

Abstract
The decreasing cost and increasing scope and power of emerging genomic technologies are reshaping the field of molecular ecology. However, many modern genomic approaches (e.g., RAD‐seq) require large amounts of high‐quality template DNA. This poses a problem for an active branch of conservation biology: genetic monitoring using minimally invasive sampling (MIS) methods. Without handling or even observing an animal, MIS methods (e.g., collection of hair, skin, faeces) can provide genetic information on individuals or populations. Such samples typically yield low‐quality and/or quantities of DNA, restricting the type of molecular methods that can be used. Despite this limitation, genetic monitoring using MIS is an effective tool for estimating population demographic parameters and monitoring genetic diversity in natural populations. Genetic monitoring is likely to become more important in the future as many natural populations are undergoing anthropogenically driven declines, which are unlikely to abate without intensive adaptive management efforts that often include MIS approaches. Here, we profile the expanding suite of genomic methods and platforms compatible with producing genotypes from MIS, considering factors such as development costs and error rates. We evaluate how powerful new approaches will enhance our ability to investigate questions typically answered using genetic monitoring, such as estimating abundance, genetic structure and relatedness. As the field is in a period of unusually rapid transition, we also highlight the importance of legacy data sets and recommend how to address the challenges of moving between traditional and next‐generation genetic monitoring platforms. Finally, we consider how genetic monitoring could move beyond genotypes in the future. For example, assessing microbiomes or epigenetic markers could provide a greater understanding of the relationship between individuals and their environment.
Keywords: conservation genetics, DNA fingerprinting, individual identification, noninvasive genetic sampling, population demography, wildlife forensics, wildlife management
1. INTRODUCTION
The current era of rapid global environmental change (Zalasiewicz, Williams, Haywood, & Ellis, 2011) is predicted to lead to a rapid loss of biodiversity (Pimm et al., 2014). To assess and mitigate the impact of this loss, many national and international organizations have established biodiversity monitoring strategies (e.g., Kurtz, Jackson, & Fisher, 2001; United Nations Environment Programme Convention on Biological Diversity SBSTTA, 2003). Key tools for biodiversity monitoring utilise methodological approaches from the field of genetic monitoring, relying on genetic tools for evaluating change (Stetz, Kendall, Vojta, & GeM, 2011). Genetic monitoring focuses on quantifying temporal changes in population genetic metrics, or other population data, generated using molecular markers (Schwartz, Luikart, & Waples, 2007). Genetic monitoring can be used to estimate many biological parameters of interest, including demographic parameters such as abundance, vital rates, occupancy, hybridization, disease status; population genetic parameters including genetic diversity, structure and effective population size; and increasingly, responses to selective pressures such as exploitation (e.g., trophy hunting) and climate change (Schwartz et al., 2007; Stetz et al., 2011). Here, we examine genetic monitoring approaches that use noninvasive (e.g., naturally shed feathers) or minimally invasive (e.g., biopsy darts, buccal swabs) samples (hereafter MIS) because wildlife ecology and conservation has benefitted greatly from the new data provided by these approaches (Beja‐Pereira, Oliveira, Alves, Schwartz, & Luikart, 2009).
Genetic monitoring using MIS approaches was first introduced in 1992 as a method to obtain genetic samples from brown bears (Ursus arctos; Höss, Kohn, Pääbo, Knauer, & Schröder, 1992; Taberlet & Bouvet, 1992; see Box 1) and to study social structure in chimpanzees (Pan troglodytes; Morin & Woodruff, 1992). MIS has become the method of choice for genetic monitoring of many vertebrate species. This is because sampling of hair, faeces, remote skin biopsies or feathers provides DNA from free‐ranging animals that can be used to identify individuals across time and space and generates genetic data without having to catch, handle or in some cases, even observe them (Beja‐Pereira et al., 2009; Schwartz et al., 2007; Waits & Paetkau, 2005). In the last 25 years, researchers have demonstrated a variety of important applications of MIS including detecting rare species (Palomares, Godoy, Piriz, O'Brien, & Johnson, 2002; Valière et al., 2003), estimating population size and other demographic parameters (Carroll et al., 2013; Kendall et al., 2009; Kohn et al., 1999; Rudnick, Katzner, Bragin, Rhodes, & DeWoody, 2005; Woodruff, Lukacs, Christianson, & Waits, 2016; Woods et al., 1999), evaluating genetic diversity and gene flow (Epps et al., 2005; Gerloff, Hartung, Fruth, Hohmann, & Tautz, 1999; Lucchini et al., 2002; Palsbøll et al., 1997), detecting movement and migration (Dixon et al., 2006; Proctor, Mclellan, Strobeck, & Barclay, 2005), evaluating social structure (Constable, Ashley, Goodall, & Pusey, 2001; Ford et al., 2011; Morin et al., 1994), detecting hybridization (Adams, Kelly, & Waits, 2003; Bohling et al., 2016; Steyer et al., 2016), monitoring disease epizootics (Kohn & Wayne, 1997; Schunck, Kraft, & Truyen, 1995), identifying diet items (De Barba et al., 2014; Höss et al., 1992; Taberlet & Fumagalli, 1996) and wildlife forensic applications (Banks, Horsup, Wilton, & Taylor, 2003; Ernest, Rubin, & Boyce, 2002; Lukoshek et al., 2009; Wasser et al., 2004).
Box 1. Brown bears (Ursus arctos) as a model system for the development of MIS approaches.
1.
Figure 1.
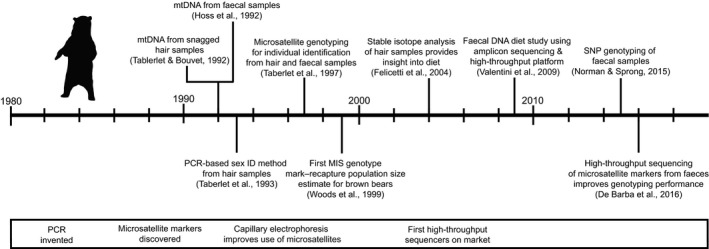
The brown bear is the most widely distributed bear species and is locally endangered at many locations across its range. The desire for alternative methods to monitor this charismatic species launched the field of noninvasive genetic sampling, and the field has kept pace with technological developments. First, Taberlet and Bouvet (1992) and Höss et al. (1992) demonstrated that mitochondrial DNA (mtDNA) sequences could be obtained from snagged hair and faecal samples, respectively. Höss et al. (1992) were also the first to demonstrate the ability to amplify diet items in scat by sequencing a 356 bp rbcL chloroplast sequence to identify the dominant plant in their diet (Photinia villosa). These were the first studies to document successful amplification of DNA from hair and faecal samples of wild species. Soon researchers were amplifying nuclear DNA to determine sex (Taberlet, Mattock, Dubois‐Paganon, & Bouvet, 1993) and for individual identification (Taberlet et al., 1997). This work was critical to the understanding of microsatellite genotyping errors and approaches for minimizing their impact on MIS data sets (Taberlet et al., 1996). MIS was then used extensively in Europe in the 1990s to obtain data on genetic diversity, genetic structure, phylogeography and minimum counts of population size (Kohn, Knauer, Stoffella, Schroder, & Paabo, 1995; Taberlet & Bouvet, 1992; Taberlet et al., 1997). In the late 1990s, North American researchers embraced MIS methods as an alternative approach for population estimation and produced the first mark–recapture population estimates using DNA extracted from brown bear hair samples collected from barbed‐wire hair snares (Mowat & Strobeck, 2000; Woods et al., 1999), which revolutionised methods for estimating population size (Boulanger, Himmer, & Swan, 2004; Kendall et al., 2009). This approach was expanded to couple stable isotope analysis of hair samples with genetic analysis to provide a new approach for noninvasively determining the number of brown bears in Yellowstone park feeding on cutthroat trout and estimating the number of fish consumed per year by bears (Felicetti et al., 2004; Haroldson et al., 2005; Teisberg et al., 2014). MIS applications have expanded to include obtaining DNA from saliva on mammalian (Farley, Talbot, Sage, Sinnott, & Coltrane, 2014) and salmonid (Wheat, Allen, Miller, Wilmers, & Levi, 2016) carcasses to conduct species and individual identification. MIS has been the main method used to track small remnant or reintroduced populations in Europe (e.g., De Barba, Waits, Garton, 2010; Karamanlidis et al., 2010), Pakistan (Bellemain, Nawaz, Valentini, Swenson, & Taberlet, 2007), western continental United States (Proctor et al., 2012; Romain‐Bondi et al., 2004) and the Gobi desert (McCarthy, Waits, & Mijiddorj, 2009; Tumendemberel et al., 2015). Brown bears have also been an important model system for the transition from genetic to genomic approaches in MIS. For example, they have been the focus of dietary metabarcoding studies (De Barba et al., 2014; Valentini et al., 2009). Recently, new approaches were developed to sequence PCR‐amplified microsatellites on an Illumina platform to obtain multilocus genotypes from brown bears (De Barba et al., 2017). This approach increased success rates by 20%–30% and decreased costs per sample by 40% compared to traditional capillary electrophoresis genotyping of microsatellite loci. Also, SNP loci have been identified for brown bears and successfully genotyped for faecal samples using the Fluidigm platform (Norman & Spong, 2015; Spitzer, Norman, Schneider, & Spong, 2016). These advancements using genomic methods provide much promise for the continued noninvasive genetic monitoring of brown bears across their range. The figure shows the timeline of the key advances in using MIS for genetic monitoring of brown bears, along with the approximate timing of some key molecular methods.
There is now a wealth of published evidence that MIS is comparable in costs or more cost‐effective (De Barba, Waits, Genovesi, et al., 2010; Solberg, Bellemain, Drageset, Taberlet, & Swenson, 2006) than traditional methods (e.g., camera trapping, tracks and signs and even trapping animals) and that collection and analysis of larger genetic sample sizes are often possible (De Barba, Waits, Garton, et al., 2010; Marucco et al., 2009; Solberg et al., 2006; Stenglein, Waits, Ausband, Zager, & Mack, 2010), prompting many wildlife managers to shift to MIS approaches. Extensive methodological and analytical development has been invested in establishing protocols to maximise success rates and minimise error rates when using these low‐quality DNA sources for genetic monitoring (Beja‐Pereira et al., 2009; Broquet & Petit, 2004; Miquel et al., 2006; Morin et al., 2010; Smith & Wang, 2014; Taberlet et al., 1996; Taberlet & Luikart, 1999; Waits & Paetkau 2005; Wang, 2016). Genetic monitoring is set to become more important in the future, largely because many vertebrate species have undergone rapid, anthropogenic population declines (Li et al., 2016) that are unlikely to abate without intensive management efforts. Fortunately, the genomic revolution of the early 2000s has given rise to a variety of more precise or more powerful molecular techniques that will make genetic monitoring even more effective in the future.
New technologies for genetic monitoring typically rely upon single nucleotide polymorphisms, or SNPs (Morin, Luikart, & Wayne, 2004). Unlike more conventional DNA markers such as microsatellites, SNPs have relatively few alleles per locus (theoretically up to four but usually only two due to low mutation rates; Glaubitz, Rhodes, & DeWoody, 2003) and often have more limited application across species than microsatellite markers, often being species‐specific. In addition, SNP loci are more prone to ascertainment bias, as they are selected because of their high polymorphism in the populations of interest but are often monomorphic in even closely related populations (Gautier et al., 2009). However, SNP‐based approaches have great potential for noninvasive genotyping as (i) large numbers of loci can be surveyed simultaneously, particularly with next‐generation sequencing or genotyping platforms, and (ii) the relative ease of scoring, analysis and modelling of SNP genotype data due to the digital/binary nature of the data. The latter point contrasts favourably with the near continuous distribution of microsatellite alleles that can be difficult to consistently characterise and thus could cause scoring errors.
For these reasons, we focus this review on recent genomic methods and platforms for producing SNP genotypes from MIS, considering factors such as development costs and error rates. We evaluate whether these new approaches will enhance our ability to investigate questions in genetic monitoring, such as estimating abundance, genetic structure and relatedness. As the field is in a period of unusually rapid transition, we also highlight the importance of legacy data sets and recommend how to address the challenges of moving between traditional and next‐generation genetic monitoring platforms. Finally, we consider how genetic monitoring could move beyond genotypes in the future. For example, assessing microbiomes could provide a greater understanding of the relationship between individuals and their environment.
2. SAMPLING AND METHODOLOGICAL CONSIDERATIONS
2.1. Sampling issues
Sampling strategies for non‐ or minimally invasive material in the natural environment depend on the research aims and objectives at hand and can be conducted randomly, opportunistically or using standardised designs. For example, sampling strategies may be designed to maximise the total number of individuals detected (typically used for minimum census estimates and population genetic studies) or to maximise recaptures using high intensity sampling over a limited geographic range (to estimate ranging behaviour or territory size for an individual or group of individuals, and to estimate population size, e.g., Rudnick, Katzner, Bragin, & DeWoody, 2008). When considering the estimation of many population genetic parameters, sampling should be designed to be random with respect to kin (this can also be addressed by post hoc data pruning, but see Waples & Anderson, 2017). It is also important to consider the temporal sampling interval, which can affect sample sizes, genotyping success rates, genotyping error rates and impact the ability to meet modelling assumptions for mark–recapture and occupancy analyses (Lonsinger, Gese, Dempsey, & Kluever, 2015; Woodruff, Johnson, & Waits, 2015).
When planning a MIS or noninvasive sampling strategy, it is important to account for patterns of social structure (random or nonrandom association of individuals), habitat‐use and availability of the material produced (e.g., faeces, urine, partially consumed food). This is, in part, because it is important to maximise sampling opportunities for elusive species, given the labour‐intensive nature of field work, but also because certain parameters (e.g., genotype capture‐recapture methods to estimate census size) require the application of assumptions about sampling that may or may not be satisfied if sampling is conducted incorrectly.
Consideration also needs to be given to the most suitable collection and storage method for the study species and sample type. For example, the time since deposition, environmental conditions, part of faeces sampled and storage medium can influence the quality of genotypes obtained, showing the importance of sampling protocol (Stenglein, De Barba, Ausband, & Waits, 2015; Wultsch, Waits, Hallerman, & Kelly, 2016).
2.2. Molecular methodologies
The human and agricultural genetics communities have already embraced SNPs for genotyping because of their myriad advantages over microsatellites (although microsatellites are still preferred by some in the human forensics field; Butler, 2015; FAO, 2015). There are many methods for genotyping thousands of SNPs, including variations on RAD‐seq (Andrews, Good, Miller, Luikart, & Hohenlohe, 2016; Baird et al., 2008) and genotyping by sequencing (Elshire et al., 2011). These approaches could be useful in MIS if sufficient DNA can be obtained (e.g., Chiou & Bergey, 2015), but these anonymous‐marker approaches often require considerably more DNA than is typically available to biologists using MIS. The DNA extracted from such samples may also contain xenobiotic environmental DNA (eDNA), often from nontarget organisms, and thus require rigorous postsequencing filtering. Furthermore, these approaches genotype far more loci than needed for individual identification and assessments of relatedness, population structure and other parameters of general interest in genetic monitoring studies and are thus economically inefficient. However, some next‐generation sequencing and advanced genotyping methods are particularly suitable for the low‐quality or quantity of DNA that are typically obtained from MIS; we broadly categorise these into SNP arrays and target enrichment methods. We highlight these methodologies in the subsequent sections, but acknowledge that significant prior sequencing and bioinformatic analyses will be required to identify loci suitable for genotyping MIS samples using these platforms (e.g., Andrews et al., 2016; De Wit et al., 2012; Elshire et al., 2011; Morin et al., 2018). We provide a brief description of the methods and their application to MIS samples, including information on error rates and approximate costings (Table 2).
2.2.1. SNP arrays
Platforms that more efficiently assess relevant numbers of SNPs include the Fluidigm SNPtype assay (Tables 1 and 2). Briefly, the Fluidigm assay uses a two‐stage amplification process with the first pair of primers amplifying the locus containing the SNP and the second pair amplifying specific alleles, integrating distinct fluorescent labels. The Fluidigm platform simultaneously genotypes up to 96 SNP loci in 96 samples, determining the SNP genotype at an individual locus by measuring the fluorescence intensity of both alleles. The fisheries community has embraced SNP genotyping assaying scores of loci with the Fluidigm platform (Bonanomi, Therkildsen, Retzel, & Berg, 2016; Campbell & Narum, 2011; Hauser, Baird, Hilborn, Seeb, & Seeb, 2011), and recently, several wildlife studies have also used this platform in a monitoring context (Table 1: Doyle et al., 2016; Kraus et al., 2015; Nussberger, Wandeler, & Camenisch, 2014). The Fluidigm SNP type assay seems to have relatively low error rates (e.g., 0.2% in DeWoody et al., 2017; 0.4% in Doyle et al., 2016; where error rates are estimated from the number of mismatches between replicates and consensus genotype; 1%–3% per allele in Kraus et al., 2015; 1.7% per locus in Nussberger et al., 2014). The low error rate is important for all aspects of molecular ecology, but particularly for inferences of individual identification, parentage and relatedness. In addition, the Fluidigm platform had a higher genotyping success rate than microsatellite genotyping in hair samples from wolf faeces (87% and 70%, respectively) and wild cat hair (80% vs. 54%) but similar success rates in brown bear hair samples (97% and 99%, respectively; after quality control; von Thaden et al., 2017).
Table 1.
Contemporary approaches for genotyping low‐quality and/or quantity DNA samples
| Reference | Platform/method | Starting material | Species | Inference |
|---|---|---|---|---|
| SNP Arrays | ||||
| Morin and Mccarthy (2007) | Ampliflour SNP genotyping | Bone | Bowhead whale (Balaena mysticetus) | Development/validation of SNP markers |
| Mesnick et al. (2011) | Ampliflour SNP and microsatellite genotyping | Skin | Sperm whale (Physeter macroephalus) | Population structure |
| Nussberger et al. (2014) | Fluidigm | Hair | European wildcat (Felis silvestris silvestris) | Validation of SNP markers and studying introgression |
| Ruegg et al. (2014) | Fluidigm | Feathers | Wilson's warbler (Cardellina pusilla) | Tracking migratory populations |
| Kraus et al. (2015) | Fluidigm | Faeces | Grey wolf (Canis lupus) | Development/validation of SNP markers |
| Norman and Spong (2015) | Fluidigm | Faeces | Brown bear (Ursus arctos) | Reconstructing pedigrees and estimating dispersal |
| Doyle et al. (2016), Katzner et al. (2016) | Fluidigm | Feathers | Golden eagle (Aquila chrysaetos) | Population structure, parentage and provenance |
| Stetz et al. (2016) | Fluidigm | Faeces | River otter (Lontra canadensis) | Development/validation of markers, population assignment |
| Spitzer et al. (2016) | Fluidigm | Faeces | Brown bear (Ursus arctos) | Pedigree and population size estimation |
| DeWoody et al., 2017; | Fluidigm | Skin | Grey whale (Eschrichtius robustus) | Individual ID and relatedness |
| von Thaden et al. (2017) | Fluidigm | Hair and faeces | European wildcat (Felis silvestris silvestris); brown bear (Ursus arctos); grey wolf (Canis lupus) | Validation and population structure analysis |
| Hoffman et al. (2012) | Illumina GoldenGate genotyping assay | Skin | Antarctic fur seal (Arctocephalus gazella) | Development/validation of markers |
| Monzón et al. (2014) | Illumina GoldenGate genotyping assay BeadXpress platform | Faeces | Coyote (Canis latrans) | Admixture and hybridization |
| Fitak et al. (2015) | MassARRAY (Sequenom) | Faeces | Pumas (Puma concolor) | Development/validation of SNP markers |
| Goossens et al. (2016) | MassARRAY (Sequenom) | Faeces | Asian elephant (Elephas maximus) | Population structure and genetic diversity, comparison of SNPs with microsatellites |
| Fabbri et al. (2012) | SNPs Pyrosequencing (Biotage), SNaPshot (ABI), Taqman (ABI) | Faeces | Grey wolf (Canis lupus) | Development/validation of markers |
| Targeted sequence capture | ||||
| Perry et al. (2010) | RNA bait capture/Illumina sequencing (Agilent's SureSelect) | Faeces | Chimpanzees (Pan troglodytes) | Validation/SNP genotyping for genetic diversity |
| Snyder‐Mackler et al. (2016) | RNA bait capture/Illumina sequencing | Faeces | Baboons (Papio papio) | Development/validation of markers, pedigree analysis |
| De Barba et al. (2017) | High‐throughput sequencing of microsatellites (Illumina MiSeq) | Faeces | Brown bear (Ursus arctos) | Development/validation of markers |
| Other examples | ||||
| Chiou and Bergey (2015) | ddRAD using FecalSeq | Faeces | Baboons (Papio papio) | Development/validation of markers |
| Russello et al. (2015) | nextRAD | Hair | American pika (Ochotona princeps) | Population structure and outlier loci analysis |
Table 2.
