

Abstract
Background
Mitotic fission is increased in pulmonary arterial hypertension (PAH), a hyperproliferative, apoptosis-resistant disease. The fission mediator, dynamin related protein 1 (Drp1) must complex with adaptor proteins to cause fission. Drp1-induced fission has been therapeutically targeted in experimental PAH. Here we examine the role of two recently discovered, poorly understood, Drp1 adapter proteins, mitochondrial dynamics protein of 49 and 51 kDa (MiD49 and MiD51) in normal vascular cells and explore their dysregulation in PAH.
Methods
Immunoblots of pulmonary artery smooth muscle cells (PASMC, control, n=6; PAH, n=8) and immunohistochemistry of lung sections (control, n=6; PAH, n=6) were used to assess the expression of MiD49 and MiD51. The effects of manipulating MiDs on cell proliferation, cell cycle, and apoptosis were assessed in human and rodent PAH PASMC using flow cytometry. Mitochondrial fission was studied by confocal imaging. A microRNA (miR) involved in the regulation of MiD expression was identified using microarray techniques and in silico analyses. The expression of circulatory miR was assessed using qRT-PCR in healthy volunteers (HV) vs PAH patients from Sheffield, UK (plasma, HV, n=29, PAH, n=27; whole blood, HV, n=11, PAH, n=14), and then confirmed in a cohort from Beijing, China (plasma, HV, n=19, PAH, n=36; whole blood, HV, n=20, PAH, n=39). This work was replicated in monocrotaline and SU5416-hypoxia, preclinical PAH models. siRNA targeting MiDs or a miR mimic were nebulized to rats with monocrotaline-induced PAH (n=4–10).
Results
MiD expression is increased in PAH PASMC, which accelerates Drp1-mediated mitotic fission, increases cell proliferation and decreases apoptosis. Silencing MiDs (but not other Drp1 binding partners, Fis1 or MFF) promotes mitochondrial fusion and causes G1-phase cell cycle arrest, through ERK1/2 and CDK4-dependent mechanism. Augmenting MiDs in normal cells causes fission and recapitulates the PAH phenotype. MiD upregulation results from decreased miR-34a-3p expression. Circulatory miR-34a-3p expression is decreased in both PAH patients and in preclinical models of PAH. Silencing MiDs or augmenting miR-34a-3p regresses experimental PAH.
Conclusion
In health, MiDs regulate Drp1-mediated fission whilst in disease, epigenetic upregulation of MiDs increases mitotic fission, which drives pathologic proliferation and apoptosis resistance. The miR-34a-3p-MiD pathway offers new therapeutic targets for PAH.
Keywords: mitochondrial dynamics, mitochondrial fission, extracellular signal regulated kinase (ERK), cyclin dependent kinase (CDK), dynamin related protein 1 (Drp1), miR-34a-3p, cell cycle
Introduction
Pulmonary arterial hypertension (PAH) is a vascular disease in which pulmonary vascular obstruction, due to vasoconstriction, inflammation, fibrosis and a proliferation/apoptosis imbalance, increases right ventricular afterload leading to fatal right ventricular failure1. Excessive cell proliferation and apoptosis resistance are important features of pulmonary artery smooth muscle cells (PASMC) in both PAH patients and rodent models of PAH. This “neoplastic phenotype” of PAH PASMC persists in cell culture and is promoted by acquired changes in mitochondrial metabolism, including a shift to uncoupled glycolysis, known as the Warburg phenomenon2, and fragmentation of the mitochondrial network3.
Mitochondria exist in dynamic networks that undergo continuous fission (division) and fusion (union)4. Altered mitochondrial dynamics in PAH result, in part, from excessive mitochondrial fission caused by increased activation of the fission-mediating GTPase, dynamin related protein 1 (Drp1)3, 5, 6. When activated, Drp1 moves from the cytosol to the outer mitochondrial membrane (OMM) where it interacts with its binding partners in a multimerization reaction that culminates in fission7–9. Coordination between mitosis and mitochondrial division ensures equitable distribution of mitochondria between daughter cells and is mediated, in part, by a shared dependence of both processes on similar kinases, including cyclin B-CDK1 (reviewed in4). Pathologic levels of Drp1 activation in both PAH and non-small cell lung cancer, the prototypic disease in which proliferation is increased and apoptosis suppressed, cause mitochondrial fragmentation and promote an imbalance of proliferation and apoptosis in both conditions10. Molecular or chemical inhibition of Drp1 regresses both diseases by causing cell cycle arrest3, 10.
In mammals, four OMM proteins act as Drp1 receptors: fission 1 (Fis1), mitochondrial fission factor (MFF), mitochondrial dynamics protein of 49 kDa (MiD49) and mitochondrial dynamics protein of 51 kDa (MiD51)11. MiD49 and MiD51 are expressed on the OMM12; however, they also exist in the cytosol, where their role is unknown13. Even the role of MiDs as promoter versus inhibitors of fission remains controversial and their role in human disease is unknown. Overexpression of either MiD49 or MiD51 increases Drp1 recruitment to the mitochondria12. In one report, MiD overexpression caused fusion13, perhaps by acting as bait for Drp1; however, in another study, overexpression enhanced fission14, perhaps by enhancing the compactness and efficiency of the fission apparatus12. The possibility that MiD49 and MiD51 might have a role in disease has not been assessed, although a recent report identified a single nucleotide polymorphism in MiD49 that was associated with adverse remodeling of small pulmonary arteries in PAH15. Moreover, the role of endogenous MiDs in normal cell biology or in human disease is largely unknown, with the few studies to date largely relying on heterologous overexpression of MiDs in cell lines.
Here, we examine the fundamental role of these newly discovered Drp1 binding partners in regulating mitochondrial dynamics and the cell cycle in normal PASMC, and describe the pathologic cause and consequences of their dysregulation in PAH. We report that the expression of both MiD49 and MiD51 is pathologically elevated in PASMC and small pulmonary arteries (PA) from PAH patients, as well as in two rodent models of disease. Super resolution confocal imaging reveals the formation of a ring-shaped macromolecular fission apparatus, comprised of MiDs and Drp1, at the site of fission. Silencing MiD expression restores mitochondrial fusion and reverses the pseudo-neoplastic phenotype of PAH PASMC by decreasing cell proliferation and increasing apoptosis in human and experimental PAH. The impact of MiD knockdown on proliferation results as a consequence of cell cycle arrest in G1 phase, suggesting that mitotic fission is controlled by a cell cycle checkpoint. Furthermore, the progression of PAH has been linked to the dysregulation of several miRs, including miR-20416, miR-12617, miR-12418, miR-2519 and miR-13819. These miRs influence disease processes in a variety of tissues, including the right ventricle, skeletal muscle and pulmonary vasculature, respectively. In our study we demonstrate that MiD expression is epigenetically upregulated by a decrease in microRNA (miR)-34a-3p, in both PAH patients and animal models. Overexpression of either MiD recapitulates the PAH phenotype in normal PASMC. In contrast, silencing MiD49 or MiD51 or augmenting a miR-34a-3p mimic, reverses established PAH in vivo, highlighting the contribution of the miR-34a-3p-MiD-Drp1 axis to the pathophysiology of human and experimental PAH and the viability of this pathway as a target for new therapeutic strategies.
Methods
The data and analytic methods will be made available to other researchers from the corresponding author upon reasonable request for purposes of reproducing the results or replicating the procedure. But we cannot make the materials available due to their scarcity (in the case of cells and tissues) or the cost (in case of therapeutic siRNAs and miRs).
Cell culture and reagents
PAH PASMC were isolated from PAH patients. Normal PASMC were isolated from control subjects or purchased from Lonza or Cell Applications Inc. PASMC lines were studied within 6 passages. PASMC were grown in Medium 231 supplemented with Smooth Muscle Growth Supplement (SMGS, Life Technologies, Carlsbad, CA, USA). Experiments were performed on 6 normal PASMC lines (50% Female, Mean age: 50.7 years) and 8 PAH PASMC lines (28% Female, Mean age: 41.2 years). Blood Outgrowth Endothelial Cells (BOEC) were isolated and cultured from PAH patients and normal individuals (Demographics in Supplementary Table 1) as previously described20. Briefly blood was collected from normal individuals (n=5) and PAH patients (n=6) by venipuncture. Peripheral blood mononuclear cell (PBMC) was isolated from the whole blood by density gradient centrifugation. PBMC were cultured in BOEC generation medium for 7–14 days for the appearance of outgrowth colonies.
