

Abstract
C–C bond formations are frequently the key steps in cofactor and natural product biosynthesis. Historically, C–C bond formations were thought to proceed by two electron mechanisms, represented by Claisen condensation in fatty acids and polyketide biosynthesis. These types of mechanisms require activated substrates to create a nucleophile and an electrophile. More recently, increasing number of C–C bond formations catalyzed by radical SAM enzymes are being identified. These free radical mediated reactions can proceed between almost any sp3 and sp2 carbon centers, allowing introduction of C–C bonds at unconventional positions in metabolites. Therefore, free radical mediated C–C bond formations are frequently found in the construction of structurally unique and complex metabolites. This review discusses our current understanding of the functions and mechanisms of C–C bond forming radical SAM enzymes and highlights their important roles in the biosynthesis of structurally complex, naturally occurring organic molecules. Mechanistic consideration of C–C bond formation by radical SAM enzymes identifies the significance of three key mechanistic factors: radical initiation, acceptor substrate activation and radical quenching. Understanding the functions and mechanisms of these characteristic enzymes will be important not only in promoting our understanding of radical SAM enzymes, but also for understanding natural product and cofactor biosynthesis.
1. Introduction
Naturally occurring organic small molecules are often characterized by their unique and complex structures. Such structures are frequently critical for their biological functions when the small molecules have to bind to target macromolecules with high specificity. In Nature, the complex and diverse structures of natural products are constructed through a series of enzyme-catalyzed reactions during their biosynthesis. One of the key steps common in most biosynthetic pathways is C–C bond formation to construct the carbon skeleton of the final natural products. Most frequently, such carbon skeleton formations are constructed by two-electron chemistry reactions requiring activation of the carbon centers that are used as nucleophiles and electrophiles. For example, polyketide synthases (PKS) are responsible for formation of the carbon skeletons of many natural products, and catalyze C–C bond formations through a Claisen condensation. In the typical PKS-catalyzed reactions, malonate or methylmalonate linked to the active site Cys residue of ketosynthase (KS) undergoes decarboxylation, and the resulting carbanion on the β-carbon attacks the thioester of a β-keto acid intermediate linked to an acyl carrier protein (ACP) or coenzyme A (CoA). The malonate or methylmalonate are inherently activated for the decarboxylation, and the thioester of the β-keto acid intermediate is activated to receive the nucleophilic attack and subsequent dissociation from ACP or CoA. Therefore, these reactions require mechanisms to both generate the activated species and protect them from the environment until the two substrates encounter each other in the enzyme active site.
In the past decade, numerous free radical-mediated C–C bond formations have been identified in many natural product and cofactor biosynthetic pathways. These reactions install C–C bonds in positions distinct from those possible by the nucleophilic mechanisms and are, therefore, important for the creation of the unique and diverse structures of the metabolites. Most of these free-radical mediated C–C bond formations are catalyzed by radical S-adenosylmethionine (SAM) enzymes.1 The radical SAM enzymes form one of the largest enzyme superfamilies with more than 113 000 annotated sequences, and more than 80 distinct demonstrated or proposed reactions. These enzymes are characterized by their ability to catalyze the reductive cleavage of SAM using oxygen sensitive, [4Fe–4S] clusters.2 Most frequently, radical SAM catalyzed reactions transiently generate a 5′-deoxyadenosyl radical (5′-dA•) or its equivalent, which abstracts an H-atom from the substrate and initiates the free-radical mediated reaction (Fig. 1). Unlike many other enzymes that use free radicals as a mechanism for catalysis, the reactions catalyzed by radical SAM enzymes are independent of molecular oxygen and can be either oxidative or redox neutral reactions, which significantly extends the scope of their reactivities. Because of this unique reactivity, radical SAM enzymes are capable of introducing C–C bonds between unactivated or minimally activated sp3 and sp2 carbon centers. As a result, the use of these enzymes in biosynthetic pathways allows installation of C–C bonds in unconventional positions, providing a means to create structural diversity in natural products and cofactors. Thus, characterization of C–C bond forming radical SAM enzymes has become important for understanding the biosynthesis of many natural products. In this review, we will summarize the roles of C–C bond forming radical SAM enzymes in natural product and cofactor biosynthetic pathways and their catalytic mechanisms.
Fig. 1.

Generalized reaction scheme for radical SAM enzymes.
2. Overview of the mechanism of C–C bond formation by radical SAM enzymes
To date, known C–C bond formations by radical SAM enzymes proceed between sp3 and sp2 carbon centers. Fig. 2 shows the generalized mechanism of C–C bond formation by a radical SAM enzyme. In general, a radical is first generated on an sp3 carbon center of the donor substrate through H-atom abstraction by 5′-dA• or its equivalent (step 1). The resulting donor substrate radical attacks an sp2 carbon center of the acceptor substrate (step 2). Eventually, the product radical is quenched to complete the catalytic cycle (step 3). Here, we will take a closer look at this generalized scheme by considering the three requirements for specific radical mediated C–C bond formation in synthetic chemistry: radical initiation, acceptor activation and radical quenching. Such comparison highlights the similarities and differences between the synthetic and enzyme catalyzed reactions, and the role of enzymes in free radical mediated C–C bond formation.
Fig. 2.

Generalized mechanism of C–C bond formation by radical SAM enzymes. Catalysis proceeds through three major steps; (1) radical initiation, (2) C–C bond formation, and (3) radical quenching. C–C bond formation may require activation of the acceptor substrate to achieve efficient and specific reactions. In some enzymes, radical quenching does not take place immediately after the C–C bond formation and the radical intermediate is used for complex radical rearrangement reactions. The C–C bonds formed are highlighted in blue ovals.
In the radical SAM enzymes, reactions are initiated by H-atom abstraction by the high energy primary radical, 5′-dA• (Fig. 2, step 1), which defines the scope of substrates for radical SAM enzymes. The bond dissociation energy (BDE) for the 5′-dA• C5′–H bond is 94–101 kcal mol−1,3 which is higher than most C–H bonds on sp3 carbon centers. Even C–H bonds of unactivated carbons of saturated hydrocarbons, such as octanoic acid (BDE 98–101 kcal mol−1 (ref. 3)) in the reaction catalyzed by lipoyl synthase,4 can be cleaved. Methane, whose C–H BDE is 104–105 kcal mol−1,3 may be about the only unacceptable substrate. Therefore, by using 5′-dA•, radical SAM enzymes can abstract H-atoms from almost any organic molecule sp3 carbon center. On the other hand, 5′-dA• is not reactive enough to abstract an H-atom from sp2 centers, which typically have BDE of >110 kcal mol−1 for weakly or non-activated sp2 centers, such as benzene and pyridine, and >98 kcal mol−1 for activated sp2 centers such as pyrimidine. The reactivity of 5′-dA•, therefore, limits the reaction of radical SAM enzymes to that between sp3 and sp2 centers, and no radical SAM enzymes characterized to date are known to catalyze the conjugation of two sp2 centers.
Currently, 5′-dA• generation by the radical SAM enzymes is thought to proceed by homolytic cleavage of the SAM C5′–S bond through a back side attack on the SAM sulfonium by the unique Fe of the [4Fe–4S]+ cluster (Fig. 3a, homolytic mechanism). This proposal is based on the conformation of SAM bound to the [4Fe–4S] clusters in the resting state of the enzymes in solution as well as in crystals, in which the SAM S is in close proximity (3–4 Å) to the unique Fe, and the Fe–S–C5′ atoms are almost collinearly aligned (Fig. 3b).5–9 The interaction between the SAM cleavage product and the [4Fe–4S] cluster was also investigated in a state that mimics a reaction intermediate using the SAM analog, Se-adenosyl-L-selenomethionine (Se-SAM), and a substrate analog, trans-dehydrolysine, which traps a radical intermediate as an allylic radical.10 This study was performed using lysine 2,3-aminomutase (LAM) a radical SAM enzyme that catalyzes reversible cleavage of SAM, which is different from the majority of radical SAM enzymes which catalyze irreversible SAM cleavage. Incubation of LAM with Se-SAM and trans-dehydrolysine resulted in the accumulation of a catalytically competent allylic radical intermediate. Characterization of this allylic radical state of LAM by selenium X-ray absorption spectroscopy revealed a relatively short distance (2.7 Å) between the selenomethionine Se and the unique Fe of the [4Fe–4S] cluster.10 Together, these observations support the close proximity of the SAM S and the unique Fe of the [4Fe–4S] cluster. The back-side attack model was also suggested based on precedents in synthetic chemical reactions in which an alkyl group on sulfonium is displaced by an aryl radical with inversion of configuration at the sulfur.11 Therefore, in sum, these observations and considerations form bases for the current consensus for the backside attack mechanism. It is important to note that there is an ~390 mV gap between the redox potentials of SAM and the radical SAM [4Fe–4S] clusters.12 Therefore, the homolytic cleavage of SAM is a thermodynamically unfavorable process.13 Significant ambiguity remains how the enzyme overcomes this redox potential gap.
Fig. 3.

Mechanism of SAM cleavage by radical SAM enzymes (a) proposed mechanism of reductive cleavage of SAM by radical SAM enzymes. SAM cleavage requires the ligation of SAM to the [4Fe–4S]2+ cluster followed by cluster reduction to the 1+ state. Two previously proposed mechanisms for the C5′–S bond cleavage (homolytic and heterolytic mechanisms) are shown. The potential SAM conformational change required for heterolytic cleavage is indicated by the dashed arrow. (b) Structure of SAM in complex with the [4Fe–4S] cluster in the active site of PLF-AE (3CB8).6 The distance between the unique Fe and the SAM sulfonium sulfur is shown by the dashed line. Note the collinear orientation of the unique Fe with the SAM sulfonium sulfur and C5′.
Recently, the presence of a previously unappreciated mechanism was suggested based on the characterization of pyruvate formate lyase activating enzyme (PFL-AE) by the Broderick and Hoffman lab.14 In this report, the authors observed a novel organometallic species with a covalent bond between the unique Fe of the [4Fe–4S] cluster in the 3+ state and C5′ of 5′-dA (5′-dA–[4Fe–4S]3+, Fig. 3a). Currently, there are two mechanistic proposals to explain this observation. In the first model, SAM is cleaved homolytically, and the resulting 5′-dA• reacts with the unique Fe of the [4Fe–4S]2+ cluster to generate 5′-dA–[4Fe–4S]3+ (Fig. 3a, homolytic mechanism). In this mechanism, 5′-dA–[4Fe–4S]3+ is considered as a means to store the highly reactive 5′-dA• until the substrate is appropriately positioned for H-atom abstraction. Alternatively, 5′-dA–[4Fe–4S]3+ is formed by a nucleophilic attack of the unique Fe of the [4Fe–4S]2+ cluster on the C-5′ of SAM (Fig. 3a, heterolytic mechanism). In this case, 5′-dA–[4Fe–4S]3+ is a reaction intermediate preceding the formation of 5′-dA•, and the C5′–S bond of SAM is cleaved heterolytically. The heterolysis would proceed regardless of the redox potential gap between SAM and the [4Fe–4S]+ cluster, and therefore provides an explanation to overcome the gap. However, the heterolysis cannot proceed from the conformation of SAM–[4Fe–4S] complex observed in the crystal structures since the SAM C-5′ is too far removed from the unique Fe. Thus, if SAM cleavage proceeds via heterolytic cleavage, SAM has to undergo a conformational change that brings the C-5′ atom close to the unique Fe of the [4Fe–4S] cluster. Regardless of the mechanism, the observation of 5′-dA–[4Fe–4S]3+ emphasizes our incomplete understanding of the mechanism of SAM cleavage or the mechanism that controls the reactivity of 5′-dA•, both of which are critical for radical SAM enzymes to catalyze radical reactions with high specificity.
Another important aspect of the radical initiation is the reduction of the [4Fe–4S] cluster from the oxidized state (2+) into the catalytically relevant reduced state (1+, Fig. 3). As discussed in this review, accumulating evidence suggests that the nature of the reductant used is important for many radical SAM enzymes to perform physiologically relevant reactions. For example, when PqqE in the PQQ cofactor biosynthesis was assayed using a non-physiological chemical reductant, the enzyme catalyzed an abortive cleavage of SAM without any reaction between 5′-dA• and the substrate (see Section 3.2). In the case of NosL during biosynthesis of the antibiotic nosi-heptide, the use of chemical reductants altered the regiospecificity of C–C bond cleavage (see Section 5.1). In both cases, the artificial activity observed was suppressed when the assays were performed using the flavodoxin–flavodoxin reductase system, which is in general considered as the physiological reductant for most radical SAM enzymes.2,15–18 On the other hand, for the radical SAM enzyme Dph2 involved in diphthamide tRNA modification formation, a specific reductase, Dph3, has been reported (see Section 6.1). These observations all suggest the significance of the physiologically relevant reductase for both in vitro mechanistic studies as well as understanding the function of radical SAM enzymes in general.
The next step in the mechanism of radical SAM catalyzed C–C bond formations is substrate radical attack onto an sp2 center of the acceptor (Fig. 2, step 2). In synthetic reactions, the specificity of radical attack is determined by several different factors related to the acceptor. The most frequently harnessed is perhaps the polar effect for acceptor substrates with electron withdrawing groups.19 In such reactions, nucleophilic radicals, or radicals with a singly occupied molecular orbital (SOMO) of relatively high energy, prefer to react with electron-deficient alkenes (Fig. 4a). In this case, the electron-withdrawing group attached to the alkene substrate lowers the energy of the lowest unoccupied molecular orbital (LUMO) thereby permitting greater overlap between the SOMO of the radical and the LUMO of the alkene. This effect is best illustrated by the reactivity of the 5-carbomethoxy-5-hexenyl radical (Fig. 4b), which undergoes cyclization via the 6-endo mode rather than the typically favored 5-exo pathway.20 Other factors could also play important roles, such as stereoelectronic or steric substituent effects, mostly in cyclic systems such as carbohydrates.21 While the significance of the enzyme active site environment on acceptor activation is not known, the inherent reactivity of the acceptor substrate has been suggested to be important for some radical SAM enzymes. For example, stereoelectronic control of radical quenching has been proposed for the reaction catalyzed by TunB during tunicamycin biosynthesis (see Section 3.4).22 In the case of PolH23 and MqnE,24 the radical on the donor substrate attacks C3′ of an enolpyruvyl functional group of the acceptor substrate that is activated with electron withdrawing carboxylic acid groups in a manner similar to the synthetic reactions (see Sections 4.1 and 5.3, respectively). Intriguingly, halogenation of the substrate in the MqnE reaction resulted in loss of the specificity of the radical attack.25 In the case of MoaA, the enzyme may activate the acceptor substrate using its auxiliary cluster (see Section 3.1). While further mechanistic investigations are needed to test such hypotheses, it is possible that acceptor activation plays a key role in the enzyme catalyzed radical reactions. It is also important to consider the reactivity of the acceptor substrate when investigating the mechanism of radical SAM enzymes, especially for those enzymes that catalyze complex rearrangement reactions.
Fig. 4.

The origin of regioselectivity in synthetic radical C–C bond formation reactions. (a) Generalized scheme for C–C bond formation with a donor substrate activated by an electron withdrawing group (EWG). (b) Stereoselectivity of radical cyclization of the 5-carbomethoxy-5-hexenyl radical.
The final step of radical mediated C–C bond formation is quenching of the radical intermediate (Fig. 2, step 3). In synthetic reactions, radical quenching almost always proceeds reductively. For example, in reactions mediated by the tributyltin radical (Bu3Sn•, Fig. 5), the reaction cycle is initiated by generation of a substrate radical (R•) by Bu3Sn• through abstraction of a halogen atom from an aryl or alkyl halide substrate (RX). R• then attacks an sp2 center of the radical acceptor substrate activated by an electron withdrawing group (EWG). The resulting radical is quenched by tributyltin hydride (Bu3SnH), yielding the product and regenerating Bu3Sn•. On the other hand, radical SAM enzymes offer more flexibility because their radical quenching can be reductive or oxidative. Therefore, the radical quenching mechanism adds diversity to the reactions catalyzed by radical SAM enzymes. Despite such unique and important reactivity, the mechanism of this step in radical SAM enzyme catalysis is largely unexplored. The only exception is that for PolH for which radical quenching was demonstrated to be mediated by a redox active Cys residue that is essential for stereospecificity of the reaction.23 In some enzymes such as MqnE,24 the radicals are not quenched immediately after C–C bond formation, and undergo further rearrangement reactions. Therefore, the mechanism of radical quenching could be used to diversify the type of C–C bond formation reactions catalyzed by radical SAM enzymes.
Fig. 5.

