

Abstract
The RNA-guided nucleases derived from the CRISPR-Cas systems in bacteria and archaea have found numerous applications in biotechnology, including genome editing, imaging, and gene regulation. However, the discovery of novel Cas nucleases has outpaced their characterization and subsequent exploitation. A key step in characterizing Cas nucleases is determining which protospacer-adjacent motif (PAM) sequences they recognize. Here, we report advances to an in vitro method based on an E. coli cell-free transcription-translation system (TXTL) to rapidly elucidate PAMs recognized by Cas nucleases. The method obviates the need for cloning Cas nucleases or gRNAs, does not require the purification of protein or RNA, and can be performed in less than a day. To advance our previously published method, we incorporated an internal GFP cleavage control to assess the extent of library cleavage as well as Sanger sequencing of the cleaved library to assess PAM depletion prior to next-generation sequencing. We also detail the methods needed to construct all relevant DNA constructs, and how to troubleshoot the assay. We finally demonstrate the technique by determining PAM sequences recognized by the Neisseria meningitidis Cas9, revealing subtle sequence requirements of this highly specific PAM. The overall method offers a rapid means to identify PAMs recognized by diverse CRISPR nucleases, with the potential to greatly accelerate our ability to characterize and harness novel CRISPR nucleases across their many uses.
Keywords: Cas9, Cas12a, PAM, sgRNA, crRNA, TXTL
1. Introduction
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-CRISPR-associated (Cas) systems have become widespread biomolecular tools. In nature, these systems act as an adaptive immune system that protects prokaryotes from mobile genetic elements such as plasmids and bacteriophage [1–5]. In addition to their fascinating role in microbial ecology and evolution, these systems have proved to be remarkably useful, and have been repurposed for numerous applications such as gene regulation, imaging, and, notably, genome editing [6–9].
CRISPR-Cas systems are unique in that targeting them to cleave or bind to a new DNA sequence requires only the expression of a new non-coding guide RNA (gRNA). Upon expression, a Cas nuclease and its gRNA form a ribonucleoprotein complex (RNP). This RNP targets and cleaves near DNA sequences (called protospacers) that are complementary to the specificity-determining region (called the spacer) of the gRNA and are flanked by a protospacer-adjacent motif (PAM) (Fig. 1A) [10]. Because the specificity of targeting derives from the spacer sequence of the gRNA base-pairing with the sequence of interest, re-engineering the target site simply involves the modification of the gRNA. In contrast, protein engineering was required to change the targeting specificity of other prior genome-editing technologies [11–13].
Fig. 1.
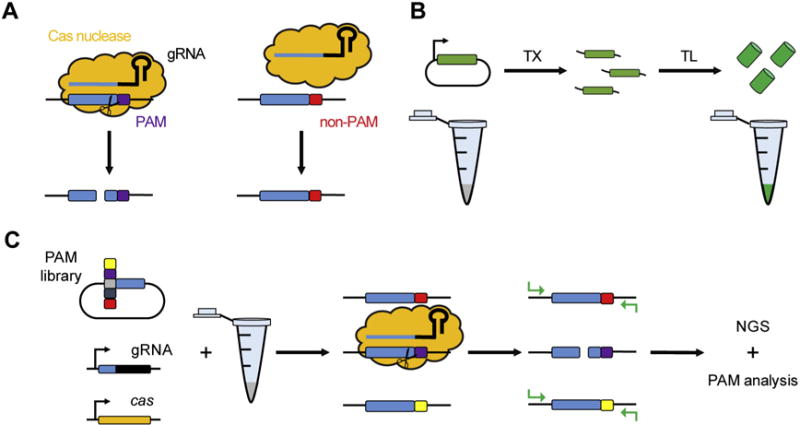
An overview of TXTL and its application for PAM determination. (A) Targeting by Cas nucleases. Targeting relies on a ribonucleoprotein complex composed of the Cas nuclease(s) and a guide RNA (gRNA) targeting DNA flanked by a PAM. Sequences lacking a PAM are ignored by the complex, even if the protospacer is perfectly complementary to the spacer portion of the gRNA. (B) Image showing the basic concept of TXTL. DNA encoding genes or non-coding RNA can be expressed in a single TXTL reaction. (C) The flow of a PAM characterization assay using TXTL. A plasmid library containing the randomized PAM sequence as well as DNA encoding a Cas nuclease and its gRNA can be added to a TXTL mix. After expression of the Cas nuclease and its gRNA, a ribonucleoprotein complex will be formed. Then the PAM libraries containing PAMs will be cleaved, while those containing a non-PAM PCR amplified with appropriate adapters and indices for analysis through a NGS platform. The PAM of the Cas nuclease will then be deciphered through a depletion analysis.
The vast majority of CRISPR-based technologies have utilized DNA-targeting Type II Cas9 nucleases [14,15] or Type V-A Cas12a (Cpf1) nucleases [16]. However, these two systems are a small subset of the stunningly diverse array of CRISPR-Cas systems found in nature. CRISPR-Cas systems are currently divided into two classes, six types, and over 30 subtypes [17], and are estimated to be present in approximately 50% and 90% of bacteria and archaea, respectively [18]. The characterization of some of these systems has revealed nucleases containing the ability to degrade (rather than simply cleave) DNA, recognize diverse PAM sequences, recognize fewer off-target sequences, and efficiently process their own gRNA arrays for multiplexing in eukaryotic systems [19–23]. Furthermore, the recent discovery of the Type VI Cas nuclease, Cas13a, was found to target RNA instead of DNA and has opened new technological opportunities based on RNA targets [19,24]. However, despite these advances, most CRISPR-Cas systems remain to be characterized and/or adapted for use in biotechnology.
Determining the PAM requirements of a CRISPR-Cas system is a critical step in its characterization. All characterized CRISPR-Cas systems, except for Type III, have required a PAM or PAM-related sequence for efficient cleavage of target DNA [14,25–27]. Conversely, identifying sequences flanked by PAMs is critical for predicting and minimizing off-target effects [28]. The PAM sequence required by a particular system varies widely in sequence length, content, and position relative to the protospacer. This variation occurs not only across higher-level (Class, Type) divisions of CRISPR-Cas systems, but also within subtypes among the species from which the Cas nuclease was isolated [23]. Furthermore, some Cas nucleases are able to utilize multiple PAM sequences with complicated relationships between the nucleotides required at different positions in the PAM [29]. Therefore, methods to characterize the PAM requirements of particular Cas nucleases play a vital role in studying their properties.
1.1. Previous methods to characterize PAMs of CRISPR-Cas systems
Previous methods have been developed to identify PAMs of various Cas nucleases. These methods include the original in silico method used to demonstrate the existence of PAMs [26], as well as the development of more recent high-throughput in vivo [16,30–34] and in vitro [16,35–38] assays. Here, we briefly describe each method. In-depth descriptions of each method have been provided elsewhere [10,39].
