

Abstract
The prolyl isomerase cyclophilin A (CypA) regulates the Jak2/Stat5 pathway, which is necessary for mammary differentiation and the pathogenesis of breast cancer. In this study, we assessed the role of this isomerase during mammary gland development and erbB2-driven tumorigenesis. Genetic deletion of CypA resulted in delayed mammary gland morphogenesis and differentiation with corresponding decrease in Jak2/Stat5 activation; mammary gland cross-transplantation confirmed this defect was epithelial in nature. Analysis of mammary stem and progenitor populations revealed significant disruption of epithelial maturation. Loss of CypA in the erbB2 transgenic mouse model revealed a marked increase in mammary tumor latency that correlated with decreased Stat5 activation, associated gene expression, and reduced epithelial cell proliferation. These results demonstrate an important role for CypA in the regulation of Jak2/Stat5-mediated biology in mammary epithelium, identifying this isomerase as a novel target for therapeutic intervention.
Keywords: Breast cancer, mammary gland biology, prolyl isomerase, prolactin receptor, signal transduction
Introduction
Identified initially as a cyclosporine A (CsA)-interacting protein(1), cyclophilin A (CypA) was subsequently recognized as a cis-trans prolyl isomerase(2). Although classically thought to assist in protein folding, a significant body of evidence has implicated cyclophilins as signaling “switches”(3). In essence, the process of isomerization of the proline imide bond induces a conformational change in the peptide substrate backbone, enabling activation/inactivation of its intrinsic enzymatic and/or functional activity. Numerous examples of the switching activity of cyclophilins have been presented, including regulatory roles in cell surface receptor(4–6), kinase(6,7), and transcription factor function(8).
Current data implicate cyclophilins both in physiologic and pathophysiologic processes in humans. Cyclophilin B contributes to collagen I folding and its loss is associated with the phenotype of osteogenesis imperfecta(9), while cyclophilin A contributes to vascular endothelial proliferation and remodeling(10). In malignancy, CypA is overexpressed in lung, pancreatic, and oral squamous cancers(11,12). At the cellular level, inhibition of CypA by pharmacologic or loss-of-function approaches resulted in decreased in vitro growth of hepatocellular, breast, and non-small cell lung cancer cells(12,13). Of note, highly significant reductions in the incidence of breast cancer were noted in a cohort of 25,000 female patients that had received CsA as a mainstay of their immunosuppressive therapy following renal or cardiac allograft(14).
The recognition of the role of CypA in the pathobiology of breast cancer arose from our observation of a critical function served by this foldase during prolactin receptor (PRLr) signaling. Classically associated with the terminal maturation of the mammary gland during pregnancy(15,16), prolactin (PRL) has also been shown to contribute to mammary oncogenesis(4,17). This is supported by the following observations: 1) Breast cancer risk is significantly elevated in women who demonstrate high levels of serum PRL(18), 2) mining of expression array databases and immunohistochemical analyses reveal marked increases in PRLr expression in malignant breast tissues across ER+/ER-/Her2+ phenotypes(19–21), and 3) in vivo gain- and loss-of-function studies targeting PRL significantly alter mammary tumor latency(22,23).
Given the significance of PRLr signaling during mammary development and the pathogenesis of breast cancer, our lab has sought to identify mechanisms that regulate the activation of PRLr-associated signaling pathways. In this context, our studies revealed that CypA, specifically its isomerase activity, was required for the phosphorylation and activation of the tyrosine kinase Jak2 following ligand binding(4,24). Like the PRLr, Jak2 has also been implicated at the in vitro and in vivo level as significantly contributing to mammary development, lactation, and oncogenesis(25–28). Mechanistically, mutagenic approaches revealed that a proline residue in the intracellular domain (P334), near the “Box 1” Jak2-binding motif, was necessary for CypA engagement. As in vitro loss-of-function approaches further confirmed the relationship between CypA and Jak2, a natural extension of these studies was to examine the effect of small molecular prolyl isomerase inhibitors such as CsA and NIM811(29,30). Treatment with these agents blocked Jak2/Stat5 activation and resulted in the in vitro inhibition of ER+ and ER- breast cancer cell proliferation, motility, invasion, and anchorage independent growth(24). Parallel in vivo studies utilizing murine xenograft models revealed that CsA treatment inhibited metastatic progression of both ER+ and ER- breast cancer cells(24). Collectively, these studies support the hypothesis that prolyl isomerase inhibition may represent a novel therapeutic strategy for human breast cancer, in part through its inhibition of Jak2 kinase activation in mammary epithelium.
Materials and Methods
Mice
The 129S6 x SvEv Ppia−/− mice previously described(6,31) were obtained from the Jackson Laboratory. Given that most mouse mammary gain-/loss-of-function models are bred into the FVB background, marker-assisted accelerated backcrossing (MAX-BAX®) was performed at Charles River Laboratories. to generate congenic FVB ppia+/− mice (>99.9% FVB background), which were used in all studies described herein. Immunoblot analysis of mouse tissues from ppia−/− confirmed the loss of CypA expression, as previously documented (see Fig 1S; (6,31)). To evaluate differences in growth, ppia+/+, ppia+/−, and ppia−/− mice were weighed twice a week for 3 weeks, and then once a week for 24 weeks with a minimum of 5 mice per cohort.
Figure 1.
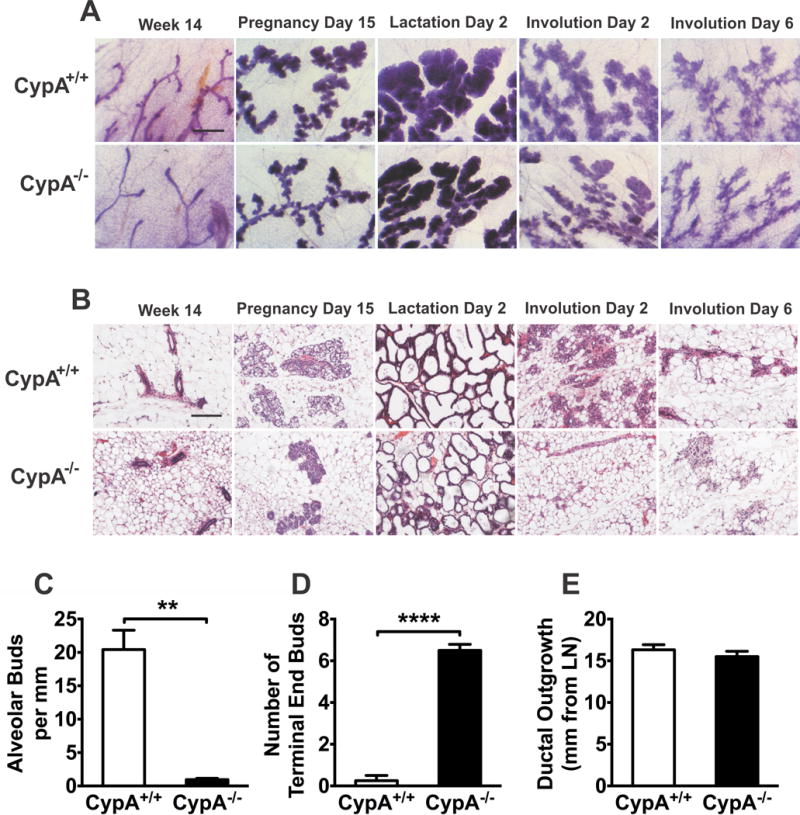
Loss of CypA results in delayed mammary morphogenesis. (a) Mammary gland whole mounts were dissected from CypA+/+ and −/− mice at the times indicated and fixed, processed, and stained with carmine alum prior to microscopy (Bar = 750 microns). (b) Paraffin-embedded inguinal mammary glands dissected at the indicated times were thin sectioned and stained with hematoxylin and eosin. (Bar = 250 microns). (c) The number of alveolar buds per mm of duct length was quantitated in CypA+/+ and −/− females at 14 weeks of development in whole mount mammary preparations (n = 4). (d) Terminal end buds were manually counted at the growth front into mammary adipose tissue in CypA+/+ and −/− mice at 14 weeks of development in whole mount mammary preparations (n = 5 each). (e) The maximal ductal outgrowth in mm from the reference point of the intra-mammary lymph node was measured at 14 weeks of age in whole mount mammary preparations from CypA+/+ and −/− mice (n = 5 each).