Immunohistochemistry
To assess localization and expression of MiDs in human (normal, n=6, PAH, n=6, Supplementary Table 1) and rodent models (MCT model: Ctrl n=5, MCT n=5; Su/Hx model: Ctrl n=3, Hx n=3, Su/Hx n=3), lung immunohistochemistry was performed with a Ventana Autostainer (Ventana Discovery XT, Ventana Medical Systems, Tucson, AZ, USA) using the company’s buffer solution and staining protocol.
Immunofluorescence staining
Immunofluorescence staining of the formalin-fixed paraffin embedded rat lung tissues was performed as previously described21. Briefly, rat lung sections were labeled with smooth muscle actin (SMA, Abcam, Cambridge MA, USA), von Willebrand factor (vWF, Dako Denmark A/S, Glostrup, Denmark) antibodies. Nuclei were labeled with DAPI (Life Technologies, Carlsbad, CA, USA).
Small interfering RNA (siRNA) treatment of PASMC
For siRNA treatment, PAH PASMC were grown to 60–80% confluence and then transfected with 25 picomole of siRNA using the Lipofectamine® RNAiMAX Transfection Reagent (Life Technologies, Carlsbad, CA). The sequences of siRNA duplexes specific for human MiD49, MiD51, Fis1, MFF, rat MiD49, MiD51 and negative control are available in the supplementary section. The knock down efficiency was assessed after 48 hours using qRT-PCR (Bio-Rad, Hercules, CA, USA) and 72 hours using immunoblotting.
Cell cycle analysis
To assess the effects of MiD expression on cell cycle progression flow cytometry was performed on cells that were initially synchronized by serum starvation. PAH PASMC were transfected with siRNA targeting MiD49 or MiD51 versus a ctrl-siRNA for 24h. After siRNA transfection, the cells were serum starved for 48 hours to synchronize the cells at G1/G0 phase. The cells were then stimulated with 20% FBS/Medium 231 for 24h. Cells were harvested, suspended in phosphate-buffered saline (PBS), and fixed with 70% ethanol (v/v) at −20°C. The fixed cells were washed twice with cold PBS and incubated with PI/RNase Staining Buffer (BD Biosciences, Franklin Lakes, NJ, USA) at room temperature for 15 minutes. The samples were analyzed through flow cytometry using a fluorescence-activated cell sorter (Beckman Coulter Inc, Brea, CA, USA).
Cell proliferation assay
Cell proliferation was quantified using the Click-iT EdU kit according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA). Measurements were made 72h following administration of siRNA or plasmid transfection.
Apoptosis Assay
Cells were grown in 100 mm dishes and were harvested and counted at 80–90% confluence. 1.5 x106 cells were electroporated (LONZA 4D-Nucleofactor, Rochester, NY, USA) with ctrl-siRNA, siMiD49 or siMiD51 using the inbuilt program, FG113 following manufacturer’s instructions. The cells were collected after 72h and stained with the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instruction. Detection and quantification of apoptotic cells were obtained by flow cytometry analysis (Beckman Coulter FC500). Apoptosis was also assessed by measuring the activity of caspase3/7 after transfecting the PAH PASMC for 48h using the Caspase-Glo 3/7 Assay Kit (Promega, Madison, WI, USA) following manufacturer’s instructions.
Quantification of mitochondrial fission
Cells grown in glass-bottom dishes (MatTek Corporation, Ashland, MA, USA) were transfected with siRNAs against either MiD49 or MiD51 or ctrl-siRNA. Cells were loaded with the mitochondrial potentiometric dye, tetramethylrhodamine (TMRM; 20 nM, 20 minutes in culture medium at 37°C; Molecular Probes, Eugene, OR, USA) or infected with Adv-mNeon Green for 48h prior to imaging. Cells were imaged with a Leica SP8 laser scanning confocal microscope using a 1.40NA, 63X oil immersion objective with × 2 digital zoom (excitation 561 nm, emission > 575 nm for TMRM and excitation 491nm, emission 505–530 nm for Adv-mNeon Green; Leica Planapo, Wetzlar, Germany). Acquired images were background subtracted, filtered (median), thresholded, and binarized to identify individual mitochondria using ImageJ (U.S. National Institutes of Health (NIH), Bethesda, MD, USA). Continuous mitochondrial structures were counted with ImageJ’s particle counting subroutine and the number was normalized to the total mitochondrial area to obtain the mitochondrial fragmentation count (MFC) for each image, a previously validated measure of mitochondrial fragmentation10. For every intervention, 8–25 randomly selected cells were imaged by a blinded microscopist and MFC was calculated. A lower MFC indicates a more fused mitochondrial network.
Machine learning
Mitochondrial morphology was further analyzed by a machine-learning based categorization using the Leica LAS X software. Briefly, every mitochondrion in each cell was divided into three categories (punctate, intermediate or filamentous) based on their morphology. Fifteen to twenty mitochondria from each group were manually identified and categorized in order to inform the machine-learning algorithm. The algorithm is calculated based on the characteristics (area, length and sphericity) of the mitochondria. Then the algorithm was applied to all the images. The percentage area of each category in each confocal image was calculated and the distribution of the three categories was compared among groups.
Mitochondrial Networking
Mitochondrial Networking was determined with mitochondrial targeted, photo-activatable green fluorescent protein (Mito-PA-GFP) using a metric called the mitochondrial networking factor (MNF). An increase in MNF indicates higher degree of mitochondrial network fusion3, 10.
Whole Cell Micropolarimetry
PASMC micropolarimetry was performed by Mitochondrial flux measured with Seahorse XFe24 (Seahorse Biosciences, North Billerica, MA, USA) in PAH PASMC after 48h of transfection with siMiD49 or siMiD51. A detailed methodology is described in the supplementary section.
miRNA microarray
To identify candidate miRs that are dysregulated in PAH we performed a miR microarray study. Total RNA from cultured PASMC of 6 PAH patients and 3 healthy subjects was extracted using the mirVana miRNA isolation kit (Ambion/Life Technologies). RNA quality was assessed by measurement of absorbance at 260nm and 280nm using a spectrophotometer (cutoff of 1.8–2.2) and by calculation of a RNA integrity number (RIN) in an Agilent Bioanalyzer (RIN > 6). Microarray experiments were carried out at the Centre for Applied Genomics at the Hospital for Sick Children (Toronto, Canada). Differential expression of miRNAs between the PAH and control cohorts was examined using the Affymetrix GeneChip miRNA 4.0 Array (Affymetrix, Santa Clara, CA, USA). RNA samples were prepared using the FlashTag Biotin HSR RNA Labeling Kit (Affymetrix) prior to hybridization at 48°C for 16–18h. Fluorescent signals were captured using the Affymetrix GeneChip Scanner 3000. Affymetrix CEL files were normalized using the robust multiarray average (RMA) algorithm within the Affymetrix Expression Console Software (version 1.4.1.46). The resulting normalized files were then analyzed within the Transcriptome analysis Console (TAC version 3.1.0.5) and a One-Way between-subject ANOVA (unpaired) was applied. These final data were then filtered to include only human miRs that satisfied a 2-fold cut-off, and P<0.05.
All data have been submitted to the NCBI Gene Expression Omnibus (GEO accession number: GSE108707).