1.1.1. In silico PAM determination
PAMs were originally identified using bioinformatics [26]. Once it was realized that CRISPR spacers were derived from mobile genetic elements, alignments of the spacers to plasmids and phage genomes revealed the existence of a motif that flanked the protospacers. This method continues to be used to identify PAMs. By aligning incorporated spacer sequences from a prokaryotic genome containing a CRISPR array to bacteriophage or plasmid sequences, PAMs of various CRISPR-Cas systems can be deciphered. However, this method is limited by the availability of identifying matching sequences; many spacers appear to recognize ‘dark matter’ [40–47]. Furthermore, the relatively small number of spacers (tens to hundreds) that can be examined this way may not be sufficient to identify weakly recognized PAMs. Finally, this method conflates the PAM requirements for acquiring a spacer and using a spacer for interference despite differences in these sequences [48]. These limitations have been overcome by assaying the ability of libraries of randomized sequences to act as PAMs.
1.1.2. In vivo PAM determination
The first in vivo PAM assay took advantage of the ability of CRISPR-Cas systems to cure bacteria of plasmids [16,30–33]. To assay for functional PAMs, E. coli cells were co-transformed with (1) a plasmid containing a randomized PAM library flanked by a unique protospacer and plasmids encoding (2) a Cas nuclease and (3) a gRNA targeting the protospacer. Cells were then selected for all three plasmids by recovering them on media containing appropriate antibiotics. Plasmids containing a PAM are cleaved and the cells that contain them could not grow, while cells containing plasmids with non-PAMs were able to propagate. By comparing the frequency of a sequence in the library before and after selection, individual PAM sequences could be identified. While this assay has been used to successfully determine the PAMs of various Cas nucleases, it is limited by the requirement of high library coverage and the potential for cells containing mutated Cas or gRNA plasmids but functional PAMs to be amplified during cell outgrowth.
Soon after, another in vivo assay, named “PAM screen achieved by NOT-gate repression” (PAM-SCANR), was developed [34]. This assay was the first to involve the binding of catalytically inactive Cas nucleases to a target sequence containing a PAM. To assess the binding event, a genetic circuit involving the LacI repressor and GFP reporter was engineered to ensure a positive, fluorescent readout of PAMs. The cells expressing GFP were sorted through fluorescence-activated cell sorting (FACS), and with subsequent plasmid preparations, library prep, and a next-generation sequencing (NGS) run, functional PAMs were identified. While this assay reduced the limitations from the previous in vivo assay, PAM-SCANR is limited by the requirement of cloning in the Cas nuclease and its gRNA of interest into the PAM-SCANR plasmid system, identifying and mutating the catalytic domains of the Cas nuclease of interest, as well as in vivo limitations to library coverage.
1.1.3. In vitro PAM determination
Previously developed in vitro assays are based on cleaving PAM library plasmids within in vitro conditions followed by either a positive [35,36,38] or negative [16,37] screen for cleavage. Negative screens involve incubating a PAM library with purified Cas nucleases and in vitro transcribed gRNAs. After an appropriate reaction time, plasmids containing PAMs were cleaved, while plasmids with non-PAMs remained intact. This depletion of PAMs was measured by preparing a high-throughput sequencing library using PCR, which does not amplify cleaved sequences. Positive screens proceed similarly, except that adapters are ligated to the free ends of cleaved sequences for NGS. While these assays give the user more control of reaction conditions and place fewer restriction on library size, they require the cloning and protein purification of each Cas nuclease in the system and in vitro transcription of gRNAs, limiting their throughput.
1.2. TXTL-based PAM determination
While the previously developed systematic (as opposed to bioinformatic) PAM assays each have their own limitations, a common disadvantage is the time required to perform them. Be they transformations, cultures, or protein purification, all of the previously developed assays involve time-intensive protocols, requiring weeks to months to complete.
To contribute to the characterization of CRISPR-Cas systems, we developed a PAM assay based on an E. coli cell-free transcription-translation system (TXTL) [49,50]. TXTL systems are generally capable of in vitro expression of RNA and proteins in a single reaction (Fig. 1B). Our TXTL platform has the added advantage of containing the native transcriptional and translational machinery (i.e. E. coli core RNA polymerase, σ70, and ribosomes) as well as all proteins (e.g. RNase III) found in E. coli, which allows for flexible use of various expression constructs and proper prokaryotic RNA processing. DNA that encodes for non-coding RNA and proteins can be added to TXTL and expressed within a few hours [51]. Compared to the previous PAM assays, this system has two key benefits: (1) expression of the necessary protein and RNA components for the PAM assay and cleavage by the RNP complex occur in the TXTL reaction, which removes the need for protein purification; and (2) linear DNA can be used to express the necessary components, which largely eliminates the need for cloning.
To perform the PAM characterization assay with TXTL, plasmid or linear DNA expressing the necessary Cas nucleases and gRNAs are added to the TXTL mix. RNA expression and protein translation by the TXTL mix result in the formation of the RNP complex (Fig. 1C). If linear DNA is used, a RecBCD inhibitor must be added to protect the DNA from degradation [52]. The gRNA is designed to target a library of plasmids containing a conserved sequence flanked by a randomized set of potential PAM sequences. Depletion of PAM sequences from the library is measured by adding the adapters and indices necessary for high-throughput sequencing using PCR to both the cleaved library and to a control library expressing a non-targeting gRNA.
Here, we used this TXTL-based assay to rapidly characterize the PAM of a Cas9 originating from the bacterium Neisseria meningitides (NmeCas9). Our assay is improved relative to the assay described in Marshall and Maxwell et al. (2018) by the following: (1) using ten variable nucleotides flanking the protospacer in the randomized PAM library, (2) describing how to generate an internal control for Cas protein activity using a fluorescent reporter plasmid, and (3) describing how to use Sanger sequencing to verify Cas protein activity. The PAM of NmeCas9 was previously characterized [31,53,54]. We chose to demonstrate our assay using this system since it has an exceptionally long PAM (consensus NNNNGATT), which represents a ‘worst-case scenario’ for characterizing a new Cas nuclease. Because the PAM of NmeCas9 is known, we also showed additional control experiments that can be used to measure the kinetics of cleavage by the nuclease in TXTL, which is possible only when a PAM for the nuclease is known.
2. Methods
Here, we describe the detailed methods to perform a PAM determination assay for any Cas nuclease of interest. All DNA used in this procedure should be suspended in molecular biology-grade, nuclease-free H2O (e.g. ThermoFisher, 10977-015) and not an elution buffer provided in DNA preparation kits unless specified. All plasmid DNA should be prepared using a midiprep (not a mini-prep) kit, eluting in nuclease-free H2O by following the protocol associated with the midiprep kit. We have found that preparing plasmid DNA using midiprep leads to more consistent experimental results. All oligos, plasmid DNA, and gene fragment sequences are reported in the Appendix section.