For explant cultures, the fourth set of mammary glands was dissected from 14- week old CypA+/+ and Cyp A−/− mice under aseptic conditions. Mammary glands were cut into 3mm3 pieces and placed into pre-warmed 37°C DMEM/F12 media with HEPES and 1% pen/strep and incubated for 18 hours at 37°C to starve cells of PRL. These tissues were then incubated in the presence or absence of 250ng/mL PRL for 15 min, followed by harvesting in ice cold PBS, prior to homogenization, lysis in Laemmli buffer, and western blot analysis.
For tumorigenesis studies, the well-recognized mouse model of transgene-driven tumorigenesis (32) utilizing the MMTV-driven rat erbB2 receptor containing the activating an mutation (Val664Glu) was employed. Mice expressing this transgene were obtained from Jackson laboratories (strain #005038) and were bred successively to obtain erbB2 x ppia+/+, x ppia+/−, and x ppia−/− offspring on the FVB background. The progeny of these crossings were palpated weekly until tumors were noted; subsequently the mice were palpated thrice weekly. Mice were sacrificed when the tumor approximated 10% of body weight, or lethargy or tumor excoriation was noted, in accordance with Northwestern University IACUC guidelines. Following sacrifice, all tumors were promptly dissected and weighed postmortem. Following fixation with 10% buffered neutral-buffered formalin, tumors were paraffin-embedded and processed for histologic and histochemical analyses. No significant differences in morphology in the resultant tumors were noted between the ppia+/+, ppia+/−, or ppia−/− x erbB2 crosses. All studies described herein were approved by the Northwestern University IACUC. For in vivo pharmacologic inhibition, Nu−/− mice were gavaged with the non-immunosuppressive, cyclophilin prolyl isomerase inhibitor NIM811 twice daily for a total dose of 100 mg/kg/d, prior to PRL stimulation.
Mammary Gland Cross-Transplants
Mammary gland cross-transplants were performed as previously described(33). Anesthetized, three-week old female recipients underwent surgical excision of their fourth mammary tree. The resected tissue was reserved for whole mount processing to verify that the entire ductal tree was removed from the fat pad. In turn, the fourth mammary gland of 5 to 10-week old virgin female donors was dissected and the 1mm piece of mammary tissue obtained was inserted into the cleared fat pad of the recipient. Recipient mice were sacrificed at 14 weeks of age for analysis of the transplanted mammary gland by whole mount analysis.
Western blot analysis
Mouse liver homogenates were analyzed by western blot analysis as previously described(24). Antibodies utilized for these studies were obtained from the following sources and used as described: anti-CypA (EMD Millipore), anti-tubulin (Life Technologies). Mouse mammary gland homogenates were also subjected to western blot analysis. Antibodies utilized for these studies were obtained from the following sources and used as described: anti-pStat5 (Cell Signaling Technologies, 1:1000), anti-Stat5 (Santa Cruz, 1:1500) diluted in TBS-T with 3% soy protein isolate (NOW Sports).
Whole mount staining
Dissected fourth mammary gland pairs were split with one gland formalin-fixed/paraffin embedded and the other processed for whole mount staining(34). Whole mount glands were fixed in 10% formalin overnight, washed, and then placed into acetone overnight to remove intra-mammary fat. Following extensive washing, the gland was digested with 1mg/ml collagenase II (Sigma) in PBS for 4 hours at 37°C. After washing, the processed gland was stained with Carmine Alum (Sigma) overnight. After rinsing, remaining connective tissue was micro-dissected to expose the mammary tree. Following dehydration, the mounted tissues were covered with Entellan New (Electron Microscopy Sciences).
Immunohistochemistry
For hematoxylin/eosin and immunohistochemical (IHC) labeling, harvested mammary glands were fixed in 10% formalin, dehydrated, and paraffin embedded. Five micron sections were obtained and stained with hematoxylin and eosin (Sigma). Immunohistochemical labeling was performed as described previously (20) All primary antibodies were obtained from the following sources and utilized at the indicated titers: cleaved-caspase-3 (Cell Signaling Technologies, 1:1000), Ki-67 (Cell Signaling Technologies, 1:300), CISH (Abcam, 1:800), phospho-Stat5a (Cell Signaling Technologies, 1:25), lactalbumin (Santa Cruz, 1:300), whey acidic protein (Santa Cruz, 1:800), beta casein (Abbiotec, 1:300). After washing, slides were incubated with SignalStain® Boost IHC Detection Reagent (Cell Signaling Technologies) for 30 min, followed by incubation with SignalStain® DAB Substrate (Cell Signaling Technologies) for 10 min. Washed slides were counterstained with hematoxylin (Gill No. 3, Sigma) and incubated with 1X Scott′s Tap Water Substitute (Sigma) for additional bluing. Slides were dehydrated, and coverslipped using Permount™ mounting medium (Electron Microscopy Sciences). Sections from at least 3 harvested mammary glands from independent mice from each time point were scored. Cleaved-caspase-3 and Ki-67 staining were expressed as percent positive cells; all others as an Allred score, namely as a function of % positive cells x mean intensity (0-3 scale).
Flow Cytometry
For enzymatic dissociation, dissected fourth mammary gland pairs from 14-week old CypA+/+ or CypA −/− female mice were incubated for 16 hrs at 37°C in 5 mL of 1X gentle collagenase/hyaluronidase (StemCell Technologies) in DMEM/F12 (1:1) medium (Life Technologies), supplemented with 5% fetal bovine serum (FBS, Atlanta Biologicals) and 1% penicillin/streptomycin (Life Technologies). After incubation and centrifugation, the resultant cell pellet was resuspended in 5 mL of a 4:1 mixture of cold ammonium chloride (StemCell Technologies) and cold Hank’s Balanced Salt Solution Modified (HBSS, Stem Cell Technologies), supplemented with 2% FBS. Following centrifugation, the cell pellet was resuspended in 5 mL pre-warmed 0.25% Trypsin-EDTA (Life Technologies), and the cell suspension was mixed by gentle pipetting with a P1000 disposable tip for 3 min. After washing, the cell pellet was resuspended in 2 mL of pre-warmed dispase (5 mg/mL, StemCell Technologies), supplemented with 200 uL DNase I (1 mg/mL, StemCell Technologies). The resulting suspension was incubated for 5 min at 37°C, diluted with 10 mL cold HBSS supplemented with 2% FBS, and filtered through a 40 μm filter. The resuspended cells were enriched for epithelial populations (i.e. Lin− cells) using the EasySep™ Mouse Epithelial Cell Enrichment Kit (StemCell Technologies), according to the manufacturer’s protocol. For fluorescent labeling, cells were pre-incubated with Mouse BD Fc Block (BD Biosciences) at 2 μg per 100 μL cell suspension for 5 min at 4°C. The cells were then incubated with BV510 rat anti-mouse CD24 (BD Biosciences), Alexa Fluor® 488 rat anti-mouse CD29 (BD Biosciences), and Alexa Fluor® BV421 hamster anti-mouse CD61 (BD Biosciences) at 0.25 μg, 1 μg, and 0.25 μg per 100 μL cell suspension, respectively, for 30 min at 4°C. After washing, propidium iodide was added to the labeled cells at 50 ng per 100 μL cell suspension. Cells were analyzed on the BD LSRFortessa-X20™ (BD Biosciences), using BD FACSDiva™ software for data acquisition and analysis at the VCU Massey Cancer Center Flow Cytometry Shared Resource Core. To characterize CD61+/29+ populations as previously described (35), intact cells were gated from debris and aggregates using forward and side scatter; dead cells were eliminated using propidium iodide exclusion. Dual parameter CD29 vs. CD61 analysis was then performed on cells identified as CD24+.