General scheme of the cycle of radical mediated C–C bond formation in synthetic reactions mediated by the tributyltin radical (Bu3Sn•). Bu3Sn• abstracts a halogen atom from an aryl or alkyl halide substrate (RX) and generates the substrate radical (R•), which then attacks an sp2 center of the radical acceptor substrate activated by an electron withdrawing group (EWG). The resulting radical is quenched by tributyltin hydride (Bu3SnH), yielding the product and regenerating Bu3Sn•.
Since the C–C bond forming radical SAM enzymes are in the early stage of functional characterization, detailed mechanisms are not known for most of them. However, we consider the three mechanistic aspects described above to be critical for understanding the mechanisms of radical mediated C–C bond formations. Thus, in this review, at least one of those three mechanistic aspects will be highlighted for each radical SAM enzyme discussed. Through these discussions, we aim to summarize the current understanding of the mechanism by which radical SAM enzymes control the fate of their free radical reactions.
3. The SPASM–twitch family
The SPASM–twitch family is the largest family of radical SAM enzymes. Its members contain extended C-terminal domains that harbor one or two auxiliary [4Fe–4S] clusters,26,27 and the family is divided into two subfamilies. The SPASM subfamily was defined based on the C-terminal extension domain found in radical SAM enzymes involved in the maturation of sub-tilosin A, pyrroloquinoline quinone, anaerobic sulfatase, and mycofactocin. All SPASM C-terminal domains contain two auxiliary [4Fe–4S] clusters.26,27 Members of the Twitch subfamily, which are distinct in terms of primary amino acid sequence, also harbor a C-terminal domain that is structurally homologous to the SPASM family except that it is truncated. As a result, Twitch C-terminal domains contain only one [4Fe–4S] cluster. An increasing number of enzymes in the SPASM–twitch family have been determined to catalyze C–C bond formations.
One of the key mechanistic paradigms for all SPASM–twitch family members is the redox function of their auxiliary [4Fe–4S] clusters. In general, functional characterization of the auxiliary clusters has been difficult for two major reasons. First, in many SPASM–twitch family members, with some exceptions from the recently characterized members,28,29 the auxiliary clusters are critical for the stability of the proteins, and mutations in the Cys ligands frequently result in insoluble protein expression.30,31 Second, the radical intermediates of the radical SAM enzyme reactions are transient and frequently kinetically masked by the preceding slow steps such as SAM cleavage, which precludes characterization of the [4Fe–4S] clusters in the state relevant to catalysis. Therefore, although redox functions are frequently proposed for these auxiliary clusters, very little evidence for those functions is available. The best characterized system is BtrN, which catalyzes oxidation of a hydroxyl group into a ketone during the conversion of 2-deoxy-scyllo-inosamine (DOIA, Fig. 6a) into 3-amino-2,3-dideoxy-scyllo-inosose (amino-DOI) during the biosynthesis of the antibiotic butirosin.32 This enzyme catalyzes oxidation of a DOIA hydroxyl group to the amino-DOI ketone via H-atom abstraction from the DOIA C-1 to yield a protonated ketyl radical intermediate33 that is oxidatively quenched by releasing a proton and an electron. This oxidative quenching was proposed to be catalyzed using the auxiliary cluster as the electron acceptor.34 However, enzymological evidence for this proposal is limited, and therefore a significant ambiguity still remains. Recent determination of the redox potentials of the Fe–S clusters in BtrN revealed that its auxiliary cluster is 255 mV more difficult to reduce than the radical SAM cluster.35 Additionally, in the BtrN crystal structure, the distance between the auxiliary cluster and DOIA C-1 is comparable to or even slightly longer than that between the radical SAM cluster and DOIA C-1 (9.5 vs. 8.6 Å, Fig. 6b).34 Thus, if the auxiliary cluster serves as the electron acceptor, there has to be a mechanism to facilitate electron transfer from the radical intermediate to the auxiliary cluster and to the terminal electron acceptor. Elucidation of the physiological redox partners may hold the key to understanding this radical quenching step. For most other SPAMS-Twitch family members, including all of those that catalyze C–C bond formation, the redox function of their auxiliary cluster(s) is unknown. As summarized below, we will discuss the function and mechanism of C–C bond formation by members of this family with particular focus on the roles of their auxiliary clusters.
Fig. 6.

Radical mediated alcohol oxidation catalyzed by BtrN. (a) The proposed mechanism of BtrN-catalyzed dehydrogenation of DOIA into amino-DOI during the biosynthesis of the aminoglycoside antibiotic butirosin.32,33 (b) Active-site structure of BtrN in complex with DOIA and SAM.34 The shortest distances between the DOIA C-1 and the radical SAM and auxiliary clusters is shown by the dashed lines.
3.1. MoaA in molybdenum cofactor biosynthesis
MoaA is one of the representative members of the Twitch subfamily and is involved in biosynthesis of the molybdenum cofactor (Moco). Moco is an enzyme cofactor essential for the catalytic functions of many redox enzymes. Unlike many other cofactors, Moco cannot be taken up as a nutrient and therefore requires de novo biosynthesis.36,37 As a consequence, the Moco biosynthetic pathway is essential for production of all the Moco-dependent enzymes. In humans, Moco biosynthesis is essential for healthy development of the brain, and genetic mutations in the Moco biosynthetic enzymes cause an inheritable and fatal metabolic disorder termed Moco deficiency (MoCD).38 More recently, Moco biosynthesis in pathogenic bacteria has been found to be critical for their ability to cause infectious diseases. In particular, pathogens such as Mycobacterium tuberculosis39 require Moco dependent nitrate reductase for the anaerobic respiration essential for survival in the low oxygen environment of the host body, and small molecule inhibitors of Moco biosynthesis have been shown to be effective for eradication of chronic tuberculosis in a mouse model.40
During Moco biosynthesis, MoaA, together with a second protein, MoaC, is responsible for the conversion of GTP into the intermediate, cyclic pyranopterin monophosphate (cPMP, 1 in Fig. 7a). The mechanism of cPMP formation was first investigated using isotope labeling experiments, which indicated that the GTP C-8 guanine base is inserted between C2′ and C3′ of the GTP ribose moiety41 (Fig. 7a). This labeling pattern is distinct from all other pterin compounds, where the GTP C-8 is hydrolyzed and lost during the early steps of biosynthesis, suggesting the presence of a distinct biosynthetic mechanism for Moco.37 The involvement of MoaA in this process was described in the late 1990s,44 and the crystal structure of MoaA was solved in 2004.45 Still, its exact function had long been unknown. Because of the unique reactivities of radical SAM enzymes in general, MoaA was initially proposed to be responsible for all the complex transformations required for formation of the pyranopterin structure of cPMP from GTP,46 and its product was assumed to be pyranopterin triphosphate (2, Fig. 7b), the phosphorylated form of cPMP.47 This proposal had become the predominant view in the field after structural characterization of MoaA and MoaC by multiple labs in the 2000s and the early 2010s.37 However, recent characterization of the MoaA product revealed significantly different functions for the two enzymes.43 In this study, the oxygen-sensitive MoaA product was isolated under strictly anaerobic conditions and structurally characterized as 3′,8-cyclo-7,8-dihydro-GTP (3′,8-cH2GTP, 3 in Fig. 7c). Purified 3′,8-cH2GTPwas shown to be converted to cPMP by recombinant MoaC with a Km value lower than 60 nM,43 and the crystal structure of MoaC in complex with 3′,8-cH2GTP provided further evidence that 3′,8-cH2GTP is the MoaC substrate.48 Together, these observations provided strong evidence that the complex rearrangement required for pyranopterin ring formation is catalyzed by MoaC, with MoaA catalyzing C–C bond formation between the GTP C3′ and C8. This finding was in sharp contrast to the conventional view of the mechanism of cPMP formation, where MoaA was thought to be solely responsible for all the complex rearrangement reactions (Fig. 7b). Thus, the functional characterization of MoaA, the C–C bond forming radical SAM enzyme, significantly changed the view of the pathway, and provided the first clear picture of the mechanism of pyranopterin backbone formation during Moco biosynthesis.
Fig. 7.

Moco biosynthesis and the mechanism of pyranopterin ring formation. (a) Overview of the Moco biosynthetic pathway. The symbols on GTP and cPMP indicate the source of the carbon and nitrogen atoms in cPMP as determined by isotope labeling studies.41,42 (b) Previously proposed functions for MoaA and MoaC, with MoaA catalyzing all the complex transformations required for the formation of the pyranopterin backbone structure of cPMP. (c) Revised functions for MoaA and MoaC, with MoaC catalyzing the complex rearrangement reactions to construct the pyranopterin structure, and MoaA catalyzing C3′-C8 bond formation.43 (d) The fate of C-8 in the biosynthesis of the other pterin containing molecules biopterin, folate and the flavins. In all these cases, the guanine C-8 is lost as formic acid.
The role of MoaA in Moco biosynthesis can be compared to the first step of the reactions catalyzed by GTP cyclohydrolase I and II in the biosynthesis of other pterin-related compounds as seen in Fig. 7d. These enzymes catalyze the conversion of GTP into dihydroneopterin triphosphate or 2,5-diamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate, respectively, with the first step of catalysis being hydrolysis of the guanine base C-8 as formic acid. This hydrolysis reaction makes the guanine base susceptible to subsequent complex rearrangement. By comparison, MoaA modifies GTP by forming a covalent bond between the C3′ and C8. The product of MoaA, 3′,8-cH2GTP (3), has a chemically labile aminal moiety that is proposed to be cleaved in the first step of the MoaC catalyzed rearrangement reaction.48 Intriguingly, MoaC does not require any cofactors, suggesting that no strong chemical activation is needed to convert 3′,8-cH2GTP into cPMP. Therefore, MoaA can be viewed as an activator of GTP to make it susceptible to the MoaC-catalyzed rearrangement.
Elucidation of the catalytic function of MoaA made this enzyme one of the first characterized C–C bond forming radical SAM enzymes, and Fig. 8a shows the proposed mechanism for MoaA catalysis.43 In this model, MoaA catalyzes abstraction of the GTP H-3′ atom using 5′-dA•. H-atom abstraction at this position has been demonstrated using deuterated GTP,43,49 and in MoaA crystal structures, the SAM C-5′ is positioned close to the C3′ of GTP,45,46 consistent with this H-3′ abstraction mechanism (Fig. 8b). The resulting C-3′ radical (4) then attacks C8 of the guanine base to form an aminyl radical intermediate (5) that is subsequently reduced by transfer of both an electron and a proton to yield 3′,8-cH2GTP (3).
Fig. 8.

The structure and mechanism of MoaA. (a) Proposed mechanism of MoaA-catalyzed C3′-C8 bond formation. (b) Model of the MoaA active site structure. SAM was modeled based on comparison to crystal structures of MoaA in complex with SAM (PDB ID, 1TV8) and with GTP (PDB ID, 2FB3). Reprinted with permission from J. Am. Chem. Soc., 2013, 135, 7019–7032. Copyright 2013 American Chemical Society.
The crystal structure of MoaA revealed the presence of two [4Fe–4S] clusters in the active site45 (Fig. 8b). The N-terminal cluster is the canonical radical SAM cluster CXXXCXXC motif, whereas the C-terminal cluster is the auxiliary cluster with three Cys ligands and the N1 of the GTP guanine base as the Fe ligands (Fig. 8b).46,50 This binding mode was proposed to alter the tautomerization of the guanine base from the normally favored keto tautomer to the enol tautomer (Fig. 9a) based on modeling of the enol tautomer into the crystal structure.50 However, the effects of such tautomerization on the catalytic mechanism of MoaA was not clear. Here, we propose an alternative model, in which binding of GTP to the C-terminal [4Fe–4S] cluster facilitates protonation at N7 (Fig. 9b), which should facilitate attack by the C-3′ radical at C-8 and subsequent radical quenching. The effect of metal binding to N1 of guanine has been studied using complexes of Pt(II) with N9-methyl or N9-ethyl guanine (Fig. 9c).51,52 These complexes exhibit significantly increased pKas at N7 of 4–5. If [4Fe–4S] clusters have similar effects, the binding of GTP to the C-terminal [4Fe–4S] cluster would facilitate protonation at N-7 (Fig. 9b). Since neither protonation of O-6 nor the enol tautomer were observed in the Pt(II) complex,51 it is likely that the guanine base is in the keto tautomer. The location of O-6 in the keto form is consistent with H-bonding interaction with the two catalytically essential Arg residues, R266 and R268. Similar H-bonding interaction between O-6 and the aminogroup of ethylenediamine was observed in the X-ray crystal structure of N9-methylguanine in complex with Pt(II) (Fig. 9c).51,52 Protonation of N-7 of GTP in the MoaA active site at the physiological pH likely requires additional assistance from the environment around N-7. While the MoaA crystal structure does not reveal specific residues around N-7 that could stabilize the protonated form,45,46 recent studies revealed that the C-terminal tail, which is disordered in the crystal structures, is likely serving as part of the active site.53 Therefore, the C-terminal carboxylate may provide an anion that stabilizes the protonated form of N-7. Based on these considerations, we propose the tautomerization and protonation state of the GTP guanine base in the MoaA active site to be that shown in Fig. 9b. Protonation at N-7 would make C-8 more electron deficient, facilitating attack of the C-3′ radical at C-8. In addition, protonation at this position eliminates the need for a proton transfer during reduction of the aminyl radical intermediate. Therefore, this model provides explanation to multiple aspects of the mechanism of MoaA catalysis.
Fig. 9.