2.1. Creating DNA required for PAM assay
Materials
Plasmid CSM160
Gene fragment CSM-GB089
Oligos CSMpr1308-1311
Oligos Chi6.FWD/REV
NEBuilder HiFi DNA Assembly Cloning Kit (NEB, E5520S)
Midiprep kit (e.g. Zymo Research DNA Clean & Concentrate-5, D4013)
Q5 Hot Start High-Fidelity DNA Polymerase (NEB, M0493S)
DpnI (20,000 units/ml; NEB, R0176)
LB media (e.g. ThermoFisher, 10855-001)
LB + chloramphenicol agar plates (e.g. bioWORLD, 30627011-1)
LB supplemented with 25 mg/ml chloramphenicol
Chloramphenicol (e.g. MilliporeSigma, R4408)
DNA purification kit (e.g. Genesee Scientific, 11-302)
NEB 5-alpha electrocompetent cells (NEB, C2989K)
DNA ladder (e.g. NEB, N3200S)
Optional materials
Oligos T7.FWD/REV (optional for expressing Cas nucleases using linear DNA)
Oligos to add the PAM-library protospacer flanked by a known PAM in the reporter plasmid
Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) to add the PAM-library protospacer flanked by a known PAM in the reporter plasmid
Plasmid P70a-deGFP (Addgene, #40019) to create a reporter plasmid for rate-of-cleavage control reactions
Protocol
2.1.1. Annealing Chi6 DNA
To successfully express linear DNA in TXTL, inhibitors of the RecBCD complex such as GamS or DNA containing Chi sites must be added [52]. The latter is particularly useful because it is inexpensive and easily obtained. For the experiments described below, we added Chi6 DNA (DNA containing six χ sites) whenever linear DNA was added to the reaction. To make a 50 mM stock of Chi6 DNA, 50 ml of 100 mM Chi6.FWD and Chi6.REV oligos suspended in nuclease-free H2O were mixed in a PCR tube and annealed in a thermocycler using the program in Table 1.
Table 1.
Thermocycler program used to anneal oligos to create Chi6 DNA.
| Temperature (°C) | Time (s) |
|---|---|
| 95 | 300 |
| Reduce temperature by 1 °C | 15 |
| REPEAT 79x | |
| 4–10 | ∞ |
2.1.2. Creating DNA to express Cas nucleases
To prepare DNA for expressing your Cas nuclease of interest, three options are available.
First, the Cas nuclease can be expressed from a bacterial expression vector with a constitutive or inducible promoter (pBAD18, pET, etc.). Use standard cloning techniques to generate these constructs. Prepare the plasmid DNA using a midiprep kit. Measure the concentration of the plasmid using standard techniques. If the concentration of the DNA is less than 20 nM, the plasmid can be concentrated using a DNA purification kit.
Second, the Cas nuclease can be expressed from linear DNA encoding the Cas nuclease under the control of a T7 promoter through gene synthesis or through PCR amplification of the gene with an extended 5′ primer. A T7 promoter is used because: (1) it is short enough to allow for straightforward PCR amplification, and (2) the T7 polymerase can transcribe mRNA efficiently from linear DNA. Cas nucleases could likely be expressed from linear DNA using a constitutive σ70 promoter, but we have not investigated this possibility.
Expressing Cas nucleases from linear DNA is useful for at least two reasons. (1) We have found that some Cas nucleases are almost impossible to clone into vectors containing a prokaryotic promoter suitable for expression in TXTL due to cell toxicity. However, we have been able to readily clone these proteins into vectors that lack promoters. (2) This method can also be used to prepare DNA for expressing the Cas nuclease in TXTL directly from genomic DNA containing the cas gene-of-interest.
To generate linear DNA with the Cas nuclease being expressed from a T7 promoter from either genomic DNA encoding the cas gene or from a plasmid with the cas gene lacking a promoter, design primers as shown in the Appendix (see TJpr371/372). These primers add the T7 promoter, Shine-Dalgarno sequence, and a T7 terminator to the 5′ and 3′ ends of the cas gene. These primers will need to be adapted by altering their binding sequence for each Cas nuclease of interest. Use these primers to perform PCR using Taq or the Q5 polymerase. Verify that the PCR yields a single bright band at the expected molecular weight using gel electrophoresis using a small sample of the PCR. Purify the DNA from the remaining PCR using a DNA purification kit and elute in nuclease-free H2O.
To synthesize linear DNA expressing the Cas nuclease, design the gene fragment to include: (1) a binding site for CSMpr1105, (2) the T7 promoter, (3) a strong ribosomal binding site, (4) the sequence of the cas gene, (5) a transcription terminator (preferably from T7), and (6) a binding site for CSMpr1106. Lyophilized gene fragments can be resuspended in nuclease-free H2O to 20–40 nM and added directly to TXTL. If a large number of reactions are needed, use CSMpr1105/1106 to PCR amplify the gene fragment using either Taq or Q5 polymerase. See the sequence “Cas-GB” in the appendix for a gene fragment expressing FnCas12a.
Note that if the Cas nuclease is expressed from a T7 promoter, the plasmid P70a-T7RNAP must be added to the TXTL mixture to a final concentration of 0.2 nM as described below for expression from linear DNA. If expressed from an arabinose-inducible promoter, arabinose must be added to the TXTL mix to 20 mM. If the Cas nuclease is repressed by LacI (as is the case for pET vectors), IPTG must be added to the TXTL mix to 0.5 mM.
2.1.3. Creating DNA to express gRNAs
Similar to the Cas nucleases, gRNAs can be expressed from either plasmid or linear DNA. We generally express gRNAs from linear DNA, so we only describe that method in detail here. Otherwise, clone a spacer similar to the one in CSM-GB191 into a suitable gRNA expression vector of interest and prepare the plasmid DNA using a midiprep kit as above.
To express the gRNA, order a gene fragment (IDT, Eurofins, etc.) containing the following required elements: (1) a binding site for CSMpr1105, (2) a constitutive promoter (e.g. sigma 70 promoter), (2) a sequence expressing the appropriate ncRNA needed to target the Cas nuclease to the appropriate sequence in the randomized PAM library, (3) a transcription terminator, and (4) a binding site for CSMpr1106. An annotated sequence (CSM-GB191 and CSM-GB019) is provided in the appendix that was used to target Cas9 and FnCas12a, which use a 5′ and 3′ PAM, respectively. If it is unknown whether the Cas protein of interest recognizes a 5′ or 3′ PAM, gRNA sequences targeting both a 3′ and 5′ PAM should be designed.
Resuspend the gene fragment in nuclease-free H2O and normalize to 20 nM. If a large number of reactions are needed, use CSMpr1105/1106 to amplify the gene fragment using either Taq or Q5 polymerase, then use a DNA purification kit to resuspend the amplified DNA in nuclease-free H2O.
Along with the gRNA targeting the PAM library, another gRNA construct should be ordered as a negative control that does not target any sequence in the PAM library or the reporter construct. An annotated sequence (CSM-GB019) expressing a non-targeting Spy-Cas9 sgRNA is provided in the appendix.
Note that, if required, a tracrRNA can also be expressed in TXTL using a gene fragment with the same elements as those described above.