Statistics
Differences between group means were determined by unpaired Student's t-test assuming equal variances, with the exception of the immunoblot quantitation, for which a One-way ANOVA with the Holm-Sidak correction for multiple comparisons was performed. For all, and statistical difference defined as p < 0.05, using GraphPad Prism V6.0g (GraphPad Software, Inc., La Jolla, CA).
Results
Since the pathways regulating mammary gland development can also provide understanding of the mechanisms utilized during oncogenesis, it was reasoned that genetic ablation of the CypA locus might provide additional insight into the in vivo function of this isomerase in the normal and malignant development of the mammary gland. While a non-conditional CypA−/− mouse had been previously described to be resistant to the effects of CsA(31), no study to date had examined the loss of CypA function during mammary development and oncogenesis. Given our precedent data, we hypothesized that this CypA−/− mouse should demonstrate decreased in vivo Jak2/Stat5 signaling and delayed/altered mammary development and tumorigenesis. To facilitate its use with standard genetic mouse models of mammary neoplasia, the non-conditional CypA−/− mouse was back-bred to congenicity onto the FVB background. Confirmation of the loss of CypA in the resultant progeny was confirmed at the level of protein expression by immunoblot analysis (Fig. S1a). CypA−/− x FVB mice (hereafter referred to as CypA−/− mice) were viable, demonstrating normal estrous cycles, fertility, and litter size with expected Mendelian ratios, consistent with prior reports (31).
The effect of CypA loss on mammary gland development, however, was significant, as observed by both whole mount and histologic analysis. Demonstrated in Figures 1a-c, these analyses reveal a marked loss of alveolar budding in the mammary glands of 14 wk old virgin CypA−/− mice, and was comparable to that seen with the PRLr−/− (36) and Stat5−/− (37,38) mice. Morphometric analysis of these glands, quantitatively confirmed significant reductions in the number of alveolar buds (Fig. 1c). In parallel, a persistence of terminal end buds was noted (Fig. 1d), while their penetration into the mammary fat pad was not altered (Fig. 1e). Interestingly, while alveolar budding was profoundly decreased by CypA deletion (Fig. 1c), the number of secondary ductal branches was not altered (not shown). Occasional duct ectasia was also noted in the CypA−/− females. Altered development of the mammary gland of the CypA−/− mice persisted through pregnancy and lactation. Alveologenesis proper was also significantly delayed in the Cyp−/− females both at day 15 of pregnancy (P15) and day 2 of lactation (L2). The lack of luminal clearing and epithelial secretory granules in the CypA−/− glands at P15 was also readily evident (Fig 1, a+b). In addition, while all CypA+/+ mice at L2 demonstrated a 90-100% effacement of the mammary fat with alveolar epithelium, all CypA−/− females revealed only a 50-80% replacement (Fig 1 a+b; Supplemental Fig. 1B). Finally, a far faster involution of the mammary epithelium was noted in CypA−/− females (Fig. 1, a + b) at both involution days 2 and 6 (I2 and I6). As a note, the loss of only a single allele, i.e. the CypA+/− mouse, did not result in any discernable change in mammary morphology, as the mammary development of the heterozygous females paralleled that of the homozygous littermates.
Given the significant morphologic alterations noted in the mammary glands of CypA−/− females, it was reasoned that the functional differentiation of the lactating mammary gland may also be altered. Indeed, both the PRLr−/− and Stat5−/− mice were incapable of supporting lactation(15,39). To that end, quantitative and qualitative assessments of milk production were made. To assess for quantitative alteration in milk production, litters of entirely CypA+/− progeny were bred and nursed from their corresponding CypA+/+ or −/− mothers. Litters were normalized down to n=5 and weighed twice a week. No statistically significant differences in litter weight or ability to thrive were detected (not shown). Qualitative assessment of milk protein constituents in mammary glands from P15 and L2 females utilized immunohistochemical analysis. There was no statistically significant alteration of either beta casein or whey acidic protein at the level of immunohistochemistry (Fig. S2). As seen in Figs 2a and S2a, however, a highly significant decrease the expression of lactalbumin in the mammary glands of CypA−/− females was observed at both P15 and L2. Thus, the effects of loss of CypA on both quantitative and qualitative milk production do not appear as severe as noted for both the PRLr−/− and Stat5−/− mice.
Figure 2.
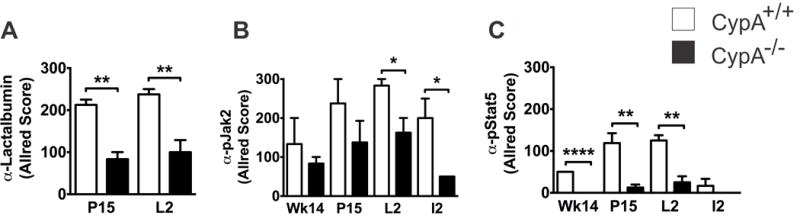
CypA−/− mice demonstrate decreased lactalbumin (panel A) expression and attenuation of Jak2 (panel B) and Stat5 (panel C) activation during pregnancy and lactation. Paraffin-embedded sections of inguinal mammary glands were immunohistochemically labelled for milk proteins with the designated antibodies at indicated times. Given the heterogeneity of IHC labeling in terms of intensity and positivity, quantitation was performed using Allred scoring. Differences in IHC labeling with anti-whey acidic protein or anti-beta casein between CypA +/+ and −/− tissues were not statistically significant For all studies a minimum of 3 mice were quantitated for each stain, at each time point; * p < 0.05; ** p < 0.01; **** p < 0.001. Selected intervals include: 14 wk virgin (Wk14), pregnancy day 15 (P15), lactation day 2 (L2), and involution day 2 (I2). Corresponding micrographs can be found in Supplemental Figure 2.
Our lab has previously shown that CypA facilitates PRL-driven Jak2/Stat5 signaling in vitro in breast cancer cells via its prolyl isomerase activity on the PRLr/Jak2 complex(24). To examine whether the developmental morphologic and qualitative lactation changes noted in the CypA−/− mammary gland was related to alterations in Jak2/Stat5 signaling, immunohistochemical analysis of CypA+/+ and CypA−/− mammary glands with anti-phospho-Jak2 and anti-phospho-Stat5 (anti-pJak2 and anti-pStat5, respectively) was performed at P15 and L2 (Fig 2b + c, and S2b+c). At both time points, significant reductions in pJak2 and pStat5 expression in the CypA−/− mice were noted. Taken as a whole, these data revealed that the developmental alterations noted in the CypA−/− mammary glands were associated with significant decreases in the phosphorylation/activation status of Jak2 and Stat5.
CypA−/− females demonstrated normal estrous cycles and fecundity suggesting an intact hypothalamic-pituitary-ovarian hormonal axis, as well as normal serum PRL, GH, and IGF1 levels (as determined by the National Hormone and Pituitary Program; not shown). Nevertheless, the changes found in the non-conditional CypA−/− mouse could be secondary to its impact on either or both of the endocrine and stromal axes. To exclude these possibilities, mammary gland cross-transplants were harvested at 14 weeks of age, following clearing of the mammary fat pad of recipients of their native mammary gland at 3 weeks of age. Analysis of the cross-transplanted mammary glands by whole mount analysis (Fig. 3a) was striking. Similar levels of alveolar budding were noted when comparing the control transplant (e.g., CypA+/+ epithelium into a CypA+/+ stromal fat pad) to the heterologous transplant of CypA+/+ epithelium into CypA−/− stromal fat pad. In contrast, a highly significant reduction in the number of alveolar buds was observed when the CypA−/− epithelium was transplanted into the CypA+/+ stromal fad. These data confirmed that the developmental defects noted in the mammary glands of CypA−/− females was the result of a primary epithelial defect, and not the result of endocrine hormones or the stromal microenvironment.