Selective summary of characteristics of next‐generation sequencing platforms that could be suitable for low‐quality or quantity DNA templates frequently obtained during MIS projects. Costings are provided in euros (€)
| Platform | Development cost | Run cost | Effort (after DNA extraction) | Information | Error rate | DNA required | References |
|---|---|---|---|---|---|---|---|
| Fluidigm | €4300 for oligos to query 96 SNPs, access to Fluidigm system | €1250 for genotyping 96 individuals at 96 SNPs | PCR | SNP genotype | ~1% A | Nanograms | Doyle et al. (2016), Katzner et al. (2016), DeWoody et al. (2017) |
| Amplifluor | €2200 for oligos for 96 loci, access to qPCR machine | €250 for genotyping 96 samples at 96 loci, based on 20 loci multiplexa | PCR and analysis of qPCR results | SNP genotype | 1.4% B | Nanograms | Mesnick et al. (2011) |
| MassARRAY | €2600 for oligos for 96 loci, assuming two alleles per locus, access to MassARRAY system | €777 (384 well format, €8.09 per sample) to €1,376 (96 well format, €14.33 per sample) to genotype 96 individuals at 96 SNPs (24‐loci multiplex) | Multiplex PCR step, clean‐up step, primer extension step and another clean‐up step, run on compact mass spectrometer | SNP genotypes | Faecal sample error rate: 24‐loci multiplex 9%; 42‐loci multiplex error rate: 25% A | Nanograms (10 ng per multiplex reaction recommended) | Goossens et al. (2016) |
| GT‐seq | <€9000: primary cost is oligos but a pilot study of the markers is suggested, high‐throughput sequencing run | €3.43 per sample, based on example where 2068 samples were genotyped at 192 loci | For each of the 22 × 96‐well plates there were two PCR steps and one normalization step | SNPs; could be extended to haplotypes | 0.01%C | Nanograms (10 ng for first PCR minimum recommended concentration) | Campbell et al. (2015), N. Campbell, pers. comm. |
| Microsatellite sequencing | Primary costs are optimisation and validation study, as well as oligonucleotides | €2470 to sequence 96 samples at 14 loci replicated eight times, or €3.20 per replicated PCR product | Multiplex PCR, purification and quantification of pooled PCR product and sequencing run | Microsatellite genotypes | Good quality reference hair: allelic dropout (ADO): 3.9%, false allele rate (FA): 0.3%, Noninvasively collected low‐quality hair: ADO: 10.6%, FA 0.8%; Low‐quality faecal samples: ADO: 13.7%, FA: 0.8%C | Not quantified in study, but estimated to be in range of nanograms | De Barba et al. (2017), M. De Barba, pers. comm. |
Error rate reported is based on replicate genotypingA or calculated per alleleB or per locusC.
Based on purchase of 5,000 assay kit.
A technologically similar, fluorescence‐based platform, Amplifluor SNP genotyping system, has been shown to be highly sensitive with low‐quality/quantity samples: there was a high level of genotyping success with as few as 10 DNA templates per assay (Morin & Mccarthy, 2007). Mesnick et al. (2011) used eight microsatellite loci and 38 Amplifluor SNP loci to investigate the population structure of North Pacific sperm whales (Physter macrocephalus). The Amplifluor SNP loci had a comparable error rate (1.4% per allele) to the microsatellite loci (0.9% per allele) in this study (Tables 1 and 2).
In contrast to the fluorescence‐based platforms, the MassARRAY platform uses mass spectrometry to determine SNP alleles. The key difference is the use of the primer extension or iPLEX reaction, which incorporates one mass‐modified nucleotide, depending on the allele and assay design, enabling the detection of single base or small insertion/deletion polymorphisms. A compact mass spectrometer (Sequenom) is then used to infer genotypes based on the position of the peaks in the spectra, corresponding to different alleles at different loci (Gabriel, Ziaugra, & Tabbaa, 2009). The platform has potential for MIS samples: in a recent study, the MassARRAY system successfully genotyped a higher proportion of puma scat samples (59.8%) than a conventional microsatellite genotyping approach (39.9%), with no significant difference in error rates between the methods (Fitak, Naidu, Thompson, & Culver, 2015). However, another study that used both microsatellite genotyping and MassARRAY assays to genotype Bornean elephant blood and faeces found a lower rate of genotyping success and higher error rates for the SNP platform in faecal samples (Goossens et al., 2016). The authors found a trade‐off between genotyping success and multiplexing level, with smaller multiplexes having greater success (Table 1, Goossens et al., 2016), and suggested that the issue could be related to the lower quality of faecal DNA.
2.2.2. Target enrichment methods
The aim of target enrichment is to selectively capture genomic regions of interest before high‐throughput sequencing. Target enrichment methods can be a highly sensitive way of selectively and reproducibly obtaining genomic data. Genomic regions can be selectively targeted using PCR, as well as in‐solution or array‐based methods. PCR‐based methods are suitable for MIS as they typically require only small amounts of starting material and, by utilizing multiplex PCR and combinatorial barcoding techniques, can be cost‐effective. One such method is GT‐seq (Campbell, Harmon, & Narum, 2015; Table 2), which has been used to genotype steelhead trout (Oncorhynchus mykiss) to assess abundance, migration timing and stock composition (Hess et al., 2016; Matala et al., 2016). GT‐seq is essentially a massively multiplexed two‐step PCR reaction; in the first PCR reaction, SNP loci are amplified in a multiplex PCR, and in the second reaction, sequencing adaptors and unique identifiers (barcodes) are added to each sample. After Illumina sequencing, the barcodes are used to separate reads into samples, using a custom bioinformatics pipeline (Campbell et al., 2015). The GT‐seq method appears to have a low error rate; the method had a 99.9% concordance rate with genotypes generated with the Fluidigm platform. The method may require additional optimization for low‐quality/quantity DNA samples, although it works well with sheared DNA templates, success rates drop off when DNA concentrations <10 ng/μl (N. Campbell, pers. comm.).
Another targeted PCR approach has focused on the use of high‐throughput sequencing to generate microsatellite genotypes (e.g., De Barba et al., 2017). This approach typically involves PCR amplification of microsatellite loci, followed by multiplexing and high‐throughput sequencing (e.g., De Barba et al., 2017; Vartia et al., 2016). The potential advantages over conventional microsatellite genotyping includes identification of length homoplasy (which can be high, e.g., identified in 38 of 53 loci; Vartia et al., 2016) and cost‐effectiveness at higher numbers of samples and/or markers (Darby, Erickson, Hervey, & Ellis‐Felege, 2016). High‐throughput sequencing of microsatellite genotypes also has the benefit of rapidly generating consensus genotypes using bioinformatic analysis pipelines, either from whole‐genome (e.g., Kistler et al., 2017) or amplicon data (e.g., Suez et al., 2016; Zhan et al., 2017). Furthermore, this approach could have the advantage of linking into legacy data sets if the same sets of loci can be used in the new and traditional microsatellite genotyping platforms. However, optimization and validation steps are required to move microsatellite genotyping on to a new sequencing platform (e.g., De Barba et al., 2017), which can be technically challenging. The application of this microsatellite genotyping with high‐throughput sequencing to MIS studies has been limited thus far. However, De Barba et al. (2017) found that a set of microsatellite loci optimised for high‐throughput sequencing increased the yield and accuracy of genotypes generated from faecal samples, compared with metrics previously reported for genotyping microsatellites from faecal samples with capillary electrophoresis.
DNA capture methods, in conjunction with high‐throughput sequencing, have been used to investigate phylogenetic questions (e.g., Hancock‐Hanser et al., 2013), but the application of such methods to within‐population studies has been limited thus far. One successful example was the use of custom biotin‐tagged RNA baits to capture genomic DNA from faecal samples from 62 wild baboons (Papio papio). The enriched libraries were sequenced with Illumina HiSeq and provided sufficient genomic markers to undertake pedigree reconstruction (Snyder‐Mackler et al., 2016). Another study, using bait captures generated from the Agilent SureSelect system, successfully sequenced more than 1.5 Mb of nuclear DNA and the entire mitochondrial genome from chimpanzee faeces (Perry, Marioni, Melsted, & Gilad, 2010). These studies highlight the potential of bait capture approaches, both custom and using a commercial provider, in a genetic monitoring context. Such approaches could be aided by the use of novel methods that enrich samples for endogenous DNA, such as FecalSeq (Chiou & Bergey, 2015).
2.3. Data analysis
SNP array platforms have proprietary software packages that are used to score genotypes and often provide a degree of confidence in genotype calls (e.g., Sequenom platform). Such automated calling is not always accurate, and it is recommended that researchers visually check the data for error. This is particularly true for noninvasively collected samples, which can have higher error rates (e.g., Bayerl et al., 2017). Target capture approaches that use high‐throughput sequencing tend to have custom bioinformatics pipelines (e.g., Campbell et al., 2015). However, the major steps are similar between studies and include filtering of reads based on quality scores and demultiplexing reads into samples and loci. Genotyping is then conducted using custom bioinformatics tools and information such as the relative frequency and read depths of sequences likely to be alleles versus PCR/sequencing artefacts (Campbell et al., 2015; De Barba et al., 2017).
2.4. Quality control
Genotype data are imperfect and subject to missing genotypes (errors of omission) as well as erroneous genotypes (errors of commission; Faria et al., 2011). Missing and erroneous genotypes can be due to many possible causes, such as suboptimal genotyping protocols, limited DNA quantity and quality, contamination and human error (Bonin et al., 2004; Pompanon, Bonin, Bellemain, & Taberlet, 2005). MIS data are especially problematic due to the low DNA quality and quantity, and can incur a high rate of error.
Missing and erroneous genotypes affect many genetic analyses, yielding potentially biased and imprecise results and, in turn, incorrect conclusions. Broadly speaking, analyses that use genotype data are more severely impacted than analyses that use allele frequency data. For example, genetic differentiation, measured by FST (Wright, 1931) and evaluated by several estimators (Nei, 1973; Weir & Cockerham, 1984), is determined by marker allele frequencies. As missing and erroneous genotypes do not substantively change allele frequencies, such errors tend to have small effects on FST. In contrast, genotype‐based analyses, such as inferences of identity, relatedness and relationship, are strongly influenced by data quality. Ignoring or underestimating genotyping errors can lead to false parentage exclusions (Dakin & Avise, 2004; Wang, 2010), false sibship exclusions (Wang, 2004), false exclusion of duplicated individuals and thus overestimation of population size (Creel et al., 2003; Waits & Leberg, 2000).
The impact of missing and erroneous genotypes also depends on how they are distributed among loci and among individuals. The best scenario is a uniform distribution, such that no specific loci and no specific individuals are too problematic to be useful. However, with MIS samples, missing and erroneous genotypes are usually clustered among individuals because the sample DNA quality and quantity can differ substantially among samples, and error rates have been shown to vary considerably across loci (Broquet & Petit, 2004; Campbell et al., 2015; Gagneux, Boesch, & Woodruff, 1997; Paetkau, 2003).
A source of error common to both microsatellites and SNP genotypes is allelic dropout (Bayerl et al., 2017; Gagneux et al., 1997). This is where a heterozygous genotype may be incorrectly typed as a homozygote. Allelic dropout is generally caused by random effects that result in missing one of the two alleles at a diploid locus. It is strongly correlated with lower coverage (5–20×; Nielsen, Paul, Alberechtsen, & Song, 2011) for SNPs from next generation sequencing (NGS). Loci can also have null alleles, which produce no observable phenotype (Dakin & Avise, 2004). Thus, null allele homozygotes would be scored as missing data, whereas a null allele heterozygote would be scored (erroneously) as a homozygote of the observable allele. Traditionally, a single best genotype is reported for an individual at a locus. The large uncertainties of such called SNP genotypes mean that erroneous results could be produced, such as an overestimation of inbreeding (Vieira, Fumagalli, Albrechtsen, & Nielsen, 2013) and biased estimates of relatedness (Vieira, Albrechtsen, & Nielsen, 2016), just as it can in standard genetic markers such as microsatellites (Bonin et al., 2004).
The best practice now is to call all possible genotypes at a SNP locus with corresponding likelihoods that summarise the quality and evidence of the reads data, as well as incorporating information on population‐level allele frequencies (Nielsen et al., 2011). Using genotype likelihoods to account for uncertainties at the individual genotype level, an appropriately designed programme can yield unbiased and accurate estimates of parameters such as inbreeding and relatedness (Vieira et al., 2013, 2016), even when the average coverage is very low, and thus, the genotype data are highly uncertain (Buerkle & Gompert, 2013). Buerkle and Gompert (2013) show that partitioning the sequencing effort maximally among individuals and obtaining approximately one read per locus and individual (1× coverage) yields the most information about a population. More statistical methods urgently need to be adapted or developed to take advantage of genotype likelihoods. One obstacle is computational burden, which increases enormously by considering three possible rather than a single genotype at each locus for each individual, although increasingly sophisticated algorithms and parallelization may mitigate this issue.
The fundamental strategy for improving data quality is by enhancing DNA quantity and quality, reducing contamination, improving PCR protocols (or NGS coverage), employing good laboratory practices and other technical improvements that are beyond the scope of this review (for more information: Bonin et al., 2004; Morin et al., 2010; Paetkau, 2003; Pompanon et al., 2005; Waits & Paetkau, 2005). As with microsatellite genotyping (Bonin et al., 2004), the best practice is to report error rates or genotype likelihoods from SNP genotype studies. There are two categories of mistyping rate estimation. One category is based on duplicated genotype data (i.e., an individual is genotyped independently multiple times at a locus), measuring the consistency of repeated genotypes (e.g., Broquet & Petit, 2004) or estimating the error rates of repeated genotypes (e.g., Johnson & Haydon, 2007; Zhan, Zheng, Bruford, Wei, & Tao, 2010). These methods generally overestimate the mistyping rate of the final genotype data set, because repeated genotyping allows for the detection and elimination of such errors in the final consensus genotypes. This has been a common method for reporting genotype error rates in many SNP array studies (Table 2).
The second category for estimating mistyping rates is based on the final consensus genotypes and is accomplished by examining the genotype against either the Hardy–Weinberg equilibrium (e.g., Hosking et al., 2004) or the Mendelian segregation law in a known (e.g., Sobel, Papp, & Lange, 2002) or reconstructed pedigree (e.g., Wang & Santure, 2009). The former is effective only in detecting null alleles and allelic dropouts that can cause directional deviations from Hardy–Weinberg proportions (i.e., an excess of homozygotes), but is ineffective for mistypings that do not cause detectable distortions, such as false alleles. This error estimation approach can have low power (e.g., Cox, 2006), and relies on the absence of confounding factors, such as strong selection, inbreeding and population structure. Some methods have been developed to make joint estimates of null allele frequencies and inbreeding (e.g., Hall, Mercer, Phillips, Shaw, & Anderson, 2012). How well such methods work has not been thoroughly evaluated, however.
Pedigree, either known or inferred, can be used in likelihood methods to detect erroneous genotypes and to estimate mistyping rate at each locus (Sobel et al., 2002; Wang, 2009). These methods can be used to infer null allele rates, allelic dropout rates and false allele rates and are highly robust to the violations of some common assumptions such as random mating and the absence of inbreeding population structure. Such mistyping estimation methods, together with data missing rates, measure data quality. More importantly, these methods allow downstream analyses to effectively filter out the noises in extracting information from the genotype data and in arriving at robust and accurate analysis results (e.g., Kalinowski, Taper, & Marshall, 2007; Wang, 2004).
3. QUESTIONS AND METRICS THAT CAN BE INVESTIGATED WITH MIS
The power of genetic monitoring using MIS is the range of questions that can be addressed. Here, we discuss how environmental samples can be used to address broad questions, such as species occupancy range, and how individual‐level MIS samples (e.g., feathers, faeces) can be used to estimate individual‐ and population‐level parameters, such as vital rates and population genetic parameters.
3.1. Environmental samples
3.1.1. Occupancy and range
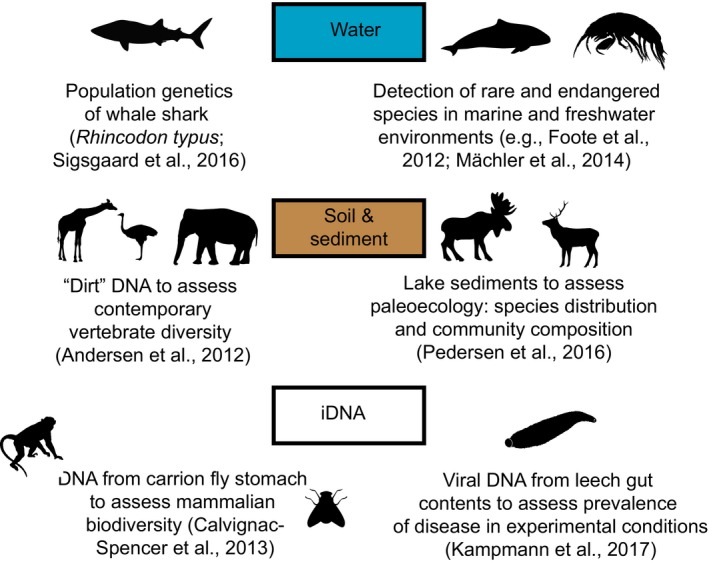
Species and site occupancy and presence/absence analysis relies on information needed to avoid biased estimates; quantifying detection rates and especially understanding whether a target species is present, but undetected (e.g., MacKenzie, Nichols, Hines, Knutson, & Franklin, 2003). Molecular data can augment these studies, enabling more accurate detection even at very low levels of occupancy using environmental samples and DNA barcoding (e.g., Boothroyd, Mandrak, Fox, & Wilson, 2016) or faecal samples of uncertain species identity (e.g., Faria et al., 2011; Palomares et al., 2002; Stanton et al., 2016), although its use is again severely constrained by DNA quality considerations. For example, Stanton et al. (2016) assayed faecal samples from an unsurveyed region in the Democratic Republic of Congo for the presence of okapi (Okapia johnstoni). Of the 24 faecal samples detected, only 12 yielded DNA but of these six were identified as okapi and these yielded four mitochondrial haplotypes (hence allowing the inference of minimally four individuals being present). Advances in environmental DNA (eDNA) analysis are enhancing our ability to examine past and present distribution and diversity of various species and communities (see Box 2).
Box 2. Environmental DNA (eDNA) in the genetic monitoring context.
1.