2.1.4. Creating a reporter plasmid
If a PAM that is recognized by a Cas nuclease is known (e.g. if at least one PAM can be identified bioinformatically), it is helpful to create a reporter plasmid to monitor the rate of cleavage by the Cas nuclease and to optimize expression conditions. Cleavage anywhere in a reporter plasmid in TXTL leads to quenching of the reporter due to RecBCD-mediated degradation [52]. By inserting the PAM library protospacer flanked by a known PAM upstream of the promoter driving the expression of GFP, the efficiency and rate of cleavage by the Cas nuclease can be determined (Fig. 2A). Note that the reporter plasmid can still be used even if the Chi6 RecBCD inhibitor is added because the inhibitor is eventually degraded by RecBCD in approximately 5 h [51,52]. Once the Chi6 DNA is degraded, RecBCD can begin degrading cleaved reporter plasmids. Indeed, Fig. 2A was generated using a gRNA and Cas nuclease expressed from linear DNA. Although it is likely that the reporter gene activity lags behind the actual cleavage of DNA (e.g. library cleavage), it still provides an estimate of cleavage time, especially for Cas nucleases that cleave DNA slowly in TXTL. In the method below, a reporter plasmid is added to each reaction in order to monitor the activity of the Cas nuclease as it cleaves the randomized PAM library.
Fig. 2.
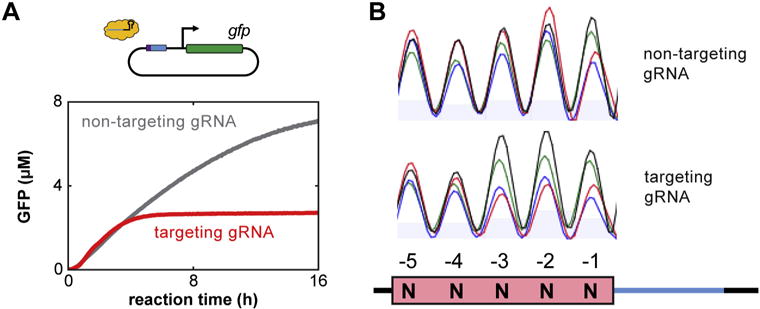
Data analysis for assessing cleavage by a Cas nuclease in TXTL. (A) A representative GFP cleavage analysis tracking fluorescence of GFP over time is shown. The red and gray lines indicate TXTL reactions containing a Cas nuclease with a gRNA targeting the GFP reporter or a non-targeting gRNA, respectively. At earlier times, GFP fluorescence is similar for both reactions. After a few hours, the reaction containing the GFP-targeting gRNA cleaves the reporter plasmid, and GFP production ceased, while in the reaction containing the non-targeting gRNA GFP continues being produced for ~16 h. (B) An example comparison between two trace files obtained from Sanger sequencing. The images show chromatograms obtained by PCR amplifying directly from two TXTL reactions. Each reaction was conducted with the FnCas12a nuclease, a 5-nt randomized PAM library, and either the non-targeting (top) or targeting (bottom) gRNA. Cleavage with the targeting gRNA shows noticeable depletion of nucleotides C and T in the −2 and −3 positions relative to cleavage with the non-targeting gRNA. The inconsistent nt abundance in the non-targeting gRNA sample can be attributed to varying frequencies of each sequence in the PAM library. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We have used the Q5 Site-Directed Mutagenesis Kit to insert a protospacer and PAM recognized by the Cas nuclease of interest into the plasmid P70a-deGFP to create reporter plasmids. An example annotated plasmid sequence (pTJ247) that we created to monitor cleavage of NmeCas9 is provided in the appendix. To create similar plasmids, use the Q5 Site-Directed Mutagenesis Kit to insert the PAM-library protospacer sequence of the appropriate length (labeled “mut protospacer” in pTJ247) at position 247 in the attached P70a-deGFP plasmid map flanked by a PAM that can be recognized by the Cas nuclease. The primers used to create pTJ247 from p70a-deGFP using the Q5 Site-Directed Mutagenesis Kit are listed in the appendix (TJpr373/374).
2.1.5. Creating the randomized PAM library
To create a randomized PAM library, we used the NEBuilder HiFi DNA Assembly Cloning Kit. We used restriction-enzyme free cloning to prevent any nucleotide biases in the randomized region that would result from cleavage by a restriction enzyme.
Note that this is the only step in our protocol that requires cloning, and that it only needs to be performed once in order to assay a large number of Cas nucleases. Purified 10 N library plasmids are available from the authors on request, contingent on availability.
Set up two separate PCRs using Q5 Hot Start High-Fidelity DNA Polymerase (Example reaction conditions in Table 2).
Reaction 1: Template is 1 ng of CSM160, primers CSMpr1308/1309, Tanneal = 72 °C, text = 41 s, 1.64kB expected product size.
Reaction 2: 10 ng of CSM-GB089, primers CSMpr1310/1311, Tanneal = 72 °C, text = 12 s, 0.48kB expected product size.
Confirm that each reaction has a product of the appropriate size by running the PCR products on a 1% agarose gel with a DNA ladder.
Add 1 ml of DpnI to the PCR product from Reaction 1, gently mix, and incubate at 37 °C for 1 h to digest the plasmid DNA.
Use the protocol from the NEBuilder HiFi DNA Assembly Cloning Kit to assemble the PCR products. For a negative control reaction, omit the PCR product generated from Reaction 2 to estimate the background.
Purify the two assembled reactions using a DNA purification kit and elute in as small of a volume of Tris-HCl pH 8 as possible. Transform 1μL of the purified reaction into 50 μL of NEB 5-alpha electrocompetent cells using the associated protocol. Recover the cells according to the associated protocol.
After recovery of the cells, make 1:10 serial dilutions of the two reactions and plate on LB + Chloramphenicol agar to estimate the number of transformants.
Incubate the plates at 37 °C and back-dilute the remaining recovered cells (1:50) in LB + Chloramphenicol media and grow overnight at 37 °C.
The total transformants from the assembly should be approximately 1–2 million and a ~30,000:1 plasmid-to-background ratio is expected.
Make glycerol stocks of the library as desired.
Midiprep the remaining overnight culture for subsequent use in the PAM library cleavage assay described below.
Table 2.
Components and thermocycler program used for PCR amplification using Q5 Hot Start High-Fidelity DNA Polymerase. An in-depth protocol is packaged with the polymerase.
| Component | Volume (μl) | Temperature (°C) | Time (s) |
|---|---|---|---|
| Nuclease-free H2O | 32.5 | 98 | 30 |
| 5x Q5 reaction buffer | 10 | 98 | 10 |
| 10 mM dNTPs | 1 | Tanneal | 30 |
| 10 μM FWD primer | 2.5 | 72 | text |
| 10 μM REV primer | 2.5 | Repeat 25x | |
| Template DNA | 1 | 72 | 120 |
| Q5 Hot Start High-Fidelity DNA Polymerase | 0.5 | 4–10 | ∞ |
2.2. PAM library cleavage in TXTL
This section describes how to use TXTL to determine the PAM requirements of Cas nucleases. We assume that four Cas nucleases will be assayed and that each Cas nuclease is expressed from a pET vector. If other expression vectors are used, then the master mix should be adjusted appropriately to ensure that the cas gene is transcribed (e.g. by adding arabinose if it is expressed from a pBAD vector). Note that if fewer than four Cas nucleases are being assayed, the master mix assembled in Step 3 can be re-frozen and thawed one time without loss of activity.