Figure 3.
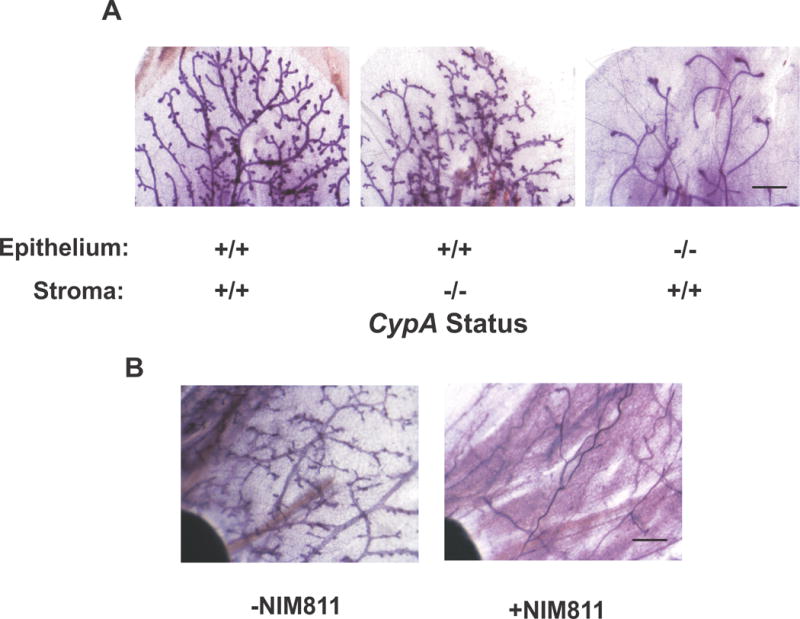
Complementary studies with CypA+/+ and −/− mammary gland cross-transplants and pharmacologic inhibition of CypA with NIM811 reveal that the defect in mammary gland development CypA−/− mice is epithelial and is also noted when CypA is inhibited following initial mammary development. (a) Cross-transplant of CypA+/+ and −/− mammary epithelium and stroma reveals that the defect in mammary alveolar budding in CypA−/− mice is epithelial. Following mammary ductal clearance at three weeks of age, epithelial cross-transplants were performed from mice aged 5-10 weeks of age. Analysis of the resultant mammary tree occurred by whole mount analysis when the recipient mice were 14 weeks of age. Bar = 750 microns. (b) Pharmacologic inhibition of CypA with NIM811 results in defective alveolar budding as seen in the mammary glands of CypA−/− mice. Nu−/− mice were treated with the CypA inhibitor, NIM811 (an analog of cyclosporine A). Mammary glands from virgin mice were harvested at week 14 after 6 weeks of treatment with NIM811 (100 ug/kg/day po) or control carrier and processed for whole mount. (Bar = 1.5mm).
To further confirm that the prolyl isomerase activity per se of CypA was necessary for normal mammary development 8-week old virgin mice were treated for 6 wks with the non-immunosuppressive inhibitor of cyclophilin prolyl isomerase activity, NIM811(30). As clearly seen in Figure 3b, NIM811-treated mice demonstrated a marked paucity of alveolar budding in the mammary gland, comparable to that seen in comparably aged Cyp−/− females. Taken together with the cross-transplant data, these findings argue that it is the prolyl isomerase activity of CypA within the mammary epithelium that is necessary for appropriate mammary alveologenesis.
Given the reduction in endogenous levels of pJak2 and pStat5 in CypA−/− mice (Fig. 2 and S2) during pregnancy and lactation, periods in the reproductive cycle demonstrating high levels of serum PRL, it was conjectured that PRL-induced signaling in a non-lactating mouse mammary gland should also be reduced. To test this hypothesis, 20-week virgin females were injected intraperitoneally with PRL, and after 30 minutes the mammary glands were harvested and prepared for immunohistochemical labeling and immunoblot analysis with anti-phospho-Jak2 or pStat5 antibodies. As hypothesized, the PRL-induced expression of both pJak2 and pStat5 in the CypA−/− mammary glands was profoundly reduced (Fig. 4A). Further confirmation of the reduction of PRL-induced Stat5 phosphorylation at the biochemical level was obtained by anti-pStat5 analysis of PRL-stimulated CypA+/+ and CypA−/− mammary explants (Fig. 4B + C).
Figure 4.
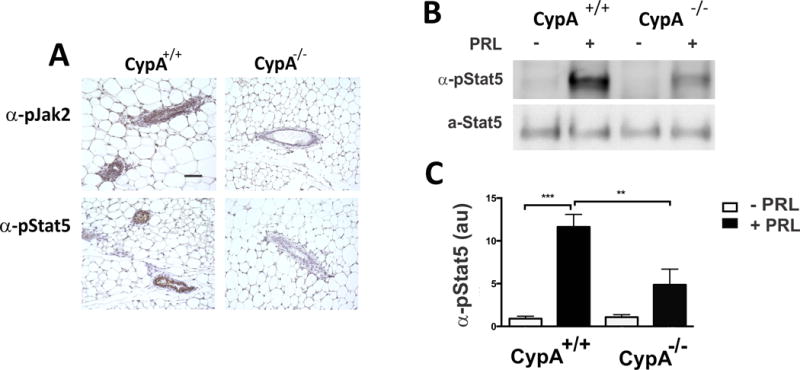
Loss of CypA results in decreased PRL-induced pJak2 and pStat5 at the immunohistochemical (Panel A) and immunoblot (Panels B + C) levels. Thirty minutes following PRL stimulation (10 ug PRL, IP), mouse mammary glands from Cyp+/+ and −/− mice (20-week virgins) were harvested, paraffin-embedded and immunostained with anti-pJak2 or –pStat5 antibodies (Bar = 100 microns) as shown in Panel A. in Panels B + C, tissue explants from CypA+/+ and CypA−/− mice were stimulated for 15 min with PRL, prior to harvest, homogenization, and immunoblot analysis with anti-pStat5 antibodies. Quantitation of pStat5 immunoblot analysis in Panel B is presented in Panel C; n=3 mice, **p < 0.01; ***p < 0.005.
Alterations in mammary gland morphology and differentiation have been linked to perturbations in the hierarchical organization of the mammary epithelium, consisting of identifiable stem, progenitor, and mature luminal cell populations. These relationships have been made evident in part through the use of loss-of-function mouse models for the transcription factors Gata-3 (35), Runx2 (40), and Elf5 (41). Given that loss of Jak2(42) and Stat5(38,39,43) function are also associated with alterations in mammary morphology, it was reasoned that the CypA−/− mouse might also demonstrate alterations to its epithelial hierarchy. To that end, flow cytometry on freshly disassociated, lin−, CD24+ cells was utilized to quantitate stem (CD29hi/CD61hi), progenitor (CD29lo/CD61hi), and mature luminal (CD29lo/CD61lo) populations in 14-week old virgin CypA+/+ and −/− mice, using previously described and validated methodology (35). These analyses (Fig. 5a) revealed significant differences between the CypA+/+ and −/− mice, as a 15-fold increase in the stem and a 34-fold increase in the progenitor populations were observed in the CypA−/− mice. Correspondingly, the CypA−/− mice also demonstrated a significant decrease in percentage of mature luminal cells. Disruption of the normal mammary development by genetic loss-of-function has frequently been associated with decreased cellular proliferation in the mammary gland (35,40), and is associated with delayed morphological development and a decrease in the volume occupied by the epithelial compartment. To assess whether a similar phenomenon was occurring in the CypA−/− mice both anti-Ki67 and -cleaved caspase-3 immunohistochemical analyses were performed on the mammary glands from 14-week old virgin mice (Figs. 5b and S3). These analyses revealed that a profound decrease in proliferation occurred in the mammary epithelium of the CypA−/− mice, whereas the rates of apoptosis between the CypA+/+ and CypA−/− mice were comparable. Taken together, these findings indicate that loss of CypA results in delayed maturation of stem and progenitor populations into mature luminal cells that is manifest in a decrease in observable proliferation in the mammary epithelium.