In silico analysis of miRNAs binding to MiD49 and MiD51
In order to identify which miRNAs can bind to MiD49 and MiD51, in silico analysis using Target Scan 7.0 and miRDB were used. Out of the 25 downregulated miRNAs, we identified miR-34a-3p as the only predicted regulator of MiD51. Although none of the downregulated miRNAs were predicted to regulate MiD49, analysis using nucleotide BLAST identified potential miR-34a-3p target sites within the 3′-UTR of MiD49 and MiD51. 3′-UTR sequences of MiD49 and MiD51 were obtained from the University of California, Santa Cruz (UCSC) Gene Sorter (http://genome.ucsc.edu/). The nucleotide sequence of hsa-miR-34a-3p (Accession # MIMAT0004557) was extracted from miRBase Release 21 (http://www.mirbase.org).
miR-binding luciferase reporter assay
To validate the binding of candidate miRs to the MiD mRNA we performed a binding assay using (3′-UTR) of MiD49 or MiD51 and miR-34a-3p. Reporter plasmids containing the untranslated region (3′-UTR) of MiD49 or MiD51 were obtained from GeneCopoeia (Rockville, MD). HEK293A cells were co-transfected with either MiD49 or MiD51 reporter plasmids together with miR-ctrl or miR-34a-3p using Lipofectamine 3000 and RNAi MAX transfection reagents (Life Technologies, Carlsbad, CA). Luciferase activity assay was performed as previously described19.
Human blood sample
Studies of circulatory miR expression of PAH patients versus healthy volunteers (HV) was performed at the University of Sheffield, UK and FuWai Hospital, Beijing, China. The collection of patient blood samples was coordinated through the Sheffield NIHR Clinical Research Facility. The study was approved by the Local Research Ethics Committee and The Sheffield Teaching Hospitals Foundation Trust Observational Cardiovascular Biobank (08/H1308/193 and STH15222), Sheffield, UK as well as the institutional review board of FuWai Hospital, Beijing, China. Informed consent was obtained from each subject before enrolment. Within each cohort HV vs PAH patients were matched for age and sex.
Animal Studies
All protocols were approved by the Queen’s University and Laval University Animal Care Committees.
MCT PAH model
MCT-induced PAH model in rats was created by subcutaneous injection of 60mg/kg of MCT, as previously described21. PAH was allowed to develop for 7 days prior to intervention. Experiments were performed 3-weeks following MCT injection. Randomization and blinding were included in the study design for the duration of the protocol including analysis.
SU5416/hypoxia PAH model
This model was created as previously described22. Experiments were performed 3-weeks following removal from the hypoxic chamber.
siMiD49 or siMiD51 or miR-34a-3p mimic therapy
Briefly, 1 nmole of siMiD49 or siMiD51 or ctrl-siRNA and 5nmole of miR-34a-3p or miR-control was administered 1 week post MCT injection to the anesthetized rat by nebulization in 50μl saline using an aerosol nebulizer (AG-AL7000STD, Kent Scientific, Torrington, Connecticut, USA). siRNAs were procured from Integrated DNA Technology (Coralville, IA, USA). miR-34a-3p and miR-control were purchased from Life Technologies. Both the siRNAs against MiDs and the ctrl-siRNA were modified for a higher stability for in vivo studies.
Hemodynamics
mPAP, RVSP, CO were measured using closed-chest technique under isofluorane anesthesia (2.5% enriched O2; 1.5ml/min), as previously described21.
Statistical analyses
Quantitative data are presented as the mean ± SEM. Intergroup differences were assessed using Students’ t-test (unpaired or paired) or one-way ANOVA as appropriate. For in vivo studies, Šídák’s multiple comparisons test was used to determine statistical significance of multiple comparisons. Statistical analyses were performed using the GraphPad Prism 7.0d software package. A P < 0.05 was considered statistically significant.
Results
Increased expression of MiD49 or MiD51 in PAH
MiD49 and MiD51 expression were increased in PASMC (Fig. 1A, Supplementary Fig. 1A) and blood outgrowth endothelial cells (BOECs; Supplementary Fig. 1B, Supplementary Table 1) from PAH patients. Confocal microscopy of PAH PASMC localized some of this increased expression to the mitochondria. Stimulated emission depletion (STED) super-resolution microscopy, which has a resolution 5-times greater than confocal microscopy (50nm), revealed MiDs within the macromolecular fission apparatus at the constriction point of the mitochondria, adjacent to Drp1 (Fig. 1B). The ring-shaped macromolecular fission apparatus is found at points of fission in normal and PAH PASMC mitochondria. In PAH, we observed more MiDs at the focal area of fission and noted that the fission apparatus appeared “tighter” (i.e. more dense) than in normal PASMC. However, endogenous MiDs are also found in the cytosol, as previously observed13. Immunohistochemistry of lungs from PAH patients (Fig. 1C, Supplementary Table 1) and rats with monocrotaline (MCT) and Sugen5416/Hypoxia (Su/Hx)-induced PAH (Fig. 1D and Supplementary Fig. 1C) confirmed the increased expression of MiD49 and MiD51 in the media (PASMC) and intima (pulmonary artery endothelial cells) of diseased pulmonary arterioles.
Fig. 1. Pathological upregulation of MiD49 and MiD51 in human and experimental PAH.


(A) Representative immunoblots and densitometry demonstrating increased protein expression of MiD49 and MiD51 in human PAH PASMC (n=6) vs normal human PASMC (n=3). β-actin was used as the loading control (*P < 0.05).
(B) Confocal images showing higher expression of MiD49 and MiD51 in PAH PASMC. STED super-resolution images showing association of MiDs with mitochondria and Drp1. Staining used in the images to create colors: mitochondria (red, MitoTracker™ Deep Red), MiD49, MiD51 (green) and Drp1 (cyan) in normal and PAH PASMC. Scale bar: 10 μm for the confocal images and 1 μm for the STED images.
(C) Representative images and quantification of immunohistochemistry demonstrating increased expression of MiD49 and MiD51 protein (brown) in the media and intima of small pulmonary arteries from human PAH lungs vs control lungs. 11–14 distal pulmonary arteries, <200μm in diameter from 6 subjects per group (***P < 0.001; n=11–14/subject). Scale bar: 25 μm.
(D) Representative images and quantification of immunohistochemistry demonstrating increased expression of MiD49 and MiD51 (brown) in the media and intima of distal pulmonary arteries from MCT PAH rats. 8–14 distal pulmonary arteries, <150 μm in diameter from 5 animals per group (***P < 0.001). Scale bar: 25 μm.
Silencing MiD49 or MiD51 promotes mitochondrial fusion
As reported previously3, PAH PASMC exhibits a fragmented mitochondrial network, which was also observed in PAH BOECs (Supplementary Fig. 1D–E). Resonant scanning showed that effective knockdown of either MiD (Supplementary Fig. 2A–B) led to increased mitochondrial motion (Supplementary movies 1–3). Knocking down MiDs also inhibited mitochondrial fission (Fig. 2A) (reduced mitochondrial fragmentation count, MFC)10 and increased percentage area of filamentous mitochondria (measured by machine-learning) (Fig. 2B). Furthermore, silencing MiDs increased mitochondrial fusion, evident as an increase in the mitochondrial networking factor (MNF). Increased MNF reflects a more rapid diffusion of photoactivated GFP within the mitochondrial matrix (Fig. 2C–D). Overexpression of either MiD in PASMC from control subjects led to mitochondrial fission, creating a phenotype similar to that seen in PAH PASMC (Fig. 2E–F). However, MiD overexpression failed to cause mitochondrial fragmentation in Drp1 knockout (KO) mouse embryonic fibroblasts (MEFs; Supplementary Fig. 3A–B), indicating that Drp1 is required for MiD-mediated fission. Interestingly no significant alteration in the expressions of fusion mediators Mfn1, Mfn2 and OPA1 was observed by silencing MiD49 in PAH PASMC. Silencing MiD51 reduced the expression of the fusion-mediators, Mfn1 and OPA1 (Supplementary Fig. 4A–C).
Fig. 2. MiD49 or MiD51 regulates mitochondrial network, cell proliferation and apoptosis.


(A) Mitochondrial fragmentation in PAH PASMC is reversed by silencing of MiD49 or MiD51. Representative images of mitochondrial networks of normal PASMC and PAH PASMC stained with the potentiometric dye TMRM (red). PAH PASMC were transfected with the specified siRNA, infected with Adv-mNeon Green and imaged after 48h following infection. Mitochondria were color coded by their morphology: red: punctate; green: intermediate; purple: filamentous. Scale bar: 10μm.