We assume that a reporter plasmid was constructed in Section 2.1.4 in order to provide an internal fluorometric control for Cas nuclease cleavage, and we assume that each Cas nuclease being assayed recognizes a protospacer of the same length and can recognize the PAM cloned into the reporter plasmid. This is true when assaying closely related Cas homologs. However, if the Cas nucleases being assayed do not recognize the same PAM, a different reporter plasmid should be added to each reaction expressing that Cas nuclease, and the TXTL recipe should be adjusted accordingly.
If a reporter plasmid is not used either because no PAM is known that is recognized by the Cas nuclease, or for some other reason, then the assay need not be carried out in a plate reader. We also assume that the Cas nuclease is active at 30 °C, and that 3 nM of DNA expressing the Cas nuclease is sufficient for expression. See Section 2.1.4 for troubleshooting Cas nuclease cleavage in TXTL.
Materials
myTXTL In vitro TX-TL protein expression kit [55] (Arbor Biosciences, 507096)
50 nM randomized PAM library
30 nM pET vector expressing the Cas nuclease of interest
20 nM DNA expressing a gRNA targeting the protospacer in the PAM library
20 nM DNA expressing a non-targeting gRNA
50 μM Chi6 DNA
96 well V-bottom plates (e.g. MilliporeSigma, CLS3357)
Cap mat (e.g. MilliporeSigma, CLS3080)
20 nM p70a-T7RNAP
50 nM reporter plasmid
50 mM IPTG (e.g. ThermoFisher, R1171)
Fluorescent plate reader capable of measuring GFP (e.g. BioTek H1)
Protocol
Thaw a tube of myTXTL on ice. Ensure that all the liquid in the tube is at the bottom of the tube by briefly centrifuging if necessary.
Add the contents of Table 3 into a thawed TXTL tube to assemble the master mix. Mix by gently vortexing.
Aliquot 9.6 μL of the master mix to eight separate Eppendorf tubes for each of the eight reactions described in Table 4.
Add 1.2 μL each of the gRNA and Cas nuclease DNA as shown in Table 4 to each reaction. Mix each reaction by pipetting. This brings each reaction to a final volume of 12 μL.
Carefully load two 5 μL sub-reactions per reaction in the bottom of a 96 well V-bottom plate (A).
Seal the plate using a cap mat. Ensure that no well has a loose seal, which would lead to evaporation of the reactions or potentially damaging the plate reader.
Load the plate into a plate reader pre-warmed to 30 °C and measure GFP fluorescence (Ex 485 nm, Em 528 nM) kinetically every 10 min.
Incubate for up to 16 h in the plate reader. (B)
- Remove and pool the 5 μL sub-reactions for each condition, and store the samples at −20 °C until ready for NGS library preparation.
- Differences in the geometry of the TXTL droplet at the bottom of the plate seem to have a large effect on kinetics of the TXTL reaction. Care should be taken to load the reactions onto the plate uniformly.
- Shorter incubation times can be used, although we have found that extended incubation does not affect the PAMs identified by the assay. The internal fluorescent control is helpful to decide how long to incubate the reaction if a shorter incubation time is desired. When the fluorescence in the wells containing the targeting gRNA stops increasing, but the fluorescence in the wells containing the non-targeting gRNA continues to increase, cleavage is complete and the TXTL reaction can be stopped
Table 3.
Recipe for TXTL master mix for PAM determination assay for Cas nucleases expressed from pET vectors. The master mix does not include DNA encoding the Cas nuclease and the gRNA. Note that if the PAM of the Cas nuclease of interest is unknown or if no reporter plasmid was constructed during step 2.1.4, then the reporter plasmid does not need to be added. Note depending on how the Cas nucleases are expressed, the IPTG and/or the p70a-T7RNAP may be omitted. *Final concentration accounts for the extra volume from the addition of the DNA constructs encoding the Cas nuclease and the gRNA.
| Component | Volume (μl) | Final Conc.* | Stock Conc. |
|---|---|---|---|
| myTXTL extract | 75 | ||
| Randomized PAM library | 1 | 0.5 nM | 50 nM |
| Reporter plasmid from step 2.1.4 | 1 | 0.5 nM | 50 nM |
| p70a-T7RNAP | 0.5 | 0.2 nM | 40 nM |
| IPTG | 0.5 | 0.5 mM | 100 mM |
| Chi6 DNA | 2 | 1 μM | 50 μM |
Table 4.
An example set of reactions to characterize the PAM requirements of four Cas nucleases prepared using the master mix in Table 3. Each Cas nuclease is incubated with the PAM library and either a targeting or a non-targeting gRNA.
| Reaction condition | DNA 1 | DNA 2 |
|---|---|---|
| 1 | Cas nuclease A | Targeting gRNA A |
| 2 | Cas nuclease A | Non-targeting gRNA |
| 3 | Cas nuclease B | Targeting gRNA B |
| 4 | Cas nuclease B | Non-targeting gRNA |
| 5 | Cas nuclease C | Targeting gRNA C |
| 6 | Cas nuclease C | Non-targeting gRNA |
| 7 | Cas nuclease D | Targeting gRNA D |
| 8 | Cas nuclease D | Non-targeting gRNA |
2.3. Assessing cleavage of the PAM library
Prior to the NGS library preparation, we recommend Sanger sequencing to evaluate Cas nuclease cleavage (Fig. 2B). This is a quick and inexpensive way to verify that the Cas nuclease was expressed and is active in TXTL. It also can provide an estimate of which PAMs are recognized by the Cas nuclease. Note that this provides another method to assess Cas nuclease activity in TXTL if a reporter plasmid cannot be constructed. If the Cas nuclease was expressed and active in TXTL, then a depletion of particular nucleotides will be observed in the reaction expressing the targeting gRNA relative to the reaction expressing the non-targeting gRNA.
Materials
Q5 Hot Start High-Fidelity DNA Polymerase
100 bp ladder (e.g. NEB, N3231S)
Oligos TJpr416/417
DNA purification kit
Protocol
Make a 1:10 dilution of each TXTL reaction in nuclease-free H2O.
Set up a PCR reaction for each TXTL reaction using the standard Q5 Polymerase protocol (see Table 2 for an example) using primers TJpr416/TJpr417, Tanneal = 67 °C, text = 20 s, using the 1:10 diluted TXTL reaction as the template DNA, and supplemented with 4% DMSO (e.g. 1 μl for a 25 μl reaction).
Run the PCR product(s) on a 1% agarose gel by mixing 2 μl 6x loading dye, 5 μl nuclease-free H2O, and 5 μl PCR product on parafilm and loading 10 μl on the gel to ensure amplification (~500 bp product).