Figure 5.
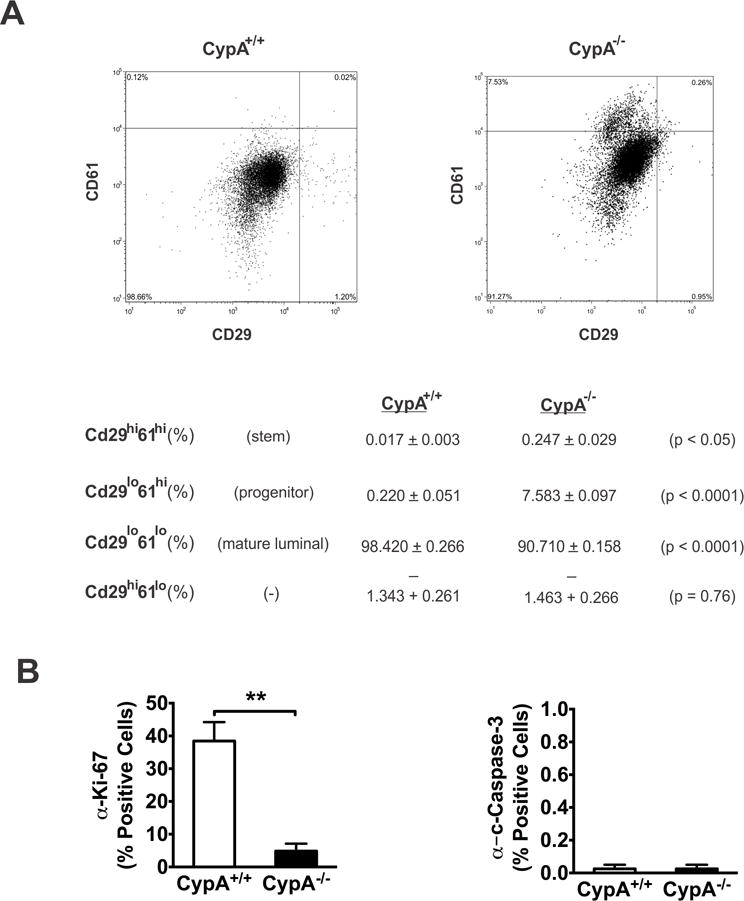
Flow cytometric analysis of CypA−/− mice reveals inhibited mammary stem/progenitor differentiation into mature luminal epithelium. (a) Lin- mouse mammary gland cells were labeled with anti-CD24, -CD29, and -CD61 antibodies. The CD24+ population was used for analysis of stem/progenitor/mature luminal population analysis with anti-CD61/-29, as per (35). A minimum of 10,000 cells were analyzed. Representative histograms are presented in the upper panels; the lower table demonstrates quantitation of the stem (CD61hi29hi), progenitor (CD61hi29lo), and mature luminal (CD61lo29lo) populations from CypA+/+ and CypA−/− mice (n = 3). (b) Loss of CypA inhibits epithelial proliferation, but does not alter apoptosis. Immunohistochemical labeling of paraffin-embedded, week 14 mammary glands with anti-Ki67 or –cleaved caspase 3 (CC3) was quantitated for % cells positive for Ki67 or CC3 staining. *p < 0.05, **p < 0.01. Corresponding immunohistochemical micrographs are presented in Supplemental Figure 3.
Many mouse loss-of-function models that produce altered mammary development, also demonstrate delayed or disrupted tumorigenesis when crossed into the appropriate line expressing a transgenic mammary oncogene (44–46). We hypothesized that mammary tumorigenesis driven by a mammary oncogene, such as ErbB2/neu would be significantly delayed by its introduction into the CypA−/− background. We based this hypothesis on the following observations: 1) Significant delays in alveolar budding with the CypA−/− mouse were noted (Figs. 1A-C); 2) Jak2-mediated signaling has been found to influence erbB2 transduction and erbB2/neu-driven mammary tumorigenesis(27); and 3) Our data revealing that CypA regulates Jak2 activation in breast cancer cell lines(24), in the developing mammary gland (see Figs. 2 and 4), and in human breast cancer xenografts(24). To that end, CypA+/− x FVB and erbB2 x FVB mice were mated, and after re-mating of select progeny, CypA+/+ and CypA−/− (xFVB) female littermates were followed for mammary tumor development for 86 weeks. As presented in the Kaplan-Meier curves in Figure 6A, a highly significant, two-fold delay in tumor latency was noted in the CypA−/− females (CypA+/+ median latency = 190d vs. CypA−/− = 475d). In parallel, survival of the CypA−/− females was significantly enhanced (CypA+/+ = 245d vs. CypA−/− = 480d). Parallel studies with CypA+/− x erbB2 females showed no increase in tumor latency or survival, paralleling the curves of the CypA+/+ mice. Interestingly, the time from observation of the initial primary tumor to euthanasia was only modestly increased between CypA+/+ and −/− mice (CypA+/+ = 51d versus CypA−/− = 70d), suggesting that cyclophilins primarily contribute to the progression of the precursor lesion into the primary invasive tumor, and secondarily promote the growth of the primary tumor per se. This was further substantiated by analysis of mammary tumor multiplicity in the mouse cohorts at time of euthanasia/death; CypA+/+ mice (9.75 ± 1.65; mean tumors/mouse ± SEM) demonstrated significantly more (p < 0.005) discrete carcinomatous foci than did CypA−/− mice (1.75 ± 0.25)
Figure 6.
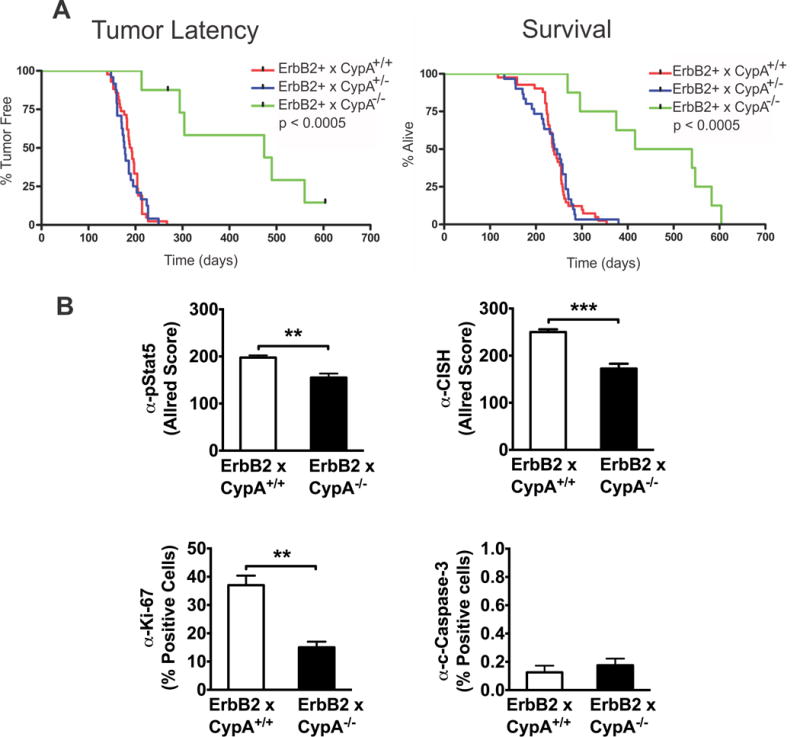
CypA deletion markedly inhibits ErbB2 mammary tumorigenesis, while concomitantly decreasing the intratumoral activated Stat5 and proliferation. Upper panels. CypA−/− x ErbB2 mice demonstrate significantly delayed tumor latency and enhanced survival. Kaplan-Meier plots of tumor-free survival (p < 0.0005) and overall survival (p < 0.0005) reveal highly significant differences between CypA+/+ and −/− mice when bred into the ErbB2 mouse model of mammary tumorigenesis. ErbB2 x CypA+/+, +/−, and −/− cohorts were generated (n>8 per group) and palpated 3x/week for tumor formation. Mice were euthanized when tumor size = 2cm or when impaired mobility, tumor excoriation, or moribund behavior became apparent. Lower panels. CypA−/− x ErbB2 tumors demonstrate lower levels of activated Stat5, Stat5-responsive CISH expression, and proliferation, with no change in apoptosis. Formalin-fixed, paraffin embedded tumors from CypA+/+ and −/− mice were immunohistochemically labelled with anti-pStat5, -CISH, -Ki-67, or –CC3. n = 3, **p < 0.01, ***p<0.005. Corresponding immunohistochemical micrographs are presented in Supplemental Figure 4.
