

Abstract
Alzheimer’s disease (AD) is characterized by slow, progressive neurodegeneration leading to severe neurological impairment, but current drug development efforts are limited by the lack of robust, human-based disease models. Amyloid-β (Aβ) is known to play an integral role in AD progression as it has been shown to interfere with neurological function. However, studies into AD pathology commonly apply Aβ to neurons for short durations at non-physiological concentrations to induce an exaggerated dysfunctional phenotype. Such methods are unlikely to elucidate early-stage disease dysfunction, when treatment is still possible, since damage to neurons by these high concentrations is extensive. In this study, we investigated chronic, pathologically-relevant Aβ oligomer concentrations to induce an electrophysiological phenotype that is more representative of early AD progression compared to an acute high-dose application in human cortical neurons. The high, acute oligomer dose resulted in severe neuronal toxicity as well as upregulation of tau and phosphorylated tau. Chronic, low-dose treatment produced significant functional impairment without increased cell death or accumulation of tau protein. This in vitro phenotype more closely mirrors the status of early-stage neural decline in AD pathology and could provide a valuable tool to further understanding of early-stage AD pathophysiology and for screening potential therapeutic compounds.
Keywords: Alzheimer’s disease, human, chronic, electrophysiology, Aβ oligomers
TOC image
Introduction
Alzheimer’s disease (AD) is a worldwide pandemic estimated to have affected 26.6 million people in 2006. By the year 2050, this figure is projected to quadruple without pharmacological intervention1. Given the increased rate of diagnosis and the rising expense associated with long-term personal care, the cost of treating AD is estimated to reach $1 trillion in the U.S. alone by 20502. Only five FDA-approved medications are currently on the market in the U.S. Though these medications are widely prescribed to treat the cognitive symptoms of AD, they do not affect the underlying disease progression and there is debate over their effectiveness3–7. In spite of the significant progress in elucidating the biological mechanisms of AD8,9, no practical treatments exist which prevent or significantly delay its progression.
Contributing to the dearth of effective medications for treating AD is the lack of robust, biomimetic models that accurately recapitulate the pathophysiology of the condition in human tissue. Without a reliable humanized model, progress in therapy development and testing is seriously hindered. Current animal models, including a wide variety of species10,11, poorly represent complex human neurochemistry and neurodegeneration12. More than three dozen drugs have failed in AD clinical trials despite positive pre-clinical data from over two dozen transgenic models10. With recent advances in the development of robust, neuronal human induced pluripotent stem cell (hiPSC) lines13–15, there is an opportunity to develop a human-based in vitro disease model for AD which may lead to better translation of lab research to clinical results.
In an attempt to recapitulate AD’s in vivo pathophysiology in a controlled in vitro environment, investigators have studied the role of amyloid beta (Aβ) in disrupting neural function by applying various forms of the protein to cultured neurons. Amyloid-β is known to be a crucial causative protein in AD development16,17 Disease progression is linked to accumulation of Aβ and formation of plaques and phosphorylated tau tangles, leading to reduced neuronal activity and eventually cell death9,18. Evidence indicates that the most toxic forms of Aβ are not the aggregated fibrils commonly associated with AD pathology, or even its monomeric form, but that low-weight, soluble Aβ (1–42) oligomers are the most harmful agents of synaptic dysfunction and neurodegeneration19–27 Specifically, dimers of Aβ have been shown to have the most profound effect16,28. However, it is unclear what the role of this protein is in the earliest stages of AD pathology. Moreover, poor translation of in vitro studies to the clinic29 suggests current Aβ-based models offer poor representations of the disease30. Studies commonly use acute doses of non-physiological Aβ concentrations to achieve neuronal impairment in vitro22,31–35. This is problematic because opposing effects have been reported when investigating higher than physiologic concentrations of Aβ and may confound conclusions drawn from these studies36. Furthermore, the high rates of cell death and expression of tau tangles in response to such treatments appear to be a better representative of late-stage AD37,38 Targeting therapeutics to treatment of this late-stage phenotype may be inappropriate since neurological damage is already severe, and likely to be irreversible39. Moreover, acute treatment with nonphysiologically relevant doses of monomers and oligomers to promote rapid neurodegeneration is fundamentally unrepresentative of AD progression in vivo, and thus poorly suited to mechanistic studies of early disease states. We have previously shown that acute application of low physiological doses of Aβ oligomers have functional effects without significant cell death in cultured rat hippocampal neurons, which could be ameliorated by curcumin40, but no studies of low dose treatment of Aβ oligomers have been done with human cells derived from iPSCs, and certainly no chronic studies have been performed.
Based on these results we hypothesized that chronic exposure of human neurons to physiologically representative doses of Aβ oligomers41–43 would produce an in vitro phenotype that may more closely represents early-stage AD pathology, and therefore a more appropriate model for evaluating novel AD therapeutics. To investigate this hypothesis, we evaluated both high dose acute and low dose acute and chronic application of Aβ oligomers on human cortical neurons derived from iPSCs. Electrophysiological and metabolic analysis of the treatment models indicate significant functional impairment and decreased viability with acute high doses of Aβ, but chronic low doses produce functional impairment without affecting cell survival. In addition, immunocytochemical analysis suggests tau expression is upregulated in cells exposed to the acute high dose, but not in cells given the chronic low dose treatment. We believe these results establish an in vitro model that may be more indicative of early AD pathology.
