found predominantly in the SVZ of the control cortex (Supplemental Fig. S6A, left). In contrast, TBR2+ IPs were frequently observed in the SVZ as well as the intermediate zone (IZ) and cortical plate (CP) (Supplemental Fig. S6A, right, asterisks), where ectopic PAX6+ RGPs were located, in the Pard3 cKO cortex. Interestingly, the relative ratio of TBR2+ IPs to PAX6+ RGPs was significantly lower in the Pard3 cKO cortex than in the control cortex across the embryonic stages (Supplemental Fig. S6B), indicating an overall reduction in IP generation by RGPs lacking PARD3. Collectively, these results suggest that PARD3 removal drastically alters the mitotic behavior and organization of RGPs, leading to dynamic changes in the number and localization of RGPs as well as IPs.
Pard3 deletion promotes RGP symmetric proliferation at the early neurogenic phase
To further dissect progenitor behavior that may be responsible for altered neurogenesis and enlarged cortex with massive heterotopia in the Pard3 cKO brain, we examined the cell cycle exit index of cortical progenitors prior to E15.5, when a progressive increase in RGPs was observed (Fig. 3). A single pulse of EdU was administered at E13.5, and brains were extracted at E14.5 and stained for the proliferative marker Ki67 (Fig. 4A). We analyzed the fraction of EdU+ cells that were Ki67− (i.e., exited the cell cycle) and found that there was a significant reduction in cell cycle exit in the Pard3 cKO cortex compared with the control (Fig. 4B).
Figure 4.

Pard3 deletion promotes RGP symmetric proliferation at early neurogenic phase. (A) Representative confocal images of E14.5 control and Pard3 cKO cortices stained for proliferating marker Ki67 (green) and EdU (red) and counterstained with DAPI (blue). A schematic protocol of the cell cycle exit assay is shown at the top. High-magnification images (dashed squares) are shown at the bottom. Arrows indicate Ki67+/EdU+ progenitors, and arrowheads indicate Ki67−/EdU+ post-mitotic cells. Bars: top, 30 µm; bottom, 5 µm. (B) Quantification of the cell cycle exit index in control and Pard3 cKO cortices at E14.5. n = 3 per genotype; unpaired two-tailed t-test with Welch's correction. In the box and whisker plot, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum. (C) Representative images of enhanced green fluorescent protein (EGFP)-expressing retrovirus-labeled two-cell clones stained with EGFP (green), PAX6 (red), and Ki67 (gray) in E14.5 control and Pard3 cKO cortices. A schematic protocol of the in vivo RGP division pattern assay is shown at the top. High-magnification images of representative clones (dashed rectangles) are shown at the right. Dashed polygons indicate the cell bodies of EGFP-expressing cells. Note that the clone in the control cortex is composed of one bipolar PAX6+/Ki67+ RGP and one multipolar PAX6−/Ki67− neuron, whereas the clone in the Pard3 cKO cortex is composed of two PAX6+/Ki67+ RGPs with a multipolar morphology. Bars: left, 30 µm; middle, 15 µm; right, 5 µm. (D) Representative 3D reconstruction images of a cortical hemisphere with all EGFP-labeled clones in control (top) and Pard3 cKO (bottom) embryos. Colored lines represent the pial surface and the lateral ventricle. (Green filled circles) RGPs; (yellow filled circles) IPs; (red filled circles) neurons (N). (E) The percentages of EGFP-labeled RGP–IP/neuron (green), IP–IP or neuron–neuron (red), and RGP–RGP (blue) clones in control (n = 4) or Pard3 cKO (n = 4) embryos at E14.5 (χ2 test).
The reduced rate of cell cycle exit is consistent with an overproduction of RGPs (Fig. 3) and also suggests a change in the division mode of RGPs. To test this, we performed in vivo clonal analysis to directly examine the division mode of RGPs (Fig. 4C). We injected serially diluted low-titer retroviruses expressing enhanced green fluorescent protein (EGFP) into the lateral ventricle at E13.5 and recovered the brains at E14.5 for analysis. To assess the division mode of labeled RGPs, brains were serially sectioned and stained with antibodies against PAX6 and Ki67. We identified all sparsely labeled cell pairs in the cortices that originated from individual dividing RGPs by three-dimensional (3D) reconstruction (Fig. 4C,D; Supplemental Movies S1, S2). In these experiments, PAX6+/Ki67+, PAX6−/Ki67+, and PAX6−/Ki67− cells corresponded to RGPs, IPs, and neurons, respectively. As expected, the majority of cell pairs in the control cortex contained a bipolar RGP and a multipolar IP or neuron (Fig. 4C [top], D [top], E; Supplemental Movie S1), indicating asymmetric neurogenic division. In contrast, the vast majority of cell pairs in the Pard3 cKO cortex contained two RGPs (Fig. 4C [bottom], D [bottom], E; Supplemental Movie S2), indicating symmetric proliferative division. Notably, the RGP pairs observed in the Pard3 cKO cortex often exhibited a multipolar morphology and were located outside the VZ (Fig. 4C, bottom; Supplemental Fig. S3D). Together, these results suggest that PARD3 removal leads to a switch in RGP division mode from asymmetric neurogenic division to symmetric proliferative division. This division mode switch would account for the progressive overproduction of RGPs at the early phase of cortical neurogenesis and the concomitant loss of early-born deep layer neurons.
Pard3 deletion promotes RGP differentiation and accelerates neurogenesis at the late neurogenic phase
While the production of early-born neurons was reduced due to abnormal symmetric proliferation of RGPs, the cortical volume was increased with excessive late-born neurons in the Pard3 cKO brain. These results indicate an accelerated neurogenesis at the late embryonic stage. To test this, we examined the embryonic production of SATB2+ neurons (Fig. 5A). At E15.5, the density of SATB2+ neurons in the Pard3 cKO cortex was significantly lower than that in the control cortex (Fig. 5A [left], B). In sharp contrast, at E17.5, the density of SATB2+ neurons in the Pard3 cKO cortex was substantially higher than that in the control cortex (Fig. 5A [right], B). This accelerated increase in SATB2+ neurons between E15.5 and E17.5 in the Pard3 cKO cortex occurred predominantly below the CP (the future HC), whereas the rate of increase in the CP (the future NC) was similar to that in the control cortex (Fig. 5C). An accelerated production of CTIP2+ neurons was also observed in the Pard3 cKO cortex (Supplemental Fig. S7). Together, these results suggest that PARD3 removal leads to an accelerated neurogenesis at the late neurogenic phase, predominantly producing superficial layer neurons that occupy the future HC.
Figure 5.