(B) Silencing of MiD49 or MiD51 reduces mitochondrial fission. Mitochondrial fragmentation was quantified by mitochondrial fragmentation count (MFC) and percentage area of punctate, intermediate and filamentous mitochondria of each image (***P < 0.001; n=15/group).
(C) Mitochondrial network is restored in PAH PASMC by silencing of MiD49 or MiD51. Representative images of the photoactivation experiments confirmed the increase in mitochondrial network in PAH PASMCs co-transfected with specified siRNA and mitochondrial matrix targeted green fluorescent protein (mito-PAGFP) plasmid for 48 h. The cells were also loaded with TMRM (red). Scale bar: 10μm.
(D) Silencing of MiD49 or MiD51 increases mitochondrial networking factor (MNF). Mitochondrial network is quantified by determining mitochondrial networking factor (MNF) which is increased in PAH PASMC following silencing of MiD49 or MiD51 (*P < 0.05, **P < 0.01; n=5/group; AU: arbitrary unit).
(E) Augmenting MiD49 or MiD51 in normal human PASMC induces mitochondrial fission. Representative images of mitochondrial networks of normal human PASMC transfected with the specified plasmid. Cells were loaded with TMRM (red). Scale bar: 10μm.
(F) Augmentation of MiD49 or MiD51 significantly increases mitochondrial fragmentation (*P < 0.05, ***P < 0.001; n=15/group).
(G) Proliferation of PAH PASMC is inhibited by silencing MiD49 or MiD51. Cell proliferation was analyzed 72h post-transfection (*P < 0.05, **P < 0.01; n=3/group).
(H) Proliferation of normal PASMC is increased by overexpressing MiD49 or MiD51. Cell proliferation was analyzed 72h post-transfection (*P < 0.05, **P < 0.01; n=3/group).
(I) Silencing of MiD49 or MiD51 induces cell cycle arrest in the G1/G0 phase. PAH PASMC was transfected with siMiD49 or siMiD51 for 24h, serum starved for 48h, and then serum stimulated for 24h. Cell cycle analyses were performed by flow cytometry following propidium iodide (PI) staining (**P < 0.01; n=3/group).
(J) Silencing of MiD49 or MiD51 increases baseline apoptosis. PAH PASMCs were labeled with Annexin VFITC and PI and assessed by flow cytometry analyses 72h post-transfection (*P < 0.05, **P < 0.01; n=3/group).
MiDs regulate proliferation, cell cycle progression, apoptosis and oxidative mitochondrial respiration
Knocking down MiD49 or MiD51 significantly decreased cell proliferation in PASMC from both PAH patients (Fig. 2G) and control subjects (Supplementary Fig. 5), without altering the expression of other Drp1 binding partners (Supplementary Fig. 6A). Of note, silencing other Drp1 receptors (MFF and Fis1) (Supplementary Fig. 6B–C) failed to inhibit cell proliferation (Supplementary Fig. 6D–E) or alter mitochondrial morphology (Supplementary Fig. 6F–H). In contrast, overexpression of MiD49 or MiD51 in control human PASMC increased cell proliferation (Fig. 2H). Reduced proliferation in response to siRNA mediated knockdown of MiD49 or MiD51 was accompanied by G1/G0 cell cycle arrest (Fig. 2I). In agreement with these findings, MiD knockdown in PAH PASMC resulted in increased basal apoptosis rates (Fig. 2J), which were accompanied by increased expression of the apoptotic mediator, Bak, and decreased phosphorylation of the cell survival-prompting kinase, Akt (Supplementary Fig. 7A). In addition, silencing MiD49 (but not MiD51) increased baseline oxygen consumption in PAH PASMC (Supplementary Fig. 7B) and upregulated the mitochondrial calcium uniporter (MCU), consistent with reversal of the Warburg effect.
Similar to human PAH PASMC, PASMC isolated from MCT PAH and Su/Hx PAH rats retain their hyperproliferative phenotype in culture and have a fragmented mitochondrial network (Supplementary Fig. 8A–C). siRNA mediated knockdown of MiD49 decreased cell proliferation in normal rat PASMC, MCT PAH PASMC and Su/Hx PAH PASMC. However, in the siMiD51 treatment group, despite induction of fusion, inhibition of cell proliferation was observed in normal rat PASMC and MCT PAH PASMC but not the Su/Hx PAH PASMC (Supplementary Fig. 8D–G). Consistent with the findings in human PAH PASMC, siMiDs inhibited mitochondria fission in both MCT and Su/Hx PASMC (Supplementary Fig. 8H–K).
Silencing MiDs inhibits cell proliferation and mitochondrial fission by inhibiting CDK4 and PDGF-ERK1/2 signaling
We next investigated the mechanistic pathway that connects MiDs with cell proliferation and mitochondrial fission in PASMC from human and experimental PAH. Extracellular-signal-regulated kinases (ERK) promotes phosphorylation of Drp1 at serine 616 (ser616)23, 24 and increases cell proliferation25. Knockdown of MiD49 or MiD51 in PAH PASMC reduced the Drp1 ser616 phosphorylation (Fig. 3A, Supplementary Fig. 9A) and decreased ERK1/2 phosphorylation in PAH PASMC stimulated with platelet derived growth factor (PDGF; Fig. 3B, Supplementary Fig. 9B). Moreover, silencing MiDs downregulated the expression of the PDGF receptors α and β (Fig. 3C) and inhibited Raf-1 phosphorylation (Supplementary Fig. 9C) suggesting that silencing MiDs inhibit mitochondrial fission and cell proliferation, in part, by inhibition of the PDGF-Raf-1-ERK1/2 signaling pathway.
Fig. 3. Silencing MiDs modulates molecular mediators that promote Drp1-induced mitochondrial fission and cell proliferation.

(A–F) Silencing MiD49 or MiD51 inhibits phosphorylation of Drp1Ser616 and reduces activation of ERK1/2 and CDK4 while reducing expression of PDGF receptors. Representative images of the immunoblots and the densitometries of the expressions of (A) p-Drp1ser616, (B) p-ERK1/2, (C) PDGF receptors α and β, (D) p-CDK4Thr172, (E) p21Waf1 and (F) p27Kip1. PAH PASMCs were transfected with siMiD49 or siMiD51. Cells were harvested for immunoblot analyses after 48h of transfection. β-actin was used as the loading control (*P < 0.05, **P < 0.01, ***P < 0.001; n=3–4/group).
Silencing MiDs repressed the expression and activity of cyclin dependent kinase 4 (CDK4) by inhibiting its phosphorylation at threonine 172 (Thr172) (Fig. 3D, Supplementary Fig. 9D–E). Furthermore, silencing MiDs increased expression of the CDK4 inhibitors p21Waf1 and p27Kip1 (Fig. 3E–F, Supplementary Fig. 9F). These results suggest that silencing of MiDs cause G1 phase arrest by the inhibition of CDK4 activity. Conversely, silencing of MiD49 or MiD51 failed to impair the activity of CDK2, the mediator of G1-S phase transition (Supplementary Fig. 9G).
Downregulation of miR-34a-3p mediates MiD49 and MiD51 upregulation in PAH
A comparison of miRNA expression profiles in PASMC from PAH patients and normal controls (Fig. 4A) identified 25 miRNAs that were significantly downregulated in PAH (Supplementary Table 2). In silico analysis of these miRNAs identified potential miR-34a-3p target sites within the 3′-UTR of MiD49 and MiD51 (Fig. 4B). Decreased expression of miR-34a-3p in PAH PASMC was confirmed by qRT-PCR (Fig. 4C). Binding of miR-34a-3p to the 3′-UTR of MiD49 and MiD51 was confirmed using a luciferase reporter fused to the 3′-UTR of the MiD49 or MiD51 gene (Fig. 4D). Co-transfection of HEK293A cells with miR-34a-3p mimic and the reporter constructs decreased luciferase activity, confirming binding of the miR to the 3′-UTR of MiD49 or MiD51. Downregulation of miR-34a-3p was also found in the whole blood of MCT and Su/Hx PAH rats (Supplementary Fig. 10A). Two temporally and geographically discrete cohorts of different ethnicity showed significant decrease in miR-34a-3p expression in whole blood and plasma from IPAH patients compared to the age- and sex-matched healthy volunteers (Fig. 4E, Supplementary Table 2). The downregulation of plasma miR-34a-3p identified patients with PAH (Fig. 4F) but did not predict disease-related mortality (data not shown). Similar findings were noted in the blood of both preclinical PAH models (Supplementary Fig. 10A).