Possible effects of the ligation of GTP to the C-terminal cluster of MoaA. (a) Previously proposed pyrimidinol tautomer of guanine base. (b) Our proposal for the structure of the activated form of GTP in the active site of MoaA. (c) Reported structure of Pt(II)–N9-methylguanine complex.
Theoretically, the C-terminal [4Fe–4S] cluster could also serve as an electron donor.43 However, there is currently very little experimental evidence to support such a role for this cluster. For it to serve as the electron donor, the cluster would have to be reduced to the 1+ state. Our preliminary EPR characterization of wt-MoaA suggests that the N- and C-terminal clusters can be reduced by sodium dithionite in ~1 : 2 ratio (unpublished results), indicating that the C-terminal cluster is more easily reduced. Considering the reported redox potentials for the [4Fe–4S] clusters in radical SAM enzymes,12,54 the redox potential of MoaA’s C-terminal cluster is likely between −0.4 and −0.6 V. Together with the previous report showing that the 5′,8-cyclo-deoxyadenosine aminyl radical can be reduced by the reduced form of methylviologen (E° = −0.45 V),55 the estimated redox potential of the C-terminal cluster is sufficiently reductive to reduce the aminyl radical. However, it is currently unknown if the C-terminal cluster is in the reduced form during catalysis.
Overall, these mechanistic considerations suggest that the MoaA active site likely provides the environment for both activation of the guanine base for attack by the C3′ radical as well as quenching of the putative aminyl radical. Therefore, MoaA could serve as a very good model system to understand how enzymes activate radical acceptor substrates to catalyze highly specific radical mediated C–C bond formations.
3.2. PqqE in pyrroloquinonline quinone biosynthesis56
PqqE is one of the founding members of the SPASM subfamily, and is responsible for the first step in biosynthesis of the cofactor pyrroloquinonline quinone (PQQ, see Fig. 10b for structure). PQQ is found in many dehydrogenases that catalyze oxidation of alcohols and aldose sugars mostly in Gram-negative bacteria.57 In particular, a membrane bound PQQ-dependent glucose dehydrogenase acts in non-glycolytic energy production in the periplasm of many Gram-negative bacteria.58,59 This enzyme catalyzes oxidation of D-Glc and other aldose sugars into their corresponding lactones, while reducing the orthoquinone of PQQ to its quinol form. The reduced PQQ is then re-oxidized by release of two electrons used to reduce ubiquinone followed by ATP production.59 Isolated PQQ has a midpoint redox potential of +90 mV at pH 7, which is much more oxidative than flavin (−210 mV) or nicotinamide (−320 mV), but comparable to that of ubiquinone (+66 mV at pH 7).60 The role of PQQ in mammals is much more controversial. While mammals do not produce PQQ, they acquire PQQ from their diet, and PQQ has been detected in mammalian blood, milk and tissues. In mouse models, exclusion of PQQ from the diet for several generations results in infertility.58 More recent studies suggest a role for PQQ in mitochondrial biogenesis.61 While these studies indicate a requirement for low levels of PQQ in a mammalian diet, the full range of mammalian targets and precise mechanism of action remain to be elucidated.
Fig. 10.
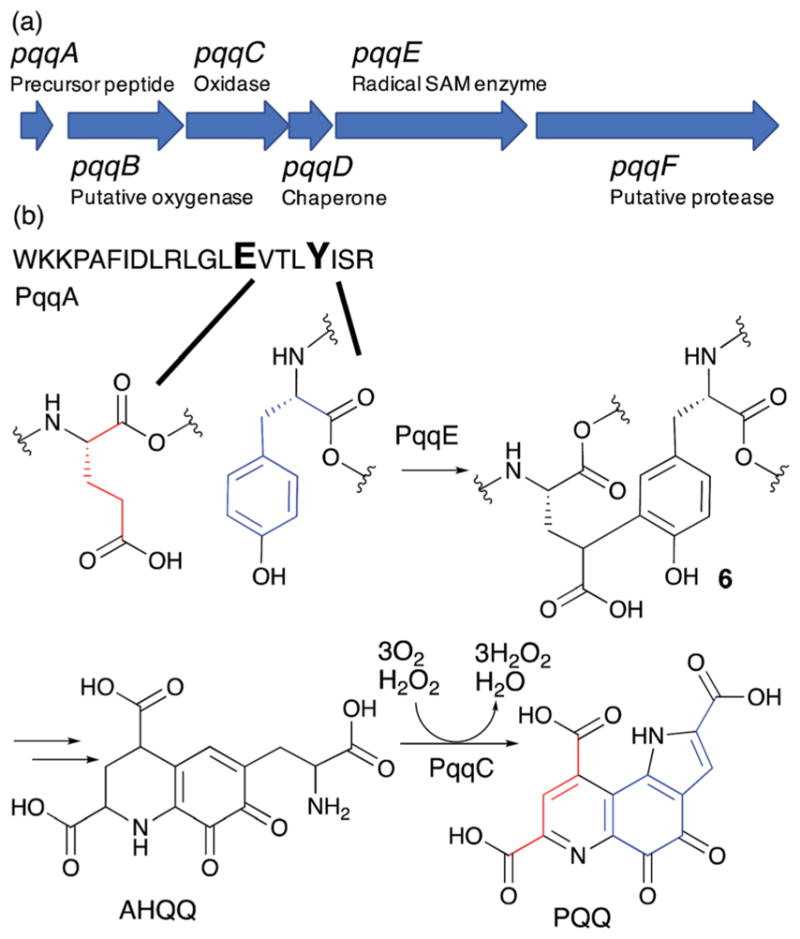
PQQ biosynthetic pathway. (a) PQQ biosynthetic gene cluster. (b) Formation of PQQ from the conserved glutamate and tyrosine of the PqqA peptide. AHQQ is an isolated intermediate that is further modified by the cofactorless oxidase, PqqC, to yield PQQ.
PQQ is biosynthesized from the Glu and Tyr62,63 residues of the PqqA precursor peptide through the actions of the products of five genes in the PQQ gene cluster (Fig. 10a). While it was known that 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid (AHQQ) is an intermediate of PQQ biosynthesis (Fig. 10b),64–66 the mechanism by which AHQQ is formed from the precursor peptide, PqqA, had long been unknown. Recently, PqqE, a radical SAM enzyme, was found to act on PqqA to catalyze C–C bond formation between the γ position of the glutamate and the C-3 of the tyrosine to yield compound 6 (Fig. 10b).56 Intriguingly, PqqE’s C–C bond formation activity requires the presence of the chaperone protein PqqD.56 Sequences homologous to PqqD have been found in biosynthetic pathways for many ribosomally synthesized and post-translationally modified peptides (RiPPs).67 These sequences were later identified as RiPP precursor peptide recognition elements (RREs) that bind the leader peptide of RiPPs and facilitate insertion of the amino acid to be modified into the active site of the enzyme.68 In most RiPP pathways, the RRE sequence is a part of the biosynthetic enzyme. However, PqqD is a stand-alone RRE with a Kd of 0.13–0.39 μM for its interaction with PqqA, 10–12 μM for its interaction with PqqE, and 4.5 μM for the PqqA–PqqD–PqqE ternary complex.67 Recent NMR structural characterization of PqqD revealed that its PqqA binding sites are comparable to that of RREs in other RiPPs biosynthetic enzymes.69
Successful reconstitution of PqqE activity was achieved using a flavodoxin (FldA)/flavodoxin reductase (FNR) system with NADPH as a reductant. Chemical reductants such as titanium(III) citrate or sodium dithionite do not yield crosslinked PqqA, and only result in abortive cleavage of SAM.70 While it is not known why the chemical reductants do not work for this system, one possibility is the need for a match in the redox potentials between the reductant and the radical SAM [4Fe–4S] clusters. As described below, it is possible that the auxiliary cluster may serve as an electron acceptor during the cross-linking reaction (see below), and therefore, its redox state could be critical for catalysis. In assays of PqqE from Methylobacterium extorquens, FldA from other organisms such as E. coli or Azotobacter vinelandii, was used because the physiological reductant for PqqE is not yet known. Considering the reported low turnover number of M. extorquens PqqE in these assays (4% after 24 h), identification of the physiological reductant is likely an important step toward better understanding of the mechanism of PqqE.
Based on the structures of the substrate and the product, a possible mechanism for PqqE catalysis is shown in Fig. 11.56 In this model, PqqE first abstracts a H-atom from the gamma position of glutamate. The resulting radical 7 then attacks the C3 position of tyrosine. Subsequent deprotonation could occur at the phenol OH (mechanism A) or 3-position (mechanism B), likely depending on the availability of the general base. This deprotonation accompanies release of an electron. The identity of the electron acceptor in this reaction remains ambiguous, although the auxiliary [4Fe–4S] cluster may serve as the electron acceptor.
Fig. 11.

Possible mechanisms for PqqE-catalyzed crosslinking of the Glu and Tyr residues of PqqA. Mechanism B was previously proposed by the Klinman lab.56
3.3. Streptide biosynthesis enzymes
RiPPs are an important class of natural products with a wide range of biological activities and diverse chemical structures that are frequently characterized by a cyclic structure that prevents their digestion by peptidases and enhances their stability.71,72 Currently, multiple different strategies have been characterized for the cyclization of these compounds, including thioether, ester, and amide linkages, and nitrogenous heterocycles. Recently, a subclass of RiPPs (such as streptide isolated from Streptococcus thermophilus) with a macrocyclic structure containing a unique Lys–Trp C–C crosslink has been reported (Fig. 12).73 These streptide-related RiPPs are produced by many streptococci, and their production is regulated by a quorum sensing cascade involving a short hydrophobic peptide (SHP) streptococcal pheromone and the Rgg transcriptional regulator. The exact biological functions of streptide and related RiPPs are unknown.
Fig. 12.

Biosynthesis of streptide. The amino acid sequences of representative RiPP precursor peptides are shown with the core peptide sequence highlighted in red. The Lys and Trp residues that undergo crosslinking are shown in bold. The C–C bond formed by the radical SAM enzyme StrB is highlighted by the blue ovals.
Studies by the Seyedsayamdost lab revealed that the Lys–Trp C–C bond of streptide (blue highlight in Fig. 12) is formed by the radical SAM enzyme StrB,73 a member of the SPASM subclass of the radical SAM enzymes (Fig. 12). Purified and reconstituted StrB contains three [4Fe–4S] clusters; one [4Fe–4S] cluster bound to the canonical radical SAM motif and the other two being the auxiliary clusters associated with the SPASM motif.73 StrB was shown to form the C–C bond between the lysine β-carbon and the C-7 side chain of the Trp residue in its precursor peptide, StrA.73 The Seyedsayamdost and Ando labs solved the crystal structure of SuiB (a close ortholog of StrB) from Streptococcus suis.74 SuiB uses the precursor peptide SuiA, a homolog of StrA (Fig. 12), as a substrate.75 Three structures of SuiB were solved; one without any ligand, one with SAM, and one with both SAM and SuiA. The SuiB N-terminal domain adopts a typical RRE motif, which is thought to be responsible for recognition of the SuiA leader peptide. However, in the crystal structure, this leader peptide was primarily interacting with the catalytic barrel of SuiB formed by its radical SAM and SPASM domains. While the SuiA core peptide is disordered in the structure, computer simulation of the core peptide structure suggested that the binding mode of the leader peptide is consistent with positioning of the core sequence in the SuiB active site.74 Thus, binding of the leader peptide to the catalytic barrel of SuiB, rather than its RRE domain, is likely relevant to SuiB catalysis.
The mechanism of Lys–Trp C–C bond formation by StrB was first investigated using an StrA peptide substrate containing a [2H8]Lys residue, where incorporation of the deuterium atom into the 5′-dA product was observed.73 Subsequent studies using Streptococcus agalactiae AgaB, another ortholog of StrB, and analogs of its corresponding AgaA substrate peptide provided insights into the mechanism of the radical quenching step.75 In this study, the side chains of the Lys and Trp residues that undergo crosslinking were varied. The study revealed that while the amino group of Lys was essential for catalysis, replacement of the nitrogen atom of the Trp indole with sulfur was accepted, suggesting that the indole ring nitrogen is not essential. However, a phenyl ring was not accepted as a substrate, suggesting that the ring size is critical for the catalysis. Based on these observations, a catalytic mechanism for AgaB was proposed whereby it abstracts an H-atom from the β-position of Lys and the resulting radical 8 attacks the Trp C7. The resulting Trp radical 9 is oxidized by release of an electron and a proton. Two mechanisms were proposed for this step as seen in Fig. 13.74 In one case (mechanism A), a base residue in the active site abstracts a proton at the 7′-position.75 In the second case (mechanism B), the amino group of the AgaA peptide substrate Lys residue serves as a general base.75 In both cases, one of the auxiliary clusters was proposed as the electron acceptor based on the structure and the previous proposals for other SPASM family members.
Fig. 13.

Two proposed radical mechanisms for the radical SAM enzyme AgaB.
3.4. TunB in tunicamycin biosynthesis
TunB, a putative SPASM family member, is involved in the key C–C bond formation during biosynthesis of tunicamycins (Fig. 14a), representatives of a class of fatty acyl nucleoside antibiotics produced by several species of actinobacteria. Tunicamycins exhibit potent antibacterial activity by inhibiting bacterial translocase I (MraY) which catalyzes the conversion of myramyl peptide 1-phosphate onto a C51 lipid during peptidoglycan formation in bacterial cell walls.76 Tunicamycins also inhibit GlcNAc-1-P transferases which are involved in the biosynthesis of wall teichoic acids (teichoic acids that are covalently bound to peptidoglycan),77 a cell wall structure found in Gram-negative bacteria that is essential for the virulence of many pathogenic bacteria. Unfortunately, in mammals, tunicamycins disrupt N-linked protein glycosylation by inhibiting dolichol phosphate GlcNAc-1-P transferase, and also inhibit protein palmitoyltransferases. As a result, tunicamycins are extremely cytotoxic, limiting their use as therapeutic agents. Therefore, an understanding of their biosynthesis is important for the discovery of tunicamycin derivatives with less toxicity.
Fig. 14.

Biosynthesis of tunicamine-containing antibiotics. (a) Structures of a representative tunicamycin, corynetoxin and streptoviridin. The 11-carbon tunicamine structure is highlighted in red, and the unique C5′–C6′ in the blue ovals. (b) Proposed tunicamycin biosynthetic pathway. The only experimentally demonstrated steps are those catalyzed by TunA and TunF. TunB is proposed to catalyze free radical-mediated C–C bond formation between UDP-6-deoxy-5,6-ene-GalNAc (11) and uridine to form the tunicamine backbone of UDP-N-acetyltunicamine-uracil (12).
Tunicamycins are structurally characterized by the presence of the unique 11-carbon dialdose core tunicamine (red highlight in Fig. 14) conserved among antibiotics such as the streptoviridins, corynetoxins, and mycospocidins (Fig. 14a). Feeding experiments using isotopically labeled precursors indicate that tunicamine is biosynthesized from uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) and uridine. Conjugation of these two precursors requires formation of a unique C5′–C6′ bond (Fig. 14a). Initially, this bond was proposed to form via nucleophilic attack of a carbanion generated on the C6″ of a UDP-GlcNAc derivative on the C-5′ of uridine-5′-aldehyde.78 However, identification of the tunicamycin gene cluster (tun), which included the tunB gene coding for a putative radical SAM enzyme, led to a different proposal, where TunB is responsible for the formation of the C5′–C6′ bond in UDP-N-acetyltunicamine-uracil (12)79 (Fig. 14b). This proposal was supported by subsequent functional characterization of TunA and TunF, with TunA being shown to catalyze the dehydration of UDP-GlcNAc into 10 and TunF catalyzing subsequent epimerization at C-4″ of 10 to form UDP-6-deoxy-5,6-ene-GalNAc (11, Fig. 14b). Furthermore, tunicamycin producing bacteria lacking the tunB gene was found to accumulate a compound with mass identical to 11, consistent with the proposed function of TunB.22 Based on the amino acid sequence homology of TunB with radical SAM enzymes, this transformation was proposed to proceed by the radical mechanism shown in Fig. 15,22,79 whereby TunB abstracts a H-atom from C-5′ of uridine, and the resulting C5′ radical attacks C6″ of 11. The resulting C-7′ radical intermediate (13) is then reductively quenched to yield the product 12. The regio- and stereoselectivity of this reductive radical quenching was rationalized based on stabilization of the C7′ radical by an overlap of the C7′ SOMO radical and the LUMO of C8′–O8′, as well as quasi–homo–anomeric effects (Fig. 15), where the addition of a H-atom from the axial side is favoured.22 To date, the catalytic function of TunB has not been demonstrated in vitro, and no mechanistic studies have been performed. It would be of mechanistic interest to know whether the enzyme active site provides additional effects on the regio- and stereo-specificity of the reaction.
Fig. 15.

Proposed mechanism for the TunB catalysis. The catalytic function of TunB was proposed based on the accumulation of 11 in the culture of a tunicamycin producer lacking the tunB gene,22 and TunB gene activity has not been reconstituted in vitro.
4. Radical SAM enzymes with N-terminal cofactor binding domains
An increasing number of radical SAM enzymes have been found to harbor an N-terminal domain that binds cobalamin or other cofactors. For example, RimO catalyzes thiomethylation of ribosomal protein S12 using persulfides bound to an auxiliary [4Fe–4S] cluster in its N-terminal UPF0004 domain.80 A structurally homologous N-terminal domain is also found in B12-dependent radical SAM enzymes where it binds the cobalamin cofactor.81 While most functionally characterized members of this group so far are responsible for methylation of unactivated carbon or phosphorus centers, recent studies have revealed that some of these enzymes are responsible for carbon skeleton formations where the roles of B12 or the N-terminal domain itself are still unclear. Using the representative members, NikJ/PolH, OxsB and BchE, we will discuss the mechanism of C–C bond formation in this family.
4.1. NikJ and PolH in the biosynthesis of C-5′ extended nucleoside antibiotics
NikJ and PolH are unique members of this group as they do not require cobalamin, an auxiliary [4Fe–4S] cluster or any other cofactors for their reported catalytic functions. Still, these enzymes harbor an N-terminal extension that exhibits weak homologies to UPF0004 and the cobalamin binding domains. The function of this NikJ/PolH N-terminal domain is currently unknown. NikJ catalyzes C–C bond formation during biosynthesis of the nikkomycins while PolH performs the same function during polyoxin biosynthesis. Nikkomycins and polyoxins are representative members of the peptidyl nucleoside (PN) class of antifungal natural products. PNs exhibit highly selective antifungal activities without any detectable toxicity to plants and animals by inhibiting chitin synthase, an enzyme required for the formation of chitin in the fungal cell wall.82 Because of their potency and selectivity, PNs have been used for agricultural purposes, and are currently under clinical investigation for human use.83
The pharmacophore of PNs is the C5′-modified nucleoside, aminohexuronic acid (AHA (17), Fig. 16), which mimics the uridine moiety of UDP-GlcNAc, the substrate for chitin synthase. AHA is formed from UMP and phosphoenol pyruvate (PEP) via the C5′-extended high-carbon nucleoside, octosyl acid (OA, 15), or its derivative octosyl acid 5′-phosphate (OAP, 16). The characteristic bicyclic structure of OA has also been found in other antifungal nucleoside natural products such as the ezomycins and malayamycins (Fig. 17a). Originally, the OA structure was thought to be formed via a 5′-aldehyde intermediate and attack of PEP C3′ on the aldehyde to form the C–C bond (Fig. 16, lower reaction).84 However, in the 2000’s, NikO in the nikkomycin pathway, followed by PolA in the polyoxin pathway, were found to catalyze the conjugation of PEP and UMP to yield 3′-enolpyruvyl UMP (EP-UMP, 14).85,86 This observation was unexpected and left the mechanism of C5′–C6′ bond formation in OA ambiguous.
Fig. 16.

Biosynthesis of nikkomycins and polyoxins. The characteristic C5′-extension in the PN antifungal natural products is highlighted with blue ovals. AHA is biosynthesized through EP-UMP and OAP. The original proposal for C5′–C6′ formation via a hypothetical uridine 5′-aldehyde intermediate is also shown in the lower reactions.
Fig. 17.