Pard3 deletion promotes RGP symmetric differentiation and accelerates neurogenesis at the late neurogenic phase. (A) Representative confocal images of control and Pard3 cKO cortices stained for layer II/III/V neuronal marker SATB2 (red) and counterstained with DAPI (blue) at E15.5 (left) and E17.5 (right). Asterisks indicate SATB2+ neurons located below the CP (i.e., the future “HC”). Bar, 30 µm. (B) Quantification of the number of SATB2+ cells per 200-µm radial column of the control (blue; n = 3) and Pard3 cKO (red; n = 4) embryos at E15.5 and E17.5 (unpaired two-tailed t-test with Welch's correction). Data are presented as mean ± SEM. (C) Quantification of the ratio of SATB2+ cells at E17.5 versus E15.5 per 200-µm radial column in the CP (left) and below the CP (right; i.e., the future “HC”) of the control (n = 3) and Pard3 cKO (n = 4) embryos (unpaired two-tailed t-test with Welch's correction). In the box and whisker plot, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum. (D) Representative confocal images of EGFP-expressing retrovirus-labeled two-cell clones stained with EGFP (green), PAX6 (red), and Ki67 (gray) in E16.5 control and Pard3 cKO cortices. A schematic protocol of the in vivo RGP division pattern assay is shown at the top. High-magnification images of representative clones (dashed rectangles) are shown at the right. Dashed polygons indicate the cell bodies of EGFP-expressing cells. Note that the clone in the control cortex is composed of one bipolar PAX6+/Ki67+ RGP and one multipolar PAX6−/Ki67− neuron, whereas the clone in the Pard3 cKO cortex is composed of two PAX6−/Ki67+ IPs. Bars: left, 30 µm; middle in contro, top in Pard3 cKO, 15 µm; right in control, bottom in Pard3 cKO, 5 µm. (E) Representative 3D reconstruction images of a cortical hemisphere with all EGFP-labeled clones in control (top) and Pard3 cKO (bottom) embryos. Colored lines represent the pial surface and the lateral ventricle. (Green filled circles) RGPs; (yellow filled circles) IPs; (red filled circles) neurons (N). (F) The percentages of EGFP-labeled RGP–IP/neuron (green), IP–IP or neuron–neuron (red), and RGP–RGP (blue) clones in control (n = 4) or Pard3 cKO (n = 4) embryos at E16.5 (χ2 test).
To further understand the cellular basis of the accelerated neurogenesis, we examined the division mode of RGPs in vivo at the late neurogenic stage (i.e., after E15.5) by performing in vivo clonal analysis (Fig. 5D). In the control cortex, the vast majority of sparsely labeled cell pairs contained a RGP and an IP or neuron (Fig. 5D [top], E [top], F; Supplemental Movie S3), indicative of asymmetric neurogenic division. In contrast, in the Pard3 cKO cortex, a large fraction of sparsely labeled cell pairs contained two IPs or two neurons (Fig. 5D [bottom], E [bottom], F ; Supplemental Movie S4), indicative of symmetric differentiation/neurogenic division. Together, these results suggest that PARD3 removal results in a switch in RGP division mode from asymmetric neurogenic division to symmetric differentiation division at the late neurogenic phase. Notably, PARD3 removal also led to a randomization of the mitotic spindle orientation of dividing RGPs at or away from the VZ surface (Supplemental Fig. S8), indicating that PARD3 regulates the mitotic spindle orientation of RGPs. Given the initial overproduction of RGPs, this division mode switch would lead to an accelerated and excessive production of late-born neurons that largely contribute to the heterotopia and enlarged cortex.
Temporally distinct alterations in RGP division coincide with HIPPO signaling
Our results showed that PARD3 removal leads to temporally distinct changes in RGP mitotic behavior, raising the intriguing question about the underlying mechanism of the differences. Recent studies in cell cultures suggest that cell polarity proteins, including PARD3, regulate the HIPPO pathway (Lv et al. 2015; Zhang et al. 2016), a conserved signaling pathway that controls cell proliferation and tissue development (Pan 2010; Yu and Guan 2013). To test whether HIPPO signaling is associated with abnormal behavior of RGPs in the Pard3 cKO cortex, we stained cortical sections with an antibody against YAP, a major downstream effector of the HIPPO pathway (Fig. 6; Pan 2010; Yu and Guan 2013). Suppression of HIPPO signaling typically leads to elevated YAP expression and/or reduced YAP phosphorylation and thereby promotes cell proliferation, whereas activation of HIPPO signaling usually results in decreased YAP expression and/or increased YAP phosphorylation and facilitates cell differentiation (Pan 2010; Yu and Guan 2013).
Figure 6.

Pard3 deletion leads to temporally distinct alterations in the level of HIPPO signaling. (A) Representative confocal images of E13.5 control and Pard3 cKO cortices stained for PAX6 (green) and HIPPO pathway effector YAP (red) and counterstained with DAPI (blue). High-magnification images (dashed rectangles) of RGPs in the VZ (arrows; areas 1) and extra-VZ (filled arrowheads; areas 2) are shown at the right. Open arrowheads indicate YAP expression in the blood vessel, which exhibits no obvious change between control and Pard3 cKO cortices. High-magnification images, including those of individual RGPs (dashed lines), are shown at the right. Note the increased nuclear YAP signals in RGPs in the VZ and extra-VZ upon Pard3 deletion. Bars: left, 30 µm; top right, 15 µm; bottom right, 2.5 µm. (B) Representative confocal images of E13.5 control and Pard3 cKO cortices stained for PAX6 (green) and phospho-YAP (red) and counterstained with DAPI (blue). Bars: left, 30 µm; right, 15 µm. (C) Quantification of YAP staining signal intensity per RGP in the VZ and extra-VZ of the control and Pard3 cKO cortices at E13.5. n = 3 brains per genotype; unpaired two-tailed t-test with Welch's correction. (A.U.) Arbitrary unit. (D) Quantification of phospho-YAP staining signal intensity per RGP in the VZ and extra-VZ of the control and Pard3 cKO cortices at E13.5. n = 3 brains per genotype; unpaired two-tailed t-test with Welch's correction. (E) Representative confocal images of E17.5 control and Pard3 cKO cortices stained for PAX6 (green) and YAP (red) and counterstained with DAPI (blue). Bars: left, 30 µm; right, 15 µm. (F) Quantification of YAP staining signal intensity per RGP in the VZ and extra-VZ of the control and Pard3 cKO cortices at E17.5. n = 3 brains per genotype; unpaired two-tailed t-test with Welch's correction. For all box and whisker plots, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum.
Interestingly, compared with the control, the expression of YAP in RGPs, including in the nucleus, was increased (Fig. 6A,C)—whereas the expression of phosphorylated YAP (pYAP) in RGPs was significantly decreased (Fig. 6B,D)—in the Pard3 cKO cortex at E13.5. These results suggest that PARD3 removal in RGPs leads to decreased HIPPO activity at the early neurogenic stage, which would promote excessive RGP proliferation. In contrast, compared with the control, YAP expression in RGPs was substantially reduced in the Pard3 cKO cortex at the late neurogenic phase (Fig. 6E,F), consistent with accelerated differentiation/neurogenesis at this stage. Notably, no obvious change in the expression of YAP or pYAP in the blood vessels was observed (Fig. 6A,B,E, open arrowheads). Together, these results suggest that PARD3 removal causes temporally distinct changes in HIPPO signaling in RGPs that coincide with the alterations in their mitotic behavior.