Fig. 4. miR-34a-3p is decreased in PAH and is a negative regulator of MiD49 and MiD51.

(A) miRNA expression profiling in PAH and normal PASMCs. Volcano plot showing expression change of miRNAs in PAH relative to control samples. Each dot represents one probe set. Red: reduction; green: increase; n=3 for normal PASMC and n=6 for PAH PASMC.
(B) In silico prediction of miR-34a-3p targeting MiD49 and MiD51. (i) The putative binding site of miR-34a-3p on the 3′-UTR of MiD49 as predicted by nucleotide BLAST. (ii) Nucleotide sequences of MiD49 3′-UTR and miR-34a-3p. The predicted binding site is highlighted in red. Nucleotide positions are indicated in parentheses. (iii) The two putative binding sites of miR-34a-3p on the 3′-UTR of MiD51. (iv) Nucleotide sequences of MiD51 3′-UTR and miR-34a-3p. Predicted target sites are highlighted in blue. Nucleotide positions are indicated in parentheses. The expect value is calculated by BLAST to describe the number of hits one can “expect” to see by chance. The lower the expect value, the more significant the match.
(C) miR-34a-3p is decreased in PAH PASMC. Quantification of miR-34a-3p was performed by qRT-PCR (**P < 0.01; n=4 for normal PASMC and n=6 for PAH PASMC).
(D) miR-34a-3p binds to 3′-UTRs of MiD49 and MiD51. miR-34a-3p was found to repress the activity of luciferase reporter, indicating its binding to the 3′-UTRs of MiD49 and MiD51 genes (**P < 0.01; n=5 and 8 for MiD49 and MiD51, respectively).
(E) miR-34a-3p is decreased in whole blood and plasma from IPAH patients. miR-34a-3p expression from whole blood and plasma from two cohorts were normalized to U6 and miR-23-3p, respectively, and analyzed by qRT-PCR (*P < 0.05, ***P < 0.001; n=11–29 for healthy volunteer and n=14–39 for IPAH patient).
(F) miR-34a-3p identified patients with IPAH. Receiver operating characteristic (ROC) curves showing sensitivity and specificity of whole blood and plasma miR-34a-3p for differentiating patients with IPAH from healthy volunteers at the time of diagnosis (Sheffield whole blood: AUC=0.7857, P = 0.0160; Beijing whole blood: AUC=0.807, P = 0.0002; Sheffield plasma: AUC=0.7573, P = 0.0010; Beijing plasma: AUC=0.8846, P < 0.0001).
Moreover, miR-34a-3p transfection decreased the expression of MiD49 and MiD51 in human PAH PASMC (Fig. 5A). Conversely, administration of anti-miR-34a-3p increased the expression of MiD49 and MiD51 in both normal human and rat PASMC (Fig. 5B and Supplementary Fig. 10B). Augmenting miR-34a-3p fused the mitochondrial network, inhibited proliferation, and induced apoptosis in human PAH PASMC (Fig. 5C–H). miR-34a-3p mimic transfection also decreased proliferation and mitochondrial fission in PASMC from normal, MCT-PAH and Su/Hx PAH rats. (Supplementary Fig. 10C–G).
Fig. 5. Increasing expression of miR-34a-3p downregulates MiD49 and MiD51 in PAH PASMC; administering anti-miR-34a-3p upregulates MiD49 and MiD51 in normal PASMC.

(A) Overexpression of miR-34a-3p downregulates MiD49 and MiD51. Representative images of immunoblots and densitometries showing the expressions of MiD49 and MiD51 in PAH PASMC transfected with miR-34a-3p. Cells were transfected with miR-34a-3p for 72h. β-actin was used as the loading control (*P < 0.05; n=3–4/group).
(B) Anti-miR-34a-3p treatment upregulates MiD49 and MiD51 in normal PASMC. Representative images of immunoblots and densitometries showing the expressions of MiD49 and MiD51 in normal human PASMC transfected with anti-miR-34a-3p for 72h. β-actin was used as the loading control (*P < 0.05; n=4/group).
(C–H) Overexpression of miR-34a-3p inhibits mitochondrial fission, cell proliferation and induces apoptosis in PAH PASMC.
(C) Representative images of mitochondrial networks of PAH PASMC transfected with miR-34a-3p mimic. The cells were also infected with Adv-mNeon Green and imaged 48h following infection. Mitochondria were color coded by their morphology: red: punctate; green: intermediate; purple: filamentous. Scale bar: 10μm.
(D) Mitochondrial fragmentation was quantified by mitochondrial fragmentation count (MFC) and percentage of area of punctate, intermediate and filamentous mitochondria (***P < 0.001; n=16–20/group).
(E) Mitochondrial network is restored in PAH PASMC by augmenting miR-34a-3p. Representative images of the photoactivation experiments confirmed the increase in mitochondrial network in PAH PASMCs co-transfected with miR-34a-3p and mitochondrial matrix targeted green fluorescent protein (mito-PAGFP) plasmid for 48 h. The cells were also loaded with the potentiometric dye TMRM (red). Scale bar: 10μm.
(F) Quantification of mitochondrial network in PAH PASMC by augmenting miR-34a-3p. Mitochondrial network is quantified by determining mitochondrial networking factor (MNF) which is increased in PAH PAMC following transfection with miR-34a-3p mimic. (*P < 0.05; n=6/group; AU: arbitrary unit).
(G) Augmenting miR-34a-3p inhibits proliferation of PAH PASMC. Cell proliferation was analyzed 72h following miR-34a-3p mimic transfection in PAH PASMC (*P < 0.05; n=3/group).
(H) Augmenting miR-34a-3p induces apoptosis of PAH PASMC. PAH PASMC transfected with miR-34a-3p mimic. Apoptosis was assessed by measuring the activity of caspase3/7 48h following transfection with miR-34a-3p mimic (n=2 IPAH PASMC lines/group).
Therapeutic implications of the miR-34a-3p-MiD pathway in vivo
The therapeutic efficacy of silencing MiDs or augmenting miR-34a-3p was determined by nebulizing rats early in the course of MCT-induced PAH with siRNAs against either MiDs or a miR-34a-3p mimic. Treatment with either siMiD or the miR-34a-3p mimic, but not appropriate control materials, caused marked hemodynamic improvement, with decreased mean pulmonary artery pressure (mPAP), right ventricular systolic pressure (RVSP) and total pulmonary resistance (TPR), as well as increased cardiac output (CO) (Fig. 6A–B). These hemodynamic benefits were accompanied by regression of vascular obstruction, as quantified by a decrease in medial thickness in small pulmonary arteries of MCT treated rats (Fig. 6C, Supplementary Fig. 11A–B). siMiDs (Supplementary Fig. 11C–F) or miR-34a-3p (Supplementary Fig. 11G–J) administration caused no renal or hepatic toxicity.
Fig. 6. Demonstration of the therapeutic relevance of the miR-34a-3p-MiD pathway in preclinical model.


(A) Nebulized siMiDs regress monocrotaline-induced PAH (MCT PAH). Compared to control rats, MCT PAH rats had elevated PAP, RVSP and decreased CO, as determine by closed-chest right heart catheterization. siMiD49 and siMiD51 treatments were effective in decreasing PAP, RVSP and increasing CO resulting in significant decrease of TPR (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; One way ANOVA, n=4–10/group).
(B) Augmenting miR-34a-3p regresses MCT PAH. Decreased PAP, RVSP and increased CO resulting in significant decrease of TPR (n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; One-way ANOVA, n=4–10/group).
(C) Representative images of longitudinal and cross sections of small pulmonary arteries and summary data indicating the regression of PAH caused by siMiD49 or siMiD51 or by miR-34a-3p mimic was associated with a decrease in the wall thickness in MCT PAH rats. Regression of PAH was assessed by measuring % of wall thickness by immunofluorescence staining of smooth muscle actin (SMA) and von Willebrand Factor (vWF) (blue: DAPI, red: SMA, green: vWF) (***P < 0.001; One-way ANOVA, n=4–5/group). Scale bar: 50μm.