Genomic sequencing technologies are broadening the scope of eDNA studies in genetic monitoring. Researchers have demonstrated that ecological research questions can be addressed using DNA extracted from water, soil, sediments, snow, browsed foliage, as well as invertebrates (“iDNA”; Schnell et al., 2015) that feed on species of interest: some examples are illustrated below.
Figure 2.
3.2. Individual‐level samples
3.2.1. Individual identification and its application: Abundance/density
The recapture of individuals, identified by their genotype, across time and space, has allowed genetic monitoring to become a key tool in estimating abundance, density and demographic parameters in a variety of species. It has been particularly important in species that are evasive, endangered (Taberlet et al., 1997), dangerous (Kendall et al., 2009) or otherwise difficult to capture/recapture (Constantine et al., 2012), such as those that show limited variation in natural markings, reducing the usefulness of conventional identification from photographs (e.g. juvenile cetaceans, Carroll et al., 2016). The use of genetic monitoring to estimate abundance ranges from the enumeration of the number of genotypes in a region (Taberlet et al., 1997), to single‐session models (Miller, Joyce, & Waits, 2005; Petit & Valière, 2006), to occupancy (Lonsinger, Gese, Bailey, & Waits, 2017; Marucco, Avanzinelli, & Boitani, 2012), to complex mark–recapture models that integrate sex, age and reproductive status information (Carroll et al., 2013; Woodruff et al., 2016). The advent of spatial mark–recapture models (Efford, 2004, 2011; Royle & Young, 2008) has improved analytical tools for density estimates using genetic monitoring approaches (Mollet, Kéry, Gardner, Pasinelli, & Royle, 2015; Russell et al., 2012; Thompson, Royle, & Garner, 2012).
Historically, population estimation in genetic monitoring has relied on individual identification using microsatellite loci. Recognition that genotyping error, correlated with low‐quality DNA templates, can create large biases in population abundance estimates (Waits & Leberg, 2000) has required the development of methods that generate consensus genotypes from multiple PCR replicates (Taberlet et al., 1997) or models that directly incorporate genotyping error (Lukacs & Burnham, 2005; Wang, 2016). In transitioning to the genomics era, new approaches such as direct sequencing of microsatellite loci (De Barba et al., 2017) and SNP analysis will be used (Fitak et al., 2015; Kraus et al., 2015). Large panels of markers from next‐generation sequencing will allow for the more efficient identification of related individuals. This will allow the use of close kin mark–recapture models, which extend the idea of using the recapture of individuals to the recapture of close kin to estimate demographic parameters such as effective population size (Bravington, Skaug, & Anderson, 2016; Wang, 2009).
3.2.2. Other demographic parameters
Long‐term effective management of populations and species requires sound knowledge of key demographic parameters, such as survival and growth rates. The most common way to estimate such parameters is from long‐term studies that follow individuals over time (McClintock, White, Antolin, & Tripp, 2009). Long‐term MIS studies have been an effective way to estimate survival and growth rates in a range of species, by tracking individuals using their genotypes. This has been accomplished using mark–recapture models in species such as southern right whales (Eubalaena australis; Carroll et al., 2013, 2016), the dendrobatid frogs (Allobates femoralis; Ringler, Mangione, & Ringler, 2015), Māui dolphins (Cephalorhynchus hectori maui; Baker et al., 2013), brown bears (Tenan et al., 2016) and imperial eagles (Aquila heliaca; Rudnick et al., 2005). The definitive DNA marks provided by genetic monitoring can provide robust population estimates in age‐structured populations that can be difficult to observe in the wild. The difference between observational and MIS genetic population estimates can have profound impacts on demographic models and associated conservation actions (Katzner, Ivy, Bragin, Milner‐Gulland, & DeWoody, 2011).
3.2.3. Individual space use and movement
Genetic monitoring using MIS can also provide valuable information on individual space use, movement patterns and dispersal. This approach has been used to monitor population expansion and individual dispersal distances in reintroduction efforts for brown bears (De Barba, Waits, Garton, et al., 2010), grey wolves (Canis lupus; Stenglein et al., 2010) and Columbia Basin pygmy rabbits (Brachylagus idahoensis; Demay, Becker, Rachlow, & Waits, 2017), investigate connectivity between migratory habitats in humpback whales (Megaptera novaeangliae; Constantine et al., 2014; Garrigue et al., 2011), to monitor roosting movements in eagles (Rudnick et al., 2008) and to detect natural range expansion (Carroll et al., 2014; Valière et al., 2003) using microsatellites. MIS using microsatellites has also been valuable for assessing the effectiveness of corridors (Dixon et al., 2006) and evaluating potential barriers (Epps et al., 2005; Kendall et al., 2009; Proctor et al., 2005). More recently, SNPs have been utilised to estimate pedigree‐based dispersal models in brown bears (Norman & Sprong, 2015) and to infer individual provenance (i.e., identify potential migrants) based on the distribution of pairwise relatedness (DeWoody et al., 2017).
3.2.4. Relatedness and kin structure (kinship)
Since the development of relatively large panels of markers (microsatellites and more recently SNPs), those panels have been used to monitor the existing relationships between individuals of a given population, either to investigate genetic and social structure, gene flow, reconstruct pedigrees or minimise inbreeding (Caniglia, Fabbri, Galaverni, Milanesi, & Randi, 2014; Da Silva, Lalonde, Quse, Shoemaker, & Russello, 2010; Jones et al., 2002; Peters, Queller, Imperatriz‐Fonseca, Roubik, & Strassmann, 1999; Stenglein, Waits, Ausband, Zager, & Mack, 2011). Metrics generally used to measure relatedness between two individuals estimate either a summary statistic (such as coancestry coefficient and its equivalents), which would correspond to the relatedness between two individuals, or the probability that two individuals are linked with a particular relationship (parent–offspring, first cousins, self‐outbred sibs, etc.) given the data (Wang, 2011). In some cases, the reliability of relatedness estimates can be limited, especially when the population under study exhibits low genetic variation for the marker set; therefore a priori simulations should be performed to select the most appropriate estimator and assess its accuracy (Glaubitz et al., 2003; Taylor, 2015). The development of NGS tools is expected to increase the availability of high‐density panels, thus improving the reliability of estimators. It may also allow the use of new metrics, such as chromosome‐segment‐based ones, considering the measurement of coancestries based on shared segments of identity by descent, instead of averaging, marker by marker, the probability that two alleles are identical in state (De Cara, Villanueva, Toro, & Fernández, 2013).
3.3. Population genetic parameters
3.3.1. Genetic diversity
Historically, microsatellites were used with MIS to produce estimates of population genetic variation based on allelic diversity and heterozygosity. Allelic diversity, which is often high and variable among microsatellite loci, is not very informative for SNPs. This is because SNPs have comparatively few alleles, generally limited to one or two (i.e., third or fourth alleles at a locus do not materialise before one of the original two is lost due to drift or selection).
On the other hand, estimates of heterozygosity using SNP loci can be more informative than microsatellites because the additional SNP loci surveyed provide higher precision. For example, Doyle et al. (2016) surveyed 162 SNPs in golden eagles and found that mean observed heterozygosity (HO) was 0.32 ± 0.01 in juveniles whereas adult HO was 0.35 ± 0.01, a significant statistical difference consistent with expectations of viability selection. Unfortunately, the types of SNP arrays often used in MIS studies preclude the evaluation of other genetic diversity metrics that will likely be important in the future (e.g., runs‐of‐homozygosity or copy number variants, see Leroy et al., 2017). This is a factor worth considering when planning a study, as evaluating change in genetic diversity metrics over time is an important task of genetic monitoring (see Box 3).
Box 3. The importance of “delta” in genetic monitoring.
1.
Endangered species are, by definition, the subject of local, regional, national and international legislation, including the Convention on Biological Diversity (CBD). The CBD's 2020 Targets include a commitment to “minimise genetic erosion” and “safeguard genetic diversity” (Bruford, Davies, Dulloo, Faith, & Walters, 2017; Hoban et al., 2013). These commitments require a means of verification and imply a reference point from which to determine changes, or “delta,” in genetic diversity. The statistical approaches needed to evaluate changes in genetic diversity over short timescales, however, require development. Temporal genetic monitoring of species at the same location has been accomplished in a some well‐studied populations or species of high conservation concern (e.g., Italian brown bears; De Barba, Waits, Garton, et al., 2010; Māui dolphins; Baker et al., 2016) or where hybridization is a threat (e.g., red wolves and coyotes; Bohling et al., 2016).
In the absence of samples from a population over time, analysis of genetic data using single point samples can provide insights into recent demographic change (e.g., Goossens et al., 2006). However, single point estimators can have wide variance and provide inconsistent values depending on the methods chosen or model assumptions (Barker, 2011). To aid understanding of which metrics would be the most sensitive to detecting short‐term declines in genetic diversity, Hoban et al. (2014) carried out an assessment of temporal indicators of genetic erosion (sensu Aichi Target 13).
The number of alleles per genetic locus (K) outperformed all other potential indicators across all scenarios. However, the power with which to detect a decline in diversity in K varied with more samples or markers with, for example, 2500 SNPs being effective at detecting minor demographic declines after 8–10 generations (see Figure). Hoban et al. (2014) also found that statistical power to detect change improved if samples were available before the onset of decline, implying that archived and museum collections can clearly play an important role as part of monitoring programmes.
Figure 3.
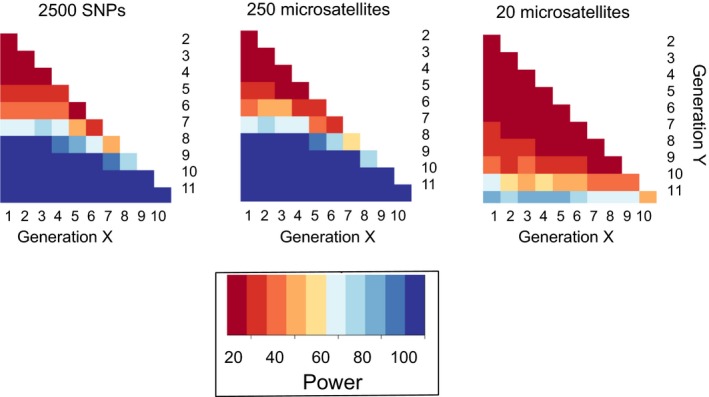
Figure: Modified from Hoban et al. (2014): Comparison of the proportion of 100 replicates (i.e., power) in which the indicator, K (alleles per locus), was significantly different between generations X and Y. The scenario simulates a population that has experienced an exponential decline of 97%, using different types and numbers of genetic markers. The darkest blue is power >0.90; power <0.50 is orange; and power <0.10 is dark red.
3.3.2. Effective population size
Populations of conservation concern are usually small and thus experience inbreeding and genetic drift that could lead to a depletion of genetic variation. The parameter effective population size measures the strength of the stochastic processes of inbreeding and genetic drift (Wright, 1931) in a population. It is defined as the size of an idealised population which would give rise to the same rate of inbreeding or drift as observed in the actual population under consideration (Caballero, 1994; Wang, Santiago, & Caballero, 2016). For wild populations where pedigree data are unavailable, marker data, generated from MIS, can be used to estimate both historical (e.g., Beerli & Felsenstein, 2001) and recent/current effective size of a population (Wang, 2016). Recent effective population size can be estimated by approaching a wide range of signals (temporal variance in allele frequency, frequency of close relatives, linkage disequilibrium, heterozygosity excess, etc.) measuring either inbreeding or genetic drift in a given time period. By consequence, depending on the data available, various approaches can be implemented, each with its own advantages and limits. For instance, linkage disequilibrium estimates of contemporary effective population size can be obtained from unlinked microsatellites or SNPs. When the linkage information between SNPs is also available, linkage disequilibrium estimates allow the inference of effective population size over past generations (Hayes, Visscher, Mcpartlan, & Goddard, 2003).
While this approach has its limitations and caveats (Palsbøll, Peery, Olsen, Beissinger, & Bérubé, 2013), MIS has been used to estimate long‐term effective population sizes in species such as southern right whales (Carroll et al., 2015) and Sumatran orangutans (Pongo abelii; Nater et al., 2013). Historical samples can provide a direct way of assessing past levels of genetic diversity and effective population size and therefore any recent changes in these metrics. Although not typically undertaken using MIS, such studies provide important management information for species of conservation concern, for example, museum specimens were used to assess historical diversity in species such as brown bears (Ursos actos; Miller & Waits, 2003) and Seychelles warbler (Acrocephalus sechellensis; Spurgin et al., 2014).
Contemporary estimates of effective population size or number of effective breeders are also a critical indication of the genetic resilience of a population (Frankham, Bradshaw, & Brook, 2014), and have been estimated with MIS for brown bears (De Barba, Waits, Garton, et al., 2010; Gonzalez et al., 2016), Hector's dolphin (Cephalorhynchus hectori; Hamner, Constantine, Mattlin, Waples, & Baker, 2017), Māui dolphin (Baker et al., 2016) and Eurasian otters (Lutra lutra; Koelewijn et al., 2010). For the purpose of genetic management of endangered species, the current or contemporary effective size is more relevant than historical or long‐term effective size (Wang, 2016).
3.3.3. Social and genetic structure
In addition to the presence/absence and censusing of individuals, additional information can be gained from MIS studies on the socio‐genetic structure of the population being surveyed. This has become a necessity in certain fields (especially in primatology) where, even if individuals can be observed and identified, invasive sampling is regarded as unethical and is often prohibited. Such studies may allow identification of social group‐mediated genetic structure and inferences on sex‐biased dispersal and how these may be modified by habitat fragmentation (e.g., Minhos et al., 2016) and/or hunting and exploitation (e.g., Ferreira da Silva et al., 2014). Understanding social structure and spatial assortment of related individuals using MIS is also an important factor underpinning the accuracy of capture–recapture molecular censusing (Miller et al., 2005; Zhan et al., 2006).
Both in socially structured and unstructured species, population boundaries may spatially coincide with a sampling area being studied using MIS methods. In such cases, it is important to know where these boundaries lie in order to infer the underlying demographic processes structuring the population(s), and to assign individuals to those populations using the correct allele frequency data. Over recent years, numerous studies have successfully investigated genetic structure in wild populations using MIS (e.g., Norman et al., 2017; Russello, Waterhouse, Etter, & Johnson, 2015; Steyer et al., 2016). Different approaches have been developed to investigate the genetic structuring of a group or population, using either multivariate analysis (Jombart, Pontier, & Dufour, 2009) or Bayesian methods for optimizing population features such as Hardy–Weinberg equilibrium (Pritchard, Stephens, & Donnelly, 2000) and even allowing for the integration of environmental and spatial data for interpretation purposes (e.g., Caye, Deist, Martins, Michel, & Francois, 2016; Guillot, Mortier, & Estoup, 2005). Further, these structure‐based approaches are relatively robust in the face of bias related to small sample size or even genotyping error (Smith & Wang, 2014).
3.3.4. Hybridization and introgression
For some species, hybridization and introgression are major threats to population and species persistence creating a need for long‐term genetic monitoring (Allendorf, Leary, Spruell, & Wenburg, 2001). Genetic monitoring approaches using MIS have been applied to detect hybridization in multiple carnivore species including grey wolves (Caniglia et al., 2014; Godinho et al., 2015; Kopaliani, Shakarashvili, Gurielidze, Qurkhuli, & Tarkhnishvili, 2014; Monzón, Kays, & Dykhuizen, 2014), Eastern wolves (Canis lycaon, Benson, Patterson, & Wheeldon, 2012), red wolves (Canis rufus; Adams et al., 2003; Bohling et al., 2016) and wildcats (Felix silvestris silvestris; Anile, Ragni, Randi, Mattucci, & Rovero, 2014; Steyer et al., 2016). The majority of these studies have used mitochondrial DNA and microsatellite markers, but a few have used SNPs to detect hybridization or monitor grey wolves (Kraus et al., 2015; Monzón et al., 2014) and hybridization between wildcats and domestic cats (Nussberger et al., 2014; Oliveira et al., 2015).
3.4. Nontarget DNA: Diet
DNA metabarcoding combined with high‐throughput sequencing has proven to be an effective genetic monitoring tool to characterise diet (Pompanon et al., 2012; Valentini et al., 2009). This method has been used to noninvasively study diet in a diverse range of species including Adelie penguins (Pygoscelis adeliae; Jarman et al., 2013), golden‐crowned sifaka (Propithecus tattersalli; Quéméré et al., 2013), subterranean rodents (Ctenomys sp.; Lopes et al., 2015), tapir (Tapirus terrestris; Hibert et al., 2013), brown bears (De Barba et al., 2014; Elfström et al., 2014; Valentini et al., 2009), golden marmots (Marmota caudata; Valentini et al., 2009), African herbivores (Kartzinel et al., 2015), Hawaiin tree snails (Achatinella spp.; O'Rorke et al., 2015; Price, O'Rorke, Amend, & Hadfield, 2017), red deer (Cervus elaphus; Fløjgaard, De Barba, Taberlet, & Ejrnæs, 2017) and leopard cats (Prionailurus bengalensis; Shehzad et al., 2012). While technical limitations mean that diet inference is typically semi‐quantitative (De Barba et al., 2014; Deagle, Chiaradia, Mcinnes, & Jarman, 2010; Pompanon et al., 2012), the ability to identify primary dietary components is useful for comparative ecological studies (although see Thomas, Deagle, Eveson, Harsch, & Trites, 2016; Thomas, Jarman, Haman, Trites, & Deagle, 2013 for advances in quatitative methods). Furthermore, metagenomic approaches whereby shotgun sequencing is used to characterise both prey and potential pathogens in faecal samples holds the potential to simultaneously characterise diet and microbiomes, while avoiding some of the earlier technical limitations (Srivathsan, Ang, Vogler, & Meier, 2016). In a broader context, assaying dietary niche through genetic monitoring techniques is likely to play a future role in determining the vulnerability of populations to disturbances (Clare, 2014) and is already aiding the restoration and relocation plans for endangered species (Price et al., 2017).
4. PAST AND FUTURE OPPORTUNITIES
4.1. Legacy data sets
The sheer abundance of microsatellite data sets associated with MIS conservation studies is impressive. Thus, it would be desirable if future monitoring efforts could tie an individual's established microsatellite DNA profile to a new SNP profile. Many individuals of long‐lived species like trees, whales, bears or eagles have already been genetically tagged using microsatellites. In an ideal world, a new DNA profile generated with SNPs would be matched to those generated previously with microsatellites. Unfortunately, this is time‐consuming and expensive because it would require SNP genotyping a reference sample for each individual or having a way to link the SNP genotype to the microsatellite genotype. In principle, it might be possible using a high‐density SNP array to genotype individuals at each microsatellite locus. However, in practice, this depends on the availability of the SNPs, the extent of linkage disequilibrium, recombination rates, nucleotide substitution rates, effective population size, as well as the practicalities of designing assays for the repetitive genomic regions that microsatellites represent. In practice, it is an easy decision to forego microsatellites and establish a new SNP array when monitoring a “new” species. There are online tools, such as the ConGress website that contains a Decision Making Tool, that can help managers to use power analyses to identify optimal methods for a MIS analysis ( http://www.congressgenetics.eu).
In those cases with extensive legacy data sets, it might make the most sense to use “microsatellite sequencing” techniques (e.g., De Barba et al., 2017) in an effort to continue surveying the same loci (albeit with a different technology), at the same time as expanding genome sampling. It may be possible to impute genotypes if sufficiently large sample sizes are available for present and past data, and both legacy and modern platforms, as is routinely carried out for individual types using different SNP panels in livestock species (e.g., Druet, Schrooten, & de Roos, 2010). As an example from cattle, the imputation of 12 microsatellite markers was conducted using a set of 982 SNPs, located within 500 kb of the targeted microsatellite markers (McClure, Mullen, & Kearney, 2014; McClure, Sonstegard, Wiggans, & Van Tassell, 2012).