HIPPO signaling regulation depends on both Pard3 deletion and NOTCH activity
Temporally distinct changes in YAP expression in Pard3-deficient RGPs indicate that additional factors are critical for HIPPO signaling regulation in response to PARD3 removal. To explore this, we examined the role of NOTCH signaling, a central signaling pathway in regulating RGP division and progeny output (Chenn and McConnell 1995; Gaiano et al. 2000; Li et al. 2003; Petersen et al. 2004; Dong et al. 2012). NOTCH signaling activity is thought to be high in RGPs to maintain their progenitor identity. To test whether NOTCH activity is related to temporally distinct changes in YAP expression and RGP mitotic behavior upon PARD3 removal, we first examined NOTCH activity in RGPs at different embryonic stages. To achieve this, we took advantage of a well-characterized NOTCH signaling activity reporter mouse line that contains the consensus NOTCH effector C promoter-binding factor 1 (CBF1; also called RBPJ) response element to direct the expression of a nuclear-localized Venus fluorescent protein (CBF:H2B-Venus) (Nowotschin et al. 2013). Using this reporter line, the endogenous NOTCH signaling activity can be inferred based on Venus expression. As expected, Venus expression was observed predominantly in the VZ, where RGPs reside (Supplemental Fig. S9A). Moreover, the Venus intensity in RGPs progressively decreased as development proceeded (Supplemental Fig. S9B), indicative of a gradual decrease in NOTCH activity. Interestingly, the decrease in Venus expression was most prominent between E14.5 and E15.5, suggesting a sharp reduction in NOTCH signaling in RGPs at E15.5 under normal conditions. Notably, this drastic decrease in NOTCH activity temporally coincides with HIPPO signaling change as well as the alteration in RGP mitotic behavior in the Pard3 cKO cortex. These results are consistent with the notion that NOTCH activity level influences HIPPO signaling and YAP expression and/or phosphorylation in RGPs in response to PARD3 removal.
To directly test this, we examined whether enhanced NOTCH signaling activity would promote YAP expression in RGPs lacking PARD3 at the late neurogenic stage. We took advantage of the conditional Notch1 intracellular domain expression allele R26-LSL-NICD, which allows Cre recombinase-dependent expression of NICD with constitutive signaling activity (Murtaugh et al. 2003). In addition, we used a tamoxifen (TM)-inducible cortical RGP-specific Cre mouse line, Emx1-CreER (Kessaris et al. 2006), to selectively remove Pard3 as well as drive NICD expression in a temporal-specific manner. To ensure effective removal of PARD3 toward the late neurogenic phase, we treated timed pregnant single- or double-mutant mice with a single dose of TM at E13.5 and examined YAP expression in the cortex at E17.5 (Fig. 7). Consistent with our previous observation, YAP expression was reduced in Pard3-deleted RGPs compared with the control (Fig. 7A [top], B). Interestingly, while YAP expression in NICD-expressing RGPs did not obviously change, it was substantially increased in Pard3-deleted and NICD-expressing RGPs, especially in those located outside the VZ (Fig. 7A [bottom], B). Together, these results suggest that PARD3 removal in conjunction with high NOTCH activity promotes YAP expression in RGPs.
Figure 7.

PARD3 and NOTCH signaling work synergistically to regulate HIPPO signaling. (A) Representative confocal images of E17.5 control (top left), Emx1-CreER;Pard3fl/fl (top right), Emx1-CreER;R26-LSL-NICD (bottom left) and Emx1-CreER;Pard3fl/fl;R26-LSL-NICD (bottom right) cortices stained for PAX6 (green) and YAP (red) and counterstained for DAPI (blue). A schematic protocol of TM induction is shown at the top. High-magnification images (dashed squares) of the RGPs in the VZ and extra-VZ (filled arrows and arrowheads and insets) are shown at the right. Open arrowheads indicate YAP signal in the blood vessel. Dashed circles indicate the cell bodies of representative RGPs in the extra-VZ. High-magnification images are shown in the insets. Note that the VZ and ectopic PAX6+ cells in the Emx1-CreER;Pard3fl/fl;R26-LSL-NICD cortex exhibit a strong YAP staining signal. Bars: left, 50 µm; right, 15 µm; inset, 5 µm. (B) Quantification of the YAP staining signal intensity per RGP in the VZ (left) and extra-VZ (right) at E17.5. n = 3 brains per genotype; unpaired two-tailed t-test with Welch's correction. (A.U.) Arbitrary unit. In the box and whisker plot, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum.
As expected on the basis of NOTCH and HIPPO signaling activities, while the number of PAX6+ RGPs was reduced in the Pard3-deleted cortex, it was increased in the NICD-expressing cortex compared with the control (Supplemental Fig. S10A,B). Importantly, the number of RGPs was further substantially increased in the Pard3-deleted and NICD-expressing cortex, especially outside the VZ (Supplemental Fig. S10A, asterisk). Notably, compared with the control, the NOTCH signaling activity did not exhibit any obvious change at the early neurogenic phase but was significantly decreased at the late neurogenic phase in the Pard3 cKO cortex (Supplemental Fig. S9C,D), indicating that NOTCH signaling does not change prior to PARD3 removal. Together, these results suggest that PARD3, NOTCH activity, and HIPPO signaling act synergistically in regulating RGP mitotic behavior.
To further test this, we examined whether the initial excessive proliferation of RGPs and the reduction in HIPPO signaling upon PARD3 removal at the early neurogenic stage were dependent on NOTCH activity. To suppress NOTCH activity, we used a conditional deletion allele of Rbpj (i.e., Cbf1) that encodes a transcriptional regulator essential for NOTCH signaling (Han et al. 2002). We generated cortex-specific single or double knockout of Pard3 and Rbpj using Emx1-Cre. Notably, while we observed a drastic overproduction and ectopic localization of PAX6+ RGPs in the Pard3 cKO cortex, there was a strong reduction of PAX6+ RGPs and a concomitant increase in TUJ1+ post-mitotic neurons in the Pard3 and Rbpj conditional double-knockout (Pard3;Rbpj cDKO) cortex, similar to that in the Rbpj cKO cortex (Supplemental Fig. S10C–E). Moreover, YAP expression level was significantly decreased in the Pard3;Rbpj cDKO cortex compared with the control (Supplemental Fig. S9E,F). Together, these results suggest that the excessive RGP proliferation and HIPPO signaling suppression at the early neurogenic phase caused by PARD3 removal depend on the relatively high NOTCH signaling.