To assess whether parallels between the developing mammary gland and erbB2-driven tumorigenesis existed in the CypA−/− mice, the levels of proliferation, apoptosis, Stat5 phosphorylation, and expression of the Stat5-responsive CISH gene were assessed by immunohistochemistry in primary tumors from CypA+/+ and Cyp−/− mice (Figs 6B and S4). As was seen in the 14-week old mammary gland, erbB2 tumors from CypA−/− mice demonstrated decreased Stat5 activation, CISH expression, and Ki67 labeling, but unchanged levels of cleaved caspase-3 labeling, when compared to CypA+/+ mice. Taken together, these data suggest that the function of signaling pathways altered by CypA loss contribute to both mammary development and tumorigenesis.
Discussion
The developmental delays in mammary gland differentiation in the CypA−/− mouse demonstrate many similarities to other loss-of-function mouse models of the PRL-PRLr-Jak2-Stat5 signaling pathway(37,38,42). The requirement for this signaling axis is required for alveolar budding and terminal end bud regression. This phenotype was also observed in the CypA−/− mouse (Fig 1) and is consistent with the role of CypA in PRLr-driven activation of Jak2(24). Correspondingly, it is not surprising that secondary ductal branching in CypA−/− mice was preserved, as this process appears to be principally driven by the actions of progesterone, as has been noted in PRL−/− females supplemented with progesterone(36,47).
Defective alveolar development proper persisted during pregnancy in the CypA−/− mouse, as a lack of luminal clearing of the alveoli and secretory granule formation in the epithelium was observed. This too has been noted in genetic deletions in the PRL-PRLr-Jak2-Stat5 pathway(37–39,42,47). What distinguishes the CypA−/− mouse from these other genetic deletion models, however, was their ability to successfully lactate and nurse their progeny, as measured by the viability and weights of CypA−/− litters. A trivial explanation for this is that the CypA−/− mouse was on a congenic FVB background (i.e. a strain related phenomenon), unlike many of the other noted genetic models. We view this possibility as unlikely, given that no difficulties in lactation or litter viability were noted in the parental line of mixed background, CypA−/− 129S6 x SvEv, either by our lab or the lab where this strain was generated(31). A more likely basis for the mammary phenotype of the CypA−/− mouse, may relate to a significant, but not complete, loss of Jak2/Stat5 signaling (Figs 2 and S2). Precedent in vitro studies by our lab have revealed a significant (70-90%) inhibition of the Jak2/Stat5 pathway in vitro following CypA loss or inhibition in breast cancer cells(24). While there were no significant differences in comparably sized litters from either CypA−/− or CypA+/+ dams, lactation in the CypA−/− mouse was not entirely normal. Morphologically, full expansion of the alveolar epithelium into the mammary fat pad during lactation did not occur in CypA−/− females (Fig 1). Similarly, unlike the PRLr−/− mouse which demonstrates a global deficiency of milk protein constituent synthesis, the loss of milk protein synthesis in the CypA−/− mouse was selective with only a significant decrease of lactalbumin expression noted in CypA−/− alveolar epithelium (Figs 2 and S2). The selective differences in milk protein expression observed may suggest that proteins such as lactalbumin require a higher level of induced Stat5 activity, not achieved in the CypA−/− mouse. Taken together, these data suggest that titration of Jak2/Stat5 function by CypA has discrete impacts on mammary development.
Precedent studies(36,47) using the non-conditional PRLr−/− mouse have revealed that the defect in alveologenesis and lactation in this model is epithelial and not stromal (or endocrine) in origin. Mammary gland cross-transplants of CypA+/+ and CypA−/− donors and recipients demonstrated a similar phenomenon, namely the developmental defects observed in CypA−/− mice are epithelial in nature. The in vivo role of CypA in epithelial differentiation was also confirmed by treatment of CypA+/+ mice with the non-immunosuppressive analog of cyclosporine A (CsA), NIM811. The non-immunosuppressive properties of NIM811 are the result of a single amino acid side chain replacement, resulting in its inability to engage calcineurin A. As a consequence, NIM811 is not immunosuppressive and has none of the other toxic side effects of CsA(30,48). Treatment of 14-week old virgin females, resulted in the inhibition of alveolar budding as was observed with the CypA−/− females (Figs 1 and 4). While the effect of NIM811 on lactation proper was not examined here, given the data presented here with the CypA−/− mouse and a report of successful lactation in a female patient receiving cyclosporine(49), we would predict that NIM811-treated mice would successfully lactate, but with alterations in milk composition and fat pad effacement as noted above. Alterations in mammary gland development, as seen in the CypA−/− mouse, have been frequently associated with delayed/inhibited maturation of the epithelial hierarchy of stem, progenitor and mature luminal cells in the mammary gland, as determined by flow cytometric and functional approaches (35,40,43). Significant expansion of the CD61hi populations were noted for both stem (CD29hi/CD61hi; 15-fold increase) and progenitor (CD29lo/CD61hi; 34-fold increase)) populations. Correspondingly, a decrease in mature luminal cells (CD29lo/CD61lo) was also noted. Taken together these observations are consistent with inhibited differentiation in CypA−/− mammary epithelium (Fig. 5), particularly at the progenitor to mature luminal level, paralleling what was seen in the above cited loss-of-function mouse models.
When crossed into an erbB2 model of transgene-driven mammary carcinogenesis (32), CypA−/− x erbB2 mice demonstrated significantly prolonged tumor latency/survival and decreased tumor multiplicity in comparison to the Cyp+/+x erbB2 cohorts (Fig. 6). This may in part result from the decreased levels of Stat5 activation, Stat5-driven gene expression, and proliferation noted in CypA−/− x erbB2 tumors (Figs. 6 and S4). However, only a modest difference in the time-to-death from first tumor observation between the CypA−/− and Cyp+/+ mice was noted, as was seen with the Jak2−/− x erbB2 mice(27). Thus, the CypA−/− and Jak2−/− mice demonstrate some similarities in their effects on tumor initiation and primary tumor growth(24,27,45). Treatment of ER+ and ER- xenografts with the prolyl isomerase inhibitor cyclosporine A (CsA), following initial tumor establishment in the absence of CsA, also had only a modest effect on primary tumor growth, however, metastasis in these treated models was profoundly inhibited(24). A significant difference, however, between the CypA−/− x erbB2 and Jak2−/− x erbB2 (45) mouse models is observed in their respective differential delays in median tumor latency in comparison to wild type x erbB2 mice (delays of 235d for CypA−/− x erbB2 vs. wt mice versus 42d for Jak2−/− x erbB2 vs. wt mice). This would suggest with respect to mammary tumorigenesis, the loss of CypA function may have a broader action, perhaps by affecting relevant signaling pathways other than those directly associated with Jak2. Indeed, while our working model of CypA action during PRLr signaling is focused on the activation of Jak2 by CypA, many downstream Jak2 targets such as AKT and MAPK (24), as well as the Tec tyrosine kinase family (7,50) are regulated by CypA activity. Taken together, our findings indicate that CypA significantly contributes to in vivo PRLr-mediated signaling, mammary gland development/differentiation and tumorigenesis, representing a novel therapeutic target at the levels of chemoprevention and the inhibition of metastatic progression.