Such imputation is likely to be far more difficult in wild species that lack pedigrees and dense marker panels. That said, it might be possible to use known or suspected relationships among individuals (e.g., full‐siblings) to leverage microsatellite‐based fingerprinting against SNP‐based fingerprinting.
4.2. Future directions
Evolving technology means that genetic monitoring of populations is expanding beyond genotypes. We broadly categorise these methods into those that will help enhance understanding of population demography, health and “functional” or adaptive genetic monitoring. The latter moves beyond using neutral alleles for individual identification and estimation of population genetic parameters to assay loci linked to processes such as inbreeding and adaptation (Table 3). Wildlife forensics is also set to benefit from technological advances (see Box 4).
Table 3.
Beyond genotypes: selected examples of the application of genomic sequencing technology to study ecology and evolution of species using minimally invasive samples
| Reference | Inference | Platform/method | Starting material | Species |
|---|---|---|---|---|
| Assessing genetic diversity | ||||
| Hans et al. (2015) | Diversity of MHC loci | Pooled PCR amplicon sequencing on Illumina MiSeq | Faeces | Gorilla (Gorilla gorilla) |
| Ang et al. (2016) | Diversity of mtDNA | Pooled PCR amplicon sequencing on Illumina HiSeq | Faeces | Tonkin snub‐nosed monkey (Rhinopithecus avunculus) |
| Sigsgaard et al. (2016) | MtDNA haplotype diversity and identity | Illumina MiSeq (bulk sequencing) | eDNA water sample | Whale shark (Rhincodon typus) |
| Health/diet/demography | ||||
| Valentini et al. (2009) | Diet | PCR amplicons sequencing 454 platform | Faeces | Golden marmots (Marmota caudata) and brown bears (Ursus arctos) |
| Shehzad et al. (2012) | Diet | Pooled PCR amplicon sequencing on Illumina | Faeces | Leopard cat (Prionailurus bengalensis) |
| Jarman et al. (2013) | Diet | Pooled PCR amplicon sequencing on Ion Torrent | Faeces | Adelie penguin (Pygoscelis adeliae) |
| Quéméré et al. (2013) | Diet | Pooled PCR amplicon sequencing on Illumina | Faeces | Golden‐crowned sifaka (Propithecus tattersalli) |
| De Barba et al. (2014) | Diet | Pooled PCR amplicon sequencing on Illumina | Faeces | Brown bear (Ursus arctos) |
| Kartzinel et al. (2015) | Diet and niche partitioning | Pooled PCR amplicon sequencing on Illumina | Faeces | Seven large mammalian herbivores |
| O'Rorke et al. (2015); Price et al. (2017) | Diet and niche partitioning, environmental restoration planning | Pooled PCR amplicon sequencing on Illumina | Faeces | Hawaiian tree snails (Achatinella spp.) |
| Srivathsan et al. (2016) | Diet and gut parasite characterization | mtDNA shotgun sequencing Illumina HiSeq | Faeces | Banded leaf monkey (Presbytis femoralis) |
| Apprill et al. (2017) | Characterization of respiratory microbiome | Pooled PCR amplicon sequencing on Illumina | Exhaled breath samples | Humpback whale (Megaptera novaeangliae) |
| Raverty et al. (2017) | Genetic monitoring of respiratory microbiome | PCR amplicon sequencing of bacterial DNA barcodes and direct culture of bacteria | Exhaled breath samples | Killer whale (Orcinus orca) |
| Polanowski et al. (2014) | Estimate of chronological age | Bisulphite conversion of PCR products and PYROMARK 24 Pyrosequencing platform sequencing (Qiagen) | Remote skin biopsy sample | Humpback whale (Megaptera novaeangliae) |
Box 4. Wildlife Forensics.
1.
Figure 4.

Image: White rhinoceros (Ceratotherium simum). Credit: J. A. DeWoody.
As the global threat of illegal wildlife trade becomes more apparent, the use of genetic and genomic tools in the fight against wildlife crime has increased substantially (Corlett, 2017; Ogden & Linacre, 2015; Staats et al., 2016). Traditional genetic tools are increasingly being applied to forensic casework involving material inherently lacking in viable genetic material, for example, microsatellite markers to locate the likely origin of seized elephant ivory (e.g., Wasser et al., 2015), and similar tools are now in routine use to enable the development of a database to allow the matching of carcasses and seized, poached African rhinoceros horn (Harper, Vermeulen, Clarke, De Wet, & Guthrie, 2013). DNA barcoding is being increasingly used in the identification of traded products to species, such as pangolin scales (Mwale et al., 2017), as well as estimating the number of whales traded in meat markets (Baker et al., 2007).
The use of genomics has, however, opened up the possibility of additional applications in the forensic field, including the development of simple, cost‐effective tools to analyse extremely problematic samples and to address questions that were otherwise statistically unattainable using standard genetic approaches. For example, it is possible to identify putrid bushmeat samples, which can be highly degraded once seized, to species level or beyond using low‐cost microarrays (e.g., Rönn et al., 2009). A landmark paper in 2011 developed a set of SNPs for investigation of false eco‐certification of exploited European fish stocks using population assignment that relies on divergent SNPs under the influence of selection in species in otherwise undifferentiated populations, where standard microsatellite‐based population assignment had proved impossible (Nielsen et al., 2012). Furthermore, portable sequencing devices, such as the MinIon (Oxford Nanopore Technologies), are starting to be used to sequence samples in field laboratory conditions (Edwards, Debbonaire, Sattler, Mur, & Hodson, 2016; Quick et al., 2016). This leads to the possibility that in the near future researchers will be able to undertake real‐time assessments of the species, and potentially population of origin, of products in markets.
4.2.1. Population demography
Estimating the chronological age of individuals through genetic monitoring would provide broader insights into population dynamics. Age classes, or the chronological age of individuals in a population, are a critical component to estimating past and future growth rates, as well as population‐level responses to biotic (e.g., prey resources) and abiotic (e.g., hunting) pressures. Conventionally, longitudinal studies that track individuals in well‐studied populations have been the only way to estimate age for many species (Clutton‐Brock & Sheldon, 2010). However, molecular age biomarkers (MAB), those derived from measurable changes in DNA or RNA abundance or sequence change, offer a new way to estimate chronological age. One MAB that held promise was telomeres, and although it has been found to work well in some bird species (e.g., Haussmann, Vleck, & Nisbet, 2003), its wider applicability has been limited (Dunshea et al., 2011). A recent paper showed that epigenetic markers can be used to estimate age in humpback whales (Polanowski, Robbins, Chandler, & Jarman, 2014), using MIS, an approach that has promise in other species (Jarman et al., 2015).
Epigenetic markers might have utility in monitoring other facets of population demography, as epigenetic changes have been linked to early life conditions (Gapp, von Ziegler, Tweedie‐Cullen, & Mansuy, 2014), reproductive maturity (Lomniczi et al., 2013), survival (Fairlie et al., 2016) and response to chemical or physical stressors (Feil & Fraga, 2012), in a variety of species. The development of epigenetic markers therefore has the potential to monitor how environmental processes can influence population demography through monitoring development and fecundity over time. However, it will require much development to apply such methods to noninvasively collected samples. Innovations in sequencing platforms that do not require bisulphite conversion to examine methylation patterns, such as PacBio (Rhoads & Au, 2015), will be useful. Studies that evaluate how the DNA degradation that often occurs in noninvasive genetic sampling impacts assay methods will also be required.
4.2.2. Monitoring health
The microbial communities living on or in multicellular organisms or “hosts,” termed microbiomes, are a rich area of study in humans and, increasingly, wild animals. Host health and fitness can be affected by the microbiome through different mechanisms: the microbiome could act directly to protect health, through competitive exclusion or by stimulating immunity, or act indirectly, by modifying metabolism or development (Bahrndorff, Alemu, Alemneh, & Lund Nielsen, 2016). For example, research has linked changes in skin bacterial microbiome with outbreaks of chytrid fungus in endangered frog populations (e.g., Jani & Briggs, 2014), and there is evidence that symbiotic bacteria on amphibian skin generate metabolites protective against the fungus (Loudon et al., 2014). Additionally, the microbiome might include known pathogens (Acevedo‐Whitehouse, Rocha‐Gosselin, & Gendron, 2010; Delgado et al., 2017): long‐term, noninvasive monitoring of the of the southern resident killer whale population in North America showed that antibiotic‐resistant bacteria were present in the respiratory microbiome of apparently healthy individuals (Raverty et al., 2017). Therefore, microbiomes could be regularly screened using MIS for the presence of both beneficial and harmful components as part of an ongoing genetic monitoring scheme. Changes in the characteristics of the microbiome over time might also be indicative of changes in the quality of the social or broader environment (Amato et al., 2013; Tung et al., 2015) and can be significantly differentiated among individuals within a population (Klein‐Jöbstl et al., 2014). Additionally, studies in model organisms have used proteomic analysis of faecal samples to noninvasively monitor host–microbe interaction during development (e.g., Young et al., 2015) and disease processes (e.g., Yau, Leong, Zeng, & Wasinger, 2013).
As the gut microbiome is closely related to diet (Amato et al., 2013; Delsuc et al., 2014), it has been suggested as a potential screening tool to identify dietary components (Bahrndorff et al., 2016). However, evidence suggests that survey methods focusing on noninvasively collected faecal samples need to carefully consider the change in microbiome linked to environmental conditions, time since deposition and focal species (Menke, Meier, & Sommer, 2015).
4.2.3. Functional or adaptive genetic monitoring
Traditional genetic monitoring has focused on presumably neutral markers to identify individuals and to assess genetic diversity. When whole‐genome data are available, investigators have the choice of using intergenic SNPs from gene deserts or of using “non‐neutral” markers derived from protein‐coding genes thought to be targets of natural selection (DeWoody et al., 2017; Doyle et al., 2016). This can be an important distinction, because the non‐neutral loci are often more sensitive indicators of population differentiation (Freamo, O'reilly, Berg, Lien, & Boulding, 2011). By combining genomic and environmental data, landscape genomics approaches can also be a powerful approach to infer and define conservation units (Funk, McKay, Hohenlohe, & Allendorf, 2012).
Only a few studies have yet investigated the possibilities of using MIS approaches for such a purpose. Russello et al. (2015) used hair samples to investigate genetic diversity and in the American pika hair, detecting several candidate gene regions which exhibited putative signatures of divergent selection for adaptation to altitude. Given the potential environment shifts related to climate change that can be expected, landscape genomics may offer useful insight to better monitor and manage wild and domestic populations.
5. CONCLUSION
Genetic monitoring with MIS has proven to be a valuable tool to monitor and manage species and populations. With increasing access to new technological advances, researchers will be able to go beyond identifying individuals to investigate their role in the ecosystem and assess population‐level dynamics. Such tools will be necessary to meet the challenges of conservation biology in a rapidly changing environment.
DISCLAIMER
The views expressed in this information product are those of the authors and do not necessarily reflect the views or policies of FAO.
CONFLICT OF INTEREST
The author declares no conflict of interest.
ACKNOWLEDGEMENTS
This work was assisted through participation in the Next generation genetic monitoring Investigative Workshop at the National Institute for Mathematical and Biological Synthesis, sponsored by the National Science Foundation through NSF Award #DBI‐1300426, with additional support from The University of Tennessee, Knoxville. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The authors would like to thank the French Government for providing for the secondment of Dr Gregoire Leroy to FAO. ELC was supported by a Marie Slodowska Curie Fellowship, (Behaviour‐Connect) funded by the EU Horizon 2020 Programme. MWB was supported by a Royal Society Wolfson research merit award. LW was supported by the University of Idaho. This research was supported in part by NSF awards 1355106 and 1357386 to AES. Animal shapes in Boxes 1 and 2 are from http://www.phylopic.org and under Public Domain and Creative Commons licences, that can be reused with the following acknowledgements: whale shark; Scarlet23 (vectorised by T. Michael Keesey), elephant; T. Michael Keesey; ostrich: Matt Martyniuk (vectorised by T. Michael Keesey).
Carroll EL, Bruford MW, DeWoody JA, et al. Genetic and genomic monitoring with minimally invasive sampling methods. Evol Appl. 2018;11:1094–1119. 10.1111/eva.12600
REFERENCES
- Acevedo‐Whitehouse, K. , Rocha‐Gosselin, A. , & Gendron, D. (2010). A novel non‐invasive tool for disease surveillance of free‐ranging whales and its relevance to conservation programs. Animal Conservation, 13(2), 217–225. 10.1111/j.1469-1795.2009.00326.x [DOI] [Google Scholar]
- Adams, J. R. , Kelly, B. T. , & Waits, L. P. (2003). Using faecal DNA sampling and GIS to monitor hybridization between red wolves (Canis rufus) and coyotes (Canis latrans). Molecular Ecology, 12, 2175–2186. 10.1046/j.1365-294X.2003.01895.x [DOI] [PubMed] [Google Scholar]
- Allendorf, F. W. , Leary, R. F. , Spruell, P. , & Wenburg, J. K. (2001). The problems with hybrids: Setting conservation guidelines. Trends in Ecology and Evolution, 16, 613–622. 10.1016/S0169-5347(01)02290-X [DOI] [Google Scholar]
- Amato, K. R. , Yeoman, C. J. , Kent, A. , Righini, N. , Carbonero, F. , Estrada, A. , … Leigh, S. R. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. The ISME Journal, 716, 1344–1353. 10.1038/ismej.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, K. , Bird, K. L. , Rasmussen, M. , Haile, J. , Breuning‐Madsen, H. , Kjær, K. H. … Willerslev, E. (2012). Meta‐barcoding of “dirt” DNA from soil reflects vertebrate biodiversity. Molecular Ecology 21(8), 1966–1979. 10.1111/j.1365-294X.2011.05261.x [DOI] [PubMed] [Google Scholar]
- Andrews, K. R. , Good, J. M. , Miller, M. R. , Luikart, G. , & Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics, 17, 81–92. 10.1038/nrg.2015.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang, A. , Srivathsan, A. , Meier, R. , Luu, T. B. , Le, Q. K. , & Covert, H. (2016). No evidence for mitochondrial genetic variability in the largest population of critically endangered Tonkin snub‐nosed monkeys in Vietnam. Primates, 57(4), 449–453. 10.1007/s10329-016-0571-x [DOI] [PubMed] [Google Scholar]
- Anile, S. , Ragni, B. , Randi, E. , Mattucci, F. , & Rovero, F. (2014). Wildcat population density on the Etna volcano, Italy: A comparison of density estimation methods. Journal of Zoology, 293(4), 252–261. 10.1111/jzo.12141 [DOI] [Google Scholar]
- Apprill, A. , Miller, C. A. , Moore, M. J. , Durban, J. W. , Fearnbach, H. , & Barrett‐Lennard, L. G. (2017). Extensive core microbiome in drone‐captured whale blow supports a framework for health monitoring. mSystems, 2, e00119–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrndorff, S. , Alemu, T. , Alemneh, T. , & Lund Nielsen, J. (2016). The microbiome of animals: Implications for conservation biology. International Journal of Genomics, 2016, 5304028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, N. , Etter, P. , Atwood, T. , Currey, M. , Shiver, A. , Lewis, Z. , … Johnson, E. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3, e3376 10.1371/journal.pone.0003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, C. S. , Cooke, J. G. , Lavery, S. , Dalebout, M. L. , Ma, Y. U. , Funahashi, N. , … Brownell, R. L. (2007). Estimating the number of whales entering trade using DNA profiling and capture‐recapture analysis of market products. Molecular Ecology, 16(13), 2617–2626. 10.1111/j.1365-294X.2007.03317.x [DOI] [PubMed] [Google Scholar]
- Baker, C. S. , Hamner, R. M. , Cooke, J. , Heimeier, D. , Vant, M. , Steel, D. , & Constantine, R. (2013). Low abundance and probable decline of the critically endangered Māui's dolphin estimated by genotype capture‐recapture. Animal Conservation, 16(2), 224–233. 10.1111/j.1469-1795.2012.00590.x [DOI] [Google Scholar]
- Baker, C. S. , Hamner, R. M. , Oremus, M. , Stanley, M. , Brown, P. , & Constantine, R. (2016). Estimating the abundance and effective population size of Māui's dolphins using microsatellite genotypes in 2010 – 11, with retrospective matching to 2001 – 07. Department of Conservation, Auckland, 1–44.