Deletion of Yap and Taz fully suppresses giant heterotopia formation
To further determine the functional role of HIPPO signaling in driving defective RGP behavior and cortical malformation in the absence of PARD3, we took advantage of the conditional deletion alleles of Yap and Taz (Reginensi et al. 2013) and generated cortical-specific single, double, or triple knockout of Pard3, Yap, or Taz using Emx1-Cre (Fig. 8; Supplemental Fig. S10). Notably, simultaneous deletion of Yap and Taz suppressed the drastic overproduction and ectopic localization of PAX6+ RGPs in the Pard3 cKO cortex at the embryonic stage (Fig. 8A,B). No obvious change in the density or localization of RGPs was observed in the Yap and Taz single- or double-cKO cortex (Fig. 8A,B). These results strongly suggest that YAP and TAZ play an essential role in mediating abnormal behavior and organization of RGPs lacking PARD3.
Figure 8.

Yap and Taz deletion suppresses ectopic RGP generation and heterotopia formation in the Pard3-deficient cortex. (A) Representative confocal images of E14.5 control, Pard3 cKO, Yap;Taz cDKO, and Pard3;Yap;Taz conditional triple-knockout (cTKO) cortices stained for PAX6 (green) and counterstained for DAPI (blue). Asterisk indicates the ectopic PAX6+ RGPs in the Pard3 cKO cortex. Bar, 30 µm. (B) Quantification of the number of PAX6+ cells per 200-µm radial column in E14.5 control, Pard3 cKO, Yap;Taz cDKO, and Pard3;Yap;Taz cTKO cortices. n = 6 brains per genotype; unpaired two-tailed t-test with Welch's correction. (C) Representative nissl staining images of P12 control, Pard3 cKO, Yap;Taz cDKO, and Pard3;Yap;Taz cTKO brain sections. (Asterisks) Giant heterotopia; (arrowheads) defective VZ surface and hydrocephalus. Bar, 2.5 mm. (D) Representative confocal images of P12 control, Pard3 cKO, Yap;Taz cDKO, and Pard3;Yap;Taz cTKO cortices stained for layer V/VI neuronal marker CTIP2 (green) and layer II–IV neuronal marker CUX1 (red) and counterstained for DAPI (blue). Bar, 70 µm. (E) Quantification of the number of CTIP2+ (top) and CUX1+ (bottom) cells per 300-µm radial column in control, Pard3 cKO, Yap;Taz cDKO, and Pard3;Yap;Taz cTKO cortices at P12. n = 6 brains per genotype; unpaired two-tailed t-test with Welch's correction. For all box and whisker plots, the center line indicates the median, the box indicates the interquartile range, and the whiskers indicate minimum and maximum.
Strikingly, in contrast to the massive heterotopia formation in the Pard3 cKO cortex, we observed no heterotopia formation in the Pard3, Yap, and Taz conditional triple-knockout (Pard3;Yap;Taz cTKO) cortex at the postnatal stage (Fig. 8C). Compared with the control, the lamination was largely normal in the Pard3;Yap;Taz cTKO cortex. There was no increase in the density of superficial layer neurons; instead, we observed a relatively small but significant reduction in the densities of both superficial and deep layer neurons in the Pard3;Yap;Taz cTKO cortex as well as in the Yap;Taz cDKO cortex (Fig. 8D,E). Consistent with a previously reported role of YAP in regulating the generation of ependymal cells that line the ventricle (Park et al. 2016), we observed a loss of ependymal cells (Supplemental Fig. S10F) and severe hyhrocephalus (Fig. 8C, arrowheads) in the Yap;Taz cDKO and Pard3;Yap;Taz cTKO brains, which may contribute to the small decrease in cortical size. Together, these results clearly suggest that cortical enlargement and massive heterotopia formation in the Pard3 cKO cortex depend on YAP and TAZ, two essential transcriptional coactivators of the HIPPO pathway.
Discussion
Proper RGP organization and division are instrumental to orderly neurogenesis and cortical development; however, the molecular control of the orderly RGP behavior in the embryonic cortex remains poorly understood. In this study, we reveal for the first time that the temporally distinct activities of PARD3 in concert with dynamic HIPPO and NOTCH signaling regulate RGP division and cortical formation. At the early neurogenic phase, when NOTCH activity is relatively high, PARD3 removal suppresses HIPPO signaling and promotes RGP symmetric proliferation, leading to an excessive production of RGPs or RG-like progenitors largely located outside the VZ at the expense of early-born deep layer neurons. In sharp contrast, at the late neurogenic phase, when NOTCH activity is relatively low, PARD3 removal enhances HIPPO signaling, reduces NOTCH activity, and facilitates RGP symmetric differentiation, resulting in an accelerated and overproduction of late-born superficial layer neurons predominantly residing below the emerging cortex. As a result, the mutant cortex is significantly enlarged with massive SBH, and the mutant animal exhibits increased seizure susceptibility. Simultaneous removal of YAP and TAZ, two key transcriptional coactivators of the HIPPO pathway, prevents RGP overproliferation and delocalization in the absence of PARD3 and fully suppresses excessive neurogenesis and giant heterotopia formation. Together, our data define a key molecular and signaling program of PARD3, HIPPO signaling, and NOTCH activity, which act in concert to ensure orderly RGP division behavior and neuronal production in the mammalian cortex.
Previous studies using shRNAs or morpholinos have implicated PARD3 in regulating neural progenitor cell division in vertebrates. However, its function appears to be contrary in different systems (i.e., zebrafish vs. mice) with regard to promoting the generation of progenitors (proliferation) (Alexandre et al. 2010; Dong et al. 2012) or neurons (differentiation) (Costa et al. 2008; Bultje et al. 2009). Interestingly, we found that PARD3 removal can facilitate either proliferation or differentiation of RGPs, depending on the developmental stage, in conjunction with HIPPO and NOTCH signaling activities. Our finding that PARD3 removal promotes RGP proliferation at the early neurogenic stage (e.g., E13.5) is essentially opposite to the previous study showing that PARD3 knockdown causes RGP premature cell cycle exit and depletion (Costa et al. 2008). The temporally distinct function of PARD3 in regulating progenitor behavior has not been suggested in the previous studies, including those in C. elegans and Drosophila. Our data indicate that PARD3 acquires complex functions in supporting RGP division and orderly neurogenesis in the mammalian cortex.
A recently study in Madin-Darby canine kidney (MDCK) epithelial or tumor cells suggested that PARD3 has a dual role in regulating HIPPO signaling, depending on the cellular context (Zhang et al. 2016). Our data suggest that NOTCH activity can be a key factor in determining the precise regulation of HIPPO signaling by PARD3. Previous genome-wide analysis has implicated YAP as a direct transcriptional target of NOTCH activity (Li et al. 2012). Notably, we found that elevated NOTCH activity alone (e.g., NICD expression) is ineffective in promoting YAP expression. On the other hand, elevated NOTCH activity together with PARD3 removal strongly promotes YAP expression. These results suggest that cellular organization such as polarity and junction formation related to PARD3 expression is critical for HIPPO signaling regulation, consistent with their functional context in tissue development and homeostasis (Pan 2010; Yu and Guan 2013). PARD3 has also been shown recently to regulate the activation of TAZ (Lv et al. 2015), the other downstream transcription coactivator of the HIPPO pathway (Pan 2010). As shown in our functional analysis, both YAP and TAZ play a critical role in PARD3-dependent regulation of RGP behavior and cortical neurogenesis.