Sanger sequence the resulting amplicons using TJpr416 or TJpr417 as the sequencing primer.
Align the trace files (.ab1) from the Sanger sequencing of the TXTL reactions containing the targeting and non-targeting gRNA with the PAM library using Benchling or similar software.
Examine the aligned trace files by zooming into the randomized sequence flanking the protospacer. Successful cleavage leads to a less random distribution of some nucleotides in the randomized area in the targeting gRNA control. An example trace file of a library cleaved by a Cas12a homolog can be seen in Fig. 2.
If the peaks from each position within the randomized PAM sequence do not look obviously different between the targeting and non-targeting gRNA reactions, troubleshooting is likely needed to increase expression and/or cleavage of the Cas nuclease. Note that a highly specific PAM would show negligible depletion by Sanger sequencing because the PAM represents a small fraction of all possible sequences.
2.4. Troubleshooting Cas nuclease cleavage in TXTL
If no cleavage is observed using either the reporter plasmid or Sanger sequencing, first check that all of the expression constructs are correct. If the expression constructs are correct, there are a few parameters that can be tuned to improve the expression of the Cas nuclease. Troubleshooting is greatly facilitated by having a fluorometric readout.
Double or quadruple the concentration of the DNA expressing the Cas nuclease.
Double or quadruple the concentration of the DNA expressing the gRNA.
Try increasing the temperature of the reaction to 37 °C or 42 °C.
Some enzymes require supplementation with cations such as Mg2+ or Ca2+. Try supplementing these trace elements to the TXTL reaction.
Out of the ~20 Cas nucleases we have assayed with this technique, we have only found two with which we were unable to observe DNA cleavage in TXTL and that did not respond to the optimization techniques above. It could be that these Cas nucleases cannot cleave DNA, or it could be that this method is broadly but not universally applicable.
2.5. NGS library preparation
Materials
TXTL reaction(s) of interest
Q5 Hot Start High-Fidelity DNA Polymerase
Oligos RL133/134/135
Oligos TJpr418/419/422/423
Nextera indexing primers
AMPureXP beads (Fisher Scientific, NC9933872)
100 bp DNA ladder
Magnetic rack (e.g. Thermofisher, AM10027)
Optional Materials
Additional Illumina index oligos if analyzing more than two reaction conditions.
This section describes the preparation of the NGS libraries from the cleaved PAM libraries. Oligos RL133/134 add Illumina Nextera adapters to the PAM library adjacent to the area of the randomized PAM sequence. These primers were previously used by Leenay and co-workers [34]. This method could be improved by incorporating random barcodes in these primers to enable the identification of PCR duplicates, but we have not found this to be necessary to yield accurate results.
Protocol
Add Nextera adapters to the cleaved PAM libraries and controls.
Set up 50 μl PCR(s) using the standard Q5 Polymerase protocol (see Table 2 for an example) using primers RL133/RL134, Tanneal = 62.9 °C, text = 7 s, using the 1:10 diluted TXTL sample as the template DNA, and supplemented with 2 μl DMSO.
On parafilm, mix 2 μl 6x loading dye and 10 μl PCR product and load onto a 1% agarose gel.
Run the gel with 10 μl 100 bp ladder at 100 V until dye travels ~80% of the gel.
Image the gel with a standard UV transilluminator. You should see a strong 200 bp band.
- Clean up the PCR product(s):
- Vortex the AMPureXP beads.
- Mix 40 μL beads with the PCR product(s) and mix well by pipetting up and down.
- Incubate the mixture(s) at room temperature for 5 min.
- Place the PCR tube(s) onto a magnetic rack and wait 2 min or until the supernatant is clear.
- With the tube(s) remaining on the magnetic rack, use a pipette to remove the clear supernatant.
- Wash the beads by adding 200 μl of fresh 80% ethanol to the tube(s) and wait 30 s. Remove the supernatant and repeat the wash step.
- After repeating wash step, remove the supernatant and let the sample(s) air dry for 10 min.
- Remove the PCR tube rack from the magnetic rack, and elute the DNA from the beads by adding 45 μl of 10 mM Tris-HCl pH 8 to the tube(s) and mixing well.
- Incubate the sample(s) for 2 min, then place the PCR tube(s) back onto the magnetic tube rack and allow the beads to separate from the liquid for additional 2 min.
- Transfer 40 μl of the supernatant to a new tube ensuring that no beads are transferred.
- Nanodrop the sample(s). The average concentration should be ~12 ng/μl.
Add Illumina indices and binding domains.
With the Nextera adapters on the PAM library, Illumina indices and binding domains can be attached using PCR. To run more than one library on a single sequencing lane, a unique pair of primers must be used for each sample in order to attach a unique pair of i5 and i7 indices to each sample. A list of the Nextera indices and the oligos that they are embedded within are provided by Illumina in their technical support document “Illumina Adapter sequences” under the file heading “Illumina Nextera Adapters.” The two pairs of indexing oligos from this table that we used to elucidate the NmeCas9 PAM are provided in the appendix (RL135 (i5) and TJpr422/423 (i7)).
Add indices to the library using PCR using the recipe and reaction conditions in Table 5. Use a unique pair of i5 and i7 primers for each reaction.
- Clean the second PCR product(s) using the same protocol as in the first cleanup, except for the following changes:
- Mix 56 μl beads with the PCR product(s) at step (2)
- Elute with 25 μl 10 mM Tris-HCl and transfer 20 μl to a new tube at steps (8–9)
Measure the final sample concentration(s) using a Nanodrop or, for more accurate quantitation, a Qubit (e.g. ThermoFisher, A30225). The average concentration(s) should be ~40 ng/μl.
Based on the measured concentrations, run ~100 ng of total DNA on a 1% gel (2 μL 6x loading dye, 100 ng PCR product, up to 10 μL nuclease-free H2O) with 10 μl 100 bp ladder. Run at 100 V until dye travels ~80% of gel.
Image the gel. The band should be at the 300 bp marker, and the intensity should be comparable to the band corresponding to the 100 ng marker.
Prepare the sample(s) to the concentrations required for sequencing (e.g. NCSU Sequencing Facility requires 10 nM in 20 μL). Note: To sequence low-complexity libraries, a high-diversity library must generally be spiked in during sequencing. Our sequencing libraries were spiked with 15% PhiX libraries before sequencing.
Table 5.