- Banks, S. C. , Horsup, A. , Wilton, A. N. , & Taylor, A. C. (2003). Genetic marker investigation of the source and impact of predation on a highly endangered species. Molecular Ecology, 12, 1663–1667. 10.1046/j.1365-294X.2003.01823.x [DOI] [PubMed] [Google Scholar]
- Barker, J. S. F. (2011). Effective population size of natural populations of Drosophila buzzatii, with a comparative evaluation of nine methods of estimation. Molecular Ecology, 20(21), 4452–4471. 10.1111/j.1365-294X.2011.05324.x [DOI] [PubMed] [Google Scholar]
- Bayerl, H. , Kraus, R. H. S. , Nowak, C. , Foerster, D. W. , Fickel, J. , & Kuehn, R. (2017). Fast and cost‐effective single nucleotide polymorphism (SNP) detection in the absence of a reference genome using semi‐deep next generation random amplicon sequencing (RAMseq). Molecular Ecology Resources, 10.1111/1755-0998.12717 [DOI] [PubMed] [Google Scholar]
- Beerli, P. , & Felsenstein, J. (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proceedings of the National Academy of Sciences of the United States of America, 98(8), 4563–4568. 10.1073/pnas.081068098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja‐Pereira, A. , Oliveira, R. , Alves, P. C. , Schwartz, M. K. , & Luikart, G. (2009). Advancing ecological understandings through technological transformations in noninvasive genetics. Molecular Ecology Resources, 9(5), 1279–1301. 10.1111/j.1755-0998.2009.02699.x [DOI] [PubMed] [Google Scholar]
- Bellemain, E. , Nawaz, M. A. , Valentini, A. , Swenson, J. E. , & Taberlet, P. (2007). Genetic tracking of the brown bear in northern Pakistan and implications for conservation. Biological Conservation, 134(4), 537–547. 10.1016/j.biocon.2006.09.004 [DOI] [Google Scholar]
- Benson, J. F. , Patterson, B. R. , & Wheeldon, T. J. (2012). Spatial genetic and morphologic structure of wolves and coyotes in relation to environmental heterogeneity in a Canis hybrid zone. Molecular Ecology, 21(24), 5934–5954. 10.1111/mec.12045 [DOI] [PubMed] [Google Scholar]
- Bohling, J. H. , Dellinger, J. , McVey, J. M. , Cobb, D. T. , Moorman, C. E. , & Waits, L. P. (2016). Describing a developing hybrid zone between red wolves and coyotes in eastern North Carolina, USA. Evolutionary Applications, 9(6), 791–804. 10.1111/eva.12388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi, S. , Therkildsen, N. O. , Retzel, A. , & Berg, R. (2016). Historical DNA documents long‐distance natal homing in marine fish. Molecular Ecology, 25, 2727–2734. 10.1111/mec.13580 [DOI] [PubMed] [Google Scholar]
- Bonin, A. , Bellemain, E. , Eidesen, P. B. , Pompanon, F. , Brochmann, C. , & Taberlet, P. (2004). How to track and assess genotyping errors in population genetics studies. Molecular Ecology, 13(11), 3261–3273. 10.1111/j.1365-294X.2004.02346.x [DOI] [PubMed] [Google Scholar]
- Boothroyd, M. , Mandrak, N. E. , Fox, M. , & Wilson, C. C. (2016). Environmental DNA (eDNA) detection and habitat occupancy of threatened spotted gar (Lepisosteus oculatus). Aquatic Conservation: Marine and Freshwater Ecosystems, 1119, 1107–1119. 10.1002/aqc.2617 [DOI] [Google Scholar]
- Boulanger, J. , Himmer, S. , & Swan, C. (2004). Monitoring of grizzly bear population trends and demography using DNA mark–recapture methods in the Owikeno Lake area of British Columbia. Canadian Journal of Zoology, 82, 1267–1277. 10.1139/z04-100 [DOI] [Google Scholar]
- Bravington, M. V. , Skaug, H. J. , & Anderson, E. C. (2016). Close‐Kin Mark‐Recapture. Statistical Science, 31(2), 259–274. 10.1214/16-STS552 [DOI] [Google Scholar]
- Broquet, T. , & Petit, E. (2004). Quantifying genotyping errors in noninvasive population genetics. Molecular Ecology, 13(11), 3601–3608. 10.1111/j.1365-294X.2004.02352.x [DOI] [PubMed] [Google Scholar]
- Bruford, M. W. , Davies, N. , Dulloo, M. , Faith, D. P. , & Walters, M. (2017). Monitoring changes in genetic diversity In Walters M. & Scholes R. J. (Eds.), The GEO handbook on biodiversity observation networks (pp. 107–128). Cham: Springer International Publishing; 10.1007/978-3-319-27288-7 [DOI] [Google Scholar]
- Buerkle, C. A. , & Gompert, Z. (2013). Population genomics based on low coverage sequencing: How low should we go? Molecular Ecology, 22, 3028–3035. 10.1111/mec.12105 [DOI] [PubMed] [Google Scholar]
- Butler, J. M. (2015). The future of forensic DNA analysis. Philosophical Transactions of the Royal Society B, 370, 20140252 10.1098/rstb.2014.0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A. (1994). Developments in the prediction of effective population size. Heredity, 73, 657–679. 10.1038/hdy.1994.174 [DOI] [PubMed] [Google Scholar]
- Calvignac‐Spencer, S. , Merkel, K. , Kutzner, N. , Kühl, H. , Boesch, C. , Kappeler, P. M. … Leendertz, F. H. (2013). Carrion fly‐derived DNA as a tool for comprehensive and cost‐effective assessment of mammalian biodiversity. Molecular Ecology 22(4), 915–924. 10.1111/mec.12183 [DOI] [PubMed] [Google Scholar]
- Campbell, N. R. , Harmon, S. A. , & Narum, S. R. (2015). Genotyping‐in‐Thousands by sequencing (GT‐seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Molecular Ecology Resources, 15(4), 855–867. 10.1111/1755-0998.12357 [DOI] [PubMed] [Google Scholar]
- Campbell, N. R. , & Narum, S. R. (2011). Development of 54 novel single‐nucleotide polymorphism (SNP) assays for sockeye and coho salmon and assessment of available SNPs to differentiate stocks within the Columbia River. Molecular Ecology Resources, 11(Suppl. 1), 20–30. 10.1111/j.1755-0998.2011.02977.x [DOI] [PubMed] [Google Scholar]
- Caniglia, R. , Fabbri, E. , Galaverni, M. , Milanesi, P. , & Randi, E. (2014). Noninvasive sampling and genetic variability, pack structure, and dynamics in an expanding wolf population. Journal of Mammalogy, 95(1), 41–59. 10.1644/13-MAMM-A-039 [DOI] [Google Scholar]
- Carroll, E. L. , Baker, C. S. , Watson, M. , Alderman, R. , Bannister, J. L. , Gaggiotti, O. E. , … Harcourt, R. (2015). Cultural traditions across a migratory network shape the genetic structure of southern right whales around Australia and New Zealand. Scientific Reports, 5, 16182 10.1038/srep16182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, E. L. , Childerhouse, S. J. , Fewster, R. , Patenaude, N. J. , Steel, D. J. , Dunshea, G. , … Baker, C. S. (2013). Accounting for female reproductive cycles in a superpopulation capture recapture framework. Ecological Applications, 23, 1677–1690. 10.1890/12-1657.1 [DOI] [PubMed] [Google Scholar]
- Carroll, E. L. , Fewster, R. , Childerhouse, S. J. , Patenaude, N. J. , Boren, L. , & Baker, C. S. (2016). First direct evidence for natal wintering ground fidelity and estimate of juvenile survival in the New Zealand southern right whale Eubalaena australis . PLoS ONE, 11, e0146590 10.1371/journal.pone.0146590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, E. L. , Rayment, W. , Alexander, A. , Baker, C. S. , Patenaude, N. J. , Steel, D. J. , … Childerhouse, S. J. (2014). Reestablishment of former wintering grounds by the New Zealand southern right whales. Marine Mammal Science, 30, 206–220. 10.1111/mms.12031 [DOI] [Google Scholar]
- Caye, K. , Deist, T. M. , Martins, H. , Michel, O. , & Francois, O. (2016). TESS3: Fast inference of spatial population structure and genome scans for selection. Molecular Ecology Resources, 16, 540–548. 10.1111/1755-0998.12471 [DOI] [PubMed] [Google Scholar]
- Chiou, K. L. , & Bergey, C. M. (2015). FecalSeq: Methylation‐based enrichment for noninvasive population genomics from feces. bioRxiv, 032870. 10.1101/032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare, E. L. (2014). Molecular detection of trophic interactions: Emerging trends, distinct advantages, significant considerations and conservation applications. Evolutionary Applications, 7(9), 1144–1157. 10.1111/eva.12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutton‐Brock, T. , & Sheldon, B. C. (2010). Individuals and populations: The role of long‐term, individual‐based studies of animals in ecology and evolutionary biology. Trends in Ecology and Evolution, 25(10), 562–573. 10.1016/j.tree.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Constable, J. L. , Ashley, M. V. , Goodall, J. , & Pusey, A. E. (2001). Noninvasive paternity assignment in Gombe chimpanzees. Molecular Ecology, 10(5), 1279–1300. 10.1046/j.1365-294X.2001.01262.x [DOI] [PubMed] [Google Scholar]
- Constantine, R. , Jackson, J. A. , Steel, D. J. , Baker, C. S. , Brooks, L. , Burns, D. , … Garrigue, C. (2012). Abundance of humpback whales in Oceania using photo‐identification and microsatellite genotyping. Marine Ecology Progress Series, 453, 249–261. 10.3354/meps09613 [DOI] [Google Scholar]
- Constantine, R. , Steel, D. J. , Allen, J. , Anderson, M. , Andrews, O. , Baker, C. S. , … Ward, J. (2014). Remote Antarctic feeding ground important for east Australian humpback whales. Marine Biology, 161, 1087–1093. 10.1007/s00227-014-2401-2 [DOI] [Google Scholar]
- Corlett, R. T. (2017). A bigger toolbox: Biotechnology in biodiversity conservation. Trends in Biotechnology, 35, 55–65. 10.1016/j.tibtech.2016.06.009 [DOI] [PubMed] [Google Scholar]
- Cox, D. G. (2006). Quantification of the power of Hardy‐Weinberg equilibrium testing to detect genotyping error. Human Heredity, 61, 10–14. 10.1159/000091787 [DOI] [PubMed] [Google Scholar]
- Creel, S. , Spong, G. , Sands, J. , Rotella, J. , Zeigle, J. , Joe, L. , … Smith, D. (2003). Population size estimation in Yellowstone wolves with error‐prone noninvasive microsatellite genotypes. Molecular Ecology, 12, 2003–2009. 10.1046/j.1365-294X.2003.01868.x [DOI] [PubMed] [Google Scholar]
- Da Silva, A. G. , Lalonde, D. R. , Quse, V. , Shoemaker, A. , & Russello, M. A. (2010). Genetic approaches refine ex situ lowland tapir (Tapirus terrestris) conservation. Journal of Heredity, 101, 581–590. 10.1093/jhered/esq055 [DOI] [PubMed] [Google Scholar]
- Dakin, E. , & Avise, J. C. (2004). Microsatellite null alleles in parentage analysis. Heredity, 93, 504–509. 10.1038/sj.hdy.6800545 [DOI] [PubMed] [Google Scholar]
- Darby, B. J. , Erickson, S. F. , Hervey, S. D. , & Ellis‐Felege, S. N. (2016). Digital fragment analysis of short tandem repeats by high‐throughput amplicon sequencing. Ecology and Evolution, 6(13), 4502–4512. 10.1002/ece3.2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Barba, M. , Miquel, C. , Boyer, F. , Mercier, C. , Rioux, D. , Coissac, E. , & Taberlet, P. (2014). DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: Application to omnivorous diet. Molecular Ecology Resources, 14, 306–323. 10.1111/1755-0998.12188 [DOI] [PubMed] [Google Scholar]
- De Barba, M. , Miquel, C. , Lobréaux, S. , Quenette, P. Y. , Swenson, J. E. , & Taberlet, P. (2017). High‐throughput microsatellite genotyping in ecology: Improved accuracy, efficiency, standardization and success with low‐quantity and degraded DNA. Molecular Ecology Resources, 17, 492–507. 10.1111/1755-0998.12594 [DOI] [PubMed] [Google Scholar]
- De Barba, M. , Waits, L. P. , Garton, E. O. , Genovesi, P. , Randi, E. , Mustoni, A. , & Groff, C. (2010). The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Molecular Ecology, 19(18), 3938–3951. 10.1111/j.1365-294X.2010.04791.x [DOI] [PubMed] [Google Scholar]
- De Barba, M. , Waits, L. P. , Genovesi, P. , Randi, E. , Chirichella, R. , & Cetto, E. (2010). Comparing opportunistic and systematic sampling methods for non‐invasive genetic monitoring of a small translocated brown bear population. Journal of Applied Ecology, 47, 172–181. 10.1111/j.1365-2664.2009.01752.x [DOI] [Google Scholar]
- De Cara, M. Á. R. , Villanueva, B. , Toro, M. Á. , & Fernández, J. (2013). Using genomic tools to maintain diversity and fitness in conservation programmes. Molecular Ecology, 22(24), 6091–6099. 10.1111/mec.12560 [DOI] [PubMed] [Google Scholar]
- De Wit, P. , Pespeni, M. H. , Ladner, J. T. , Barshis, D. J. , Seneca, F. , Jaris, H. , … Palumbi, S. R. (2012). The simple fool's guide to population genomics via RNA‐Seq: An introduction to high‐throughput sequencing data analysis. Molecular Ecology Resources, 12, 1058–1067. 10.1111/1755-0998.12003 [DOI] [PubMed] [Google Scholar]
- Deagle, B. E. , Chiaradia, A. , Mcinnes, J. , & Jarman, S. N. (2010). Pyrosequencing faecal DNA to determine diet of little penguins: Is what goes in what comes out? Conservation Genetics, 11, 2039–2048. 10.1007/s10592-010-0096-6 [DOI] [Google Scholar]
- Delgado, M. L. , Singh, P. , Funk, J. A. , Moore, J. A. , Cannell, E. M. , Kanesfsky, J. , … Scribner, K. T. (2017). Intestinal microbial community dynamics of white‐tailed deer (Odocoileus virginianus) in an agroecosystem. Microbial Ecology, 74, 496–506. 10.1007/s00248-017-0961-7 [DOI] [PubMed] [Google Scholar]
- Delsuc, F. , Metcalf, J. L. , Wegener Parfrey, L. , Song, S. J. , Gonzalez, A. , & Knight, R. (2014). Convergence of gut microbiomes in myrmecophagous mammals. Molecular Ecology, 23, 1301–1317. 10.1111/mec.12501 [DOI] [PubMed] [Google Scholar]
- Demay, S. M. , Becker, P. A. , Rachlow, J. L. , & Waits, L. P. (2017). Genetic monitoring of an endangered species recovery: Demographic and genetic trends for reintroduced pygmy rabbits (Brachylagus idahoensis). Journal of Mammalogy, 98, 350–364. 10.1093/jmammal/gyw197 [DOI] [Google Scholar]
- DeWoody, J. A. , Fernandez, N. B. , Brüniche‐Olsen, A. , Antonides, J. D. , Doyle, J. M. , San Miguel, P. , … Bickham, J. (2017). Characterization of the gray whale (Eschrichtius robustus) genome and a genotyping array based on single nucleotide polymorphisms in candidate genes. Biological Bulletin, 232, 186–197. 10.1086/693483 [DOI] [PubMed] [Google Scholar]
- Dixon, J. D. , Oli, M. K. , Wooten, M. C. , Eason, T. H. , McCown, J. W. , & Paetkau, D. (2006). Effectiveness of a regional corridor in connecting two Florida black bear populations. Conservation Biology, 20(1), 155–162. 10.1111/j.1523-1739.2005.00292.x [DOI] [PubMed] [Google Scholar]
- Doyle, J. M. , Katzner, T. E. , Roemer, G. W. , Cain, J. W. , Millsap, B. A. , McIntyre, C. L. , … DeWoody, J. A. (2016). Genetic structure and viability selection in the golden eagle (Aquila chrysaetos), a vagile raptor with a Holarctic distribution. Conservation Genetics, 17, 1307–1322. 10.1007/s10592-016-0863-0 [DOI] [Google Scholar]
- Druet, T. , Schrooten, C. , & de Roos, A. P. W. (2010). Imputation of genotypes from different single nucleotide polymorphism panels in dairy cattle. Journal of Dairy Science, 93(11), 5443–5454. 10.3168/jds.2010-3255 [DOI] [PubMed] [Google Scholar]
- Dunshea, G. , Duffield, D. , Gales, N. , Hindell, M. , Wells, R. S. , & Jarman, S. N. (2011). Telomeres as age markers in vertebrate molecular ecology. Molecular Ecology Resources, 11(2), 225–235. 10.1111/j.1755-0998.2010.02976.x [DOI] [PubMed] [Google Scholar]
- Edwards, A. , Debbonaire, A. R. , Sattler, B. , Mur, L. A. , & Hodson, A. J. (2016). Extreme metagenomics using nanopore DNA sequencing: A field report from Svalbard, 78 N. bioRxiv, 073965. 10.1101/073965. [DOI] [Google Scholar]
- Efford, M. (2004). Density estimation in live‐trapping studies. Oikos, 106, 598–610. 10.1111/j.0030-1299.2004.13043.x [DOI] [Google Scholar]
- Efford, M. (2011). Estimation of population density by spatially explicit capture – recapture analysis of data from area searches. Ecology, 92, 2202–2207. 10.1890/11-0332.1 [DOI] [PubMed] [Google Scholar]
- Elfström, M. , Davey, M. L. , Zedrosser, A. , Müller, M. , De Barba, M. , Støen, O. G. , … Swenson, J. E. (2014). Do Scandinavian brown bears approach settlements to obtain high‐quality food? Biological Conservation, 178, 128–135. 10.1016/j.biocon.2014.08.003 [DOI] [Google Scholar]
- Elshire, R. J. , Glaubitz, J. C. , Sun, Q. , Poland, J. A. , Kawamoto, K. , Buckler, E. S. , & Mitchell, S. E. (2011). A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE, 6(5), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epps, C. W. , Palsbøll, P. J. , Wehausen, J. D. , Roderick, G. K. , Ramey, R. R. , & McCullough, D. R. (2005). Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecology Letters, 8, 1029–1038. 10.1111/j.1461-0248.2005.00804.x [DOI] [Google Scholar]
- Ernest, H. , Rubin, E. , & Boyce, W. M. (2002). Fecal DNA analysis and risk assessment of mountain lion predation of bighorn sheep. Journal of Wildlife Management, 66, 75–85. 10.2307/3802873 [DOI] [Google Scholar]
- Fabbri, E. , Caniglia, R. , Mucci, N. , Thomsen, H. P. , Krag, K. , Pertoldi, C. , … Randi, E. (2012). Comparison of single nucleotide polymorphisms and microsatellites in non‐invasive genetic monitoring of a wolf population. Archives of Biological Sciences, 64(1), 320–336. [Google Scholar]
- Fairlie, J. , Holland, R. , Pilkington, J. G. , Pemberton, J. M. , Harrington, L. , & Nussey, D. H. (2016). Lifelong leukocyte telomere dynamics and survival in a free‐living mammal. Aging Cell, 15, 140–148. 10.1111/acel.12417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO . (2015). The second report on the state of the world's animal genetic resources for food and agriculture In Scherf B. D. & Pilling D. (Eds.), FAO commission on genetic resources for food and agriculture assessments, Rome, Italy: http://www.fao.org/3/a-i4787e/index.html [Google Scholar]
- Faria, P. J. , Kavembe, G. D. , Jung'a, J. O. , Kimwele, C. N. , Estes, L. D. , Reillo, P. R. , … Bruford, M. W. (2011). The use of non‐invasive molecular techniques to confirm the presence of mountain bongo Tragelaphus eurycerus isaaci populations in Kenya and preliminary inference of their mitochondrial genetic variation. Conservation Genetics, 12, 745–751. 10.1007/s10592-011-0181-5 [DOI] [Google Scholar]
- Farley, S. , Talbot, S. L. , Sage, G. K. , Sinnott, R. , & Coltrane, J. (2014). Use of DNA from bite marks to determine species and individual animals that attack humans. Wildlife Society Bulletin, 38(2), 370–376. 10.1002/wsb.391 [DOI] [Google Scholar]
- Feil, R. , & Fraga, M. F. (2012). Epigenetics and the environment: Emerging patterns and implications. Nature Reviews Genetics, 13, 97–109. 10.1038/nrg3142 [DOI] [PubMed] [Google Scholar]
- Felicetti, L. A. , Schwartz, C. C. , Rye, R. O. , Gunther, K. A. , Crock, J. G. , Haroldson, M. A. , … Robbins, C. T. (2004). Use of naturally occurring mercury to determine the importance of cutthroat trout to Yellowstone grizzly bears. Canadian Journal of Zoology, 82, 493–501. 10.1139/z04-013 [DOI] [Google Scholar]
- Ferreira da Silva, M. J. , Godinho, R. , Casanova, C. , Minhós, T. , Sá, R. , & Bruford, M. W. (2014). Assessing the impact of hunting pressure on population structure of Guinea baboons (Papio papio) in Guinea‐Bissau. Conservation Genetics, 15, 1339–1355. 10.1007/s10592-014-0621-0 [DOI] [Google Scholar]
- Fitak, R. R. , Naidu, A. , Thompson, R. W. , & Culver, M. (2015). A new panel of SNP markers for the individual identification of North American Pumas. Journal of Fish and Wildlife Management, 7(4), 13–27. [Google Scholar]
- Fløjgaard, C. , De Barba, M. , Taberlet, P. , & Ejrnæs, R. (2017). Body condition, diet and ecosystem function of red deer (Cervus elaphus) in a fenced nature reserve. Global Ecology and Conservation, 11, 312–323. 10.1016/j.gecco.2017.07.003 [DOI] [Google Scholar]
- Foote, A. D. , Thomsen, P. F. , Sveegaard, S. , Wahlberg, M. , Kielgast, J. , Kyhn, L. A. … Gilbert, M. T. P. (2012). Investigating the potential use of environmental DNA (eDNA) for genetic monitoring of marine mammals. PLoS ONE 7(8), 2–7. 10.1371/journal.pone.0041781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, M. J. , Hanson, M. B. , Hempelmann, J. A. , Ayres, K. L. , Emmons, C. K. , Schorr, G. S. , … Balcomb‐Bartok, K. (2011). Inferred paternity and male reproductive success in a killer whale (Orcinus orca) population. Journal of Heredity, 102, 537–553. 10.1093/jhered/esr067 [DOI] [PubMed] [Google Scholar]
- Frankham, R. , Bradshaw, C. J. , & Brook, B. W. (2014). Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biological Conservation, 170, 56–63. 10.1016/j.biocon.2013.12.036 [DOI] [Google Scholar]