Discussion
This study has 5 novel findings: 1) MiDs cause fission and this is increased in a human disease (PAH). 2) Inhibiting MiD expression prevents mitotic fission in health and disease. 3) MiD inhibition stops cell proliferation and increases apoptosis. 4) MiD expression is epigenetically upregulated by a decrease in miR-34a-3p, both in experimental PAH and in PAH patients. 5) These discoveries were translated into two new therapies that attenuate PAH in a rodent model (nebulized siMiD49 or siMiD51 or nebulized miR-34a-3p).
This study clarifies the role of the Drp1 binding partners, MiD49 and MiD51, in normal cell biology, while simultaneously highlighting the impact of their upregulation on the pathogenesis of PAH. In normal vascular cells, MiDs function as an important link between mitosis and mitochondrial fission. In PAH, the upregulation of MiD49 and MiD51 supports increased mitotic mitochondrial fission leading to pathological mitochondrial fragmentation, cellular proliferation and apoptosis resistance. We were able to use super resolution microcopy to image, for the first time, the macromolecular fission apparatus (Fig. 1B). We demonstrated that it contained both Drp1 and MiDs. In PAH there were qualitatively more MiDs at the site of fission and the fission apparatus appeared denser, consistent with the notion that increased MiDs focuses the constriction apparatus, thereby enhancing fission12.
Intriguingly, PAH patients displayed individual variation in terms of which of the MiDs was elevated (Fig. 1A), with patients often exhibiting increased MiD49 or MiD51 relative to control, but not both. This heterogeneity of expression was also seen at the mRNA level (data not shown). Despite this heterogeneity, PAH patients displayed a conserved, fragmented mitochondrial phenotype regardless which of the MiDs was upregulated (Fig. 2A). Overexpression of either MiD49 or MiD51 in isolation was also sufficient to reproduce the mitochondrial and proliferative phenotype of PAH in normal PASMC (Fig. 2E–F, 2H), highlighting a redundancy in the function of the two MiDs and suggesting that even healthy cells possess a sufficient basal pool of activated Drp1 to drive fission and proliferation when MiD levels increase. Our findings are at odds with Palmer et al. who suggested MiD overexpression inhibited fission, possibly by sequestering inactivated Drp114. The basis for this difference is unclear; however, our findings are robust (Fig. 2E–F).
This work advances the field from an earlier view that fission primarily reflects the expression, activity or posttranslational modification of Drp1 (reviewed in4). It is now clear the expression and function of the MiDs are critical determinants of mitochondrial dynamics and cell cycle regulation. This view is further supported by our observation that silencing MiDs is sufficient to restore mitochondrial fusion (Fig. 2A–D), slow cell proliferation (Fig. 2G) and enhance apoptosis (Fig. 2J) in PAH PASMC. In this regard, MiD49 and MiD51 are unique, since silencing other Drp1 receptor proteins, MFF or Fis1, did not alter cell proliferation or mitochondrial fission (Supplementary Fig. 6D–H). MiDs appear to have a special role in the fission that accompanies mitosis whereas Fis1 appears to be important in pathologic fission, as occurs in ischemia-reperfusion injury26. While MiDs regulate fission independently of other Drp1 binding partners (Supplementary Fig. 6A), the inability of MiDs to promote fission in MEFs lacking Drp1 (Supplementary Fig. 3) demonstrates that the availability of activated Drp1 is essential to this process. This new work adds detail to our previous discovery that inhibiting mitotic fission slows cell cycle progression. We are now showing the Drp1 binds MiDs to mediate mitotic fission. Super-resolution microscopy demonstrates the assembly of these partners at focal areas of fission, as part of the macromolecular fission apparatus (Fig. 1B). The Drp1-dependent proliferative effect of MiDs indicates that they are obligatory Drp1 binding partners in hyperproliferative disease. We did not study the effects of MiD expression on mitochondrial membrane potential.
Besides hyperproliferation and apoptosis resistance, PAH PASMC exhibit another pseudo-neoplastic phenotype by displaying perturbed oxygen sensing due to dysregulated mitochondrial redox signaling. This increases reliance on glycolysis thereby creating Warburg’s phenomenon2. We show that inhibiting MiDs increases apoptosis (Fig. 2J) and restores oxidative metabolism and MCU expression (Supplementary Fig. 7B–C), consistent with our recent discovery that the downregulation of MCU is a central contributor to both Warburg metabolism and fission in PAH19 (Supplementary Fig. 7B). This indicates that the miR-34a-3p-MiD-Drp1 pathway interacts with the miR-25- and -138-MCU pathway.
We also identified the mechanisms by which MiDs regulate fission and cell cycle progression (Fig. 7A). MiDs are Drp1 binding partners and attract more Drp1 to the OMM12. Thus, greater MiD expression is expected to enhance fission, as observed (Fig. 2E–F). However, we show that MiDs also regulate kinases that control Drp1 activity. Drp1 phosphorylation can be initiated by several kinases, including CDK1 and ERK10, 23, 24, which activate Drp1 through phosphorylation of serine 6163, 27. We found that silencing MiDs inhibited ERK1/2 (Fig. 3B) and decreased phosphorylation of Drp1 at serine 616 (Fig. 3A), supporting the dual mechanisms of MiD-induced fission. However silencing MiDs did not increase the expression of the mitochondrial fusion mediators Mfn1, Mfn2 and OPA1. Indeed, siMiD51 decreased the expression of Mfn1 and OPA1 (Supplementary Fig. 4A–C). Thus siMiD-mediated mitochondrial fusion does not result from upregulation of fusion mediators.
Fig. 7. Demonstration of the therapeutic relevance of the miR-34a-3p-MiD pathway in preclinical model.
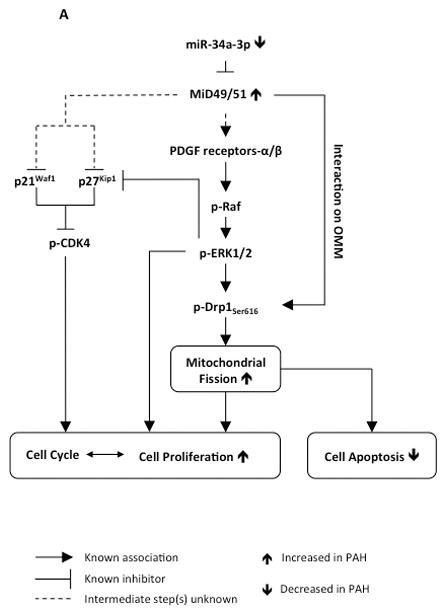

(A) Representation of the proposed miR-MiDs pathway in PAH relative to normal. (B) Decreased expression of miR-34a-3p in PAH patients leads to increased expression of the Drp1 binding partners MiD49 and MiD51 which in turn increases mitotic fission and promotes cell proliferation.
Knocking down MiDs caused cell cycle arrest at G1/G0 phase (Fig. 2I). Progression of cell cycle in early G1 phase versus the transition from G1-S phase is mediated by CDK4 versus CDK2, respectively28. The activation of CDK2 involves dephosphorylation of Tyr15 and Thr1429. Our results revealed that silencing MiDs does not inhibit CDK2 activity, as evidenced by the unchanged phosphorylation status at Tyr15 of CDK2 (Supplementary Fig. 9G). However, knocking down MiDs inhibited CDK4 activity by downregulating CDK4 expression and phosphorylation at Thr17230 (Fig. 3D, Supplementary Fig. 9D–E). Silencing MiDs also increased the expression of the CDK4 inhibitor p21Waf1 31 and p27Kip132 (Fig. 3E–F and Supplementary Fig. 9F). Our observation is supported by studies showing that ERK is a negative regulator of p27Kip133, 34. Therefore siMiDs mediated inhibition of cell cycle arrest can be explained by the inhibition of ERK1/2 activity achieved by silencing MiDs which in turn activates p27Kip1 resulting in decreased CDK4 activity. When taken together, these results show a multifactorial mechanism underlying the inhibition of mitochondrial fission and cell cycle arrest at G1 phase achieved by silencing MiDs (Figs. 2A–D, 2I, 7A). We previously showed that inhibiting Drp1 or augmenting Mfn2, both resulting in impaired mitotic fission, also results in cell cycle arrest10.