The initial excessive proliferation of RGPs upon PARD3 removal occurs at E11–E12, prior to any obvious defects in the organization of the VZ, including the integrity of the apical VZ surface. While PARD3 has been shown previously to be enriched at the VZ surface junction formed between the endfeet of neighboring RGPs (Bultje et al. 2009), our data suggest that PARD3 is not essential for the maintenance of existing junctions at the VZ surface. Moreover, our data suggest that a primary function of PARD3 is to regulate progenitor division mode and daughter cell fate specification in concert with NOTCH and HIPPO signaling activities. On the other hand, PARD3 is likely required for junction formation by newly specified RGPs. As a result, overproduced RGPs lacking PARD3 fail to form junctions at the VZ surface (i.e., be anchored to the apical junction belt of the VZ) and consequently become ectopically localized outside the VZ. As time proceeds, accumulating RGPs become ectopically located, and the junction organization at the VZ surface is progressively disrupted.
Of note, even when junction defects were observed toward the late neurogenic stage, there was no drastic global disorganization of the VZ in the Pard3 cKO cortex. This is clearly distinct from the RhoA or junction gene (e.g., Cadherin2 or α-E-Catenin) mutant cortex (Cappello et al. 2012; Gil-Sanz et al. 2014; Schmid et al. 2014) despite the fact that a similar SBH (but with no obvious cortical enlargement) is observed. Due to the severe disruption of the junction and VZ, dividing cells labeled by PHH3 in the embryonic RhoA or junction gene mutant cortex are broadly dispersed. It is unclear whether the division mode of RGPs in these mutant cortices exhibits any changes. In contrast, many dividing cells in the embryonic Pard3 cKO cortex remain aligned at the VZ surface (e.g., E15.5), as expected for dividing VZ RGPs, in addition to those ectopically localized dividing cells. Therefore, our data reveal that the initial switch in RGP division mode producing excessive ectopically localized RGPs followed by an accelerated neurogenesis triggers the formation of massive SBH and an enlarged cortex. Consistent with this, we found that simultaneous deletion of Yap and Taz suppresses the excessive production and ectopic localization of RGPs due to Pard3 deletion and completely prevents the formation of heterotopia. Our data not only demonstrate that the overproliferation of RGPs depends on HIPPO signaling but also show that RGP overproliferation is responsible for their delocalization away from the VZ and the subsequent heterotopia formation.
While the overall thickness of the NC in the Pard3 cKO brain is greatly reduced, the relative proportion of individual neuronal layers appears to be largely comparable with the control cortex. On the other hand, the HC or hetertopia is predominantly composed of late-born superficial layer neurons. These results suggest that the progenitor origins of the NC and heterotopia are different. It is intriguing that a normally laminated cortex consisting of both superficial and deep layer neurons forms above the massive heterotopia. The orderly production and migration of neurons occupying the NC likely occur in the Pard3 cKO brain before E15.5, when the VZ and radial glial fiber scaffold remain prominent. On the other hand, the accumulating RGPs in the ectopic location undergo excessive proliferation and do not effectively generate neurons initially. Remarkably, the heterotopia is completely suppressed in the Pard3;Yap;Taz cTKO brain. In accordance with this, the ectopic RGPs are greatly reduced in the Pard3;Yap;Taz cTKO cortex at the embryonic stage. These results confirm that the massive heterotopia originates from the ectopic RGPs. While cortical heterotopia has been observed previously (Cappello et al. 2012; Gil-Sanz et al. 2014; Schmid et al. 2014; Jossin et al. 2017), our study defines the distinct progenitor origin of heterotopia as well as the underlying signaling mechanism.
The initial change in RGP mitotic behavior and the adult phenotype in the Pard3 cKO cortex are reminiscent of those in HeCo mice, which carry a retrotransposon insertion in echinoderm microtubule-associated protein-like 1 (Eml1) (Croquelois et al. 2009; Kielar et al. 2014). It is intriguing that both PARD3 and EML1 are microtubule-interacting proteins (Chen et al. 2013), indicating a potential importance of microtubule regulation in RGPs for division mode control. Interestingly, EML1 mutations have been genetically linked to human megalencephaly and giant ribbon-like heterotopia with epilepsy and intellectual disability (Kielar et al. 2014). This relatively rare type of cortical malformation is distinct from the classic human SBH linked to mutations in genes encoding other microtubules or microtubule-associated proteins predominantly involved in controlling neuronal migration, such as α1-Tubulin (TUBA1A), Lissencephaly 1 (LIS1, also known as PAFAH1B1), and Doublecortin (DCX) (Bizzotto and Francis 2015). Notably, the etiology of megalencephaly in association with giant heterotopia in human patients remains largely unclear, as the cortex in HeCo mice is not obviously enlarged (normocephalic). In comparison, our study demonstrates that Pard3 deletion causes a drastic cortical malformation that is highly reminiscent of human megalencephaly with giant ribbon-like heterotopia as well as increased seizure susceptibility. Therefore, our study highlights a progenitor origin of cortical malformation linked to progenitor organization and division mode control and provides insights into the pathophysiology of a severe atypical form of human disorder.
Materials and methods
Animals
The Pard3 knockout-first (Pard3tm1a(KOMP)Wtsi) embryonic stem cell line (EPD0334_1_C04) was obtained from the International Knockout Mouse Consortium. After confirmation, embryonic stem cell clones were injected into C57BL/6J blastocysts, and the resulting chimeras were crossed with C57BL/6J females to obtain germline transmission. The knockout-first allele was converted to the conditional allele by crossing with B6.Cg-Tg(ACTFLPe) mice (The Jackson Laboratory, stock no. 005703) to excise the gene trap cassette. The resulting Pard3fl/+ conditional mice were subsequently intercrossed to generate Pard3fl/fl mice, in which exons 8 and 9 were flanked by loxP sites. Deletion of exons 8 and 9 causes a frameshift in the subsequent exons. Emx1-Cre (stock no. 005628) and Nex-Cre (Schwab et al. 2000) mice were used to delete Pard3 in the cortex. R26-LSL-NICD mice (stock no. 008159) were purchased from The Jackson Laboratory. Emx1-CreER, Rbpjfl/fl, CBF:H2B-Venus, Yapfl/fl, and Tazfl/fl (Reginensi et al. 2013) mouse lines were kindly provided by Dr. N. Kessaris (University College London, UK), Dr. B.G. Novitch (University of California at Los Angles), Dr. A.K. Hadjantonakis (Memorial Sloan Kettering Cancer Center), Dr. Alexander L. Joyner (Memorial Sloan Kettering Cancer Center), and Dr. Jeff Wrana (The Lunenfeld-Tanenbaum Research Institute, Canada), respectively. Genotyping was carried out using standard PCR protocols. The mice were maintained at the facilities of Memorial Sloan Kettering Cancer Center, and all animal procedures were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee. For timed pregnancies, the plug date was designated as E0, and the date of birth was defined as P0.