Comparison of the mechanism of biosynthesis of antifungal (a) vs. antibacterial (b) C-5′ extended nucleosides. The C5′-extension in the antifungal and antibacterial natural products is highlighted with blue and pink ovals, respectively. (a) Proposed biosynthesis of antifungal nucleosides from OAP as a common biosynthetic intermediate. OAP is formed from UMP by two enzymes, PolA and PolH. The hypothetical biosynthesis of malayamycin and ezomycin via OAP is shown by dashed arrows. (b) Biosynthesis of the caprazamycin class of antibacterial nucleosides. Here, the C5′ extension is formed through oxidation of C5′ of UMP into an aldehyde followed by aldol condensation with L-threonine to form 18.
Recently, this mystery was solved by functional characterization of the radical SAM enzymes conserved between the nikkomycin and polyoxin pathways (NikJ and PolH, respectively) (Fig. 17a). In this study, PolH was heterologously expressed in E. coli, purified, and its single [4Fe–4S] cluster reconstituted.23 Activity assays of the recombinant PolH with various uridine nucleosides and nucleotides revealed that PolH specifically catalyzes the conversion of EP-UMP into OAP (16).23 The essentially identical observation was made for NikJ.23 Detailed kinetic characterization revealed that the kinetics of both enzymes are comparable to those of the corresponding EP-UMP forming enzymes, NikO and PolA, suggesting the likely physiological relevance of the observed activity.23 Later, the polH gene was shown to be essential for polyoxin biosynthesis, and deletion of the polH gene resulted in accumulation of a shunt metabolite, 5′-enolpyruvyl uridine.87 These combined results established that NikJ and PolH are responsible for C5′–C6′ bond formation during the biosynthesis of the octosyl structure in the PN biosynthetic pathways.
The OA structure found in other antifungal nucleosides such as the malayamycins and ezomycins, is also likely to be formed by a similar free-radical dependent mechanism (Fig. 17a). In fact, when the genome sequence database was searched for homologs of PolH, most of them were found in putative operons for secondary metabolites that also code for the homologs of PolA.23 These analyses suggest that nucleoside natural products containing an OA moiety are likely more prevalent than is currently thought, and that the combination of the nikO and nikJ genes could serve as a genetic marker to identify biosynthetic gene clusters for nucleoside natural products structurally related to OA.
The characterization of NikJ and PolH revealed that the mechanism of carbon extension at C-5′ for the antifungal PNs is distinct from those reported for several different classes of antibacterial nucleoside natural products.88,89 In the case of the caprazamycin class of antibacterial nucleoside natural products (Fig. 17b), the C5′ extension proceeds via oxidation of UMP C-5′ to an aldehyde followed by an aldol condensation-type two-electron mechanism catalyzed by the PLP-dependent enzyme LipK that generates the C-5′ extended nucleoside 18 (Fig. 17b). On the other hand, functional characterization of the radical SAM enzymes, NikJ and PolH, suggested that the C-5′ extension in the biosynthesis of antifungal nucleoside natural products may proceed through a radical mechanism. The catalytic mechanism of PolH was studied using site-directed mutagenesis and biochemical and spectroscopic characterization.23 In the proposed mechanism (Fig. 18),23 PolH catalysis is initiated by abstraction of the H-5′ of EP-UMP (14) by 5′-dA•, followed by attack of the C-5′ radical (19) on C-3″ of the enolpyruvyl moiety to form a C7′ radical intermediate (20) which must then be reduced by transfer of both an electron and a proton. The key step in this mechanism is the C7′ radical quenching. As previously discussed, the mechanisms of radical quenching in radical SAM enzyme reactions are frequently ambiguous. For the SPASM–twitch family members, the auxiliary clusters were proposed as the electron donor/acceptor, but no experimental evidence is currently available. In the case of NikJ/PolH catalysis, no other metallo-cofactor is required for their activity. Thus, the involvement of a redox active amino acid residue was suspected. Ala scanning of Cys and Tyr residues conserved among homologs of PolH and NikJ identified C209 as a possible candidate. When C209 was mutated to a Ser, the enzyme produced a mixture of two products, OAP (16) and its C7′ epimer (21, Fig. 18).23 EPR characterization of the C209S mutant revealed accumulation of the C7′-centered radical intermediate (20).23 These observations suggested that the reaction proceeds through stereospecific quenching of the C7′-radical intermediate by the redox active C209 residue (Fig. 18). These studies on PolH provide the first evidence for a mechanism of radical quenching in C–C bond forming radical SAM enzymes, and demonstrate the significance of the radical quenching mechanism in the stereospecificity of the reaction.
Fig. 18.

Proposed mechanism of PolH-catalyzed C–C bond formation.
The involvement of a redox active Cys residue in the radical quenching was also reported for NeoN (Fig. 19), a radical SAM enzyme that catalyzes an irreversible epimerization at the 5-position of the glucosamine moiety of neomycin B by abstraction of a H-5′ atom followed by quenching of the C-5′ radical 22 from the opposite face of the molecule by a Cys residue.90 Mutation of this H-atom donating Cys residue resulted in rearrangement of the C-5′ radical 22 to eventually yield a carbocyclic compound 24 via a radical intermediate 23, and accumulation of a structurally uncharacterized radical intermediate (Fig. 19).90 The distinct fate of the accumulated radical as a response to mutation in the redox active Cys residue in NeoN and PolH may reflect the difference in the thermodynamic stability of the radical or the conformation of the carbohydrate radical species. The discovery of a Cys-mediated radical quenching mechanism in two functionally and structurally distinct radical SAM enzymes suggests the potential generality of the use of Cys residues in the active-sites of radical SAM enzymes to provide controlled quenching of radical intermediates during enzyme catalyzed free radical reactions.
Fig. 19.

Reactions catalyzed by NeoN.
While NeoN and PolH share a Cys-mediated radical quenching mechanism, their mechanisms of reduction of those Cys radicals appears distinct. NeoN is a member of the SPASM–twitch subfamily and harbors at least one auxiliary [4Fe–4S] cluster, which has been proposed to be responsible for Cys radical reduction through an attack of the thiyl radical either to one of the Cys ligand of the [4Fe–4S] cluster90 or to one of the Fe of the [4Fe–4S]2+ cluster.91 In contrast, PolH does not harbor any auxiliary [4Fe–4S] clusters. Instead, NikJ/PolH contain conserved Tyr and Trp residues that may be responsible for reduction of the Cys radical and transfer of the radical to the protein surface where a physiological electron donor is available. Since six of these conserved Tyr and Trp residues are found in the functionally uncharacterized N-terminal domains of PolH and NikJ, this domain may be responsible for electron transfer to the Cys209 residue from the protein surface where reductant is available from either chemical reductant in vitro or physiological reductase in vivo. Further studies are required to understand the function of the N-terminal domains in PolH and NikJ.
4.2. OxsB in oxetanocin biosynthesis
OxsB is the most recently characterized member of the family of C–C bond forming radical SAM enzymes with N-terminal cofactor binding domains. OxsB catalyzes formation of the unique four-membered ring structure of oxetanocin A (OXT-A (28), Fig. 20a), an antiviral first isolated and characterized from the fermentation broth of the Gram-positive soil bacteria Bacillus megaterium in the late 1980s.92,93 OXT-A has a unique oxetanosyl-N-glycoside structure with an adenine base,93 and has been shown to have antiviral activity against HIV,94,95 herpes simplex,92 and hepatitis B96 by inhibiting viral DNA polymerization. Mechanism of action studies indicated that incorporation of phosphorylated OXT-A into newly synthesized DNA causes chain termination.94
Fig. 20.

Biosynthesis of OXT-A. (a) Proposed biosynthetic pathway for OXT-A. (b) Possible mechanisms of OxsB catalysis. Cobalamin has been proposed to serve as the electron acceptor for the oxidative radical quenching step (mechanism A).81 Alternatively, cobalamin may react with the radical intermediate to facilitate cyclization (mechanism B).
Initial studies in B. megaterium revealed that the OXT-A biosynthetic gene cluster is encoded on a plasmid consisting of four different genes (oxsA, oxsB, oxrA and oxrB).97 Two of these genes encode biosynthetic enzymes essential for OXT-A formation: the divalent metal dependent HD-domain98 phosphohydrolase OxsA and the cobalamin-dependent radical SAM enzyme OxsB (Fig. 20a).81 Both of these genes have been heterologously expressed and characterized. Recombinant OxsA was shown to catalyze hydrolysis of phosphorylated OXT-A compounds.99 Recombinant OxsB harbored a single [4Fe–4S] cluster and hydroxocobalamin, consistent with its annotation as a cobalamin-dependent radical SAM enzyme.81 Initially, no catalytic activity was observed in OxsB assays using various nucleosides and nucleotides. However, product formation was observed when OxsB assays were performed in the presence of OxsA using mono, di, or triphosphorylated 2′-deoxyadenosine as substrates.81 The product of these reactions was characterized as dehydro-OXT-A phosphate (26, Fig. 20a). Since OxsA catalyzes the hydrolysis of dAMP, dADP and dATP,99 the function of OxsB was proposed to be conversion of dAMP into dehydro-OXT-A phosphate (Fig. 20a). The reason for the requirement of OxsA for OxsB activity is not clear, but it was proposed that the two proteins form a heterocomplex.81 Following the OxsB reaction, the aldehyde 26 has to be reduced to the alcohol 27 prior to conversion to OXT-A. This reaction was found to be catalyzed by an unidentified oxidoreductase in the lysate of B. megaterium NRS 269.81
Further characterization of OxsB demonstrated that it generates 5′-dA in the presence of a reductant.81 Using [2′-2H2]2′-dAMP as a substrate, OxsB generated mono-deuterated 5′-dA suggesting that the H-2′ is abstracted by 5′-dA•.81 Hydroxocobalamin was also found to be essential for OxsB activity, although its exact function remains unknown. Crystal structures of OxsB with [4Fe–4S] and cobalamin with and without SAM bound have been reported.81 Although the substrate bound structure was not solved, the crystal structure of OxsB in complex with SAM was used to map an approximate site of H-atom abstraction to be equidistant from both the SAM C-5′ and the cobalt of cobalamin at approximately 3.7 Å from each. The authors suggest that at this location, hydrogen atom abstraction could occur followed by a cobalamin-dependent reaction without any movement of either substrate or protein.81 Fig. 20b (mechanism A) shows the proposed mechanism of OxsB catalysis,81 where the reaction is initiated by abstraction of the H-2′ atom followed by C3′–C4′ bond cleavage. The resulting C-4′ radical (30) attacks C-2′ to form the four-membered ring radical intermediate 31, and subsequent oxidative quenching of this radical species would yield dehydro-OXT-A 5′-phosphate (26). Cobalamin was proposed as the electron acceptor for this radical quenching step. While this mechanism is chemically reasonable, it does not explain the juxtaposition of the cobalamin cofactor and the putative substrate binding site. Therefore, we consider an alternative possibility that one of the radical intermediates attacks Co(II) of cobalamin. One possible mechanistic option is described in Fig. 20b (mechanism B), where the C-4′ radical (30) attacks Co(II) of cobalamin to form a C4′–Co(III) intermediate (32) that can be collapsed into dehydro-OXT-A phosphate via nucleophilic closure of the oxetane ring by the favored 4-exo trig mechanism with heterolytic cleavage of the C4′–Co(III) bond. Further mechanistic investigations will provide insights into this fascinating radical SAM catalytic activity.
4.3. BchE in anaerobic bacteriochlorophyll biosynthesis
BchE is a founding member of the cobalamin-dependent radical SAM enzyme family100 and is involved in the biosynthesis of bacteriochlorophyll (BChl). BChl and chlorophyll (Chl) are central pigments for biological light harvesting and photosynthesis, and both are biosynthesized from protoporphyrin IX, a biosynthetic intermediate of heme. One of the key steps during BChl and Chl biosynthesis is formation of the fifth ring, the E-ring (Fig. 21), which enables harvesting of red light (>600 nm) for efficient photosynthesis. The E-ring is formed by oxidative cyclization of Mg-protoporphyrin monomethylester (MPE) to Mg-divinylprotochlorophyllide methylester (MDVP) (Fig. 21), presumably through C-131 oxidation followed by cyclization.101 Two distinct pathways for E-ring formation, which differ in oxygen dependency, have been reported.102–105 The aerobic pathway is conserved among most photosynthetic organisms from bacteria to plants, and involves the non-heme diiron monooxygenase, AcsF.106 The second, anaerobic pathway was found in photosynthetic purple bacteria, and involves BchE,107,108 a radical SAM enzyme with the cobalamin binding domain.107,109 The mechanistic dichotomy of the two pathways is apparent from isotope labeling experiments,105,110 whereby 18O from water is incorporated into the C-131-oxo when BChl is biosynthesized through the anaerobic pathway, while aerobic biosynthesis results in the incorporation of 18O from O2 and not from the solvent.
Fig. 21.
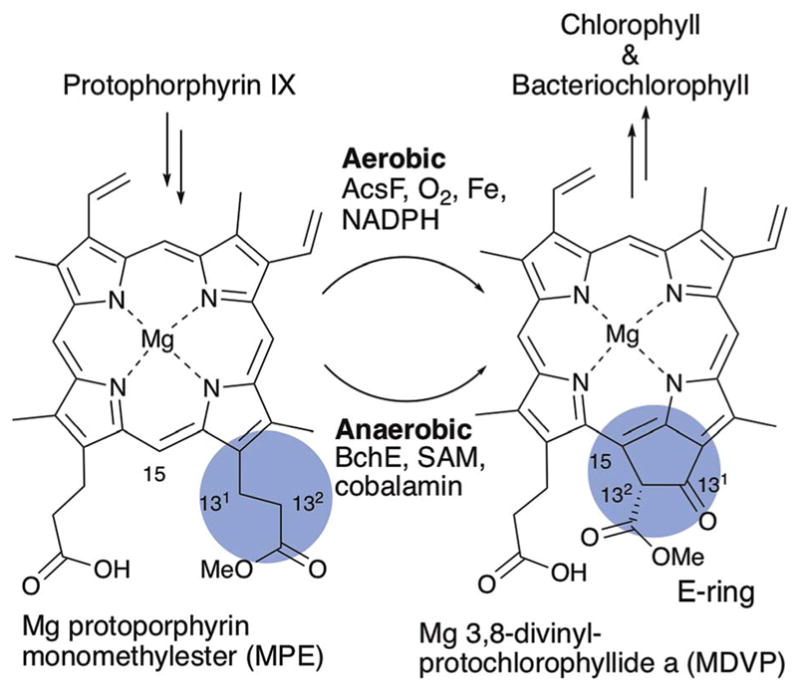
E-Ring formation during Chl and BChl biosynthesis.
The transformation of MPE to MDVP involves a C–C bond formation between C-132 and C-15 and the oxidation of C-131 from methylene to ketone. It is intriguing that all of these reactions can be catalyzed by the single radical SAM enzyme, BchE. While BchE has not been purified and its catalytic activity has never been reconstituted in vitro, the requirement of cobalamin for BchE function was supported by the observation that disruption of cobalamin biosynthetic genes blocked BChl biosynthesis and resulted in accumulation of MPE.109 BChl biosynthesis in these mutants was recovered by the addition of cobalamin to the growth medium.109 Fig. 22 shows two potential mechanisms for BchE catalysis. The first key step in these mechanisms is the oxidation of C-131. The mechanism proposed by Booker111 (Fig. 22, mechanism A) is a reaction analogous to those proposed for the cobalamin dependent radical SAM methyltransferase,112 in which 5′-dA• abstracts the H-131 atom and the resulting radical 32 attacks the hydroxyl group of hydroxocobalamin to yield hydroxy-MPE 33 and cob(II) alamin. Repeating the same hydroxylation for 33 would form a geminal diol that is equivalent to keto-MPE (34). This mechanism makes use of both cofactors; SAM and cobalamin. However, such use of hydroxocobalamin is not precedented. Alternatively, C-131 hydroxylation could proceed via sequential dehydrogenation and hydration (Fig. 22, mechanism B), where BchE abstracts the H-132 atom to generate the radical 35 that is subsequently oxidized to the olefin 36. There are precedents for similar dehydrogenation by radical SAM enzymes.113–115 Subsequent hydration by the attack of a hydroxide at C-131, which is electrophilic due to the presence of α,β-unsaturated methyl-ester, would yield hydroxy-MPE 33. Repeating the dehydrogenation of hydroxy-MPE 33 followed by keto–enol tautomerization would yield keto-MPE 34. In this dehydrogenation/hydration mechanism, cobalamin may serve as the electron acceptor similar to what has been proposed for OxsB (Fig. 20b). For either mechanism, cyclization of 34 would then likely proceed through abstraction of the H-132 atom by 5′-dA•. This H-atom abstraction position is identical to the mechanism B for the keto-MPE 34 formation, providing another advantage for the mechanism B. The resulting radical 37 would attack C-15 to form the C132–C15 bond, followed by release of a proton and an electron to give MDVP. These mechanisms predict the requirement for three equivalents of SAM to produce each molecule of MDVP. Future determination of the actual mechanism for this radical SAM enzyme will require in vitro reconstitution of BchE activity.
Fig. 22.