Supplementary Material
Supplemental Figure 1 Loss of CypA is associated delayed mammary development. A. CypA is not expressed in CypA−/− mice. Western blot analysis of mammary gland lysates from 14 wk CypA+/+ and CypA−/− virgins probed with the indicated antibodies. B. Low power photomicrographs demonstrate delay in mammary development in CypA−/− mice. Inguinal mammary glands from CypA+/+ and CypA−/− mice were dissected at the indicated times (P15, day 15 pregnancy; L2, day 2 lactation), paraffin-embedded and stained with hematoxylin and eosin prior to microscopy. (Bar = 500 microns).
Supplemental Figure 2 Immunohistochemistry of CypA+/+ and CypA−/− females at P15 and L2 demonstrate decreased levels of lactalbumin, pJak2, and pStat5 expression in CypA−/− females. No alteration in the expression of whey acidic protein or beta-casein was noted (Bar = 250 microns in Panel A; Bar = 100 microns in Panels B + C).
Supplemental Figure 3 Immunohistochemistry of CypA+/+ and CypA−/− wk 14 virgins demonstrate decreased Ki67 and unchanged cleaved caspase 3 (CC3) levels in CypA−/− females. (Bar = 250 microns).
Supplemental Figure 4 Immunohistochemistry of CypA+/+ x erbB2 and CypA−/− x erbB2 wk 14 virgins demonstrate decreased pStat5, CISH, and Ki67 and unchanged cleaved caspase 3 (CC3) in CypA−/− x erbB2 females. (Bar = 250 microns).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Significance.
Findings reveal cyclophilin A functions in normal mammary epithelial development and ErbB2-driven mammary tumorigenesis and suggest therapies targeting Cyclophilin A may be efficacious for breast cancer treatment.
Acknowledgments
Services and products in support of the research project were generated by the VCU Massey Cancer Center Flow Cytometry Shared Resource, Transgenic/Knockout Mouse Core, and the VCU Microscopy Facility, supported in part with funding from NIH-NCI Cancer Center Support Grant P30 CA016059; and by the Department of Pathology, Anatomic Pathology Research Services, at VCU Health. The assistance of Alicia Wook and Jacqueline Gribble in immunoblot optimizations is acknowledged.
This work was supported in part by grants from the NIH, R01 CA173305 and the American Association for Cancer Research/Breast Cancer Research Foundation Research grant (grant number: 10-60-26-CLEV to C. Clevenger).
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Handschumacher RE, Harding MW, Rice J, Drugge RJ. Cyclophilin: A specfic cytosolic binding protein for cyclosporin A. Science. 1984;226:544–7. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 2.Harrison RK, Stein RL. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry (Mosc) 1990;29:3813–6. doi: 10.1021/bi00468a001. [DOI] [PubMed] [Google Scholar]
- 3.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–29. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 4.Syed F, Rycyzyn MA, Westgate L, Clevenger CV. A novel and functional interaction between cyclophilin (Cyp) A and the prolactin receptor. Endocrine. 2003;20:83–9. doi: 10.1385/ENDO:20:1-2:83. [DOI] [PubMed] [Google Scholar]
- 5.Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–52. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- 6.Colgan J, Asmal M, Neagu M, Yu B, Schneidkraut J, Lee Y, et al. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity. 2004;21:189–201. doi: 10.1016/j.immuni.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Brazin KN, Mallis RJ, Fulton DB, Andreotti AH. Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin A. 2002;99:1899–904. doi: 10.1073/pnas.042529199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leverson JD, Ness SA. Point mutations in v-Myb disrupt a cyclophilin-catalyzed negative regulatory mechanism. Mol Cell. 1998;1:203–11. doi: 10.1016/s1097-2765(00)80021-0. [DOI] [PubMed] [Google Scholar]
- 9.Cabral WA, Perdivara I, Weis M, Terajima M, Bisset AR, Chang W, et al. Abnormal type I collagen post-translational modification and crosslinking in a cyclophilin B KO mouse model of recessive osteogenesis imperfecta. PLoS Genet. 2014;10:e1004465. doi: 10.1371/journal.pgen.1004465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satoh K, Matoba T, Suzuki J, O’Dell M, Nigro P, Cui Z, et al. Cyclphilin A mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation. 2008;117:3088–98. doi: 10.1161/CIRCULATIONAHA.107.756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J, Person MD, Zhu J, Abbruzzese JL, Li D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res. 2004;64:9018–26. doi: 10.1158/0008-5472.CAN-04-3262. [DOI] [PubMed] [Google Scholar]
- 12.Howard BA, Furumai R, Campa MJ, Rabbani ZN, Vujaskovic Z, Wang XF, et al. Stable RNA interference-mediated suppression of cyclophilin A diminishes non-small-cell lung tumor growth in vivo. Cancer Res. 2005;65:8853–60. doi: 10.1158/0008-5472.CAN-05-1219. [DOI] [PubMed] [Google Scholar]
- 13.Choi KJ, Piao YJ, Lim MJ, Kim JH, Ha J, Choe W, et al. Overexpressed cyclophilin A in cancer cells renders resistance to hypoxia- and cisplatin-induced cell death. Cancer Res. 2007;67:3654–62. doi: 10.1158/0008-5472.CAN-06-1759. [DOI] [PubMed] [Google Scholar]
- 14.Stewart T, Tsai S-CJ, Grayson H, Henderson R, Opelz G. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346:796–8. doi: 10.1016/s0140-6736(95)91618-0. [DOI] [PubMed] [Google Scholar]
- 15.Ormandy CJ, Binart N, Kelly PA. Mammary gland development in prolactin receptor knockout mice. J Mammary Gland Biol Neoplasia. 1997;2:355–64. doi: 10.1023/a:1026395229025. [DOI] [PubMed] [Google Scholar]
- 16.Neville MC, McFadden TB, Forsyth I. Hormonal regulation of mammary differentiation and milk secretion. J Mammary Gland Biol Neoplasia. 2002;7:49–66. doi: 10.1023/a:1015770423167. [DOI] [PubMed] [Google Scholar]
- 17.Welsch CW, Nagasawa H. Prolactin and Murine Mammary Tumorigenesis: A Review. Cancer Res. 1977;37:951–63. [PubMed] [Google Scholar]
- 18.Hankinson SE, Willett WC, Michaud DS, Manson JE, Colditz GA, Langcope C, et al. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1999;91:629–34. doi: 10.1093/jnci/91.7.629. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Clevenger CV, Minkovsky N, Kumar KG, Raghunath PN, Tomaszewski JE, et al. Stabilization of prolactin receptor in breast cancer cells. Oncogene. 2006;25:1896–902. doi: 10.1038/sj.onc.1209214. [DOI] [PubMed] [Google Scholar]
- 20.McHale K, Tomaszewski JE, Puthiyveettill R, LiVolsi VA, Clevenger CV. Altered expression of prolactin-receptor associated signaling in human breast carcinoma. Mod Pathol. 2008;21:565–71. doi: 10.1038/modpathol.2008.7. [DOI] [PubMed] [Google Scholar]
- 21.Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Expression of Prolactin and its receptor in human breast carcinoma. Endocrinology. 1997;138:5555–60. doi: 10.1210/endo.138.12.5605. [DOI] [PubMed] [Google Scholar]
- 22.Arendt LM, Rose-Hellekant TA, Sandgren EP, Schuler LA. Prolactin Potentiates Transforming Growth Factor α Induction of Mammary Neoplasia in Transgenic Mice. The American Journal of Pathology. 2006;168:1365–74. doi: 10.2353/ajpath.2006.050861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene. 2003;22:4664–74. doi: 10.1038/sj.onc.1206619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng J, Koblinski J, Dutson LB, Feeney YB, Clevenger CV. Peptidyl-prolyl isomerase regulation of Jak2 activation and the progression of human breast cancer. Cancer Res. 2008;68:7769–78. doi: 10.1158/0008-5472.CAN-08-0639. [DOI] [PubMed] [Google Scholar]