Components and thermocycler program used for the final PCR amplification of the PAM library using Q5 Hot Start High-Fidelity DNA Polymerase. Note the cycle number is reduced from 25 to 8 to reduce errors during amplification.
| Component | Volume (μl) |
Temperature (°C) |
Time (s) |
|---|---|---|---|
| nuclease-free H2O | 28.5 | 98 | 30 |
| 5x Q5 reaction buffer | 10 | 98 | 10 |
| 10 mM dNTPs | 1 | 67 | 20 |
| 10 μM FWD Nextera index (i5) | 2.5 | 72 | 10 |
| 10 μM REV Nextera index (i7) | 2.5 | repeat 8x | |
| purified PCR product | 5 | 72 | 120 |
| Q5 Hot Start High-Fidelity DNA Polymerase | 0.5 | 4–10 | ∞ |
2.6. Counting PAMs and calculating depletion
We have created a simple Python script to extract the NGS reads from PAM-SCANR libraries [34] and libraries constructed according to the method outlined above. The script looks for reads that contain the sequence flanking the variable nucleotides in the PAM library and restricts the counts to reads that have a threshold quality score at each of the variable nucleotides. The code is available as a public repository (see appendix). In addition, we have included a copy of the script in the appendix of this protocol. To use this script, follow the instructions in the readme file contained in the repository.
To calculate the depletion of each sequence in the PAM library for a Cas nuclease, we first restricted ourselves to measuring sequences that had at least 50 reads in the non-targeting control. We then normalized the counts of each sequence by the total number of reads in each library. We then calculated the quotient of counts for each sequence in the targeting library and the non-targeting library. Example R code is included in the repository along with sample output of the counting script and sample plots that can be used to represent the output of the script. PAM depletion data can also be represented using a PAM wheel as described in [10].
3. Results and discussion
To demonstrate our assay, we determined the PAM requirements of NmeCas9 expressed from linear DNA. We obtained linear DNA expressing NmeCas9 by adding a T7 promoter and transcription terminator to the NmeCas9 sequence on the plasmid DS-Nmcas (Addgene # 48646). The PAM for this particular Cas9 has previously been characterized [31,53,54]. These studies found a consensus PAM for NmeCas9 as NNNNGATT. We created a reporter plasmid for NmeCas9 by placing the, NNNNGATT PAM adjacent to the PAM library protospacer (see pTJ247 for NNNN sequence). Other PAMs with similar cleavage efficiency could have been used [29].
The GFP cleavage data from NmeCas9 (Fig. 3A) shows that the NmeCas9 RNP formed and began cleavage of the reporter plasmid by 1 h. In contrast to previous Cas nucleases we have characterized [51], NmeCas9 did not completely cleave the reporter plasmid; fluorescence continued to increase even in the targeting gRNA condition throughout the experiment. We did not observe any obvious differences between the targeting and non-targeting gRNA conditions by Sanger sequencing, which may be due to limited cleavage or the inability to observe depletion of a highly specific PAM sequence. While an incubation time of ~10 h would most likely have been sufficient, we opted for a longer reaction time to ensure adequate cleavage of the PAM library.
Fig. 3.
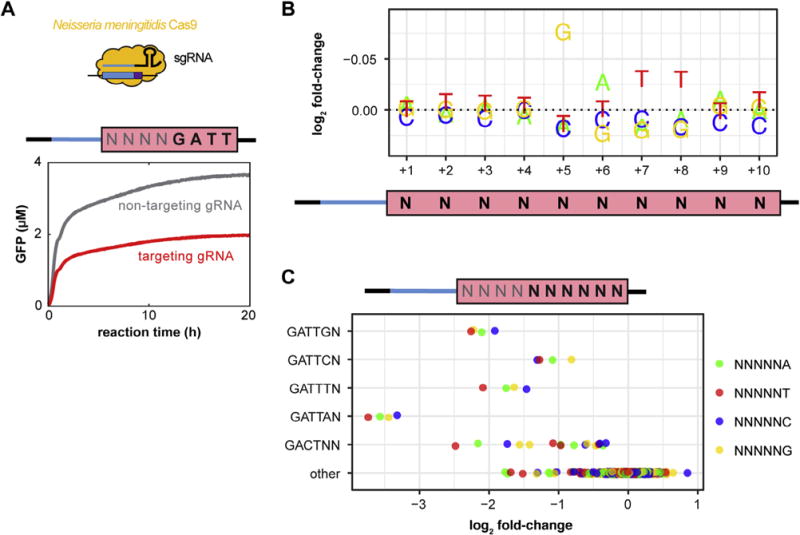
Data analysis for the TXTL-based PAM assay performed on the Cas9 from Neisseria meningitides (NmeCas9) expressed from linear DNA. (A) Fluorescence data from the GFP cleavage assay of NmeCas9 using an NNNNGATT PAM. The red and gray lines indicate TXTL reactions containing NmeCas9 with a gRNA targeting the reporter plasmid pTJ247 or a non-targeting gRNA, respectively. Both reactions had similar GFP expression rates, but after ~1 h, the reaction containing the targeting gRNA began to show a decrease in GFP production. (B) Plot showing the log2-fold change in the nucleotide frequency across the 10-nucleotide PAM library. The depletion shows the strong NNNNGATTNN with a slight bias of D in the 9th and 10th positions. Note the inverted y-axis. (C) The average fold-change of 10-mers for selected PAM motifs in the 5th-10th position is shown. While a NNNNGATTNN PAM was strong, having an A in the 9th position increases cleavage efficiency compared to the other nucleotides, and having a T in the 10th position slightly increased cleavage for NNNNGATTNN PAMs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We counted the reads mapping to each of the ten variable nucleotides in the PAM library using the script ‘count_pams.py’ with the flags ‘−n 10’ and ‘−q 10’. The former flag tells the script to look for ten variable nucleotides. The second relaxes the Phred score needed at each of the ten variable nucleotides from the default 30 to 10. We found that by relaxing the quality score we were able to increase the signal of the canonical NmeCas9 PAM because otherwise a large number of reads were rejected. This is because there were twice as many variable nucleotides in the library we used compared to previous libraries with only five variable nucleotides, which means that a Phred score of 30 was too strict. We observed similar results with a Phred cutoff score of 20, indicating that this analysis was robust to the cut-off used.
The PAM analyses for NmeCas9 showed the expected NNNNGATT PAM (see Fig. 3B and C). The fold-change plot (Fig. 3B) shows the nucleotide frequency at each position of the 10 N library. From this plot, the efficient NNNNGATT PAM was recapitulated, with the weaker PAMs of Y and W in the 7th and 8th position, respectively [29]. This plot also shows a bias of D in the 9th and 10th PAM position.
We calculated the average fold-change across all 10-mers matching selected motifs in the 5th through the 10th position (Fig. 3C). For the NNNNGATTNN motif, an ‘A’ in the 9th position increased cleavage efficiency compared to the other nucleotides. A ‘T’ in the 10th position led to the greatest cleavage for all PAMs that started with GATT. The NmeCas9 motif was relatively specific; of the motifs that matched NNNNGAYWNN, only NNNNGACTNN supported any measurable cleavage. Note that we only performed a single replicate of this PAM assay. If additional confidence that these subtle preferences reflect the real preferences of the nuclease, additional replicates would be warranted. Nevertheless, our results are consistent with previous work that observed similar PAMs when targeting DNA in mammalian cells [29]. The subtle PAM preferences for NmeCas9 could be also quantitatively assessed by cloning different PAMs flanking the NmeCas9 spacer in the reporter plasmid and repeating the reporter gene cleavage assay.