Proposed mechanism of C-131 oxidation and E-ring formation by BchE. Two possible mechanisms A and B are shown. The oxygen on C-131 of MDVP is derived from solvent or solvent exchangeable hydroxide on hydroxo cobalamin as highlighted in red bold.
5. ThiH-like enzymes
Another subset of radical SAM enzymes catalyzing C–C bond formations are those exhibiting sequence homology to ThiH. ThiH catalyzes cleavage of tyrosine into 4-hydroxytoluene and dehydroglycine during biosynthesis of the azole moiety of the cofactor thiamine (Fig. 23a).116 A similar tyrosine cleavage is catalyzed by HydG during biosynthesis of the CO and CN ligands of the hydrogenase cofactor (Fig. 23b).117 More recently, enzymes that exhibit amino acid sequence similarities with ThiH have been found to catalyze complex transformations that involve C–C bond formation reactions. Two of these, NosL and CofH, also uses aromatic amino acid as a substrate, while the other recently characterized enzymes, CofG, MqnE and MqnC, use substrates other than aromatic amino acids. Thus, the functions of these ThiH-like radical SAM enzymes are diverse. Here, we will summarize the most recent discoveries involving these ThiH-like enzymes.
Fig. 23.
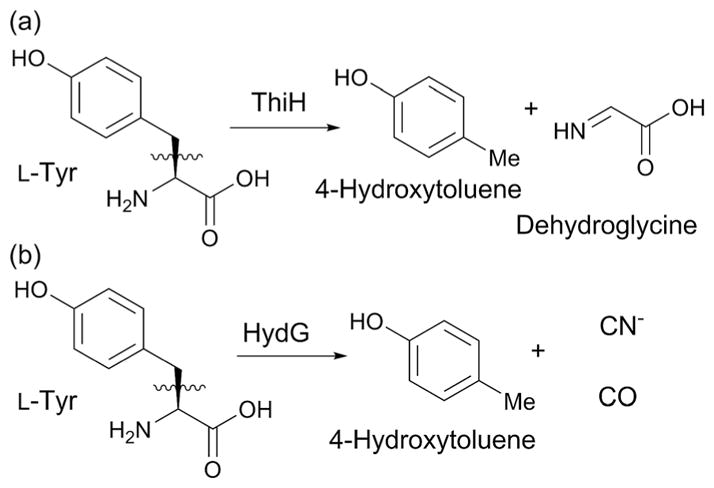
Some reactions catalyzed by ThiH-like enzymes.
5.1. NosL in the biosynthesis of nosiheptide
Thiopeptides are a subclass of RiPPs structurally characterized by macrocyclic cores consisting of a nitrogen-containing 6-membered ring, a thiazole and a dehydroamino acid. Most thiopeptides known to date exhibit antibacterial activity against Gram-positive bacteria. Among the thiopeptide antibiotics, nosiheptide (Fig. 24) possesses a unique aromatic ring, methylindolic acid (MIA), that is derived from L-Trp. The transformation of L-Trp into MIA was recently shown to be catalyzed by NosL, a ThiH-like enzyme (Fig. 24).118
Fig. 24.

Biosynthesis of nosiheptide with its MIA-derived aromatic ring shown in blue.
The catalytic mechanism of NosL was first investigated by isotope labeling experiments, which revealed that the carboxylate of MIA is derived from the carboxylate of L-Trp and that a solvent exchangeable H-atom is abstracted by 5′-dA•.118 In vitro NosL reactions with L-Trp as the substrate also produced 3-methylindole and dehydroglycine as shunt products (Fig. 24),118 which was interpreted as evidence for the Cα–Cβ bond cleavage. When the mechanism of NosL was investigated using a series of L-Trp analogs, the data suggested that H-atom abstraction occurs at the α-NH2 group.119–121 Based on these results, the mechanism shown at the top of Fig. 25 involving Cα–Cβ bond cleavage was proposed.119–121 In this mechanism, 5′-dA• is used to abstract the N–H proton and generate an amino radical (39). Subsequent cleavage of the Cα–Cβ bond yields an indole radical (40), and dehydroglycine, which are then recombined to form a hemiacetal radical intermediate (41). Release of a methanimine radical (NHCH•) and rearomatization of 42 results in MIA. The shunt product, 3-methylindole, would be produced by a reductive quenching of the radical intermediate 40, followed by rearomatization (Fig. 25, top shunt pathway).
Fig. 25.

Mechanism of NosL catalysis. (a) Proposed mechanisms of NosL reaction via Cα–Cβ bond cleavage (top) and Cα–C bond cleavage (bottom). (b) Proposed orientation of the p-orbital of the amino radical responsible for the Cα–C bond cleavage.
Recently, EPR characterization of the NosL reaction suggested a completely different mechanism that involves Cα–carboxylate (Cα–C) bond cleavage (Fig. 25a, bottom mechanism).122 In this study, EPR characterization of the NosL reaction revealed accumulation of an organic radical proposed to be a reaction intermediate based on its kinetic behavior since the amount of this species reached its maximum at 45 s and then decreased over the following 10 min. While the EPR spectra of this radical species was complicated by the presence of two overlapping signals with similar line shapes, EPR analysis using isotopically labeled Trp revealed the radicals to be two different conformers of the C-3 radical (44). Based on this observation, a new mechanism for NosL was proposed, where the amino radical 39 undergoes cleavage of the Cα–C bond to yield two intermediates, 43 and a COO(H) radical.122 The two intermediates are then recombined by attack of the COO(H) radical onto C-2 of 43 to yield the C-3 radical 44 that was detected by EPR. Radical 44 undergoes elimination of a methanimine radical (NHCH•) followed by rearomatization of 45 to yield MIA. This proposed mechanism is distinct from the previous proposal in that in this case, MIA formation by NosL involves cleavage of the Cα–C bond. In this mechanism, the Cα–Cβ bond cleavage evidenced by formation of 3-methylindole was proposed as a shunt pathway that does not yield MIA.122
The regiospecificity of NosL-catalyzed C–C bond cleavage was rationalized by electron density calculation of the amino radical intermediate 39.122 In this analysis, conformation of the amino radical 39 was first calculated based on the reported crystal structures of NosL in complex with L-Trp. Such computation resulted in two stable conformers, for which the electron density was calculated. In one of these conformers, the p-orbital of the amino radical is in parallel with the Cα–C bond (Fig. 25b), which depletes the electron density from the Cα–C bond and facilitates Cα–C bond cleavage. The other conformer had the p-orbital in parallel with the Cα–Cβ bond, which should facilitate Cα–Cβ bond cleavage. Based on these analysis, the orientation of the p-orbital of the amino radical relative to the Cα–C and Cα–Cβ bond was proposed to determine the regiospecificity of the C–C bond cleavage.122 The NosL active site environment is likely responsible for the conformation of the radical intermediate, and therefore control of the radical reactivity. Interestingly, the ratio between Cα–C and Cα–Cβ bond cleavage in the in vitro assays depends on the amount and the nature of the reductant used.118,121 When assays were performed using dithionite as the reductant, 3-methylindole was the major product, whereas the use of the flavodoxin/flavodoxin reductase system produced MIA as the major product.118,121 Therefore, it is likely that the regiospecificity of C–C bond cleavage by NosL is better controlled inside the cell with the physiological reductant.
5.2. F0 synthases (CofG/CofH and FbiC) in the biosynthesis of the F420 cofactor
CofG and CofH are homologous to each other and ThiH, and responsible for formation of the deazaflavin moiety of the F420 cofactor (Fig. 26). F420 is a redox cofactor found in bacteria,123 archaea124 and some eukaryotes.125 In some organisms, a biosynthetic precursor of F420, F0, is used as a cofactor. Both are naturally occurring deazaflavin cofactors in which the N-5 of the flavin ring is replaced with a methine. Despite their structural resemblance, the reactivities of deazaflavins are distinct from flavins. While flavins can mediate both hydride transfer and single electron transfer reactions, deazaflavins are obligate hydride donor/acceptors. In addition, the redox potential of F420 (−370 mV) is significantly lower than flavin (−210 mV) or nicotinamide (−320 mV).126 Thus, F420 is believed to act as a strong, two electron reductant in cells. While our understanding on the physiological functions of F420 and putative F420 dependent enzymes are limited, known functions include anti-TB prodrug activation,127 resistance to oxidative stress128 and biosynthesis of clinically important natural products.129,130
Fig. 26.

Biosynthesis of F420-n (n = 1–6) and riboflavin from GTP via ARP.
F420 is biosynthesized from GTP, and its initial biosynthetic steps are the same as those for riboflavin up to the point of 5-amino-6-ribitylamino-2,4-pyrimidinone (ARP) production (Fig. 26). The deazaflavin ring of F420 is then formed by a reaction between ARP and tyrosine that is catalyzed by an F0 synthase. In archaea and cyanobacteria, F0 synthase is encoded by two separate genes, cofG and cofH, while in actinobacteria, the two genes are fused as the single gene fbiC.131 The two domains of FbiC, as well as CofG and CofH are homologous to each other, and belong to the ThiH family of radical SAM enzymes.
The catalytic function of this F0 synthase was first demonstrated in vitro using FbiC from the thermophilic organism Thermobifida fusca.132 Recombinant T. fusca FbiC catalyzed the formation of F0 using ARP and tyrosine as substrates, and produced two equivalents of 5′-deoxyadenosine (5′-dA). The distinct roles of the two domains of FbiC were studied using CofG and CofH of bacterial origin.133 By performing a stepwise assay, it was determined that CofH produces a diffusible product that can be converted to F0 by CofG.133 Later, the CofH product was isolated and structurally characterized as compound 46 shown in Fig. 27a. The catalytic mechanisms of CofG and CofH were studied using a series of isotope labeling experiments. When CofH assays were performed in the presence of 80% D2O, one deuterium atom was incorporated into 5′-dA. On the other hand, CofG assays using [7,7-2H2]-46 resulted in incorporation of one of the two deuterium atoms into 5′-dA.
Fig. 27.

CofG and CofH catalysis. (a) The overall reactions catalyzed by CofH and CofG. (b) Proposed mechanism of reaction catalyzed by CofH. (c) Proposed mechanism of reaction catalyzed by CofG.
Based on these observations, a mechanism of F0 synthase was proposed as in Fig. 27b and c. In this mechanism, CofH catalyzes abstraction of the L-Tyr N–H and cleaves the Cα–Cβ bond. The resulting p-hydroxybenzyl radical 48 attacks C-5 of ARP to form radical 49. Subsequent oxidative quenching of the radical gives the CofH product (46). CofG then catalyzes H-atom abstraction from the 7-position of 46 to form the C-7 radical 50 (Fig. 27c). After that point, two distinct mechanisms have been proposed. Initially, the Begley lab proposed that the C-7 radical facilitates elimination of the 5-NH2 group as ammonia (mechanism A). The resulting cation radical 51 undergoes tautomerization and phenol deprotonation to give the C-5 radical 53, which undergoes tautomerization and C9–N6 bond formation to give the phenoxyl radical 54 which is converted to F0 by the net release of an electron and a proton. Later, the same group proposed an alternative mechanism (mechanism B in Fig. 27b), in which the C-7 radical 50 tautomerizes to 55 where attack of the radical on the imine N-6 forms the C9–N6 bond. The resulting radical 56 is oxidatively quenched and following elimination of an ammonia, yields F0. Mechanism A leaves ambiguity in the mechanism of the conversion of the phenoxyl radical intermediate 54 to F0. In mechanism B, the C-9 radical intermediate 55 would be attacking an N-6 that is not electron deficient, and undergoing 6-endo trig cyclization rather than attacking the electron deficient C-6 to undergo typically favorable 5-exo trig cyclization.134 How the enzyme might achieve excellent selectivity for N-6 over C-6 attack is of mechanistic interest, and if CofG does carry out such a reaction, there might be an acceptor activation involved in this step. Both mechanisms involve oxidative radical quenching, but the identity of the potential electron acceptor is not known.
5.3. Menaquinone biosynthesis by MqnE and MqnC
Recently, two enzymes, MqnC135 and MqnE,24 which both exhibit amino acid sequence similarity to ThiH, were reported to be responsible for bacterial menaquinone biosynthesis. These enzymes, however, do not use an aromatic amino acid as a substrate and catalyze reactions distinct from the other members of this family. Menaquinone (Fig. 28) is a lipid-soluble small molecule that serves as an electron shuttle in the bacterial electron transport chain.136 It is also an essential vitamin in humans, known as vitamin K2, and functions as a cosubstrate in the carboxylation of glutamic acid residues in proteins involved in blood coagulation137 and bone formation.138 Generally, menaquinones consist of a quinone headgroup with a hydrophobic terpene side chain which anchors the quinone to the cell membrane. Currently, two different biosynthetic pathways have been identified for production of menaquinone. The classical menaquinone biosynthesis pathway involves eight men genes and uses chorismic acid and α-ketoglutarate (α-KG) as precursors (Fig. 28a).136,139–141 A second, more recently discovered pathway, the “futalosine pathway”, was identified based on comparison of the genome sequence of bacteria that produce menaquinone, but did not contain the men genes (Fig. 28b).142 Subsequent gene disruption and complementation experiments together with feeding of radioisotopically labeled precursors and isolation of biosynthetic intermediates identified most of the biosynthetic genes and the general reaction scheme for this alternative menaquinone biosynthesis pathway (Fig. 28b).142–144
Fig. 28.

Two distinct menaquinone biosynthesis pathways. (a) Conventional menaquinone biosynthesis pathway involving the men genes. (b) Recently identified menaquinone biosynthesis pathway utilizing futalosine as the key intermediate. The radical SAM enzymes are shown in red highlights.
The futalosine pathway uses a purine nucleoside/nucleotide as a precursor in addition to chorismate, and involves formation of the unique modified nucleoside intermediate, futalosine (60).142 The enzyme responsible for formation of the C–C bond between chorismate and the purine nucleoside was not identified in the initial comparative genetics and gene knockout approach.142–144 This enzyme was eventually identified by hypothesizing the possible mechanism of futalosine formation and performing a homology search for genes in the neighborhood of the already characterized futalosine pathway genes.24 These analyses revealed that the radical SAM enzyme MqnE is frequently clustered with menaquinone biosynthetic genes. Subsequent in vitro characterization of recombinant MqnE from T. thermophiles demonstrated that MqnE catalyzes the transformation of 3-[(1-carboxyvinyl)-oxy]benzoic acid (58) into aminofutalosine (59).24 Intriguingly, MqnE uses SAM as the source of the purine nucleoside, and transfers the deoxyadenosine moiety of SAM to C-3′ of 58. The 5′-dA is therefore never formed as a product, which at the time, was unprecedented in radical SAM enzymes. Subsequently, the addition of 5′-dA to an sp2 center has been demonstrated for other radical SAM enzyme reactions using unnatural substrates. For example, NosL catalyzes the addition of 5′-dA to the unnatural substrate 2-(indolylmethyl)-acrylic acid.145 Similarly, QueE, responsible for the formation of the deazapurine ring during the biosynthesis of 7-deazapurine natural products, catalyzes the addition of 5′-dA to 6-carboxypterin, the oxidized analog of CPH4.146 Therefore, it appears that if 5′-dA• is generated in the presence of a substrate or its analogs lacking an H-atom to be abstracted, the radical is likely to attack a nearby sp2 center.
The 5′-dA addition catalyzed by MqnE, on the other hand, is distinct from the QueE and NosL reactions in that the radical intermediate undergoes a further rearrangement reaction. The mechanism of this unique rearrangement reaction was probed using halogenated substrate analogs as seen in Fig. 29.25 Use of 3′-Br/Cl–3-[(1-carboxyvinyl)-oxy]benzoic acid (63 and 64) resulted in the formation of the compound 66 as the major product (Fig. 29a) due to fast homolytic cleavage of the carbon–halogen bond β to the radical center (kC–Br = 3 × 108 s−1, and kC–Cl = 4 × 106 s−1). The same reactions also produced a minor product 68 as a result of loss in regiospecificity of the radical attack.25 Since no rearrangement products were observed, the rate constant for the rearrangement reaction was estimated as slower than 2 × 105 s−1.25
Fig. 29.

MqnE reactions with halogenated substrate analogs.
Reaction with 4-Br-3-[(1-carboxyvinyl)-oxy]benzoic acid (69) resulted in the formation of a mixture of 71 and 73 (Fig. 29b).25 In synthetic chemical reactions, aryl halide radical anions structurally similar to 70 are known to undergo dehalogenation through elimination of a bromide anion to yield a neutral aryl radical, similar to 72, followed by radical quenching via H-atom transfer. Therefore, the formation of 73 in the MqnE reaction with 69 was rationalized to be the result of formation of the aryl radical anion intermediate 70 followed by bromide elimination and radical quenching by an H-atom donor (X–H, Fig. 29b). The identity of the H-atom donor is unknown, but it is known that the H-atom is not solvent exchangeable.
Based on these observations, a catalytic mechanism in Fig. 30 was proposed,25 where MqnE reductively cleaves SAM and generate 5′-dA• which attacks the C-3′ of 3-[(1-carboxyvinyl)-oxy]benzoic acid (58). The resulting C-2′ radical 74 undergoes rearrangement via the spiroepoxide 75 and generates a radical cation intermediate. Subsequent decarboxylation and keto–enol tautomerization yields an aryl radical anion intermediate 78 that can be oxidized to yield aminofutalosine. The electron acceptor for the radical anion intermediate was proposed to be the canonical radical SAM [4Fe–4S] cluster because the amount of aminofutalosine production exceeded that of sodium dithionite in the solution.25 To our knowledge, MqnE is the only radical SAM enzyme that has yielded any experimental evidence that its canonical radical SAM cluster can serve as an electron acceptor in reactions with its physiological substrate.
Fig. 30.