- Freamo, H. , O'reilly, P. , Berg, P. R. , Lien, S. , & Boulding, E. G. (2011). Outlier SNPs show more genetic structure between two Bay of Fundy metapopulations of Atlantic salmon than do neutral SNPs. Molecular Ecology Resources, 11(Suppl. 1), 254–267. 10.1111/j.1755-0998.2010.02952.x [DOI] [PubMed] [Google Scholar]
- Funk, W. C. , McKay, J. K. , Hohenlohe, P. , & Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends in Ecology & Evolution, 27, 489–496. 10.1016/j.tree.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, S. , Ziaugra, L. , & Tabbaa, D. (2009). SNP genotyping using the sequenom massARRAY iPLEX Platform. Current Protocols in Human Genetics, Supplement 60, 2.12.1–2.12.18. [DOI] [PubMed] [Google Scholar]
- Gagneux, P. , Boesch, C. , & Woodruff, D. S. (1997). Microsatellite scoring errors associated with noninvasive genotyping based on nuclear DNA amplified from shed hair. Molecular Ecology, 6(9), 861–868. 10.1111/j.1365-294X.1997.tb00140.x [DOI] [PubMed] [Google Scholar]
- Gapp, K. , von Ziegler, L. , Tweedie‐Cullen, R. Y. , & Mansuy, I. (2014). Prospects & Overviews: Early life epigenetic programming and transmission of stress‐induced traits in mammals. BioEssays, 36, 491–502. 10.1002/bies.201300116 [DOI] [PubMed] [Google Scholar]
- Garrigue, C. , Constantine, R. , Poole, M. , Hauser, N. , Clapham, P. , Donoghue, M. , … Baker, C. S. (2011). Movement of individual humpback whales between wintering grounds of Oceania (South Pacific), 1999 to 2004. Journal of Cetacean Research and Management, 3, 275–281. [Google Scholar]
- Gautier, M. , Flori, L. , Riebler, A. , Jaffrezic, F. , Laloe, D. , Gut, I. , … Foulley, J. L. (2009). A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics, 10, 550 10.1186/1471-2164-10-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerloff, U. , Hartung, B. , Fruth, B. , Hohmann, G. , & Tautz, D. (1999). Intracommunity relationships, dispersal pattern and paternity success in a wild living community of Bonobos (Pan paniscus) determined from DNA analysis of faecal samples. Proceedings of the Royal Society B: Biological Sciences, 266, 1189–1195. 10.1098/rspb.1999.0762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaubitz, J. C. , Rhodes, E. J. , & DeWoody, J. A. (2003). Prospects for inferring pairwise relatiosnhips with single nucleotide polymorphisms. Molecular Ecology, 12, 1039–1047. 10.1046/j.1365-294X.2003.01790.x [DOI] [PubMed] [Google Scholar]
- Godinho, R. , López‐Bao, J. V. , Castro, D. , Llaneza, L. , Lopes, S. , Silva, P. , & Ferrand, N. (2015). Real‐time assessment of hybridization between wolves and dogs: Combining noninvasive samples with ancestry informative markers. Molecular Ecology Resources, 15(2), 317–328. 10.1111/1755-0998.12313 [DOI] [PubMed] [Google Scholar]
- Gonzalez, E. G. , Blanco, J. C. , Ballesteros, F. , Alcaraz, L. , Palomero, G. , & Doadrio, I. (2016). Genetic and demographic recovery of an isolated population of brown bear Ursus arctos L., 1758. PeerJ, 4, e1928 10.7717/peerj.1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens, B. , Chikhi, L. , Ancrenaz, M. , Lackman‐Ancrenaz, I. , Andau, P. , & Bruford, M. W. (2006). Genetic signature of anthropogenic population collapse in orang‐utans. PLoS Biology, 4(2), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens, B. , Sharma, R. , Othman, N. , Kun‐Rodrigues, C. , Sakong, R. , Ancrenaz, M. , … Chikhi, L. (2016). Habitat fragmentation and genetic diversity in natural populations of the Bornean elephant: Implications for conservation. Biological Conservation, 196, 80–92. 10.1016/j.biocon.2016.02.008 [DOI] [Google Scholar]
- Guillot, G. , Mortier, F. , & Estoup, A. (2005). GENELAND: A computer package for landscape genetics. Molecular Ecology Notes, 5, 712–715. 10.1111/j.1471-8286.2005.01031.x [DOI] [Google Scholar]
- Hall, N. , Mercer, L. , Phillips, D. , Shaw, J. , & Anderson, A. D. (2012). Maximum likelihood estimation of individual inbreeding coefficients and null allele frequencies. Genetics Research, 94, 151–161. 10.1017/S0016672312000341 [DOI] [PubMed] [Google Scholar]
- Hamner, R. M. , Constantine, R. , Mattlin, R. , Waples, R. , & Baker, C. S. (2017). Genotype‐based estimates of local abundance and effective population size for Hector's dolphins. Biological Conservation, 211, 150–160. 10.1016/j.biocon.2017.02.044 [DOI] [Google Scholar]
- Hancock‐Hanser, B. L. , Frey, A. , Leslie, M. S. , Dutton, P. H. , Archer, F. , & Morin, P. (2013). Targeted multiplex next‐generation sequencing: Advances in techniques of mitochondrial and nuclear DNA sequencing for population genomics. Molecular Ecology Resources, 13(2), 254–268. 10.1111/1755-0998.12059 [DOI] [PubMed] [Google Scholar]
- Hans, J. , Haubner, A. , Arandjelovic, M. , Bergl, R. A. , Funfstuck, T. , Gray, M. , … Vigilant, L. (2015). Characterization of MHC class II B polymorphism in multiple populations of wild gorillas using non‐invasive samples and next‐generation sequencing. American Journal of Primatology, 77(11), 1193–1206. 10.1002/ajp.22458 [DOI] [PubMed] [Google Scholar]
- Haroldson, M. , Gunther, K. , Reinhart, D. P. , Podruzny, S. R. , Cegelski, C. , Waits, L. P. , … Smith, J. (2005). Changing numbers of spawning cutthroat trout in tributary streams of Yellowstone Lake and estimates of grizzly bears visiting streams from DNA. Ursus, 16(2), 167–180. 10.2192/1537-6176(2005)016[0167:CNOSCT]2.0.CO;2 [DOI] [Google Scholar]
- Harper, C. K. , Vermeulen, G. J. , Clarke, A. B. , De Wet, J. I. , & Guthrie, A. J. (2013). Extraction of nuclear DNA from rhinoceros horn and characterization of DNA profiling systems for white (Ceratotherium simum) and black (Diceros bicornis) rhinoceros. Forensic Science International: Genetics, 7, 428–433. 10.1016/j.fsigen.2013.04.003 [DOI] [PubMed] [Google Scholar]
- Hauser, L. , Baird, M. , Hilborn, R. , Seeb, L. W. , & Seeb, J. E. (2011). An empirical comparison of SNPs and microsatellites for parentage and kinship assignment in a wild sockeye salmon (Oncorhynchus nerka) population. Molecular Ecology Resources, 11(Suppl. 1), 150–161. 10.1111/j.1755-0998.2010.02961.x [DOI] [PubMed] [Google Scholar]
- Haussmann, M. F. , Vleck, C. M. , & Nisbet, I. C. T. (2003). Calibrating the telomere clock in common terns, Sterna hirundo . Experimental Gerontology, 38, 787–789. 10.1016/S0531-5565(03)00109-8 [DOI] [PubMed] [Google Scholar]
- Hayes, B. J. , Visscher, P. M. , Mcpartlan, H. C. , & Goddard, M. E. (2003). Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Research, 13, 635–643. 10.1101/gr.387103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, J. E. , Ackerman, M. W. , Fryer, J. K. , Hasselman, D. J. , Steele, C. A. , Stephenson, J. J. , … Narum, S. R. (2016). Differential adult migration‐timing and stock‐specific abundance of steelhead in mixed stock assemblages. ICES Journal of Marine Science, 73, 2606–2615. 10.1093/icesjms/fsw138 [DOI] [Google Scholar]
- Hibert, F. , Taberlet, P. , Chave, J. , Scotti‐Saintagne, C. , Sabatier, D. , & Richard‐Hansen, C. (2013). Unveiling the diet of elusive rainforest herbivores in next generation sequencing era? The tapir as a case study. PLoS ONE, 8, e60799 10.1371/journal.pone.0060799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, S. , Arntzen, J. A. , Bruford, M. W. , Godoy, J. A. , Rus Hoelzel, A. , Segelbacher, G. , … Bertorelle, G. (2014). Comparative evaluation of potential indicators and temporal sampling protocols for monitoring genetic erosion. Evolutionary Applications, 7, 984–998. 10.1111/eva.12197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, S. , Hauffe, H. C. , Pérez‐Espona, S. , Arntzen, J. W. , Bertorelle, G. , Bryja, J. , … Bruford, M. W. (2013). Bringing genetic diversity to the forefront of conservation policy and management. Conservation Genetics Resources, 5(2), 593–598. 10.1007/s12686-013-9859-y [DOI] [Google Scholar]
- Hoffman, J. I. , Tucker, R. , Bridgett, S. J. , Clark, M. S. , Forcada, J. , & Slate, J. (2012). Rates of assay success and genotyping error when single nucleotide polymorphism genotyping in non‐model organisms: A case study in the Antarctic fur seal. Molecular Ecology Resources, 12(5), 861–872. 10.1111/j.1755-0998.2012.03158.x [DOI] [PubMed] [Google Scholar]
- Hosking, L. , Lumsden, S. , Lewis, K. , Yeo, A. , Mccarthy, L. , Bansal, A. , … Xu, C. (2004). Detection of genotyping errors by Hardy‐Weinberg equilibrium testing. European Journal of Human Genetics, 12, 395–399. 10.1038/sj.ejhg.5201164 [DOI] [PubMed] [Google Scholar]
- Höss, M. , Kohn, M. , Pääbo, S. , Knauer, F. , & Schröder, W. (1992). Excrement analysis by PCR. Nature, 359, 199 10.1038/359199a0 [DOI] [PubMed] [Google Scholar]
- Jani, A. J. , & Briggs, C. J. (2014). The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proceedings of the National Academy of Sciences of the United States of America, 111(47), E5049–E5058. 10.1073/pnas.1412752111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman, S. N. , McInnes, J. C. , Faux, C. , Polanowski, A. M. , Marthick, J. , Deagle, B. E. , … Emmerson, L. (2013). Adélie penguin population diet monitoring by analysis of food DNA in scats. PLoS ONE, 8, e82227 10.1371/journal.pone.0082227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman, S. N. , Polanowski, A. M. , Faux, C. E. , Robbins, J. , De Paoli‐Iseppi, R. , Bravington, M. , & Deagle, B. E. (2015). Molecular biomarkers for chronological age in animal ecology. Molecular Ecology, 24(19), 4826–4847. 10.1111/mec.13357 [DOI] [PubMed] [Google Scholar]
- Johnson, P. C. D. , & Haydon, D. T. (2007). Maximum‐likelihood estimation of allelic dropout and false allele error rates from microsatellite genotypes in the absence of reference data. Genetics, 175, 827–842. 10.1534/genetics.106.064618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , Pontier, D. , & Dufour, A.‐B. (2009). Genetic markers in the playground of multivariate analysis. Heredity, 102, 330–341. 10.1038/hdy.2008.130 [DOI] [PubMed] [Google Scholar]
- Jones, K. L. , Glenn, T. C. , Lacy, R. C. , Pierce, J. R. , Unruh, N. , Mirande, C. M. , & Chavez‐Ramirez, F. (2002). Refining the Whooping Crane studbook by incorporating microsatellite DNA and leg‐banding analyses. Conservation Biology, 16(3), 789–799. 10.1046/j.1523-1739.2002.00001.x [DOI] [Google Scholar]
- Kalinowski, S. , Taper, M. , & Marshall, T. (2007). Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Molecular Ecology, 9, 801–888. [DOI] [PubMed] [Google Scholar]
- Kampmann, M. L. , Schnell, I. B. , Jensen, R. H. , Axtner, J. , Sander, A. F. , Hansen, A. J. … Wilting, A. (2017). Leeches as a source of mammalian viral DNA and RNA ‐ a study in medicinal leeches. European Journal of Wildlife Research 63, 36 10.1007/s10344-017-1093-6 [DOI] [Google Scholar]
- Karamanlidis, A. A. , Drosopoulou, E. , de Gabriel Hernando, M. , Georgiadis, L. , Krambokoukis, L. , Pllaha, S. , … Scouras, Z. (2010). Noninvasive genetic studies of brown bears using power poles. European Journal of Wildlife Research, 56(5), 693–702. 10.1007/s10344-010-0363-3 [DOI] [Google Scholar]
- Kartzinel, T. R. , Chen, P. A. , Coverdale, T. C. , Erickson, D. L. , Kress, W. J. , Kuzmina, M. L. , … Pringle, R. M. (2015). DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. Proceedings of the National Academy of Sciences of the United States of America, 112, 8019–8024. 10.1073/pnas.1503283112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzner, T. E. , Ivy, J. A. R. , Bragin, E. A. , Milner‐Gulland, E. J. , & DeWoody, J. A. (2011). Conservation implications of inaccurate estimation of cryptic population size. Animal Conservation, 14(4), 328–332. 10.1111/j.1469-1795.2011.00444.x [DOI] [Google Scholar]
- Katzner, T. E. , Nelson, D. M. , Braham, M. A. , Doyle, J. M. , Fernandez, N. B. , Duerr, A. E. , … DeWoody, J. A. (2016). Golden Eagle fatalities and the continental‐scale consequences of local wind‐energy generation. Conservation Biology, 31(2), 406–415. [DOI] [PubMed] [Google Scholar]
- Kendall, K. C. , Stetz, J. B. , Boulanger, J. , Macleod, A. C. , Paetkau, D. , & White, G. C. (2009). Demography and genetic structure of a recovering grizzly bear population. Journal of Wildlife Management, 73, 3–17. 10.2193/2008-330 [DOI] [Google Scholar]
- Kistler, L. , Johnson, S. M. , Irwin, M. T. , Louis, E. E. , Ratan, A. , & Perry, G. H. (2017). A massively parallel strategy for STR marker development, capture, and genotyping. Nucleic Acids Research, 45(15), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein‐Jöbstl, D. , Schornsteiner, E. , Mann, E. , Wagner, M. , Drillich, M. and Schmitz‐Esser, S. (2014). Pyrosequencing reveals diverse fecal microbiota in Simmental calves during early development. Frontiers in Microbiology 5, 622 10.3389/fmicb.2014.00622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelewijn, H. P. , Pérez‐Haro, M. , Jansman, H. A. H. , Boerwinkel, M. C. , Bovenschen, J. , Lammertsma, D. R. , … Kuiters, A. T. (2010). The reintroduction of the Eurasian otter (Lutra lutra) into the Netherlands: Hidden life revealed by noninvasive genetic monitoring. Conservation Genetics, 11, 601–614. 10.1007/s10592-010-0051-6 [DOI] [Google Scholar]
- Kohn, M. , Knauer, F. , Stoffella, A. , Schroder, W. , & Paabo, S. (1995). Conservation genetics of the European brown bear ‐ a study using excremental PCR of nuclear and mitochondrial sequences. Molecular Ecology, 4, 95–103. 10.1111/j.1365-294X.1995.tb00196.x [DOI] [PubMed] [Google Scholar]
- Kohn, M. , & Wayne, R. K. (1997). Facts from feces revisited. Trends in Ecology & Evolution, 12, 223–227. 10.1016/S0169-5347(97)01050-1 [DOI] [PubMed] [Google Scholar]
- Kohn, M. , York, E. C. , Kamradt, D. , Haught, G. , Sauvajot, R. M. , & Wayne, R. K. (1999). Estimating population size by genotyping faeces. Proceedings of the Royal Society B: Biological Sciences, 266, 657–663. 10.1098/rspb.1999.0686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopaliani, N. , Shakarashvili, M. , Gurielidze, Z. , Qurkhuli, T. , & Tarkhnishvili, D. (2014). Gene flow between wolf and shepherd dog populations in Georgia (Caucasus). Journal of Heredity, 105, 345–353. 10.1093/jhered/esu014 [DOI] [PubMed] [Google Scholar]
- Kraus, R. H. S. , vonHoldt, B. , Cocchiararo, B. , Harms, V. , Bayerl, H. , Kühn, R. , … Nowak, C. (2015). A single‐nucleotide polymorphism‐based approach for rapid and cost‐effective genetic wolf monitoring in Europe based on noninvasively collected samples. Molecular Ecology Resources, 15(2), 295–305. 10.1111/1755-0998.12307 [DOI] [PubMed] [Google Scholar]
- Kurtz, J. C. , Jackson, L. E. , & Fisher, W. S. (2001). Strategies for evaluating indicators based on guidelines from the Environmental Protection Agency's Office of Research and Development. Ecological Indicators, 1, 49–60. 10.1016/S1470-160X(01)00004-8 [DOI] [Google Scholar]
- Leroy, G. , Carroll, E. L. , Bruford, M. W. , DeWoody, J. A. , Strand, A. , Waits, L. P. , & Wang, J. (2017). Next‐generation metrics for monitoring genetic erosion within populations of conservation concern. Evolutionary Applications, Online ahead of publication:, 10.1111/eva.12564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Xiang‐Yu, J. , Dai, G. , Gu, Z. , Ming, C. , Yang, Z. , … Zhang, Y.‐P. (2016). Large numbers of vertebrates began rapid population decline in the late 19th century. Proceedings of the National Academy of Sciences of the United States of America, 113, 14079–14084. 10.1073/pnas.1616804113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomniczi, A. , Loche, A. , Castellano, J. M. , Ronnekleiv, O. K. , Bosch, M. , Kaidar, G. , … Ojeda, S. R. (2013). Epigenetic control of female puberty. Nature Neuroscience, 16, 281–289. 10.1038/nn.3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsinger, R. C. , Gese, E. M. , Bailey, L. L. , & Waits, L. P. (2017). The roles of habitat and intraguild predation by coyotes on the spatial dynamics of kit foxes. Ecosphere, 8, e01749 10.1002/ecs2.1749 [DOI] [Google Scholar]
- Lonsinger, R. C. , Gese, E. M. , Dempsey, S. J. , & Kluever, B. M. (2015). Balancing sample accumulation and DNA degradation rates to optimize noninvasive genetic sampling of sympatric carnivores. Molecular Ecology Resources, 15, 831–842. 10.1111/1755-0998.12356 [DOI] [PubMed] [Google Scholar]
- Lopes, C. M. , De Barba, M. , Boyer, F. , Mercier, C. , da Silva Filho, P. J. S. , Heidtmann, L. M. , … Taberlet, P. (2015). DNA metabarcoding diet analysis for species with parapatric vs sympatric distribution: A case study on subterranean rodents. Heredity, 114(5), 525–536. 10.1038/hdy.2014.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudon, A. H. , Holland, J. A. , Umile, T. P. , Burzynski, E. A. , Minbiole, K. P. C. , & Harris, R. N. (2014). Interactions between amphibians’ symbiotic bacteria cause the production of emergent anti‐fungal metabolites. Frontiers in Microbiology, 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchini, V. , Fabbri, E. , Marucco, F. , Ricci, S. , Boitani, L. , & Randi, E. (2002). Noninvasive molecular tracking of colonizing wolf (Canis lupus) packs in the western Italian Alps. Molecular Ecology, 11, 857–868. 10.1046/j.1365-294X.2002.01489.x [DOI] [PubMed] [Google Scholar]
- Lukacs, P. M. , & Burnham, K. P. (2005). Review of capture–recapture methods applicable to noninvasive genetic sampling. Molecular Ecology, 14, 3909–3919. 10.1111/j.1365-294X.2005.02717.x [DOI] [PubMed] [Google Scholar]
- Lukoshek, V. , Funahashi, N. , Lavery, S. , Dalebout, M. , Cipriano, F. , & Baker, C. S. (2009). High proportion of protected minke whales sold on Japanese markets due to illegal, unreported or unregulated exploitation. Animal Conservation, 12, 385–395. 10.1111/j.1469-1795.2009.00302.x [DOI] [Google Scholar]
- Mächler, E. , Deiner, K. , Steinmann, P. , Altermatt, F. , Mächler, E. , Deiner, K. … Altermatt, F. (2014). Utility of environmental DNA for monitoring rare and indicator macroinvertebrate species. Freshwater Science 33, 1174–1183. 10.1086/678128 [DOI] [Google Scholar]
- MacKenzie, D. D. , Nichols, J. D. , Hines, J. E. , Knutson, M. G. , & Franklin, A. B. (2003). Estimating site occupancy, colonization, and local extinction when a species is detected imperfectly. Ecology, 84, 2200–2207. 10.1890/02-3090 [DOI] [Google Scholar]
- Marucco, F. , Avanzinelli, E. , & Boitani, L. (2012). Non‐invasive integrated sampling design to monitor the wolf population in Piemonte, Italian Alps. Hystrix, 23, 5–13. [Google Scholar]
- Marucco, F. , Pletscher, D. H. , Boitani, L. , Schwartz, M. K. , Pilgrim, K. L. , & Lebreton, J. D. (2009). Wolf survival and population trend using non‐invasive capture‐recapture techniques in the Western Alps. Journal of Applied Ecology, 46(5), 1003–1010. 10.1111/j.1365-2664.2009.01696.x [DOI] [Google Scholar]
- Matala, A. P. , Hatch, D. R. , Everett, S. , Ackerman, M. W. , Bowersox, B. , Campbell, M. , & Narum, S. (2016). What goes up does not come down: The stock composition and demographic characteristics of upstream migrating steelhead differ from post‐spawn emigrating kelts. ICES Journal of Marine Science, 73, 2595–2605. 10.1093/icesjms/fsw109 [DOI] [Google Scholar]
- McCarthy, T. M. , Waits, L. P. , & Mijiddorj, B. (2009). Status of the Gobi bear in Mongolia as determined by noninvasive genetic methods. Ursus, 20(1), 30–38. 10.2192/07GR013R.1 [DOI] [Google Scholar]
- McClintock, B. T. , White, G. C. , Antolin, M. F. , & Tripp, D. W. (2009). Estimating abundance using mark‐resight when sampling is with replacement or the number of marked individuals is unknown. Biometrics, 65, 237–246. 10.1111/j.1541-0420.2008.01047.x [DOI] [PubMed] [Google Scholar]
- McClure, M. , Mullen, M. P. , & Kearney, J. F. (2014). Application of a custom SNP chip: Microsatellite imputation, parentage SNP imputation, genomic evaluations, and across‐breed nation‐wide genetic disease prevalence with the International Beef and Dairy SNP chip. In ICAR/Interbull meeting, Berlin, Germany. May 2014 (pp. 1–13).