Tissue preparation, immunohistochemistry, confocal imaging, and quantification
Embryonic or adult mice were transcardially perfused with 4% ice-cold paraformaldehyde (PFA) in PBS (pH 7.4). Brains were dissected out and post-fixed in 4% PFA solution for 4 h at 4°C. Coronal sections were prepared at 20 µm with a cryostat (Leica Microsystem) or at 40 µm with a vibratome (Leica Microsystem). Sections were blocked in 10% serum and 0.1% Triton-X in PBS and incubated with the primary antibody overnight at 4°C. The primary antibodies used included rabbit antibody to PARD3 (1:200 for immunofluorescence and 1:1000 for Western blots [Sigma, HPA030443]), biotin-conjugated antibody to Isolectin B4 (1:500; Sigma, L2140), mouse antibody to SMI-312 (1:500; Biolegend, smi-312r), goat antibody to FOXP2 (1:200; Santa Cruz Biotechnology, sc-21069), rat antibody to CTIP2 (1:200; Abcam, ab18465), rabbit antibody to CUX1 (1:500; Santa Cruz Biotechnology, sc-13024), rabbit antibody to SATB2 (1:500; Abcam, ab92446), rabbit antibody to PAX6 (1:500; Biolegend, prb-278p), mouse antibody to PAX6 (1:50; Developmental Studies Hybridoma Bank, PAX6s), chicken antibody to GFP (1:1000; Fisher, GFP-1020), mouse antibody to Ki67 (1:200; BD Transduction Laboratories, 610968), mouse antibody to TUJ1 (1:500; Covance, MMS-435P), rabbit antibody to PCNT (1:500; Biolegend, prb-432c), mouse antibody to ZO-1 (1:500; Fisher, 33–9100), mouse antibody to N-Cadherin (1:200; BD Biosciences, 610920), rabbit antibody to β-Catenin (1:200; BD Biosciences, 610153), rat antibody to TBR2 (1:200; eBioscience, 12-4875-12), mouse antibody to YAP (1:200; Santa Cruz Biotechnology, sc-101119), rabbit antibody to phospho-YAP (1:200; Cell Signaling, 4911), mouse antibody to S100β (1:500; Thermo Fisher, MA1-25005), rabbit antibody to OLIG2 (1:500; Millipore, AB9610), rat antibody to phosphorylated histone H3 (1:200; Abcam, ab10543), rabbit antibody to BLBP (1:200; Abcam, ab32423), mouse antibody to P-Vimentin (1:500; Abcam, ab22651), rabbit antibody to PTPRZ1 (1:1000; Sigma-Aldrich, HPA015103), rabbit antibody to TNC (1:200; Abcam, ab108930), and rabbit antibody to FOXJ1 (1:200; Invitrogen, 19-9965-80). EdU staining was performed according to the manufacturer's protocol (Life Technologies). Nuclei were stained with DAPI (Sigma, D8417). Alexa fluor 488-, 546-, or 647-conjugated secondary antibodies (1:1000; Life Technologies) were used to visualize the signals of primary antibodies. For Nissl staining, the 40-µm vibratome sections were mounted onto positively charged plus slides and air-dried overnight. After the defat step, slides were submerged briefly in staining solution (5 g/L cresyl violet acetate in 0.3% acetic acid [Sigma-Aldrich]), dehydrated, and mounted. Images were acquired with a confocal microscope (Olympus, FV1000) or a brain scanner (Hamamatsu Photonics, NanoZoomer 2.0-HT), and analyzed with Volocity (ImproVision), Neurolucida (MBF Bioscience), and Photoshop (Adobe Systems).
Animals were assigned to groups according to the genotyping results. Age-matched wild-type littermates were used as the controls in all experiments. For cell number quantification, all cells positive for the corresponding markers were counted in a 200-µm width (embryonic) or 300-µm width (postnatal) columnar area from the lateral ventricle to the pial surface in similar regions of the neocortex (embryonic) or the primary somatosensory cortex (postnatal). At least three animals in each group and two sections in each hemisphere were analyzed in all experiments. Both male and female mice were used in experiments. Data are presented as mean ± SEM (standard error of the mean), and statistical differences were determined using Student's two-tailed t-test, χ2 test, ANOVA test, or nonparametric Mann-Whitney test and Kruskal-Wallis test. Statistic significance was set as P < 0.05.
Cell cycle exit analysis
To label proliferating cells, pregnant females were injected intraperitoneally with 10 mg of EdU per kilogram of body weight. At 24 h after the injection, embryonic brains were collected, sectioned, and stained with the antibodies to EdU and Ki67. The cell cycle exit index was analyzed as the fraction of EdU+/Ki67− cells among all EdU+ cells.
In vivo RGP division pattern assay
To sparsely label dividing RGPs at the VZ surface of the embryonic cortex, serially diluted low-titer replication-incompetent EGFP-expressing retroviruses were injected into the lateral ventricle of embryos in utero as described previously (Yu et al. 2009). At 24 h after the injection, embryonic brains were collected and serially sectioned using a vibratome (Leica Microsystem). Consecutive sections covering the entire cortex were collected and stained with the antibodies against EGFP, PAX6, and Ki67. The entire cortices were reconstructed to recover all EGFP-labeled cell pairs by Neurolucida (MBF Bioscience) on an upright microscope equipped with epifluorescence illumination and cooled charge-coupled device camera (Zeiss).
Seizure susceptibility analysis
Seizure susceptibility analysis was carried out as described previously (Croquelois et al. 2009). In brief, Pard3 cKO mice and control littermates at 3–4 mo of age received intraperitoneal injections of 1 mg/kg scopolamine methyl bromide (Sigma, S8502) in sterile saline 30 min prior to pilocarpine injection to reduce its peripheral cholinergic effects. Subsequently, mice were injected intraperitoneally with 300 mg of pilocarpine hydrochloride (Sigma, P6503) per kilogram of body weight in sterile saline. Mice were observed continuously for 100 min after the injection, and the most severe symptom was documented every 10 min. Seizure severity was determined by the following five stages: stage 0, normal; stage 1, single jerk; stage 2, multiple jerks; stage 3, violent convulsion; stage 4, generalized seizure; and stage 5, death. Jerks refer to an uncontrolled shake of the neck and head, convulsion refers to an uncontrolled shake of the whole body, and generalized seizure refer to the loss of consciousness followed by generalized body stiffening for 30–60 sec.