Proposed mechanism of the reaction catalyzed by MqnE.
It is noteworthy that the regiospecificity of C–C bond formation in MqnE was perturbed when 3′-Br/Cl-3-[(1-carboxyvinyl)-oxy]benzoic acid (63 and 64, Fig. 29a) was used.25 This observation demonstrates that subtle changes in the substrate structure affect the regiospecificity of the radical reaction. B12-Dependent radical SAM methyltransferase studies by the Liu147 and Eguchi148 labs demonstrated some promiscuity in the stereospecificity of H-atom abstraction by 5′-dA• or the following methyl transfer step. These examples indicate the challenge of controlling the reactivities of free radicals even within the structurally well-organized active sites of enzymes.
Following futalosine formation, generation of the bicyclic menaquinone structure requires formation of another C–C bond. This step occurs during the conversion of dehypoxanthine futalosine (DHFL) into cyclic dehypoxanthine futalosine (CDHFL), and requires another radical SAM enzyme, MqnC. Using heterologously expressed MqnC from B. halodurans, the Begley lab showed that purified MqnC was capable of converting synthetic DHFL into CDHFL (Fig. 31).135 Deuterium incorporation studies determined that the H-4 is abstracted by 5′-dA•.135 The resulting C-4 radical was proposed to attack the aromatic ring para to the carboxylate to form a C–C bond followed by deprotonation and quenching of the radical to form CDHFL. Unlike MqnE, the electron acceptor for this reaction is not known. In sum, formation of the bicyclic structure of menaquinone requires two radical SAM enzymes that catalyze two distinct C–C bond forming reactions in the newly discovered futalosine pathway.
Fig. 31.
Proposed mechanism of the reaction catalyzed by MqnC.
6. Noncanonical radical SAM enzymes
In the past decade, radical SAM enzymes that do not contain the canonical radical SAM CXXXCXXC motif have been identified. These extended superfamily members, such as Dph2 (ref. 149) and ThiC,150,151 still catalyze reductive cleavage of SAM using [4Fe–4S] clusters as the electron donor to initiate free radical chemistry. However, structural and mechanistic characterizations have revealed aspects of these enzymes that are distinct from the canonical radical SAM enzymes. To date, all of the characterized noncanonical radical SAM enzymes catalyze reactions that involve C–C bond formations critical for the construction of carbon skeletons of cofactors or protein post-translational modifications. Comparison of these enzymes with the canonical radical SAM enzymes is important for understanding the mechanism of radical mediated C–C bond formations, and this section will focus on the key mechanistic questions revealed by studies of these noncanonical radical SAM enzymes.
6.1. Dph2 in the diphthamide biosynthesis
Dph2 catalyzes transfer of the 3-amino-3-carboxypropyl (ACP) group of SAM during the posttranslational modification of a His residue in elongation factor 2 (EF-2, Fig. 32). The modified residue is called diphthamide, and important for translational fidelity.149 Diphthamide is also the target of diphtheria toxin, and thus essential for the infectious disease diphtheria in humans.152 In this disease, the pathogenic bacteria, Coryne-bacterium diphtheria, kills host cells by secreting the diphtheria toxin protein which catalyzes ADP-ribosylation of diphthamide in the cell (Fig. 32).
Fig. 32.

The diphthamide biosynthetic pathway and the mechanism of diphtheria toxicity.
Diphthamide is biosynthesized through three major steps involving the action of the products of seven genes in yeast (Dph1–7, Fig. 32). The first step involves C–C bond formation between the target His residue and the ACP group of SAM. In yeast, this first step requires the products of four genes, Dph1–4, among which Dph2 is the radical SAM enzyme responsible for transfer of the ACP group of SAM to the His residue of EF-2 (see subsequent paragraph for the functions of Dph1, 3 and 4).149 An archaeal homolog of Dph2 from Pyrococcus horikoshii (PhDph2) was used for in vitro functional and structural characterization. Despite the fact that PhDph2, like eukaryotic Dph2, does not harbor the canonical CXXXCXXC radical SAM motif, the anaerobically purified enzyme was found to contain one [4Fe–4S] cluster ligated by three Cys residues149 which was proposed to bind and catalyze the reductive cleavage of SAM. Biochemical characterization of the products of the PhDph2 reaction revealed that the enzyme produces ACP-modified EF-2 and 5′-deoxy-5′-methylthio-adenosine (MTA), with no detectable amount of 5′-dA.149 When the PhDph2 assays were performed in the presence of a large excess of the nonphysiological chemical reductant dithionite and in the absence of EF-2 substrate, PhDph2 exhibited abortive cleavage of SAM,149 suggesting that substrate binding and SAM cleavage are not very well coupled in the reported assay conditions. Characterization of this uncoupled reaction revealed two major products (Fig. 33a): homo-cysteine sulphinic acid (HSA, 81) and 2-aminobutyric acid (ABA, 82).149 Formation of these two products from SAM was explained as the result of quenching of the ACP radical either by reaction with sodium dithionite, or by transfer of a H-atom or its equivalent, respectively. Quenching of radical intermediates in radical SAM enzymes with sodium dithionite has also been reported for an active-site mutant of spore photoproduct lyase.153 Therefore, the formation of HSA in the uncoupled reaction was used as evidence for formation of the ACP radical by PhDph2.
Fig. 33.

Reactions catalyzed by Dph2. (a) The proposed mechanism of PhDph2 catalysis. (b) Reaction of PhDph2 with the SAM analog SAMCA in the presence of dithionite yielded compounds 83 and 84. Compound 84 was found to react with the [4Fe–4S]+ cluster of PhDph2 present in the solution and form a π-complex. (c) The proposed electron relay in yeast between NorW, yeast Dph3 and the Dph1-Dph2 complex.
Further studies by the Lin lab using SAM analogs demonstrated the unique reactivity of the PhDph2 [4Fe–4S] cluster. In particular, the reaction of PhDph2 with SAMCA, a SAM analog with the ACP group replaced with a 3-carboxyallyl group, generated an organometallic species (Fig. 33b).154 Originally, SAMCA was used as a potential means of trapping the ACP radical as a stable allylic radical. However, the expected radical was not observed, and instead, the reaction of PhDph2 with SAMCA produced sulfinic acids 83 and 84 as a result of quenching of the expected allylic radical by sodium dithionite. One of the resulting sulfinic acids, α-sulfinyl-3-butenoic acid (84), was found to form a π-complex between the C═C double bond of α-sulfinyl-3-butenoic acid and the unique iron of the [4Fe–4S]1+ cluster (Fig. 33b). In a separate report, PhDph2 was also shown to catalyze methyl transfer reaction from decarboxy-or deamino-SAM analogs onto one of the S atoms of the [4Fe–4S] cluster.155 Together, these observations revealed the electron rich and potentially nucleophilic nature of the PhDph2 [4Fe–4S] cluster. The unique reactivity of the PhDph2 [4Fe–4S] cluster is particularly interesting considering the distinct selectivity of SAM cleavage in Dph2 vs. 5′-dA• forming radical SAM enzymes, and should be investigated in more detail in the future.
Another unique aspect of Dph2 is the presence of its specific reductase. As described above, in yeast, the formation of ACP-modified eEF-2 requires Dph2 and three other genes (Fig. 32). Dph1 forms a hetero-dimer complex with Dph2,156 and this complex is essential for the proper folding and catalytic activity of Dph2. While the function of Dph4 is unknown, Dph3 was found to serve as the reductase of the [4Fe–4S] cluster in the Dph1–Dph2 complex.156 Dph3 is a CSL-type zinc finger protein that can exist in either a Zn2+ or Fe2+ bound form.156 The Fe2+ bound form is catalytically active, and, together with its reductase, NorW, mediates electron transfer from NADH to reduce the [4Fe–4S]2+ cluster in the Dph1–Dph2 complex (Fig. 33c).156 Intriguingly, Dph3 did not serve as an electron donor for PhDph2, suggesting a species-specific interaction between the Dph1–Dph2 complex and Dph3. In general, the [4Fe–4S] clusters of radical SAM enzymes are believed to be reduced by flavodoxins. However, these interactions between flavodoxin and radical SAM enzymes are usually not specific. Thus, the specific interaction between Dph1–Dph2 and Dph3 is unique. As we have been discussing throughout this manuscript, the elucidation of a physiological redox partner is important for functional and mechanistic characterization of radical SAM enzymes, especially for those enzymes that catalyze a large abortive SAM cleavage reaction, such as Dph2 and PqqE, or those that catalyze non-physiological shunt reactions, such as NosL. Thus, it is of interest to investigate the presence of any specific redox partners for other radical SAM enzymes.
6.2. ThiC157 and BzaA/BzaB/BzaF158
Another subfamily of radical SAM enzymes with non-canonical SAM motifs includes ThiC in thiamin biosynthesis and BzaF (or BzaA/B) in anaerobic B12 biosynthesis. These enzymes have been proposed to use 5′-dA• and catalyze complex rearrangement reactions that involve C–C bond formation. Intriguingly, ThiC and BzaF use an identical substrate, 5-aminoimidazole ribonucleotide (AIR), but produce structurally distinct products with the two reactions proposed to diverge at the C–C bond formation step. Therefore, these enzymes demonstrate the significance of regiospecificity of C–C bond formation in the outcome of the overall reactions.
6.2.1. ThiC in thiamine biosynthesis
Thiamine pyrophosphate (ThDP) is a cofactor essential for carbohydrate and amino acid metabolism, and is required for life in all organisms. In bacteria, the thiazole and pyrimidine moieties of ThDP are biosynthesized separately as thiazole monophosphate (TMP) and 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P), respectively, followed by their conjugation and phosphorylation to form the biologically active form of the cofactor, ThDP (Fig. 34).
Fig. 34.

Overview of ThDP biosynthesis in bacteria.
The pyrimidine moiety of ThDP is biosynthesized from 5-aminoimidazole ribonucleotide (AIR), an intermediate in the purine biosynthetic pathway, by the radical SAM enzyme, HMP-P synthase or ThiC (Fig. 34). The mechanism of the ThiC-catalyzed reaction was initially investigated by isotope labeling experiments carried out in vivo as well as in cell free extracts.159,160 As seen in Fig. 35a, these experiments revealed that the ribose C-2′ is converted to the C-2 methyl of the ami-noimidazole, the ribose C-4′ is inserted between C-4 and C-5 of the aminoimidazole base, and the methyl hydrogens of HMP-P are derived from ribose H-2′ and H-3′ and the solvent.
Fig. 35.

Function, structure and mechanism of ThiC. (a) The reaction catalyzed by ThiC. The fate of the AIR atoms is highlighted by balls and colors. (b) Active site structure of A. thaliana ThiC in complex with imidazole ribonucleotide and SAH. The mononuclear metal center is occupied by Zn2+. The unique Fe of the [4Fe–4S] cluster is ligated by a chloride ion. (c) Proposed mechanism of ThiC catalysis.
More recently, two groups independently reported the successful in vitro reconstitution of ThiC activity using purified recombinant ThiC from Caulobacter crescentus151 and Salmonella enterica.150,161 In both cases, anaerobically purified ThiC harbored one oxygen sensitive [4Fe–4S] cluster and required SAM and a reductant (dithionite or flavodoxin/flavodoxin reductase) for the conversion of AIR to HMP-P. Mössbauer analysis of C. crescentus ThiC revealed the presence of one Fe(II) in a four coordinate with a Cys-rich environment, and a second in a five- or six-coordination environment with hard nitrogen/oxygen ligands.151 The presence of the putative metal binding site was also observed in X-ray crystal structures of Arabidopsis thaliana ThiC (AtThiC)162 in complex with imidazole ribonucleotide, a stable analog of AIR, and S-adenosyl-L-homocystein (SAH), a competitive inhibitor of ThiC. In this crystal structure, the mononuclear center was ligated by two strictly conserved His residues (H426 and H490) and the amine and carboxylic acid of SAH (Fig. 35b). The metal was identified as Zn2+ based on EXAFS and multiwavelength anomalous difference Fourier analyses of the crystals. This metal binding site was occupied by Fe2+ when Zn2+ was omitted from the crystallization conditions, suggesting that Fe2+ may be more physiologically relevant. The importance of this metal binding site for the catalytic function of ThiC was investigated by mutagenesis of the conserved His ligands, which resulted in a 15-fold decrease in ThiC activity.
The crystal structures also revealed the presence of a [4Fe–4S] cluster ligated by three strictly conserved Cys residues (Fig. 35b).162 However, unlike the canonical radical SAM enzymes, the unique Fe site in this cluster was not ligated by SAM, but occupied by a chloride ion. Based on these observations, ThiC was proposed to bind SAM through the mononuclear metal site and not through the unique Fe of the [4Fe–4S] cluster as has been observed for the canonical radical SAM enzymes.162 However, the mechanism of SAM cleavage was proposed to proceed through a mechanism similar to the canonical radical SAM enzymes, with the C5′–S bond being homolytically cleaved. This proposal was based on the observation in the crystal structures that the C5′–S bond of SAH was collinearly aligned with one of the Cys-ligated irons of the [4Fe–4S] cluster (Fig. 35b),162 a location feasible for homolytic cleavage of the C5′–S bond through backside attack by the Cys-ligated Fe on the S atom of SAM.
The mechanism of ThiC catalysis was investigated using deuterium labeled AIR, which revealed the unique reactivity of 5′-dA•. When [5′-D2]-AIR or [4′-D]-AIR was used as the substrate, incorporation of one deuterium atom into each molecule of 5′-dA was observed (Fig. 35a).163 When [2′,3′,4′,5′-D5]-AIR was used, incorporation of two deuterium atoms into each 5′-dA molecule was observed,163 while no deuterium incorporation into 5′-dA was observed with 1′, 2′, or 3′-labeled substrates.163 Since stoichiometric amounts of 5′-dA and HMP-P were formed,163 the deuterium labeling results suggested that two hydrogen atoms, H-4′ and H-5′, are incorporated into a single 5′-deoxyadenosine during the course of the ThiC-catalyzed reaction. Based on these observations, the complex mechanism shown in Fig. 35c was proposed, in which 5′-dA• abstracts a H-atom twice, once from H-5′ and once from H-4′. In this mechanism, catalysis is initiated by the abstraction of a H-5′ atom by 5′-dA•, which induces opening of the ribofuranose ring and dissociation of the aminoimidazole base. The dissociated aminoimidazole base attacks the cation at C-4, and the resulting C-5′ neutral radical abstracts a H-atom from 5′-dA to regenerate 5′-dA•. The second H-atom abstraction takes place at the 4′ position, which triggers the ring expansion of the imidazole ring to a pyrimidine ring. Subsequent rearrangement and release of two carbon units, an electron and a proton, results in formation of HMP-P. While the mechanism behind the unique reactivity of 5′-dA• in ThiC is unknown, the unique architecture of the ThiC active site compared to the canonical radical SAM enzymes may be important for the unique reactivity of ThiC.
Finally, it is noteworthy that a narrow paramagnetic EPR signal, likely associated with an organic radical species, was observed in both C. crescentus and S. enterica ThiC.151,164 The radical-harboring S. enterica ThiC species undergoes cleavage of the peptide bond between Gly436 and His437 upon exposure to oxygen.164 This observation is reminiscent of the reactivity of Glycyl radical harboring enzymes such as pyruvate formate lyase (PFL),165 suggesting that the ThiC radical may also reside on the peptide backbone. The exact identity and the relevance of this radical to ThiC catalysis is unknown. However, considering the complexity of the ThiC reaction, it is possible that an additional free radical cofactor is involved. Characterization of such radical species may hold a key to understanding this intriguing radical enzyme.
6.2.2. BzaA/BzaB/BzaF in the anaerobic biosynthesis of the cobalamin lower ligand
Recently, homologs of ThiC were found to be involved in the vitamin B12 (cobalamin) biosynthesis pathway in anaerobic bacteria. Cobalamin is required by the majority of animals, protists, and prokaryotes, while cobalamin and all other cobamide cofactors are synthesized only by prokaryotes.166 Cobalamin is composed of a cobalt ion coordinated by a tetrapyrrolic corrin ring, an upper ligand, and a lower ligand that is covalently tethered to the corrin ring by a nucleotide loop (Fig. 36a). While the lower ligand of cobalamin is 5,6-dimethylbenzimidazole (DMB), different structures are found in other cobamide compounds,167 and distinct cobamide biosynthetic pathways have been reported for aerobic and anaerobic organisms. In aerobic organisms, DMB is formed by the BluB enzyme, which converts reduced flavin mononucleotide (FMNH2) to DMB by an oxygen-dependent mechanism.168 DMB formation in the anaerobic pathway, on the other hand, remained elusive until recently.
Fig. 36.