Though E. coli TXTL can rapidly characterize CRISPR-Cas systems, it does have some limitations. First, the temperature requirement for TXTL reactions is between 25 and 42 °C. While this range allowed for efficient cleavage using NmeCas9 and a number of other Cas nucleases we have worked with, this limit would restrict the analysis of other CRISPR-Cas systems that may require a higher temperature for efficient expression and/or cleavage. A TXTL environment also restricts the analysis of gRNA functionality in eukaryotic systems as it lacks the biological machinery native to those organisms (post-transcriptional/translational modifications, etc.). Finally, TXTL reactions lose activity after ~16 h. This may limit the use our assay for Cas nucleases that are weakly expressed or are unstable in TXTL.
4. Conclusions
Previous PAM characterization efforts have been hindered by the time required to perform existing assays. We have developed a method to characterize the PAM requirements of Cas nucleases that is much faster than previous methods. As myTXTL is commercially available for use, any lab or institution has access to use this powerful tool to characterize CRISPR-Cas systems. TXTL can also be used for other applications such as protein expression or, potentially, assessing gRNA activity.
Supplementary Material
Acknowledgments
We thank Erik Sontheimer for providing the plasmid encoding the NmeCas9.
Funding
Funding for this project was provided through DARPA (contract HR0011-16-C-01-34 to V.N. and C.L.B.), the NIH (1R35GM119561-01 to C.L.B.), and the US Department of Education and the GAANN fellowship (P200A160061 to T.J.).
Appendix A
The raw NGS data for the PAM assay with the NmeCas9 can be found under accession # GSE108357 in the NCBI gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/). All DNA sequences used in this work, as well as the data obtained from the gfp cleavage experiments can be found in the supplemental materials. The code used for the processing of the NGS data is in the Supplementary information and can also be found in the public repository: https://bitbucket.org/csmaxwell/crispr-txtl-pam-counting-script/.
Appendix B. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.ymeth.2018.02.016.
Footnotes
Conflicts of Interest.
C.L.B. is a co-founder and scientific advisory board member of Locus Biosciences and submitted provisional patent applications on CRISPR technologies. The Noireaux laboratory receives research funds from Arbor Biosciences, a distributor of the myTXTL cell-free protein expression kit.
References
- 1.Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 2.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrangou HPR, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 4.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in Staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo ML, Mullis AS, Leenay RT, Beisel CL. Repurposing endogenous type i CRISPR-Cas systems for programmable gene repression. Nucl Acids Res. 2015;43:674–681. doi: 10.1093/nar/gku971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human henome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leenay RT, Beisel CL. Deciphering communicating, and engineering the CRISPR PAM. J Mol Biol. 2017;429:177–191. doi: 10.1016/j.jmb.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouet JMP, Smih F. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urnov FD, Rebar EJ, Holmes MC, Zhang H Steve, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 13.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- 16.Zetsche B, Gootenberg JS, Abudayyeh OO, Regev A, Koonin EV, Zhang F, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van Der Oost J. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, Degennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, Van Der Oost J, Zhang F. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–34. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westra ER, van Erp PBG, Künne T, Wong SP, Staals RHJ, Seegers CLC, Bollen S, Jore MM, Semenova E, Severinov K, de Vos WM, Dame RT, de Vries R, Brouns SJJ, van der Oost J. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ, Joung JK. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JHD, Tjian R, Doudna JA. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 27.Shah SA, Erdmann S, Mojica FJM, Garrett RA. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013;10:891–899. doi: 10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucl Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CM, Cradick TJ, Bao G. The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Mol Ther. 2016;24:645–654. doi: 10.1038/mt.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elmore J, Deighan T, Westpheling J, Terns RM, Terns MP. DNA targeting by the type I-G and type I-A CRISPR-Cas systems of Pyrococcus furiosus. Nucl Acids Res. 2015;43:10353–10363. doi: 10.1093/nar/gkv1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh JRJ, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, Beisel CL. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol Cell. 2016;62:137–147. doi: 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol. 2015;16:253. doi: 10.1186/s13059-015-0818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, Nakane T, Ishitani R, Hatada I, Zhang F, Nishimasu H, Nureki O. Structure and engineering of Francisella novicida Cas9. Cell. 2016;164:1–12. doi: 10.1016/j.cell.2016.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karvelis T, Gasiunas G, Siksnys V. Methods for decoding Cas9 protospacer adjacent motif (PAM) sequences: a brief overview. Methods. 2017;121–122:3–8. doi: 10.1016/j.ymeth.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Bolotin A, Quinquis B, Sorokin A, Dusko S, Ehrlich Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 41.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 42.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 43.Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol. 2008;200–207 doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- 44.van Houte S, Buckling A, Westra ER. Evolutionary ecology of prokaryotic immune mechanisms. Microbiol Mol Biol Rev. 2016;80:745–763. doi: 10.1128/MMBR.00011-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.England WE, Whitaker RJ. Evolutionary causes and consequences of diversified CRISPR immune profiles in natural populations. Biochem Soc Trans. 2013;41:1431–1436. doi: 10.1042/BST20130243. [DOI] [PubMed] [Google Scholar]
- 46.Childs LM, England WE, Young MJ, Weitz JS, Whitaker RJ. CRISPR-induced distributed immunity in microbial populations. PLoS One. 2014;9:e101710. doi: 10.1371/journal.pone.0101710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, Koonin EV. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio. 2017;8:e01397–17. doi: 10.1128/mBio.01397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue C, Seetharam AS, Musharova O, Severinov K, Brouns SJJ, Severin AJ, Sashital DG. CRISPR interference and priming varies with individual spacer sequences. Nucl Acids Res. 2015;43:10831–10847. doi: 10.1093/nar/gkv1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garamella J, Marshall R, Rustad M, Noireaux V. The all E. coli TX-TL toolbox 2.0: A platform for cell-free synthetic biology. ACS Synth Biol. 2016;5:344–355. doi: 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- 50.Shin J, Noireaux V. An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS Synth Biol. 2012;1:29–41. doi: 10.1021/sb200016s. [DOI] [PubMed] [Google Scholar]
- 51.Marshall R, Maxwell C, Collins SP, Jacobsen T, Luo ML, Begemann MB, Gray BN, January E, Singer A, He Y, Beisel CL, Noireaux V. Rapid and scalable characterization of CRISPR technologies using an E. coli cell-free transcription-translation system. Mol Cell. 2018;69:146–157. doi: 10.1016/j.molcel.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marshall R, Maxwell CS, Collins SP, Beisel CL, Noireaux V. Short DNA containing chi sites enhances DNA stability and gene expression in E. coli cell-free transcription–translation systems. Biotechnol Bioeng. 2017;114:2137–2141. doi: 10.1002/bit.26333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucl Acids Res. 2014;42:2577–2590. doi: 10.1093/nar/gkt1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hou Z, Zhang Y, Propson NE, Howden SE, Chu L-F, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. PNAS. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 2013:e50762. doi: 10.3791/50762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.