- McClure, M. , Sonstegard, T. , Wiggans, G. , & Van Tassell, C. P. (2012). Imputation of microsatellite alleles from dense SNP genotypes for parental verification. Frontiers in Genetics, 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke, S. , Meier, M. , & Sommer, S. (2015). Shifts in the gut microbiome observed in wildlife faecal samples exposed to natural weather conditions: Lessons from time‐series analyses using next‐generation sequencing for application in field studies. Methods in Ecology and Evolution, 6(9), 1080–1087. 10.1111/2041-210X.12394 [DOI] [Google Scholar]
- Mesnick, S. L. , Taylor, B. L. , Archer, F. I. , Martien, K. K. , Treviño, S. E. , Hancock‐Hanser, B. L. , … Morin, P. (2011). Sperm whale population structure in the eastern and central North Pacific inferred by the use of single‐nucleotide polymorphisms, microsatellites and mitochondrial DNA. Molecular Ecology Resources, 11(Suppl. 1), 278–298. 10.1111/j.1755-0998.2010.02973.x [DOI] [PubMed] [Google Scholar]
- Miller, C. R. , Joyce, P. , & Waits, L. P. (2005). A new method for estimating the size of small populations from genetic mark‐recapture data. Molecular Ecology, 14(7), 1991–2005. 10.1111/j.1365-294X.2005.02577.x [DOI] [PubMed] [Google Scholar]
- Miller, C. R. , & Waits, L. P. (2003). The history of effective population size and genetic diversity in the Yellowstone grizzly (Ursus arctos): Implications for conservation. Proceedings of the National Academy of Sciences of the United States of America, 100, 4334–4339. 10.1073/pnas.0735531100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhos, T. , Chikhi, L. , Sousa, C. , Vicente, L. M. , Ferreira da Silva, M. , Heller, R. , … Bruford, M. W. (2016). Genetic consequences of human forest exploitation in two colobus monkeys in Guinea Bissau. Biological Conservation, 194, 194–208. 10.1016/j.biocon.2015.12.019 [DOI] [Google Scholar]
- Miquel, C. , Bellemain, E. , Poillot, C. , Bessière, J. , Durand, A. , & Taberlet, P. (2006). Quality indexes to assess the reliability of genotypes in studies using noninvasive sampling and multiple‐tube approach. Molecular Ecology Notes, 6(4), 985–988. 10.1111/j.1471-8286.2006.01413.x [DOI] [Google Scholar]
- Mollet, P. , Kéry, M. , Gardner, B. , Pasinelli, G. , & Royle, J. A. (2015). Estimating population size for capercaillie (Tetrao urogallus L.) with spatial capture‐recapture models based on genotypes from one field sample. PLoS ONE, 10, e0129020 10.1371/journal.pone.0129020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzón, J. , Kays, R. , & Dykhuizen, D. (2014). Assessment of coyote‐wolf‐dog admixture using ancestry‐informative diagnostic SNPs. Molecular Ecology, 5, 182–197. 10.1111/mec.12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin, P. , Foote, A. , Hill, C. , Simon‐bouhet, B. , Lang, A. , & Louis, M. (2018). SNP Discovery from single and multiplex genome assemblies of non‐model organisms. Next Generation Sequencing, 1712, 113–144. 10.1007/978-1-4939-7514-3 [DOI] [PubMed] [Google Scholar]
- Morin, P. , Luikart, G. , & Wayne, R. K. (2004). SNPs in ecology, evolution and conservation. Trends in Ecology & Evolution, 19, 208–216. 10.1016/j.tree.2004.01.009 [DOI] [Google Scholar]
- Morin, P. , Martien, K. , Archer, F. , Cipriano, F. , Steel, D. J. , Jackson, J. A. , & Taylor, B. L. (2010). Applied conservation genetics and the need for quality control and reporting of genetic data used in fisheries and wildlife management. Journal of Heredity, 101, 1–10. 10.1093/jhered/esp107 [DOI] [PubMed] [Google Scholar]
- Morin, P. , & Mccarthy, M. (2007). Highly accurate SNP genotyping from historical and low‐quality samples. Molecular Ecology Notes, 7(6), 937–946. 10.1111/j.1471-8286.2007.01804.x [DOI] [Google Scholar]
- Morin, P. , Moore, J. J. , Chakraborty, R. , Jin, L. , Goodall, J. , & Woodruff, D. S. (1994). Kin selection, social structure, gene flow, and the evolution of chimpanzees. Science, 265, 1193–1201. 10.1126/science.7915048 [DOI] [PubMed] [Google Scholar]
- Morin, P. , & Woodruff, D. (1992). Paternity exclusion using multiple hypervariable microsatellite loci amplified from nuclear DNA of hair cells In Martin R., Dixson A., & Wickings E. (Eds.), Paternity in Primates: Genetic tests and theories (pp. 63–81). Basel: Karger. [Google Scholar]
- Mowat, G. , & Strobeck, C. (2000). Estimating population size of grizzly bears using hair capture, DNA profiling, and mark‐ recapture analysis. Journal of Wildlife Management, 64, 183–193. 10.2307/3802989 [DOI] [Google Scholar]
- Mwale, M. , Dalton, D. L. , Jansen, R. , De Bruyn, M. , Pietersen, D. , Mokgokong, P. S. , & Kotzé, A. (2017). Forensic application of DNA barcoding for identification of illegally traded African pangolin scales. Genome, 60, 272–284. 10.1139/gen-2016-0144 [DOI] [PubMed] [Google Scholar]
- Nater, A. , Arora, N. , Greminger, M. P. , Van Schaik, C. P. , Singleton, I. , Wich, S. A. , … Krützen, M. (2013). Marked population structure and recent migration in the critically endangered sumatran orangutan (Pongo abelii). Journal of Heredity, 104, 2–13. 10.1093/jhered/ess065 [DOI] [PubMed] [Google Scholar]
- Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America, 70(12), 3321–3323. 10.1073/pnas.70.12.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, E. E. , Cariani, A. , Mac Aoidh, E. , Maes, G. E. , Milano, I. , Ogden, R. , … Carvalho, G. R. (2012). Gene‐associated markers provide tools for tackling illegal fishing and false eco‐certification. Nature Communications, 3, 851 10.1038/ncomms1845 [DOI] [PubMed] [Google Scholar]
- Nielsen, R. , Paul, J. , Alberechtsen, A. , & Song, Y. (2011). Genotype and SNP calling from next‐generation sequencing data. Nature Reviews Genetics, 12, 443–452. 10.1038/nrg2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, A. J. , & Spong, G. (2015). Single nucleotide polymorphism‐based dispersal estimates using noninvasive sampling. Ecology and Evolution, 5(15), 3056–3065. 10.1002/ece3.1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, A. J. , Stronen, A. V. , Fuglstad, G. A. , Ruiz‐Gonzalez, A. , Kindberg, J. , Street, N. R. , & Spong, G. (2017). Landscape relatedness: Detecting contemporary fine‐scale spatial structure in wild populations. Landscape Ecology, 32, 181–194. 10.1007/s10980-016-0434-2 [DOI] [Google Scholar]
- Nussberger, B. , Wandeler, P. , & Camenisch, G. (2014). A SNP chip to detect introgression in wildcats allows accurate genotyping of single hairs. European Journal of Wildlife Research, 60(2), 405–410. 10.1007/s10344-014-0806-3 [DOI] [Google Scholar]
- Ogden, R. , & Linacre, A. (2015). Wildlife forensic science: A review of genetic geographic origin assignment. Forensic Science International: Genetics, 18, 152–159. 10.1016/j.fsigen.2015.02.008 [DOI] [PubMed] [Google Scholar]
- Oliveira, R. , Randi, E. , Mattucci, F. , Kurushima, J. D. , Lyons, L. , & Alves, P. C. (2015). Toward a genome‐wide approach for detecting hybrids: Informative SNPs to detect introgression between domestic cats and European wildcats (Felis silvestris). Heredity, 115, 195–205. 10.1038/hdy.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rorke, R. , Cobian, G. M. , Holland, B. S. , Price, M. R. , Costello, V. , & Amend, A. S. (2015). Dining local: The microbial diet of a snail that grazes microbial communities is geographically structured. Environmental Microbiology, 17, 1753–1764. 10.1111/1462-2920.12630 [DOI] [PubMed] [Google Scholar]
- Paetkau, D. (2003). An empirical exploration of data quality in DNA‐based population inventories. Molecular Ecology, 12, 1375–1387. 10.1046/j.1365-294X.2003.01820.x [DOI] [PubMed] [Google Scholar]
- Palomares, F. , Godoy, J. A. , Piriz, A. , O'Brien, S. J. , & Johnson, W. E. (2002). Faecal genetic analysis to determine the presence and distribution of elusive carnivores: Design and feasibility for the Iberian lynx. Molecular Ecology, 11, 2171–2182. 10.1046/j.1365-294X.2002.01608.x [DOI] [PubMed] [Google Scholar]
- Palsbøll, P. J. , Allen, J. , Bérubé, M. , Clapham, P. J. , Feddersen, T. P. , Hammond, P. S. , … Oien, N. (1997). Genetic tagging of humpback whales. Nature, 388, 767–769. 10.1038/42005 [DOI] [PubMed] [Google Scholar]
- Palsbøll, P. J. , Peery, M. Z. , Olsen, M. T. , Beissinger, S. R. , & Bérubé, M. (2013). Inferring recent historic abundance from current genetic diversity. Molecular Ecology, 22, 22–40. 10.1111/mec.12094 [DOI] [PubMed] [Google Scholar]
- Pedersen, M. W. , Ruter, A. , Schweger, C. , Friebe, H. , Staff, R. A. , Kjeldsen, K. K. … Willerslev, E. (2016). Postglacial viability and colonization in North America's ice‐free corridor. Nature 537, 45–49. 10.1038/nature19085 [DOI] [PubMed] [Google Scholar]
- Perry, G. , Marioni, J. , Melsted, P. , & Gilad, Y. (2010). Genomic‐scale capture and sequencing of endogenous DNA from feces. Molecular Ecology, 19, 5332–5344. 10.1111/j.1365-294X.2010.04888.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J. M. , Queller, D. C. , Imperatriz‐Fonseca, V. L. , Roubik, D. W. , & Strassmann, J. E. (1999). Mate number, kin selection and social conflicts in stingless bees and honeybees. Proceedings of the Royal Society B: Biological Sciences, 266(1417), 379 10.1098/rspb.1999.0648 [DOI] [Google Scholar]
- Petit, E. , & Valière, N. (2006). Estimating population size with noninvasive capture‐mark‐recapture data. Conservation Biology, 20(4), 1062–1073. 10.1111/j.1523-1739.2006.00417.x [DOI] [PubMed] [Google Scholar]
- Pimm, S. L. , Jenkins, C. N. , Abell, R. , Brooks, T. M. , Gittleman, J. L. , Joppa, L. N. , … Sexton, J. O. (2014). The biodiversity of species and their rates of extinction, distribution, and protection. Science, 344, 987. [DOI] [PubMed] [Google Scholar]
- Polanowski, A. M. , Robbins, J. , Chandler, D. , & Jarman, S. N. (2014). Epigenetic estimation of age in humpback whales. Molecular Ecology Resources, 14, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompanon, F. , Bonin, A. , Bellemain, E. , & Taberlet, P. (2005). Genotyping errors: Causes, consequences and solutions. Nature Reviews Genetics, 6, 847–859. 10.1038/nrg1707 [DOI] [PubMed] [Google Scholar]
- Pompanon, F. , Deagle, B. E. , Symondson, W. O. C. , Brown, D. S. , Jarman, S. N. , & Taberlet, P. (2012). Who is eating what: Diet assessment using next generation sequencing. Molecular Ecology, 21, 1931–1950. 10.1111/j.1365-294X.2011.05403.x [DOI] [PubMed] [Google Scholar]
- Price, M. R. , O'Rorke, R. , Amend, A. S. , & Hadfield, M. G. (2017). Diet selection at three spatial scales: Implications for conservation of an endangered Hawaiian tree snail. Biotropica, 49, 130–136. 10.1111/btp.12339 [DOI] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, M. F. , Mclellan, B. N. , Strobeck, C. , & Barclay, R. M. R. (2005). Genetic analysis reveals demographic fragmentation of grizzly bears yielding vulnerably small populations. Proceedings of the Royal Society B: Biological Sciences, 272, 2409–2416. 10.1098/rspb.2005.3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, M. F. , Paetkau, D. , McLellan, B. N. , Stenhouse, G. B. , Kendall, K. C. , MacE, R. D. , … Strobeck, C. (2012). Population fragmentation and inter‐ecosystem movements of grizzly bears in Western Canada and the Northern United States. Wildlife Monographs, 180, 1–46. 10.1002/wmon.6 [DOI] [Google Scholar]
- Quéméré, E. , Hibert, F. , Miquel, C. , Lhuillier, E. , Rasolondraibe, E. , Champeau, J. , … Chikhi, L. (2013). A DNA metabarcoding study of a primate dietary diversity and plasticity across its entire fragmented range. PLoS ONE, 8, e58971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick, J. , Loman, N. J. , Duraffour, S. , Simpson, J. T. , Severi, E. , Cowley, L. , … Carroll, M. W. (2016). Real‐time, portable genome sequencing for Ebola surveillance. Nature, 530, 228–232. 10.1038/nature16996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raverty, S. A. , Rhodes, L. D. , Zabek, E. , Eshghi, A. , Cameron, C. E. , Hanson, M. B. , & Schroeder, J. P. (2017). Respiratory microbiome of endangered southern resident killer whales and microbiota of surrounding sea surface microlayer in the Eastern North Pacific. Scientific Reports, 7, 394 10.1038/s41598-017-00457-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads, A. , & Au, K. F. (2015). PacBio sequencing and its applications. Genomics, Proteomics and Bioinformatics, 13, 278–289. 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringler, E. , Mangione, R. , & Ringler, M. (2015). Where have all the tadpoles gone? Individual genetic tracking of amphibian larvae until adulthood. Molecular Ecology Resources, 15(4), 737–746. 10.1111/1755-0998.12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romain‐Bondi, K. A. , Wielgus, R. B. , Waits, L. P. , Kasworm, W. F. , Austin, M. , & Wakkinen, W. (2004). Density and population size estimates for North Cascade grizzly bears using DNA hair‐sampling techniques. Biological Conservation, 117(4), 417–428. 10.1016/j.biocon.2003.07.005 [DOI] [Google Scholar]
- Rönn, A. , Andrés, O. , López‐Giráldez, F. , Johnsson‐Glans, C. , Verschoor, E. , Domingo‐Roura, X. … Bosch, M. (2009). First generation microarray‐system for identification of primate species subject to bushmeat trade. Endangered Species Research 9, 133–142. 10.3354/esr00191 [DOI] [Google Scholar]
- Royle, J. A. , & Young, A. (2008). A hierarchical model for spatial capture–recapture data. Ecology, 89, 2281–2289. 10.1890/07-0601.1 [DOI] [PubMed] [Google Scholar]
- Rudnick, J. A. , Katzner, T. E. , Bragin, E. A. , & DeWoody, J. A. (2008). A non‐invasive genetic evaluation of population size, natal philpatry, and roosting behaviour of non‐breeding eastern imperial eagles (Aquila heliaca) in central Asia. Conservation Genetics, 9, 667–676. 10.1007/s10592-007-9397-9 [DOI] [Google Scholar]
- Rudnick, J. A. , Katzner, T. E. , Bragin, E. A. , Rhodes, O. E. , & DeWoody, J. A. (2005). Using naturally shed feathers for individual identification, genetic parentage analyses, and population monitoring in an endangered Eastern imperial eagle (Aquila heliaca) population from Kazakhstan. Molecular Ecology, 14, 2959–2967. 10.1111/j.1365-294X.2005.02641.x [DOI] [PubMed] [Google Scholar]
- Ruegg, K. C. , Anderson, E. C. , Paxton, K. L. , Apkenas, V. , Lao, S. , Siegel, R. B. , … Smith, T. B. (2014). Mapping migration in a songbird using high‐resolution genetic markers. Molecular Ecology, 23(23), 5726–5739. 10.1111/mec.12977 [DOI] [PubMed] [Google Scholar]
- Russell, R. E. , Royle, J. A. , Desimone, R. , Schwartz, M. K. , Edwards, V. , Pilgrim, K. , & McKelvey, K. S. (2012). Estimating abundance of mountain lions from unstructured spatial sampling. The Journal of Wildlife Management, 76, 1551–1561. 10.1002/jwmg.412 [DOI] [Google Scholar]
- Russello, M. A. , Waterhouse, M. D. , Etter, P. D. , & Johnson, E. A. (2015). From promise to practice: Pairing non‐invasive sampling with genomics in conservation. PeerJ, 3, e1106 10.7717/peerj.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell, I. B. , Sollmann, R. , Calvignac‐Spencer, S. , Siddall, M. E. , Yu, D. W. , Wilting, A. , & Gilbert, M. T. P. (2015). iDNA from terrestrial haematophagous leeches as a wildlife surveying and monitoring tool – prospects, pitfalls and avenues to be developed. Frontiers in Zoology, 12(1), 24 10.1186/s12983-015-0115-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunck, B. , Kraft, W. , & Truyen, U. (1995). A simple touch‐down polymerase chain reaction for the detection of canine parvovirus and feline panleukopenia virus in feces. Journal of Virological Methods, 55, 427–433. 10.1016/0166-0934(95)00069-3 [DOI] [PubMed] [Google Scholar]
- Schwartz, M. K. , Luikart, G. , & Waples, R. S. (2007). Genetic monitoring as a promising tool for conservation and management. Trends in Ecology and Evolution, 22, 25–34. 10.1016/j.tree.2006.08.009 [DOI] [PubMed] [Google Scholar]
- Shehzad, W. , Riaz, T. , Nawaz, M. A. , Miquel, C. , Poillot, C. , Shah, S. A. , … Taberlet, P. (2012). Carnivore diet analysis based on next‐generation sequencing: Application to the leopard cat (Prionailurus bengalensis) in Pakistan. Molecular Ecology, 21, 1951–1965. 10.1111/j.1365-294X.2011.05424.x [DOI] [PubMed] [Google Scholar]