Anaerobic biosynthesis of the lower ligand of cobalamin. (a) Structure of cobalamin. DMB lower ligand is highlighted in blue, the upper ligand in red. (b) The DMB biosynthetic pathway identified in E. limosum. (c) Results of isotope labeling experiments for DaBzaF. (d) Comparison of the proposed mechanisms for ThiC and BzaF.
Early isotope-labeling studies, performed mainly in the anaerobic bacterium Eubacterium limosum, suggested that anaerobic biosynthesis of DMB is an offshoot of the purine biosynthetic pathway.167 Recently, a set of genes for the anaerobic biosynthesis of DMB and other benzimidazoles was found.158 In this study, candidate genes were sought in the genome of E. limosum downstream to a cobalamin riboswitch. Of the 13 riboswitch sequences, one was located at the 5′ end of a putative operon containing five genes of unknown function in addition to a homolog of cobT, which encodes an enzyme necessary for the attachment of a lower ligand base to a cobamide precursor. This putative gene cluster was heterologously expressed in E. coli and demonstrated to be responsible for DMB biosynthesis.158 Subsequent gene knockout studies and characterization of accumulated biosynthetic intermediates successfully delineated the function of each gene in this cluster (Fig. 36b).158 In addition, AIR was proposed as a starting compound based on the similarity of BzaA to ThiC and the observation that deletion of the purK gene responsible for formation of AIR in the purine biosynthetic pathway abolished DMB biosynthesis.158 Based on these observations, the pathway for DMB biosynthesis shown in Fig. 36b was proposed.158 In this pathway, the first two steps are catalyzed by the two ThiC homologs, BzaA and BzaB, which construct the benzimidazole structure from AIR, with subsequent methylations and deoxygenation yielding DMB.
Involvement of a ThiC homolog in benzimidazole formation was demonstrated using Desulfuromonas acetoxidans BzaF (DaBzaF),169 a paralog of BzaA and BzaB. This operon contains only one copy of a ThiC homolog, DaBzaF, and is missing the rest of the bza genes.169 Still, recombinant DaBzaF alone was sufficient for the transformation of AIR into 5-hydroxybenzimidazole (5-OHBza), and produced near stoichiometric amounts of 5′-dA relative to 5-OHBza.169 The mechanism of this transformation was probed using a series of isotopically labeled AIR (Fig. 36c).169 In sharp contrast to the ThiC reaction, the AIR H-4′ was not abstracted by 5′-dA•, but was transferred to the 4′ position of 5-OHBza. From the reported results, it is not clear if each 5′-dA• generated by DaBzaF abstracts a H-atom once or twice. Nevertheless, a mechanism consistent with the isotope labeling pattern was proposed (Fig. 36d), in which DaBzaF catalyzes H-atom abstraction only once. The proposed mechanism for DaBzaF is identical to that for ThiC until dissociation of the aminoimidazole base, but differs by the position of C–C bond formation between the aminoimidazole base and ribose. The origin of the alteration in the regiospecificity of C–C bond formation is currently unknown. Comparison of the active site structures of DaBzaF and A. thaliana ThiC suggested a similar active site architecture with only four out of 21 amino acid residues in the first shell not conserved.169 Thus, it is likely that a subtle change in the active site structure/environment has a significant impact on the reaction specificity possibly at the H-atom abstraction step or during substrate activation. Further characterization of the structures and mechanisms of these two enzymes is needed to understand how they control the regio-specificity of C–C bond formation and therefore the fate of the overall reaction.
7. Conclusion
C–C bond forming radical SAM enzymes have emerged in the past decade. Their characterizations not only demonstrate interesting and novel reactivities of enzymes with free radicals, but also reveal many key steps in the biologically and medicinally important biosynthetic pathways. Considering the large size of the radical SAM superfamily and the addition of new, noncanonical radical SAM enzymes, it is easily conceivable that more radical SAM enzymes will be found in the future that catalyze unique and important C–C bond forming reactions. While the functional characterization of these enzymes has been in progress, their catalytic mechanisms are much less understood. In this review, we proposed three key aspects to consider regarding the mechanism of radical SAM enzymes: (1) the mechanism of radical initiation, (2) the mechanism of substrate activation, and (3) the mechanism of radical quenching. The C–C bond forming radical SAM enzymes discussed in this review provide excellent examples to highlight the importance of all three of these mechanistic aspects. For example, precisely controlled radical initiation is important for the specificities of reactions between the substrates and highly reactive 5′-dA• in all radical SAM enzymes. The nature of the reductant for the [4Fe–4S] cluster appears to be critical for the proper function of many radical SAM enzymes such as NosL, PqqE and Dph2. Another important question about the mechanism of radical initiation is raised by the regioselectivity difference between 5′-dA• forming enzymes and Dph2, the ACP• forming enzyme. The mechanism of substrate activation is likely important in MoaA and TunB as well as in MqnE that loses its specificity when halogenated substrate analogs are used, and the significance of radical quenching has been demonstrated for NikJ/PolH. Understanding the details of the mechanisms by which radical SAM enzymes control the fate of free radical reactions is important for future engineering of these enzymes. Such possibilities are demonstrated by PolH and NeoN, where mutation of the radical quenching Cys residue results in formation of altered products. Furthermore, the fact that BzaF and ThiC produce distinct products from the identical substrate using only subtly different active site structures highlights the importance of understanding the mechanism of radical reactivity control and the potential for future engineered radical catalysis in enzyme active sites. In sum, the C–C bond forming radical SAM enzymes provide an exciting platform for the discovery of novel enzyme catalyzed reactions as well as the future development of engineered enzyme catalysts.
Acknowledgments
8. Funding source statement
This work was supported by the Duke University Medical Center and National Institute of General Medical Sciences R01 GM112838 and R01 GM115729 (to K. Y.).
We thank Dr Margot Wuebbens for proofreading the manuscript and providing feedbacks.
Biographies
 Kenichi Yokoyama received his Ph.D. in 2008 from Tokyo Institute of Technology under the supervision of Professor Tadashi Eguchi, where he studied the aminoglycoside antibiotics biosynthesis. He then moved to MIT as a postdoc in the lab of Professor JoAnne Stubbe, where he studied the mechanism of a long-range radical propagation catalyzed by ribonucleotide reductase. In 2011, he started his independent career in the Department of Biochemistry at Duke University Medical Center as an assistant professor. His current research interests are the functions and mechanisms of enzymes involved in the biosynthesis and biological functions of natural products and cofactors.
Kenichi Yokoyama received his Ph.D. in 2008 from Tokyo Institute of Technology under the supervision of Professor Tadashi Eguchi, where he studied the aminoglycoside antibiotics biosynthesis. He then moved to MIT as a postdoc in the lab of Professor JoAnne Stubbe, where he studied the mechanism of a long-range radical propagation catalyzed by ribonucleotide reductase. In 2011, he started his independent career in the Department of Biochemistry at Duke University Medical Center as an assistant professor. His current research interests are the functions and mechanisms of enzymes involved in the biosynthesis and biological functions of natural products and cofactors.
 Edward A. Lilla received his B. Sc. in Biochemistry and Molecular Biology from the University of Georgia in 2011. In fall of the same year, he joined the Department of Biochemistry at Duke University Medical Center as a Ph.D. student. His graduate research in the Yokoyama lab mainly focused on the mechanism of antifungal nucleoside biosynthesis, in which his major achievement was the discovery of novel C–C bond forming radical SAM enzymes, PolH, and NikJ. He obtained a Ph.D. degree in 2017 and is now working for KBI Biopharma Inc.
Edward A. Lilla received his B. Sc. in Biochemistry and Molecular Biology from the University of Georgia in 2011. In fall of the same year, he joined the Department of Biochemistry at Duke University Medical Center as a Ph.D. student. His graduate research in the Yokoyama lab mainly focused on the mechanism of antifungal nucleoside biosynthesis, in which his major achievement was the discovery of novel C–C bond forming radical SAM enzymes, PolH, and NikJ. He obtained a Ph.D. degree in 2017 and is now working for KBI Biopharma Inc.
Footnotes
9. Conflicts of interest
No competing financial interests have been declared.
References
- 1.Landgraf BJ, McCarthy EL, Booker SJ. Annu Rev Biochem. 2016;85:485–514. doi: 10.1146/annurev-biochem-060713-035504. [DOI] [PubMed] [Google Scholar]
- 2.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo Y-R. Handbook of Bond Dissociation Energies in Organic Compounds. CRC Press; Washington, D.C: 2003. [Google Scholar]
- 4.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Marletta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 5.Vey JL, Drennan CL. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Proc Natl Acad Sci U S A. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 8.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. Proc Natl Acad Sci U S A. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolet Y, Amara P, Mouesca JM, Fontecilla-Camps JC. Proc Natl Acad Sci U S A. 2009;106:14867–14871. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA. Biochemistry. 2000;39:15668–15673. doi: 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- 11.Kampmeier JA. Biochemistry. 2010;49:10770–10772. doi: 10.1021/bi101509u. [DOI] [PubMed] [Google Scholar]
- 12.Wang SC, Frey PA. Biochemistry. 2007;46:12889–12895. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horitani M, Shisler K, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, Broderick JB. Science. 2016;352:822–825. doi: 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P. Biochem Biophys Res Commun. 1993;197:792–797. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 16.Bianchi V, Reichard P, Eliasson R, Pontis E, Krook M, Jornvall H, Haggardljungouist E. J Bacteriol. 1993;175:1590–1595. doi: 10.1128/jb.175.6.1590-1595.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crain AV, Broderick JB. Biochim Biophys Acta, Proteins Proteomics. 2013;1834:2512–2519. doi: 10.1016/j.bbapap.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanyal I, Gibson KJ, Flint DH. Arch Biochem Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 19.Srikanth GSC, Castle SL. Tetrahedron. 2005;61:10377–10441. [Google Scholar]
- 20.Della EW, Kostakis C, Smith PA. Org Lett. 1999;1:363–365. [Google Scholar]
- 21.Giese B. Angew Chem, Int Ed. 1989;28:969–980. [Google Scholar]
- 22.Wyszynski FJ, Lee SS, Yabe T, Wang H, Gomez-Escribano JP, Bibb MJ, Lee SJ, Davies GJ, Davis BG. Nat Chem. 2012;4:539–546. doi: 10.1038/nchem.1351. [DOI] [PubMed] [Google Scholar]
- 23.Lilla EA, Yokoyama K. Nat Chem Biol. 2016;12:905–907. doi: 10.1038/nchembio.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahanta N, Fedoseyenko D, Dairi T, Begley TP. J Am Chem Soc. 2013;135:15318–15321. doi: 10.1021/ja408594p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi S, Mahanta N, Fedoseyenko D, Williams H, Begley TP. J Am Chem Soc. 2017;139:10952–10955. doi: 10.1021/jacs.7b04209. [DOI] [PubMed] [Google Scholar]
- 26.Grell TAJ, Goldman PJ, Drennan CL. J Biol Chem. 2015;290:3964–3971. doi: 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haft DH, Basu MK. J Bacteriol. 2011;193:2745–2755. doi: 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barr I, Stich TA, Gizzi AS, Grove TL, Bonanno JB, Latham JA, Chung T, Wilmot CM, Britt RD, Almo SC, Klinman JP. Biochemistry. 2018;57:1306–1315. doi: 10.1021/acs.biochem.7b01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruender NA, Wilcoxen J, Britt RD, Bandarian V. Biochemistry. 2016;55:2122–2134. doi: 10.1021/acs.biochem.6b00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hänzelmann P, Hernandez HL, Menzel C, Garcia-Serres R, Huynh BH, Johnson MK, Mendel RR, Schindelin H. J Biol Chem. 2004;279:34721–34732. doi: 10.1074/jbc.M313398200. [DOI] [PubMed] [Google Scholar]
- 31.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J Am Chem Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 33.Yokoyama K, Ohmori D, Kudo F, Eguchi T. Biochemistry. 2008;47:8950–8960. doi: 10.1021/bi800509x. [DOI] [PubMed] [Google Scholar]
- 34.Goldman PJ, Grove TL, Booker SJ, Drennan CL. Proc Natl Acad Sci U S A. 2013;110:15949–15954. doi: 10.1073/pnas.1312228110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiocco SJ, Grove TL, Booker SJ, Elliott SJ. J Am Chem Soc. 2015;137:8664–8667. doi: 10.1021/jacs.5b03384. [DOI] [PubMed] [Google Scholar]
- 36.Mendel RR, Schwarz G. Coord Chem Rev. 2011;255:1145–1158. [Google Scholar]
- 37.Leimkuhler S, Wuebbens MM, Rajagopalan KV. Coord Chem Rev. 2011;255:1129–1144. doi: 10.1016/j.ccr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwarz G, Mendel RR, Ribbe MW. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 39.Williams M, Mizrahi V, Kana BD. Crit Rev Microbiol. 2014;40:18–29. doi: 10.3109/1040841X.2012.749211. [DOI] [PubMed] [Google Scholar]
- 40.Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, Hassani M, Huszar S, Trefzer C, Ma Z, Kaneko T, Mdluli KE, Franzblau S, Chatterjee AK, Johnson K, Mikusova K, Besra GS, Futterer K, Jacobs WR, Jr, Schultz PG. Proc Natl Acad Sci U S A. 2013;110:E2510–E2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuebbens MM, Rajagopalan KV. J Biol Chem. 1995;270:1082–1087. doi: 10.1074/jbc.270.3.1082. [DOI] [PubMed] [Google Scholar]
- 42.Rieder C, Eisenreich W, O’Brien J, Richter G, Gotze E, Boyle P, Blanchard S, Bacher A, Simon H. Eur J Biochem. 1998;255:24–36. doi: 10.1046/j.1432-1327.1998.2550024.x. [DOI] [PubMed] [Google Scholar]
- 43.Hover BM, Loksztejn A, Ribeiro AA, Yokoyama K. J Am Chem Soc. 2013;135:7019–7032. doi: 10.1021/ja401781t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiss J, Christensen E, Kurlemann G, Zabot MT, Dorche C. Hum Genet. 1998;103:639–644. doi: 10.1007/s004390050884. [DOI] [PubMed] [Google Scholar]
- 45.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hänzelmann P, Schindelin H. Proc Natl Acad Sci U S A. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanaujia SP, Jeyakanthan J, Nakagawa N, Balasubramaniam S, Shinkai A, Kuramitsu S, Yokoyama S, Sekar K. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:821–833. doi: 10.1107/S0907444910019074. [DOI] [PubMed] [Google Scholar]
- 48.Hover BM, Tonthat NK, Schumacher MA, Yokoyama K. Proc Natl Acad Sci U S A. 2015;112:6347–6352. doi: 10.1073/pnas.1500697112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mehta AP, Hanes JW, Abdelwahed SH, Hilmey DG, Hänzelmann P, Begley TP. Biochemistry. 2013;52:1134–1136. doi: 10.1021/bi3016026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lees NS, Hänzelmann P, Hernandez HL, Subramanian S, Schindelin H, Johnson MK, Hoffman BM. J Am Chem Soc. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frommer G, Mutikainen I, Pesch FJ, Hillgeris EC, Preut H, Lippert B. Inorg Chem. 1992;31:2429–2434. [Google Scholar]
- 52.Vanderveer JL, Vandenelst H, Reedijk J. Inorg Chem. 1987;26:1536–1540. [Google Scholar]
- 53.Hover BM, Yokoyama K. J Am Chem Soc. 2015;137:3352–3359. doi: 10.1021/ja512997j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinckley GT, Frey PA. Biochemistry. 2006;45:3219–3225. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chatgilialoglu C, Guerra M, Mulazzani QG. J Am Chem Soc. 2003;125:3839–3848. doi: 10.1021/ja029374d. [DOI] [PubMed] [Google Scholar]