MRI
Ex vivo MRI of 4% PFA fixed mouse brain specimens was performed on a horizontal 7 Tesla MR scanner (BrukerBiospin, Billerica) with a triple-axis gradient system. Images were acquired using a quadrature volume excitation coil (72-mm diameter) and a receive-only four-channel phased array cryogenic coil. The specimens were imaged with skull intact and placed in a syringe filled with Fomblin to prevent tissue dehydration. High-resolution diffusion MRI data were acquired using a modified 3D diffusion-weighted gradient and spin-echo (DW-GRASE) sequence (Wu et al. 2013) with the following parameters: echo time (TE)/repetition time (TR) = 33/400 msec, two signal averages, field of view (FOV) = 16 mm × 12.8 mm × 18 mm, resolution = 0.125 mm × 0.125 mm × 0.2 mm, two nondiffusion weighted image (b0), 10 diffusion directions, and b = 2000 sec/mm2. The total imaging time was ∼1 h for each specimen. From the diffusion MRI data, the average diffusion-weighted images (DWIs) were used to manually segment the entire brains. Diffusion tensors (Basser and Jones 2002) were calculated using the log-linear fitting method implemented in DTIStudio (http://www.mristudio.org) at each pixel. The fractional anisotropy (Basser and Jones 2002) maps generated from the diffusion tensor data provided strong white matter and gray matter contrasts and were used to segment the cortex and heterotopia. Structural volumes were calculated based on the results of manual segmentation.
Supplementary Material
Acknowledgments
We thank Dr. N. Kessaris, Dr. B.G. Novitch (University of California at Los Angeles), Dr. A.K. Hadjantonakis (Memorial Sloan Kettering Cancer Center), Dr. Nicoletta Kessaris (University College London, UK), Dr. A.L. Joyner (Memorial Sloan Kettering Cancer Center), Dr. K.V. Anderson (Memorial Sloan Kettering Cancer Center), and Dr. J. Wrana (Lunenfeld-Tanenbaum Research Institute, Canada) for providing the Emx1-CreER, Rbpjfl/fl, CBF:H2B-Venus, Emx1-CreER, Yapfl/fl, and Tazfl/fl mouse lines, respectively, and the members of the Shi laboratory for valuable discussion and input. This work was supported by grants from the National Institutes of Health (R01DA024681 and R01NS085004 to S.-H.S., and P30CA008748 to Memorial Sloan Kettering Cancer Center for the Core Facilities), the Human Frontier Science Program (RGP0053/2014 to S.-H.S.), the New York State Stem Cell Science grant (N13G-232 to S.-H.S.), and the Howard Hughes Medical Institute (S.-H.S.).
Author contributions: W.A.L. and S.-H.S. conceived the project. W.A.L. performed most of the experiments and analyses. W.A.L. generated the conditional Pard3 mutant mouse line with the help of S.C. and Z.L. Z.L. provided retroviruses and helped with mouse colony management. C.H.L. and J.Z. performed MRI analysis. G.M., W.B.D., and M.E.R. advised on phenotypic characterization related to human cortical malformation. W.A.L. and S.-H.S. wrote the paper with input from all other authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.313171.118.
References
- Alexandre P, Reugels AM, Barker D, Blanc E, Clarke JD. 2010. Neurons derive from the more apical daughter in asymmetric divisions in the zebrafish neural tube. Nat Neurosci 13: 673–679. [DOI] [PubMed] [Google Scholar]
- Angevine JB Jr, Sidman RL. 1961. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192: 766–768. [DOI] [PubMed] [Google Scholar]
- Anthony TE, Klein C, Fishell G, Heintz N. 2004. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron 41: 881–890. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. 2012. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135: 1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser PJ, Jones DK. 2002. Diffusion-tensor MRI: theory, experimental design and data analysis - a technical review. NMR Biomed 15: 456–467. [DOI] [PubMed] [Google Scholar]
- Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Menard A, Afanassieff M, Huissoud C, Douglas RJ, Kennedy H, et al. 2013. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 80: 442–457. [DOI] [PubMed] [Google Scholar]
- Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. 2004. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol 20: 593–618. [DOI] [PubMed] [Google Scholar]
- Bizzotto S, Francis F. 2015. Morphological and functional aspects of progenitors perturbed in cortical malformations. Front Cell Neurosci 9: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultje RS, Castaneda-Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi SH. 2009. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 63: 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello S, Bohringer CR, Bergami M, Conzelmann KK, Ghanem A, Tomassy GS, Arlotta P, Mainardi M, Allegra M, Caleo M, et al. 2012. A radial glia-specific role of RhoA in double cortex formation. Neuron 73: 911–924. [DOI] [PubMed] [Google Scholar]
- Chen S, Chen J, Shi H, Wei M, Castaneda-Castellanos DR, Bultje RS, Pei X, Kriegstein AR, Zhang M, Shi SH. 2013. Regulation of microtubule stability and organization by mammalian Par3 in specifying neuronal polarity. Dev Cell 24: 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, McConnell SK. 1995. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82: 631–641. [DOI] [PubMed] [Google Scholar]
- Chenn A, Zhang YA, Chang BT, McConnell SK. 1998. Intrinsic polarity of mammalian neuroepithelial cells. Mol Cell Neurosci 11: 183–193. [DOI] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T, Gotz M. 2008. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135: 11–22. [DOI] [PubMed] [Google Scholar]
- Croquelois A, Giuliani F, Savary C, Kielar M, Amiot C, Schenk F, Welker E. 2009. Characterization of the HeCo mutant mouse: a new model of subcortical band heterotopia associated with seizures and behavioral deficits. Cereb Cortex 19: 563–575. [DOI] [PubMed] [Google Scholar]
- Doe CQ, Fuerstenberg S, Peng CY. 1998. Neural stem cells: from fly to vertebrates. J Neurobiol 36: 111–127. [PubMed] [Google Scholar]
- Dong Z, Yang N, Yeo SY, Chitnis A, Guo S. 2012. Intralineage directional Notch signaling regulates self-renewal and differentiation of asymmetrically dividing radial glia. Neuron 74: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25: 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz SA, Kelava I, Vogt J, Wilsch-Brauninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Distler W, Nitsch R, et al. 2010. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci 13: 690–699. [DOI] [PubMed] [Google Scholar]
- Florio M, Huttner WB. 2014. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194. [DOI] [PubMed] [Google Scholar]
- Gabel LA, Manglani M, Ibanez N, Roberts J, Ramos RL, Rosen GD. 2013. Differential seizure response in two models of cortical heterotopia. Brain Res 1494: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G. 2000. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26: 395–404. [DOI] [PubMed] [Google Scholar]
- Gal JS, Morozov YM, Ayoub AE, Chatterjee M, Rakic P, Haydar TF. 2006. Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J Neurosci 26: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Postiglione MP, Krieger TG, Hernandez L, Wang C, Han Z, Streicher C, Papusheva E, Insolera R, Chugh K, et al. 2014. Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell 159: 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Moreno F, Vasistha NA, Trevia N, Bourne JA, Molnar Z. 2012. Compartmentalization of cerebral cortical germinal zones in a lissencephalic primate and gyrencephalic rodent. Cereb Cortex 22: 482–492. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Rakic P. 2013. Cortical evolution: judge the brain by its cover. Neuron 80: 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Sanz C, Landeira B, Ramos C, Costa MR, Muller U. 2014. Proliferative defects and formation of a double cortex in mice lacking Mltt4 and Cdh2 in the dorsal telencephalon. J Neurosci 34: 10475–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. 2002. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22: 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz M, Stoykova A, Gruss P. 1998. Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 21: 1031–1044. [DOI] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. 2013. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci 14: 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, Ikuta K, Honjo T. 2002. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 14: 637–645. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. 2010. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464: 554–561. [DOI] [PubMed] [Google Scholar]