- 25.Caffarel MM, Zaragoza R, Pensa S, Green AR, Watson CJ. Constitutive activation of Jak2 in mammary epithelium elevates Stat5 signaling, promotes alveologenesis and resistance to cell death, and contributes to tumorigenesis. Cell Death Differ. 2012;19:511–22. doi: 10.1038/cdd.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakamoto K, Creamer BA, Triplett AA, Wagner K-U. The Janus kinase 2 (Jak2) is required for expression and nuclear accumulation of Cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:2218–32. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]
- 27.Sakamoto K, Lin WC, Triplett AA, Wagner KU. Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: Implications for cancer prevention and therapy. Cancer Res. 2009;69:6642–50. doi: 10.1158/0008-5472.CAN-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner K-U, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initation, and progression. J Mammary Gland Biol Neoplasia. 2008;13:93–104. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- 29.Huai Q, Kim H-Y, Liu Y, Zhao Y, Mondragon A, Liu JO, et al. Crystal Structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. ProcNatlAcadSciUSA J1 - PNAS. 2002;99:12037–42. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciechomska I, Legat M, Golab J, Wesoloska A, Kurzag Z, Makeiwica A, et al. Cyclosporine A and its non-immunosuppressive derivative NIM811 induce apoptosis of malignant melanoma cells in in vitro and in vivo studies. Int J Cancer. 2005;117:59–67. doi: 10.1002/ijc.21153. [DOI] [PubMed] [Google Scholar]
- 31.Colgan J, Asmal M, Yu B, Luban J. Cyclophilin A-Deficient Mice Are Resistant to Immunosuppression by Cyclosporine. J Immunol. 2005;174:6030–8. doi: 10.4049/jimmunol.174.10.6030. [DOI] [PubMed] [Google Scholar]
- 32.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–15. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 33.Brill B, Boecher N, Groner B, Shemanko CS. A sparing procedure to clear the mouse mammary fat pad of epithelial components for transplantation analysis. Lab Anim. 2008;42:104–10. doi: 10.1258/la.2007.06003e. [DOI] [PubMed] [Google Scholar]
- 34.Plante I, Stewart MK, Laird DW. Evaluation of mammary gland development and function in mouse models. J Visualized Exp. 2011;21:1–5. doi: 10.3791/2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asselin-Labat M-L, Sutherland K, Barker H, Thomas R, Shackleton M, Forrest NC, et al. Gata-3 is an essential regulator of mammary gland morphogenesis and luminal cell differentiation. Nat Cell Biol. 2007;9:201–9. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 36.Lee HJ, Ormandy CJ. Interpaly between progesterone and prolactin in mammary development. Mol Cell Endocrinol. 2012;357:101–7. doi: 10.1016/j.mce.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 37.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner K-U, Oka T, et al. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar development. JCB. 2001;155:531–42. doi: 10.1083/jcb.200107065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Robinson GW, Wagner K-U, Garrett L, Wyhshaw-Boris A, Henninghausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–86. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 40.Owens TW, Rogers RL, Best SA, Ledger A, Mooney AM, Ferguson A, et al. Runx2 is a novel regulator of mammary epithelial cell fate in development and breast cancer. Cancer Res. 2014;74:5277–86. doi: 10.1158/0008-5472.CAN-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oakes SR, Naylor MJ, Asselin-Labat ML, Blazek KD, Gardiner-Garden M, Hilton HN, et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev. 2008;22:581–6. doi: 10.1101/gad.1614608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shillingford JM, Miyoshi K, Robinson EL, Grimm SL, Rosen JM, Neubauer H, et al. Jak2 is an essential tyrosine kinase involved pregnancy-mediated development of mammary secretory epithelium. Mol Endocrinol. 2002;16:563–70. doi: 10.1210/mend.16.3.0805. [DOI] [PubMed] [Google Scholar]
- 43.Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW, et al. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes Dev. 2009;23:2382–7. doi: 10.1101/gad.1840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4355–39. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- 45.Sakamoto H, Triplett AA, Schuler LA, Wagner KU. Janus kinase 2 is required for the initation but not maintenance of prolactin-induced mammary cancer. Oncogene. 2010;29:5359–69. doi: 10.1038/onc.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen Q, Brown PH. Transgenic mouse models for the prevention of breast cancer. Mutat Res. 2005;576:93–110. doi: 10.1016/j.mrfmmm.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 47.Brisken C, Kaur S, Chavarria TE, Binart N, Sutherland RL, Weinberg R, et al. Prolactin controls mammary gland development via direct and indirect mechanisms. Dev Biol. 1999;210:96–106. doi: 10.1006/dbio.1999.9271. [DOI] [PubMed] [Google Scholar]
- 48.Rosenwirth B, Billich A, Datema R, Donatsch P, Hammerschmid F, Harrison R, et al. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM811, a nonimmunosuppresive cyclosporine analog. Antimicrob Agents Chemother. 1994;38:1763–72. doi: 10.1128/aac.38.8.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osadchy A, Koren G. Cyclosporine and lactation: when the mother is willing to breastfeed. Ther Drug Monit. 2011;33:147–8. doi: 10.1097/FTD.0b013e318208e3a4. [DOI] [PubMed] [Google Scholar]
- 50.Andreotti AH, Bunnell SC, Feng S, Berg L, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–7. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Loss of CypA is associated delayed mammary development. A. CypA is not expressed in CypA−/− mice. Western blot analysis of mammary gland lysates from 14 wk CypA+/+ and CypA−/− virgins probed with the indicated antibodies. B. Low power photomicrographs demonstrate delay in mammary development in CypA−/− mice. Inguinal mammary glands from CypA+/+ and CypA−/− mice were dissected at the indicated times (P15, day 15 pregnancy; L2, day 2 lactation), paraffin-embedded and stained with hematoxylin and eosin prior to microscopy. (Bar = 500 microns).
Supplemental Figure 2 Immunohistochemistry of CypA+/+ and CypA−/− females at P15 and L2 demonstrate decreased levels of lactalbumin, pJak2, and pStat5 expression in CypA−/− females. No alteration in the expression of whey acidic protein or beta-casein was noted (Bar = 250 microns in Panel A; Bar = 100 microns in Panels B + C).
Supplemental Figure 3 Immunohistochemistry of CypA+/+ and CypA−/− wk 14 virgins demonstrate decreased Ki67 and unchanged cleaved caspase 3 (CC3) levels in CypA−/− females. (Bar = 250 microns).
Supplemental Figure 4 Immunohistochemistry of CypA+/+ x erbB2 and CypA−/− x erbB2 wk 14 virgins demonstrate decreased pStat5, CISH, and Ki67 and unchanged cleaved caspase 3 (CC3) in CypA−/− x erbB2 females. (Bar = 250 microns).