- 56.Barr I, Latham JA, Iavarone AT, Chantarojsiri T, Hwang JD, Klinman JP. J Biol Chem. 2016;291:8877–8884. doi: 10.1074/jbc.C115.699918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen YQ, Bonnot F, Imsand EM, RoseFigura JM, Sjolander K, Klinman JP. Biochemistry. 2012;51:2265–2275. doi: 10.1021/bi201763d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anthony C. Antioxid Redox Signaling. 2001;3:757–774. doi: 10.1089/15230860152664966. [DOI] [PubMed] [Google Scholar]
- 59.Duine JA. J Biosci Bioeng. 1999;88:231–236. doi: 10.1016/s1389-1723(00)80002-x. [DOI] [PubMed] [Google Scholar]
- 60.Urban PF, Klingenberg M. Eur J Biochem. 1969;9:519–525. doi: 10.1111/j.1432-1033.1969.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 61.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. J Biol Chem. 2010;285:142–152. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Houck DR, Hanners JL, Unkefer CJ. J Am Chem Soc. 1988;110:6920–6921. [Google Scholar]
- 63.Vankleef MAG, Duine JA. FEBS Lett. 1988;237:91–97. doi: 10.1016/0014-5793(88)80178-9. [DOI] [PubMed] [Google Scholar]
- 64.Velterop JS, Sellink E, Meulenberg JJM, David S, Bulder I, Postma PW. J Bacteriol. 1995;177:5088–5098. doi: 10.1128/jb.177.17.5088-5098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Magnusson OT, Toyama H, Saeki M, Schwarzenbacher R, Klinman JP. J Am Chem Soc. 2004;126:5342–5343. doi: 10.1021/ja0493852. [DOI] [PubMed] [Google Scholar]
- 66.Toyama H, Chistoserdova L, Lidstrom ME. Microbiology. 1997;143:595–602. doi: 10.1099/00221287-143-2-595. [DOI] [PubMed] [Google Scholar]
- 67.Latham JA, Iavarone AT, Barr I, Juthani PV, Klinman JP. J Biol Chem. 2015;290:12908–12918. doi: 10.1074/jbc.M115.646521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans RL, Latham JA, Xia YL, Klinman JP, Wilmot CM. Biochemistry. 2017;56:2735–2746. doi: 10.1021/acs.biochem.7b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu SP, King D, Britt RD, Klinman JP. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goeransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Mueller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Suessmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McIntosh JA, Donia MS, Schmidt EW. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schramma KR, Bushin LB, Seyedsayamdost MR. Nat Chem. 2015;7:431–437. doi: 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davis KM, Schramma KR, Hansen WA, Bacik JP, Khare SD, Seyedsayamdost MR, Ando N. Proc Natl Acad Sci U S A. 2017;114:10420–10425. doi: 10.1073/pnas.1703663114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schramma KR, Seyedsayamdost MR. ACS Chem Biol. 2017;12:922–927. doi: 10.1021/acschembio.6b01069. [DOI] [PubMed] [Google Scholar]
- 76.Winn M, Goss RJ, Kimura K, Bugg TD. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 77.Campbell J, Singh AK, Maria JPS, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. ACS Chem Biol. 2011;6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Price NPJ, Tsvetanova B. J Antibiot. 2007;60:485–491. doi: 10.1038/ja.2007.62. [DOI] [PubMed] [Google Scholar]
- 79.Chen WQ, Qu DJ, Zhai LP, Tao MF, Wang YM, Lin SJ, Price NPJ, Deng ZX. Protein Cell. 2010;1:1093–1105. doi: 10.1007/s13238-010-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forouhar F, Arragain S, Atta M, Gambarelli S, Mouesca JM, Hussain M, Xiao R, Kieffer-Jaquinod S, Seetharaman J, Acton TB, Montelione GT, Mulliez E, Hunt JF, Fontecave M. Nat Chem Biol. 2013;9:333–338. doi: 10.1038/nchembio.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bridwell-Rabb J, Zhong A, Sun HG, Drennan CL, Liu H-w. Nature. 2017;544:322–326. doi: 10.1038/nature21689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Munro CA. Adv Appl Microbiol. 2013;83:145–172. doi: 10.1016/B978-0-12-407678-5.00004-0. [DOI] [PubMed] [Google Scholar]
- 83.Shubitz LF, Trinh HT, Perrill RH, Thompson CM, Hanan NJ, Galgiani JN, Nix DE. J Infect Dis. 2014;209:1949–1954. doi: 10.1093/infdis/jiu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Isono K, Sato T, Hirasawa K, Funayama S, Suzuki S. J Am Chem Soc. 1978;100:3937–3939. [Google Scholar]
- 85.Chen W, Huang T, He X, Meng Q, You D, Bai L, Li J, Wu M, Li R, Xie Z, Zhou H, Zhou X, Tan H, Deng Z. J Biol Chem. 2009;284:10627–10638. doi: 10.1074/jbc.M807534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ginj C, Ruegger H, Amrhein N, Macheroux P. ChemBioChem. 2005;6:1974–1976. doi: 10.1002/cbic.200500208. [DOI] [PubMed] [Google Scholar]
- 87.He N, Wu P, Lei Y, Xu B, Zhu X, Xu G, Gao Y, Qi J, Deng Z, Tang G, Chen W, Xiao Y. Chem Sci. 2017;8:444–451. doi: 10.1039/c6sc01826b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang ZY, Chi XL, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J, Van Lanen SG. J Biol Chem. 2011;286:7885–7892. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barnard-Britson S, Chi X, Nonaka K, Spork AP, Tibrewal N, Goswami A, Pahari P, Ducho C, Rohr J, Van Lanen SG. J Am Chem Soc. 2012;134:18514–18517. doi: 10.1021/ja308185q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kudo F, Hoshi S, Kawashima T, Kamachi T, Eguchi T. J Am Chem Soc. 2014;136:13909–13915. doi: 10.1021/ja507759f. [DOI] [PubMed] [Google Scholar]
- 91.Benjdia A, Guillot A, Ruffie P, Leprince J, Berteau O. Nat Chem. 2017;9:698–707. doi: 10.1038/nchem.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shimada N, Hasegawa S, Harada T, Tomisawa T, Fujii A, Takita T. J Antibiot. 1986;39:1623–1625. doi: 10.7164/antibiotics.39.1623. [DOI] [PubMed] [Google Scholar]
- 93.Nakamura H, Hasegawa S, Shimada N, Fujii A, Takita T, Iitaka Y. J Antibiot. 1986;39:1626–1629. doi: 10.7164/antibiotics.39.1626. [DOI] [PubMed] [Google Scholar]
- 94.Izuta S, Shimada N, Kitagawa M, Suzuki M, Kojima K, Yoshida S. J Biochem. 1992;112:81–87. doi: 10.1093/oxfordjournals.jbchem.a123870. [DOI] [PubMed] [Google Scholar]
- 95.Seki J, Shimada N, Takahashi K, Takita T, Takeuchi T, Hoshino H. Antimicrob Agents Chemother. 1989;33:773–775. doi: 10.1128/aac.33.5.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ueda K, Tsurimoto T, Nagahata T, Chisaka O, Matsubara K. Virology. 1989;169:213–216. doi: 10.1016/0042-6822(89)90057-3. [DOI] [PubMed] [Google Scholar]
- 97.Morita M, Tomita K, Ishizawa M, Takagi K, Kawamura F, Takahashi H, Morino T. Biosci, Biotechnol, Biochem. 1999;63:563–566. doi: 10.1271/bbb.63.563. [DOI] [PubMed] [Google Scholar]
- 98.Aravind L, Koonin EV. Trends Biochem Sci. 1998;23:469–472. doi: 10.1016/s0968-0004(98)01293-6. [DOI] [PubMed] [Google Scholar]
- 99.Bridwell-Rabb J, Kang G, Zhong A, Liu HW, Drennan CL. Proc Natl Acad Sci U S A. 2016;113:13750–13755. doi: 10.1073/pnas.1613610113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong YS, Castelfranco PA, Goff DA, Smith KM. Plant Physiol. 1985;79:725–729. doi: 10.1104/pp.79.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walker CJ, Mansfield KE, Smith KM, Castelfranco PA. Biochem J. 1989;257:599–602. doi: 10.1042/bj2570599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nasrulhaq-Boyce A, Griffths WT, Jones OT. Biochem J. 1987;243:23–29. doi: 10.1042/bj2430023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porra RJ, Schafer W, Gad’on N, Katheder I, Drews G, Scheer H. Eur J Biochem. 1996;239:85–92. doi: 10.1111/j.1432-1033.1996.0085u.x. [DOI] [PubMed] [Google Scholar]
- 105.Porra RJ, Schafer W, Katheder I, Scheer H. FEBS Lett. 1995;371:21–24. doi: 10.1016/0014-5793(95)00854-3. [DOI] [PubMed] [Google Scholar]
- 106.Pinta V, Picaud M, Reiss-Husson F, Astier C. J Bacteriol. 2002;184:746–753. doi: 10.1128/JB.184.3.746-753.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang ZM, Bauer CE. J Bacteriol. 1990;172:5001–5010. doi: 10.1128/jb.172.9.5001-5010.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ouchane S, Steunou AS, Picaud M, Astier C. J Biol Chem. 2004;279:6385–6394. doi: 10.1074/jbc.M309851200. [DOI] [PubMed] [Google Scholar]
- 109.Gough SP, Petersen BO, Duus JO. Proc Natl Acad Sci U S A. 2000;97:6908–6913. doi: 10.1073/pnas.97.12.6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Porra RJ, Urzinger M, Winkler J, Bubenzer C, Scheer H. Eur J Biochem. 1998;257:185–191. doi: 10.1046/j.1432-1327.1998.2570185.x. [DOI] [PubMed] [Google Scholar]
- 111.Booker SJ. Curr Opin Chem Biol. 2009;13:58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Booker SJ. Curr Opin Chem Biol. 2009;13:58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J Am Chem Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 114.Fang Q, Peng J, Dierks T. J Biol Chem. 2004;279:14570–14578. doi: 10.1074/jbc.M313855200. [DOI] [PubMed] [Google Scholar]
- 115.Layer G, Verfurth K, Mahlitz E, Jahn D. J Biol Chem. 2002;277:34136–34142. doi: 10.1074/jbc.M205247200. [DOI] [PubMed] [Google Scholar]
- 116.Challand MR, Martins FT, Roach PL. J Biol Chem. 2010;285:5240–5248. doi: 10.1074/jbc.M109.056606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Driesener RC, Challand MR, McGlynn SE, Shepard EM, Boyd ES, Broderick JB, Peters JW, Roach PL. Angew Chem, Int Ed. 2010;49:1687–1690. doi: 10.1002/anie.200907047. [DOI] [PubMed] [Google Scholar]
- 118.Zhang Q, Li YX, Chen DD, Yu Y, Duan LA, Shen B, Liu W. Nat Chem Biol. 2011;7:154–160. doi: 10.1038/nchembio.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ji XJ, Li YZ, Jia YL, Ding W, Zhang Q. Angew Chem, Int Ed. 2016;55:3334–3337. doi: 10.1002/anie.201509900. [DOI] [PubMed] [Google Scholar]
- 120.Ji XJ, Li YZ, Ding W, Zhang Q. Angew Chem, Int Ed. 2015;54:9021–9024. doi: 10.1002/anie.201503976. [DOI] [PubMed] [Google Scholar]
- 121.Bhandari DM, Xu H, Nicolet Y, Fontecilla-Camps JC, Begley TP. Biochemistry. 2015;54:4767–4769. doi: 10.1021/acs.biochem.5b00764. [DOI] [PubMed] [Google Scholar]
- 122.Sicoli G, Mouesca JM, Zeppieri L, Amara P, Martin L, Barra AL, Fontecilla-Camps JC, Gambarelli S, Nicolet Y. Science. 2016;351:1320–1323. doi: 10.1126/science.aad8995. [DOI] [PubMed] [Google Scholar]
- 123.Boshoff HI, Barry CE., III Nat Rev Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 124.Graham DE, White RH. Nat Prod Rep. 2002;19:133–147. doi: 10.1039/b103714p. [DOI] [PubMed] [Google Scholar]
- 125.Glas AF, Maul MJ, Cryle M, Barends TR, Schneider S, Kaya E, Schlichting I, Carell T. Proc Natl Acad Sci U S A. 2009;106:11540–11545. doi: 10.1073/pnas.0812665106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Walsh C. Acc Chem Res. 1986;19:216–221. [Google Scholar]
- 127.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., III Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Purwantini E, Mukhopadhyay B. Proc Natl Acad Sci U S A. 2009;106:6333–6338. doi: 10.1073/pnas.0812883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nakano T, Miyake K, Endo H, Dairi T, Mizukami T, Katsumata R. Biosci, Biotechnol, Biochem. 2004;68:1345–1352. doi: 10.1271/bbb.68.1345. [DOI] [PubMed] [Google Scholar]
- 130.Coats JH, Li GP, Kuo MS, Yurek DA. J Antibiot. 1989;42:472–474. doi: 10.7164/antibiotics.42.472. [DOI] [PubMed] [Google Scholar]
- 131.Choi KP, Kendrick N, Daniels L. J Bacteriol. 2002;184:2420–2428. doi: 10.1128/JB.184.9.2420-2428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Decamps L, Philmus B, Benjdia A, White R, Begley TP, Berteau O. J Am Chem Soc. 2012;134:18173–18176. doi: 10.1021/ja307762b. [DOI] [PubMed] [Google Scholar]
- 133.Philmus B, Decamps L, Berteau O, Begley TP. J Am Chem Soc. 2015;137:5406–5413. doi: 10.1021/ja513287k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beckwith ALJ. Chem Soc Rev. 1993;22:143–151. [Google Scholar]
- 135.Cooper LE, Fedoseyenko D, Abdelwahed SH, Kim SH, Dairi T, Begley TP. Biochemistry. 2013;52:4592–4594. doi: 10.1021/bi400498d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nowicka B, Kruk J. Biochim Biophys Acta, Bioenerg. 2010;1797:1587–1605. doi: 10.1016/j.bbabio.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 137.Cranenburg ECM, Schurgers LJ, Vermeer C. Thromb Haemostasis. 2007;98:120–125. [PubMed] [Google Scholar]
- 138.Plaza SM, Lamson DW. Alternative Med Rev. 2005;10:24–35. [PubMed] [Google Scholar]
- 139.Bentley R, Meganathan R. Microbiol Rev. 1982;46:241–280. doi: 10.1128/mr.46.3.241-280.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Meganathan R. Vitam Horm. 2001;61:173–218. doi: 10.1016/s0083-6729(01)61006-9. [DOI] [PubMed] [Google Scholar]
- 141.Dairi T, Kuzuyama T, Nishiyama M, Fujii I. Nat Prod Rep. 2011;28:1054–1086. doi: 10.1039/c0np00047g. [DOI] [PubMed] [Google Scholar]
- 142.Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H, Dairi T. Science. 2008;321:1670–1673. doi: 10.1126/science.1160446. [DOI] [PubMed] [Google Scholar]
- 143.Seto H, Jinnai Y, Hiratsuka T, Fukawa M, Furihata K, Itoh N, Dairi T. J Am Chem Soc. 2008;130:5614–5615. doi: 10.1021/ja710207s. [DOI] [PubMed] [Google Scholar]
- 144.Li X, Apel D, Gaynor EC, Tanner ME. J Biol Chem. 2011;286:19392–19398. doi: 10.1074/jbc.M111.229781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ji XJ, Li YZ, Xie LQ, Lu HJ, Ding W, Zhang Q. Angew Chem, Int Ed. 2016;55:11845–11848. doi: 10.1002/anie.201605917. [DOI] [PubMed] [Google Scholar]
- 146.Bruender NA, Grell TAJ, Dowling DP, McCarty RM, Drennan CL, Bandarian V. J Am Chem Soc. 2017;139:1912–1920. doi: 10.1021/jacs.6b11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim HJ, Liu YN, McCarty RM, Liu HW. J Am Chem Soc. 2017;139:16084–16087. doi: 10.1021/jacs.7b09890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sato S, Kudo F, Kim SY, Kuzuyama T, Eguchi T. Biochemistry. 2017;56:3519–3522. doi: 10.1021/acs.biochem.7b00472. [DOI] [PubMed] [Google Scholar]
- 149.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891–896. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martinez-Gomez NC, Downs DM. Biochemistry. 2008;47:9054–9056. doi: 10.1021/bi8010253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Nat Chem Biol. 2008;4:758–765. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Collier RJ. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- 153.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-De-Choudens S, Fontecave M, Atta M. J Biol Chem. 2008;283:36361–36368. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dong M, Horitani M, Dzikovski B, Pandelia ME, Krebs C, Freed JH, Hoffman BM, Lin H. J Am Chem Soc. 2016;138:9755–9758. doi: 10.1021/jacs.6b04155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dong M, Horitani M, Dzikovski B, Freed JH, Ealick SE, Hoffman BM, Lin H. J Am Chem Soc. 2017;139:5680–5683. doi: 10.1021/jacs.7b01712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, Lin H. J Am Chem Soc. 2014;136:1754–1757. doi: 10.1021/ja4118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Nat Chem Biol. 2008;4:758–765. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hazra AB, Han AW, Mehta AP, Mok KC, Osadchiy V, Begley TP, Taga ME. Proc Natl Acad Sci U S A. 2015;112:10792–10797. doi: 10.1073/pnas.1509132112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lawhorn BG, Mehl RA, Begley TP. Org Biomol Chem. 2004;2:2538–2546. doi: 10.1039/B405429F. [DOI] [PubMed] [Google Scholar]
- 160.Newell PC, Tucker RG. Biochem J. 1968;106:279–287. doi: 10.1042/bj1060279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Palmer LD, Downs DM. J Biol Chem. 2013;288:30693–30699. doi: 10.1074/jbc.M113.500280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fenwick MK, Mehta AP, Zhang Y, Abdelwahed SH, Begley TP, Ealick SE. Nat Commun. 2015;6:6480. doi: 10.1038/ncomms7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chatterjee A, Hazra AB, Abdelwahed S, Hilmey DG, Begley TP. Angew Chem, Int Ed Engl. 2010;49:8653–8656. doi: 10.1002/anie.201003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Martinez-Gomez NC, Poyner RR, Mansoorabadi SO, Reed GH, Downs DM. Biochemistry. 2009;48:217–219. doi: 10.1021/bi802154j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wagner AFV, Frey M, Neugebauer FA, Schafer W, Knappe J. Proc Natl Acad Sci U S A. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Roth JR, Lawrence JG, Bobik TA. Annu Rev Microbiol. 1996;50:137–181. doi: 10.1146/annurev.micro.50.1.137. [DOI] [PubMed] [Google Scholar]
- 167.Renz P. Biosynthesis of the 5,6-dimethylbenzimidazole moiety of cobalamin and of the other bases found in natural corrinoids. John Wiley & Sons, Inc; New York: 1999. [Google Scholar]
- 168.Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. Nature. 2007;446:449–453. doi: 10.1038/nature05611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mehta AP, Abdelwahed SH, Fenwick MK, Hazra AB, Taga ME, Zhang Y, Ealick SE, Begley TP. J Am Chem Soc. 2015;137:10444–10447. doi: 10.1021/jacs.5b03576. [DOI] [PMC free article] [PubMed] [Google Scholar]