- Hartfuss E, Galli R, Heins N, Gotz M. 2001. Characterization of CNS precursor subtypes and radial glia. Dev Biol 229: 15–30. [DOI] [PubMed] [Google Scholar]
- Hatten ME. 1999. Central nervous system neuronal migration. Annu Rev Neurosci 22: 511–539. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. 2004. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci 101: 3196–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, Repic M, Knoblich JA. 2015. Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 16: 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY. 2001. Asymmetric cell division in the Drosophila nervous system. Nat Rev Neurosci 2: 772–779. [DOI] [PubMed] [Google Scholar]
- Johnson K, Wodarz A. 2003. A genetic hierarchy controlling cell polarity. Nat Cell Biol 5: 12–14. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Lee M, Klezovitch O, Kon E, Cossard A, Lien WH, Fernandez TE, Cooper JA, Vasioukhin V. 2017. Llgl1 connects cell polarity with cell–cell adhesion in embryonic neural stem cells. Dev Cell 41: 481–495.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, Garcia-Cairasco N, Bueno-Junior LS, Leite JP. 2014. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat 10: 1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava I, Reillo I, Murayama AY, Kalinka AT, Stenzel D, Tomancak P, Matsuzaki F, Lebrand C, Sasaki E, Schwamborn JC, et al. 2012. Abundant occurrence of basal radial glia in the subventricular zone of embryonic neocortex of a lissencephalic primate, the common marmoset Callithrix jacchus. Cereb Cortex 22: 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemphues K. 2000. PARsing embryonic polarity. Cell 101: 345–348. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. 2006. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielar M, Tuy FP, Bizzotto S, Lebrand C, de Juan Romero C, Poirier K, Oegema R, Mancini GM, Bahi-Buisson N, Olaso R, et al. 2014. Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human. Nat Neurosci 17: 923–933. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. 2008. Mechanisms of asymmetric stem cell division. Cell 132: 583–597. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. 2009. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32: 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, Anton ES. 2012. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139: 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Wang D, Shen Q, Schonemann MD, Gorski JA, Jones KR, Temple S, Jan LY, Jan YN. 2003. Inactivation of Numb and Numblike in embryonic dorsal forebrain impairs neurogenesis and disrupts cortical morphogenesis. Neuron 40: 1105–1118. [DOI] [PubMed] [Google Scholar]
- Li Y, Hibbs MA, Gard AL, Shylo NA, Yun K. 2012. Genome-wide analysis of N1ICD/RBPJ targets in vivo reveals direct transcriptional regulation of Wnt, SHH, and hippo pathway effectors by Notch1. Stem Cells 30: 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv XB, Liu CY, Wang Z, Sun YP, Xiong Y, Lei QY, Guan KL. 2015. PARD3 induces TAZ activation and cell growth by promoting LATS1 and PP1 interaction. EMBO Rep 16: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M. 2000. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 127: 5253–5263. [DOI] [PubMed] [Google Scholar]
- Manabe N, Hirai S, Imai F, Nakanishi H, Takai Y, Ohno S. 2002. Association of ASIP/mPAR-3 with adherens junctions of mouse neuroepithelial cells. Dev Dyn 225: 61–69. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. 2003. Cell migration in the forebrain. Annu Rev Neurosci 26: 441–483. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, Ogawa M. 2001. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron 31: 727–741. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. 2003. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci 100: 14920–14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. 2001. Neurons derived from radial glial cells establish radial units in neocortex. Nature 409: 714–720. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. 2004. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144. [DOI] [PubMed] [Google Scholar]
- Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis AK. 2013. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev Biol 13: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park R, Moon UY, Park JY, Hughes LJ, Johnson RL, Cho SH, Kim S. 2016. Yap is required for ependymal integrity and is suppressed in LPA-induced hydrocephalus. Nat Commun 7: 10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Krauss S, Zhong W. 2004. Continuing role for mouse Numb and Numbl in maintaining progenitor cells during cortical neurogenesis. Nat Neurosci 7: 803–811. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. 2015. Molecular identity of human outer radial glia during cortical development. Cell 163: 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. 1971. Guidance of neurons migrating to the fetal monkey neocortex. Brain Res 33: 471–476. [DOI] [PubMed] [Google Scholar]
- Rakic P. 1988. Specification of cerebral cortical areas. Science 241: 170–176. [DOI] [PubMed] [Google Scholar]
- Rakic P. 2003. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex 13: 541–549. [DOI] [PubMed] [Google Scholar]
- Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS, Pawson T, Wrana J, McNeill H. 2013. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet 9: e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reillo I, de Juan Romero C, Garcia-Cabezas MA, Borrell V. 2011. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex 21: 1674–1694. [DOI] [PubMed] [Google Scholar]
- Ross ME, Walsh CA. 2001. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci 24: 1041–1070. [DOI] [PubMed] [Google Scholar]
- Schmid MT, Weinandy F, Wilsch-Brauninger M, Huttner WB, Cappello S, Gotz M. 2014. The role of α-E-catenin in cerebral cortex development: radial glia specific effect on neuronal migration. Front Cell Neurosci 8: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, et al. 2000. Neuronal basic helix–loop–helix proteins (NEX and β2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci 20: 3714–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Nakamura K, Okamoto K, Kaneko T. 2001. Radial glia is a progenitor of neocortical neurons in the developing cerebral cortex. Neurosci Res 41: 51–60. [DOI] [PubMed] [Google Scholar]
- Thomsen ER, Mich JK, Yao Z, Hodge RD, Doyle AM, Jang S, Shehata SI, Nelson AM, Shapovalova NV, Levi BP, et al. 2016. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat Methods 13: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tsai JW, LaMonica B, Kriegstein AR. 2011. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci 14: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrin F, Manent JB, Cardoso C, Represa A. 2015. Causes and consequences of gray matter heterotopia. CNS Neurosci Ther 21: 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Huttner WB. 2003. Asymmetric cell division during neurogenesis in Drosophila and vertebrates. Mech Dev 120: 1297–1309. [DOI] [PubMed] [Google Scholar]
- Wu D, Xu J, McMahon MT, van Zijl PC, Mori S, Northington FJ, Zhang J. 2013. In vivo high-resolution diffusion tensor imaging of the mouse brain. Neuroimage 83: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Guan KL. 2013. The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YC, Bultje RS, Wang X, Shi SH. 2009. Specific synapses develop preferentially among sister excitatory neurons in the neocortex. Nature 458: 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wang S, Wang S, Qiao J, Zhang L, Zhang Z, Chen Z. 2016. Dual function of partitioning-defective 3 in the regulation of YAP phosphorylation and activation. Cell Discov 2: 16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.