In addition to these effects on cell cycle kinases, silencing MiDs also downregulated PDGF receptors α and β (Fig. 3C). PDGF is a potent mitogen, which triggers cell proliferation, migration, transformation and survival 35–37, all processes relevant to the pathogenesis of PAH. Indeed, PDGF receptors are activated in PAH38 and inhibitors of PDGF receptor, such as Imatinib, have demonstrated therapeutic benefit in animal models and in some patients38, 39. PDGF binding to the PDGF receptors activates the MAPK signal transduction cascade resulting in the phosphorylation and activation of ERK1/2. Activated ERK1/2 then translocates to the nucleus and enhances the expression of transcription factors related to cell proliferation40. It is likely that siMiD-induced downregulation of PDGF receptors contributes to the observed inhibition of cell proliferation via the MAPK signaling pathway as evidenced by the inhibition of the two members of the MAPK signaling pathway, Raf-1 and ERK1/2 (Fig. 3B–C, Supplementary Fig. 9B–C) (see proposed pathway schematic Fig. 7A). Although interaction between MiDs and PDGF receptors remains speculative, it is important to note that MiDs are located on the outer mitochondrial membrane and they possess free cytosolic domains responsible for Drp1 binding13, 14. Furthermore, mitochondria are highly motile, dynamic organelles. Indeed the outer mitochondrial membrane protein Mfn2 interacts with Ras, which is also located on cell membrane, just like PDGF receptors41, 42. Therefore, it is not unlikely that MiDs may directly interact with PDGF receptors.
The current study identifies depressed miR-34a-3p expression as the primary driver of pathologic MiD upregulation in PAH. In general, upregulation of a miR suppresses its target species whilst downregulation of the miR, as in the case of miR-34a-3p in PAH PASMC, would be expected to permit increase expression of its targets (MiD49 and MiD51). The ability to increase (Fig. 5B, Supplementary Fig. 10B) or decrease (Fig. 5A) MiDs through manipulation of miR-34a-3p confirms the central role that this miR plays in the mitochondrial fission phenotype of PAH. Further certainty of the predicted role of miR-34a-3p in regulating MiDs comes from our direct experimental confirmation that the miR indeed binds and inhibits the 3′-UTR of both MiD49 and MiD51 (Fig. 4F).
Although the role of miR-34a-3p is not well established in human disease, its complementary strand, miR-34a-5p, has been implicated in many human cancers43–45 and in PAH, where its suppression has been linked to the PDGF receptor α-mediated hyperproliferation of human PASMCs46 and vascular remodeling in response to chronic hypoxia47. Additional reports have shown that miR-34-3p is co-expressed with its complementary 5p strand in cancer cells; however, it targets different groups of transcripts48. Intriguingly, the downregulation of miR-34a-3p expression in PAH may relate to the hypermethylation of the CpG island in the promoter region of the MIR34A gene. This is supported by the studies with several cancers which displayed CpG methylation of the promoter of the precursor MIR34A and subsequent loss of miR-34a-5p expression49, 50.
Here, we demonstrate a consistent downregulation of miR-34a-3p in three PAH patient cohorts and two rodent models of PAH (Supplementary Fig. 10A). Decreased miR-34a-3p expression was observed in PASMC from Canadian subjects (Fig. 4C) as well as in the whole blood and plasma collected from UK and China cohorts (Fig. 4D). Because circulating levels of miR-34-3p are depressed in PAH patients, which correlates with expression in the PASMC itself, this miR also has potential as a diagnostic test for PAH. The expression of circulatory miR-34a-3p predicted the presence of PAH but did not predict clinical outcomes (Fig. 4E), suggesting that larger sample sizes may be required to confirm the usefulness of this miR as a biomarker for PAH.
Our in vitro data, demonstrate that inhibition of MiDs causes a sustained state of mitochondrial fusion (Fig. 2A–D), which decreases PASMC proliferation (Fig. 2G and Supplementary Figs. 5, 8E–G) and promotes apoptosis (Fig. 2J). These findings provide the biologic plausibility for the therapeutic targeting of MiDs or augmenting miR-34a-3p in PAH. We confirmed these therapeutic benefits by nebulizing siMiDs and miR-34a-3p in vivo and demonstrating the ability of these approaches to regress experimental PAH and reduce cell proliferation in vivo (Fig. 6A–C). These findings are consistent with our previous observations that creating a state of sustained mitochondrial fusion, whether by inhibiting Drp13 or augmenting the fusion mediator, mitofusin-222, arrests cell proliferation and regresses PAH.
Limitations
There are few reports of the function or localization of MiD49 and 51. It has been reported that both MiDs are exclusively located at the outer mitochondrial membrane51. This study relied on heterologous overexpression of MiDs. However, we noted that MiDs are also expressed in the cytosol. This is consistent with a prior report, which noted that while MiD51 was predominantly a mitochondrial protein, it also existed in the cytosol13. Extra-mitochondrial MiD may reflect the balance between levels of MiD and activated Drp1 or the existence of a splice variant/isoform of MiDs that targets the MiD to an extra mitochondrial location. Perhaps, like Drp1 itself, the MiDs may be involved in the division of other organelles, such as lysosomes. These questions merit future study.
In conclusion, excessive mitochondrial fission, which results in mitochondrial fragmentation, is a new hallmark of proliferative diseases, including PAH and cancer3, 10. We have shown that expression of the Drp1 binding partners, MiD49 and MiD51, are increased in PAH and that this increase drives pathological mitochondrial fission, cell proliferation and apoptosis resistance. To our knowledge, this is the first description of a role for dysregulated expression of MiD49 and MiD51 in human disease and clarifies the role of these Drp1 binding partners in normal cell biology. We also report increased MiD49 and MiD51 expression in the endothelium of diseased pulmonary arteries, as well as in BOECs isolated from PAH patients (Fig. 1C and Supplementary Fig. 1B). Although the role of MiDs in the endothelium was not examined in detail in the current study, the mitochondria in PAH BOECs are also fragmented (Supplementary Fig. 1D–E), indicating that similar mechanisms may be driving altered cellular proliferation and apoptosis-resistance in this compartment of the vascular wall. Our work identifies decreased expression of miR-34a-3p as the cause of upregulated MiD49 and MiD51 in human PAH and highlights the potential value of this miR, as a novel biomarker. Together, these studies identify the miR-34-3p-MiD-Drp1 axis as an important driver of the mitochondrial and cellular phenotype of PAH and a new target for therapeutic intervention. A schematic representation of the proposed role of miR-34a-3p in regulating MiD49 and MiD51 in PAH is provided in Fig. 7B.
Supplementary Material
CLINICAL PERSPECTIVE.
What is New?
We identify a key role for MiD49 and MiD51, two novel mitochondrial binding partners for dynamin related protein 1 (Drp1), in PAH.
Pathological elevation of MiDs in pulmonary artery smooth muscle cells (PASMC) and endothelial cells, in human and experimental PAH, accelerates mitotic fission and supports rapid cell proliferation.
MiD expression is epigenetically upregulated by decreased expression of microRNA-34a-3p.
MiDs cause fission in a Drp1-dependent manner. Silencing MiDs in PAH increases mitochondrial fusion; conversely, overexpressing MiDs in normal cells causes fission.
Silencing MiDs causes cell cycle arrest (through an ERK1/2 and CDK4-dependent mechanism), decreases cell proliferation rate and increases apoptosis.
Clinical Implications
We identify a decrease of miR-34a-3p expression in the preclinical models of PAH as well as reduced circulatory miR-34a-3p expression in patient cohorts. miR-34a-3p may be a biomarker of PAH.
MiDs are similarly dysregulated in PAH blood outgrowth endothelial cells.
We translated our cellular discoveries into two new therapies that regress PAH. In experimental models, nebulizing miR-34a-3p or siMiDs regresses pulmonary hypertension
The epigenetic acceleration of mitotic fission through the miR-34a-3p-MiD axis promotes PAH’s cancer-like deep phenotype and is a novel therapeutic target.