- Sigsgaard, E. E. , Nielsen, I. B. , Bach, S. S. , Lorenzen, E. D. , Robinson, D. P. , Knudsen, S. W. , … Thomsen, P. F. (2016). Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nature Ecology & Evolution, 1, 4 10.1038/s41559-016-0004 [DOI] [PubMed] [Google Scholar]
- Smith, O. , & Wang, J. (2014). When can noninvasive samples provide sufficient information in conservation genetics studies? Molecular Ecology Resources, 14(5), 1011–1023. [DOI] [PubMed] [Google Scholar]
- Snyder‐Mackler, N. , Majoros, W. H. , Yuan, M. L. , Shaver, A. O. , Gordon, J. B. , Kopp, G. H. , … Tung, J. (2016). Efficient genome‐wide sequencing and low‐coverage pedigree analysis from noninvasively collected samples. Genetics, 203(2), 699–714. 10.1534/genetics.116.187492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel, E. , Papp, J. C. , & Lange, K. (2002). Detection and integration of genotyping errors in statistical genetics. American Journal of Human Genetics, 70, 496–508. 10.1086/338920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg, K. H. , Bellemain, E. , Drageset, O.‐M. M. , Taberlet, P. , & Swenson, J. E. (2006). An evaluation of field and non‐invasive genetic methods to estimate brown bear (Ursus arctos) population size. Biological Conservation, 128(2), 158–168. 10.1016/j.biocon.2005.09.025 [DOI] [Google Scholar]
- Spitzer, R. , Norman, A. J. , Schneider, M. , & Spong, G. (2016). Estimating population size using single‐nucleotide polymorphism‐based pedigree data. Ecology and Evolution, 6(10), 3174–3184. 10.1002/ece3.2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurgin, L. G. , Wright, D. J. , van der Velde, M. , Collar, N. J. , Komdeur, J. , Burke, T. , & Richardson, D. S. (2014). Museum DNA reveals the demographic history of the endangered Seychelles warbler. Evolutionary Applications, 7, 1134–1143. 10.1111/eva.12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivathsan, A. , Ang, A. , Vogler, A. P. , & Meier, R. (2016). Fecal metagenomics for the simultaneous assessment of diet, parasites, and population genetics of an understudied primate. Frontiers in Zoology, 13(1), 17 10.1186/s12983-016-0150-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats, M. , Arulandhu, A. J. , Gravendeel, B. , Holst‐Jensen, A. , Scholtens, I. , Peelen, T. , … Kok, E. (2016). Advances in DNA metabarcoding for food and wildlife forensic species identification. Analytical and Bioanalytical Chemistry, 408, 4615–4630. 10.1007/s00216-016-9595-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton, D. W. G. , Hart, J. , Vosper, A. , Kumpel, N. F. , Wang, J. , Ewen, J. G. , & Bruford, M. W. (2016). Non‐invasive genetic identification confirms the presence of the Endangered okapi Okapia johnstoni south‐west of the Congo River. Oryx, 50, 134–137. 10.1017/S0030605314000593 [DOI] [Google Scholar]
- Stenglein, J. L. , De Barba, M. , Ausband, D. E. and Waits, L. P. (2010) Impacts of sampling location within a faeces on DNA quality in two carnivore species. Molecular Ecology Resources 10, 109–114. 10.1111/j.1755-0998.2009.02670.x [DOI] [PubMed] [Google Scholar]
- Stenglein, J. L. , Waits, L. P. , Ausband, D. E. , Zager, P. , & Mack, C. M. (2010). Efficient, noninvasive genetic sampling for monitoring reintroduced wolves. Journal of Wildlife Management, 74(5), 1050–1058. 10.2193/2009-305 [DOI] [Google Scholar]
- Stenglein, J. L. , Waits, L. P. , Ausband, D. E. , Zager, P. , & Mack, C. M. (2011). Estimating gray wolf pack size and family relationships using noninvasive genetic sampling at rendezvous sites. Journal of Mammalogy, 92(4), 784–795. 10.1644/10-MAMM-A-200.1 [DOI] [Google Scholar]
- Stetz, J. B. , Kendall, K. C. , Vojta, C. D. , & Genetic Monitoring (GeM) Working Group (2011). Genetic monitoring for managers: A new online resource. Journal of Fish and Wildlife Management, 2, 216–219. 10.3996/082011-JFWM-048 [DOI] [Google Scholar]
- Stetz, J. B. , Smith, S. , Sawaya, M. A. , Ramsey, A. B. , Amish, S. J. , Schwartz, M. K. , & Luikart, G. (2016). Discovery of 20,000 RAD‐SNPs and development of a 52‐SNP array for monitoring river otters. Conservation Genetics Resources, 8(3), 299–302. 10.1007/s12686-016-0558-3 [DOI] [Google Scholar]
- Steyer, K. , Kraus, R. H. S. , Mölich, T. , Anders, O. , Cocchiararo, B. , Frosch, C. , … Nowak, C. (2016). Large‐scale genetic census of an elusive carnivore, the European wildcat (Felis s. silvestris). Conservation Genetics, 17, 1183–1199. 10.1007/s10592-016-0853-2 [DOI] [Google Scholar]
- Suez, M. , Behdenna, A. , Brouillet, S. , Graça, P. , Higuet, D. , & Achaz, G. (2016). MicNeSs: Genotyping microsatellite loci from a collection of (NGS) reads. Molecular Ecology Resources, 16(2), 524–533. 10.1111/1755-0998.12467 [DOI] [PubMed] [Google Scholar]
- Taberlet, P. , & Bouvet, J. (1992). Génétique de l'Ours brun des Pyrenees (Ursus arctos): Premiers résultats. Comptes Rendus de l'Académie Des Sciences, 314, 15–21. [Google Scholar]
- Taberlet, P. , Camarra, J. J. , Griffin, S. , Uhrés, E. , Hanotte, O. , Waits, L. P. , … Bouvet, J. (1997). Noninvasive genetic tracking of the endangered Pyrenean brown bear population. Molecular Ecology, 6, 869–876. 10.1111/j.1365-294X.1997.tb00141.x [DOI] [PubMed] [Google Scholar]
- Taberlet, P. , & Fumagalli, L. (1996). Owl pellets as a source of DNA for genetic studies of small mammals. Molecular Ecology, 5(2), 301–305. 10.1111/j.1365-294X.1996.tb00318.x [DOI] [PubMed] [Google Scholar]
- Taberlet, P. , Griffin, S. , Goossens, B. , Questiau, S. , Manceau, V. , Escaravage, N. , … Bouvet, J. (1996). Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Research, 24(16), 3189–3194. 10.1093/nar/24.16.3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P. , & Luikart, G. (1999). Non‐invasive genetic sampling and individual identification. Biological Journal of the Linnean Society, 68, 41–55. 10.1111/j.1095-8312.1999.tb01157.x [DOI] [Google Scholar]
- Taberlet, P. , Mattock, H. , Dubois‐Paganon, C. , & Bouvet, J. (1993). Sexing free‐ranging brown bears Ursus arctos using hairs found in the field. Molecular Ecology, 2(6), 399–403. 10.1111/j.1365-294X.1993.tb00033.x [DOI] [PubMed] [Google Scholar]
- Taylor, H. R. (2015). The use and abuse of genetic marker‐based estimates of relatedness and inbreeding. Ecology and Evolution, 5(15), 3140–3150. 10.1002/ece3.1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teisberg, J. E. , Haroldson, M. A. , Schwartz, C. C. , Gunther, K. A. , Fortin, J. K. , & Robbins, C. T. (2014). Contrasting past and current numbers of bears visiting Yellowstone cutthroat trout streams. Journal of Wildlife Management, 78(2), 369–378. 10.1002/jwmg.667 [DOI] [Google Scholar]
- Tenan, S. , Iemma, A. , Bragalanti, N. , Pedrini, P. , De Barba, M. , Randi, E. , … Genovart, M. (2016). Evaluating mortality rates with a novel integrated framework for nonmonogamous species. Conservation Biology, 30, 1307–1319. 10.1111/cobi.12736 [DOI] [PubMed] [Google Scholar]
- Thomas, A. C. , Deagle, B. E. , Eveson, J. P. , Harsch, C. H. , & Trites, A. W. (2016). Quantitative DNA metabarcoding: Improved estimates of species proportional biomass using correction factors derived from control material. Molecular Ecology Resources, 16, 714–726. 10.1111/1755-0998.12490 [DOI] [PubMed] [Google Scholar]
- Thomas, A. C. , Jarman, S. N. , Haman, K. H. , Trites, A. W. , & Deagle, B. E. (2013). Improving accuracy of DNA diet estimates using food tissue control materials and an evaluation of proxies for digestion bias. Molecular Ecology, 23, 3706–3718. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/24102760 [DOI] [PubMed] [Google Scholar]
- Thompson, C. , Royle, J. A. , & Garner, J. (2012). A framework for inference about carnivore density from unstructured spatial sampling of scat using detector dogs. The Journal of Wildlife Management, 76, 863–871. 10.1002/jwmg.317 [DOI] [Google Scholar]
- Tumendemberel, O. , Proctor, M. , Reynolds, H. , Boulanger, J. , Luvsamjamba, A. , Tserenbataa, T. , … Paetkau, D. (2015). Gobi bear abundance and inter‐oases movements, Gobi Desert. Mongolia. Ursus, 26(2), 129–142. 10.2192/URSUS-D-15-00001.1 [DOI] [Google Scholar]
- Tung, J. , Barreiro, L. B. , Burns, M. B. , Grenier, J. C. , Lynch, J. , Grieneisen, L. E. , … Archie, E. A. (2015). Social networks predict gut microbiome composition in wild baboons. ELife, 2015(4), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations Environment Programme Convention on Biological Diversity SBSTTA . (2003). Monitoring and indicators: Designing national‐level monitoring programmes and indicators. Montreal, Canada: United Nations. [Google Scholar]
- Valentini, A. , Miquel, C. , Nawaz, M. A. , Bellemain, E. , Coissac, E. , Pompanon, F. , … Taberlet, P. (2009). New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: The trnL approach. Molecular Ecology Resources, 9(1), 51–60. 10.1111/j.1755-0998.2008.02352.x [DOI] [PubMed] [Google Scholar]
- Valière, N. , Fumagalli, L. , Gielly, L. , Miquel, C. , Lequette, B. , Poulle, M. L. , … Taberlet, P. (2003). Long‐distance wolf recolonization of France and Switzerland inferred from non‐invasive genetic sampling over a period of 10 years. Animal Conservation, 6, 83–92. 10.1017/S1367943003003111 [DOI] [Google Scholar]
- Vartia, S. , Villanueva‐Cañas, J. L. , Finarelli, J. , Farrell, E. D. , Collins, P. C. , Hughes, G. M. , … Carlsson, J. (2016). A novel method of microsatellite genotyping‐by‐sequencing using individual combinatorial barcoding. Royal Society Open Science, 3(1), 150565 10.1098/rsos.150565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, F. , Albrechtsen, A. , & Nielsen, R. (2016). Estimating IBD tracts from low coverage NGS data. Bioinformatics, 32, 2096–2102. 10.1093/bioinformatics/btw212 [DOI] [PubMed] [Google Scholar]
- Vieira, F. , Fumagalli, M. , Albrechtsen, A. , & Nielsen, R. (2013). Estimating inbreeding coefficients from NGS data: Impact on genotype calling and allele frequency estimation. Genome Research, 23, 1852–1861. 10.1101/gr.157388.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Thaden, A. , Cocchiararo, B. , Jarausch, A. , Jüngling, H. , Karamanlidis, A. A. , Tiesmeyer, A. , … Muñoz‐Fuentes, V. (2017). Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Scientific Reports, 7, 10768 10.1038/s41598-017-10647-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waits, L. P. , & Leberg, P. L. (2000). Biases associated with population estimation using molecular tagging. Animal Conservation, 3, 191–199. 10.1111/j.1469-1795.2000.tb00103.x [DOI] [Google Scholar]
- Waits, L. P. , & Paetkau, D. (2005). Noninvasive genetic sampling tools for wildlife biologists: A review of applications and recommendations for accurate data collections. Journal of Wildlife Management, 64(9), 1419–1433. 10.2193/0022-541X(2005)69[1419:NGSTFW]2.0.CO;2 [DOI] [Google Scholar]
- Wang, J. (2004). Sibship reconstruction from genetic data with typing errors. Genetics, 166, 1963–1979. 10.1534/genetics.166.4.1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. (2009). A new method for estimating effective population sizes from a single sample of multilocus genotypes. Molecular Ecology, 18, 2148–2164. 10.1111/j.1365-294X.2009.04175.x [DOI] [PubMed] [Google Scholar]
- Wang, J. (2010). Effects of genotyping errors on parentage exclusion analysis. Molecular Ecology, 19(22), 5061–5078. 10.1111/j.1365-294X.2010.04865.x [DOI] [PubMed] [Google Scholar]
- Wang, J. (2011). Unbiased relatedness estimation in structured populations. Genetics, 187, 887–901. 10.1534/genetics.110.124438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. (2016). Individual identification from genetic marker data: Developments and accuracy comparisons of methods. Molecular Ecology Resources, 16(1), 163–175. 10.1111/1755-0998.12452 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Santiago, E. , & Caballero, A. (2016). Prediction and estimation of effective population size. Heredity, 117, 193–206. 10.1038/hdy.2016.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , & Santure, A. W. (2009). Parentage and sibship inference from multilocus genotype data under polygamy. Genetics, 181(4), 1579–1594. 10.1534/genetics.108.100214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S. , & Anderson, E. (2017). Purging putative siblings from population genetic datasets: A cautionary view. Molecular Ecology, 26, 1211–1224. 10.1111/mec.14022 [DOI] [PubMed] [Google Scholar]
- Wasser, S. K. , Brown, L. , Mailand, C. , Mondol, S. , Clark, W. , Laurie, C. , & Weir, B. S. (2015). Genetic assignment of large seizures of elephant ivory reveals Africa's major poaching hotspots. Science, 349, 84–87. 10.1126/science.aaa2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser, S. K. , Shedlock, A. M. , Comstock, K. , Ostrander, E. A. , Mutayoba, B. , & Stephens, M. (2004). Assigning African elephant DNA to geographic region of origin: Applications to the ivory trade. Proceedings of the National Academy of Sciences of the United States of America, 101, 14847–14852. 10.1073/pnas.0403170101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. , & Cockerham, C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38, 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wheat, R. E. , Allen, J. M. , Miller, S. D. L. , Wilmers, C. C. , & Levi, T. (2016). Environmental DNA from residual saliva for efficient noninvasive genetic monitoring of brown bears (Ursus arctos). PLoS ONE, 11, e0165259 10.1371/journal.pone.0165259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff, S. P. , Johnson, T. R. , & Waits, L. P. (2015). Evaluating the interaction of faecal pellet deposition rates and DNA degradation rates to optimize sampling design for DNA‐based mark–recapture analysis of Sonoran pronghorn. Molecular Ecology Resources, 15, 843–854. 10.1111/1755-0998.12362 [DOI] [PubMed] [Google Scholar]
- Woodruff, S. P. , Lukacs, P. M. , Christianson, D. , & Waits, L. P. (2016). Estimating Sonoran pronghorn abundance and survival with fecal DNA and capture‐recapture methods. Conservation Biology, 30, 1102–1111. 10.1111/cobi.12710 [DOI] [PubMed] [Google Scholar]
- Woods, J. G. , Paetkau, D. , Lewis, D. , McLellan, B. N. , Proctor, M. , & Strobeck, C. (1999). Genetic tagging of free‐ranging black and brown bears. Wildlife Society Bulletin, 27(3), 616–627. [Google Scholar]
- Wright, S. (1931). Evolution in mendelian populations. Genetics, 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wultsch, C. , Waits, L. P. , Hallerman, E. M. and Kelly, M. J. (2015) Optimizing collection methods for noninvasive genetic sampling of neotropical felids. Wildlife Society Bulletin 39, 403–412. 10.1002/wsb.540 [DOI] [Google Scholar]
- Yau, Y. , Leong, R. W. , Zeng, M. , & Wasinger, V. C. (2013). Proteomics and metabolomics in inflammatory bowel disease. Journal of Gastroenterology and Hepatology (Australia), 28(7), 1076–1086. 10.1111/jgh.12193 [DOI] [PubMed] [Google Scholar]
- Young, J. C. , Pan, C. , Adams, R. M. , Brooks, B. , Banfield, J. F. , Morowitz, M. J. , & Hettich, R. L. (2015). Metaproteomics reveals functional shifts in microbial and human proteins during a preterm infant gut colonization case. Proteomics, 15(20), 3463–3473. 10.1002/pmic.201400563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalasiewicz, J. , Williams, M. , Haywood, A. , & Ellis, M. (2011). The Anthropocene: A new epoch of geological time? Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 369(1938), 835–841. 10.1098/rsta.2010.0339 [DOI] [PubMed] [Google Scholar]
- Zhan, X. , Li, M. , Zhang, Z. , Goossens, B. , Chen, Y. , Wang, H. , … Wei, F. (2006). Molecular censusing doubles giant panda population estimate in a key nature reserve. Current Biology, 16(12), 451–452. 10.1016/j.cub.2006.05.042 [DOI] [PubMed] [Google Scholar]
- Zhan, X. , Zheng, X. , Bruford, M. W. , Wei, F. and Tao, Y. (2010) A new method for quantifying genotyping errors for noninvasive genetic studies. Conservation Genetics 11, 1567 10.1007/s10592-009-9950-9 [DOI] [Google Scholar]
- Zhan, L. , Paterson, I. G. , Fraser, B. A. , Watson, B. , Bradbury, I. R. , Nadukkalam Ravindran, P. , … Bentzen, P. (2017). Megasat: Automated inference of microsatellite genotypes from sequence data. Molecular Ecology Resources, 17(2), 247–256. 10.1111/1755-0998.12561 [DOI] [PubMed] [Google Scholar]