This study further implicates dysregulation of mitochondrial dynamics as a therapeutic target in human and experimental PAH.
Acknowledgments
Source of Funding
This study was supported in part by U.S. National Institutes of Health (NIH) grants NIH 1R01HL113003-01A1 (S.L.A.) and NIH 2R01HL071115-08 (S.L.A.), Canada Foundation for Innovation (S.L.A.), Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine (S.L.A.), the American Heart Association (A.H.A.) (S.L.A.), the William J. Henderson Foundation (S.L.A.), and Canadian Vascular Network Scholar Award (AD). A British Heart Foundation Senior Basic Science Research Fellowship (FS/13/48/30453, AL). The collection of patient samples was supported by National Institute for Health Research (NIHR) Sheffield Clinical Research Facility. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
The authors thank Dr. Mads Breum Larsen (University of Pittsburg, USA) for providing the Adv-mNeon Green.
Footnotes
Disclosures:
None.
References
- 1.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–66. doi: 10.1161/CIRCULATIONAHA.108.847707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–8. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 3.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–51. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 5.Cribbs JT, Strack S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 2009;457:231–53. doi: 10.1016/S0076-6879(09)05013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–70. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- 7.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–74. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
- 10.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–86. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. 2013;288:27584–93. doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–78. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–73. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, Farber-Eger EH, Sheng Q, Shyr Y, Harrell FE, Newman JH, Brittain EL. Clinical and Biological Insights Into Combined Post- and Pre-Capillary Pulmonary Hypertension. J Am Coll Cardiol. 2016;68:2525–2536. doi: 10.1016/j.jacc.2016.09.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Cote J, Simard MJ, Bonnet S. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–48. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potus F, Malenfant S, Graydon C, Mainguy V, Tremblay E, Breuils-Bonnet S, Ribeiro F, Porlier A, Maltais F, Bonnet S, Provencher S. Impaired angiogenesis and peripheral muscle microcirculation loss contribute to exercise intolerance in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;190:318–28. doi: 10.1164/rccm.201402-0383OC. [DOI] [PubMed] [Google Scholar]
- 18.Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, Velegala S, Seeger W, McKinsey TA, Sucharov CC, Stenmark KR. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res. 2014;114:67–78. doi: 10.1161/CIRCRESAHA.114.301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am J Respir Crit Care Med. 2017;195:515–529. doi: 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ormiston ML, Toshner MR, Kiskin FN, Huang CJ, Groves E, Morrell NW, Rana AA. Generation and Culture of Blood Outgrowth Endothelial Cells from Human Peripheral Blood. J Vis Exp. 2015:e53384. doi: 10.3791/53384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay E, Nadeau V, Paradis R, Graydon C, Wong R, Johnson I, Paulin R, Lajoie AC, Perron J, Charbonneau E, Joubert P, Pibarot P, Michelakis ED, Provencher S, Bonnet S. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation. 2015;132:932–43. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 22.Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, Han M, Haney CR, Chen CT, Sharp WW, Archer SL. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:865–78. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–51. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, Lopez-Garcia C, Torres J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 2016;7:11124. doi: 10.1038/ncomms11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. 2009;8:1168–75. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, Chen KH, Hong Z, Sharp WW, Kutty S, Archer SL. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J Mol Med (Berl) 2017;95:381–393. doi: 10.1007/s00109-017-1522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 28.Satyanarayana A, Kaldis P. A dual role of Cdk2 in DNA damage response. Cell Div. 2009;4:9. doi: 10.1186/1747-1028-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11:3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato JY, Matsuoka M, Strom DK, Sherr CJ. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14:2713–21. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skildum AJ, Mukherjee S, Conrad SE. The cyclin-dependent kinase inhibitor p21WAF1/Cip1 is an antiestrogen-regulated inhibitor of Cdk4 in human breast cancer cells. J Biol Chem. 2002;277:5145–52. doi: 10.1074/jbc.M109179200. [DOI] [PubMed] [Google Scholar]
- 32.Ray A, James MK, Larochelle S, Fisher RP, Blain SW. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29:986–99. doi: 10.1128/MCB.00898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kress TR, Raabe T, Feller SM. High Erk activity suppresses expression of the cell cycle inhibitor p27Kip1 in colorectal cancer cells. Cell Commun Signal. 2010;8:1. doi: 10.1186/1478-811X-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–39. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 35.De Donatis A, Comito G, Buricchi F, Vinci MC, Parenti A, Caselli A, Camici G, Manao G, Ramponi G, Cirri P. Proliferation versus migration in platelet-derived growth factor signaling: the key role of endocytosis. J Biol Chem. 2008;283:19948–56. doi: 10.1074/jbc.M709428200. [DOI] [PubMed] [Google Scholar]
- 36.Langley RR, Fan D, Tsan RZ, Rebhun R, He J, Kim SJ, Fidler IJ. Activation of the platelet-derived growth factor-receptor enhances survival of murine bone endothelial cells. Cancer Res. 2004;64:3727–30. doi: 10.1158/0008-5472.CAN-03-3863. [DOI] [PubMed] [Google Scholar]
- 37.Yu J, Deuel TF, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta -induced phenotypic transformation. J Biol Chem. 2000;275:19076–82. doi: 10.1074/jbc.M910329199. [DOI] [PubMed] [Google Scholar]
- 38.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, Pascoe S. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182:1171–7. doi: 10.1164/rccm.201001-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Prywes R. Activation of the c-fos enhancer by the erk MAP kinase pathway through two sequence elements: the c-fos AP-1 and p62TCF sites. Oncogene. 2000;19:1379–85. doi: 10.1038/sj.onc.1203443. [DOI] [PubMed] [Google Scholar]
- 41.Chen KH, Dasgupta A, Ding J, Indig FE, Ghosh P, Longo DL. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 2014;28:382–94. doi: 10.1096/fj.13-230037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–83. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 43.Ye J, Li L, Feng P, Wan J, Li J. Downregulation of miR-34a contributes to the proliferation and migration of laryngeal carcinoma cells by targeting cyclin D1. Oncol Rep. 2016 doi: 10.3892/or.2016.4823. [DOI] [PubMed] [Google Scholar]
- 44.Almeida AL, Bernardes MV, Feitosa MR, Peria FM, Tirapelli DP, Rocha JJ, Feres O. Serological under expression of microRNA-21, microRNA-34a and microRNA-126 in colorectal cancer. Acta Cir Bras. 2016;31(Suppl 1):13–8. doi: 10.1590/S0102-86502016001300004. [DOI] [PubMed] [Google Scholar]
- 45.Shi H, Zhou S, Liu J, Zhu J, Xue J, Gu L, Chen Y. miR-34a inhibits the in vitro cell proliferation and migration in human esophageal cancer. Pathol Res Pract. 2016;212:444–9. doi: 10.1016/j.prp.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 46.Wang P, Xu J, Hou Z, Wang F, Song Y, Wang J, Zhu H, Jin H. miRNA-34a promotes proliferation of human pulmonary artery smooth muscle cells by targeting PDGFRA. Cell Prolif. 2016;49:484–93. doi: 10.1111/cpr.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, Ishizaki T. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol. 2011;300:L753–61. doi: 10.1152/ajplung.00286.2010. [DOI] [PubMed] [Google Scholar]
- 48.Huang CJ, Nguyen PN, Choo KB, Sugii S, Wee K, Cheong SK, Kamarul T. Frequent co-expression of miRNA-5p and -3p species and cross-targeting in induced pluripotent stem cells. Int J Med Sci. 2014;11:824–33. doi: 10.7150/ijms.8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 50.Schmid G, Notaro S, Reimer D, Abdel-Azim S, Duggan-Peer M, Holly J, Fiegl H, Rossler J, Wiedemair A, Concin N, Altevogt P, Marth C, Zeimet AG. Expression and promotor hypermethylation of miR-34a in the various histological subtypes of ovarian cancer. BMC Cancer. 2016;16:102. doi: 10.1186/s12885-016-2135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. 2016;129:2170–81. doi: 10.1242/jcs.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.