Results
Cell morphology and viability
In all conditions, phase-contrast microscopy revealed cells which appeared healthy and had extensive neuritic outgrowth over the 4-week culture period. Phase images indicated similar gross morphology across all conditions, with no obvious aberrant morphological changes upon addition of Aβ (Figure 1A). This was expected as profound morphological changes were not reported at the concentrations utilized herein44. Assessment of cell viability by MTT assay yielded no significant differences between control and treatment with 10 nM Aβ, both acute and chronic (p > 0.05. Figure 1B). However, treatment with 5 μM Aβ significantly reduced cell viability compared to control cultures (p = 0.005). In 5 μM Aβ-treated cultures, viability dropped by 35.66% (± 5.75). Viability in acute and chronic 10 nM treated cultures were 103.93% (± 2.83) and 99.07% (± 3.96) respectively compared to untreated controls (Figure 1B), and differences were not significant (p = 0.986 and p= 0.141, respectively). These results indicate that chronic exposure to a pathological concentration of Aβ was insufficient to induce significant levels of cell death over a 4-week culture period. Acute Aβ treatment, with concentrations not representative of levels reported in human tissue, led to significant cell death after just 48 hours in culture.
Figure 1. Treatment of hiPSC derived neurons with physiological doses of Aβ does not induce changes in gross cell morphology or viability.
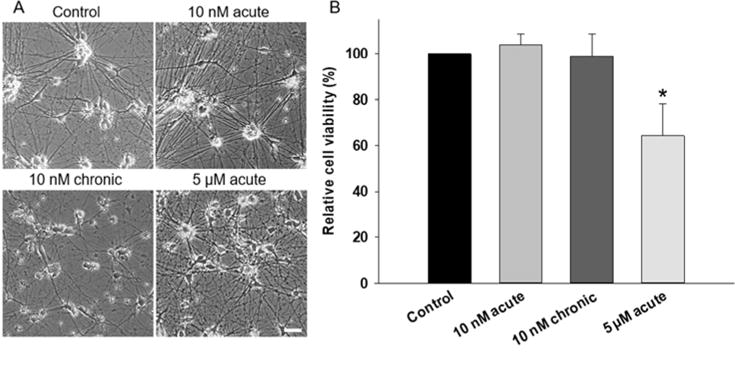
A) Representative phase contrast images of hiPSC-derived neurons comparing untreated, chronic, and acute Aβ treated cells at 30 DIV. Scale = 50 μm. B) Viability of hiPSC-derived neurons determined by MTT assay at 30 DIV following treatment with Aβ. Acute 5 μM treatment significantly reduced cell viability (P < 0.05) whereas acute and chronic 10 nM treatments had no significant effect on cell survival.
Electrophysiological function
Representative electrophysiological recordings of control and treated neurons (Figure 2A,B,C) highlight the major differences in electrophysiological function of the acute vs chronic low dose application of Aβ oligomers (quantified in Figure 3, all values reported in Supplemental Table 1). The resting membrane potential of the cultured neurons was significantly reduced in 5 μM Aβ-treated cultures compared to untreated controls (p = 0.034, Figure 3A). Conversely, no significant difference in resting membrane potential was observed for either acute or chronic 10 nM treatments (p = 0.980 and p = 0.309, respectively). Inward sodium and outward potassium currents were significantly reduced in the 5 μM Aβ-treated cultures compared to the controls (p < 0.05) but were not significantly different among control and 10 nM-treated cultures (p > 0.05, Figure 3B,C).
Figure 2. Electrophysiological evaluation of untreated, acute and chronic Aβ-treated hiPSC-derived neurons.
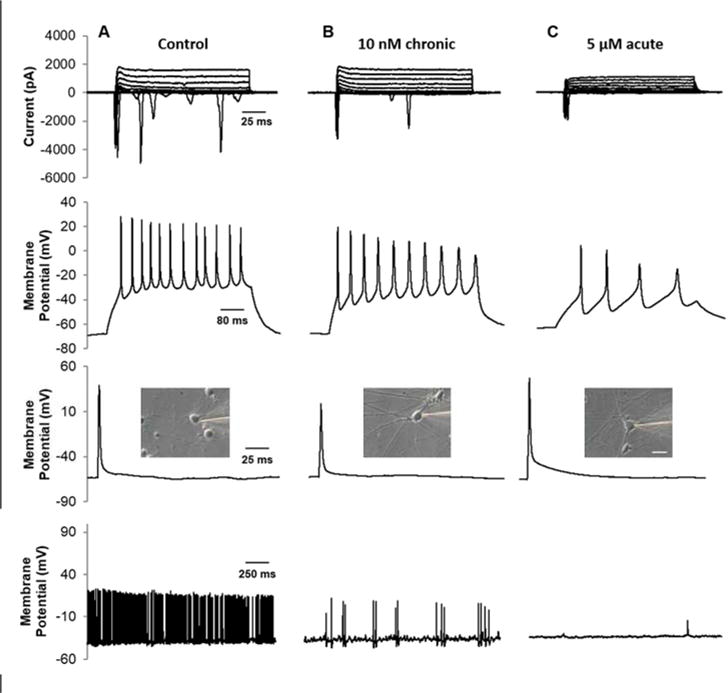
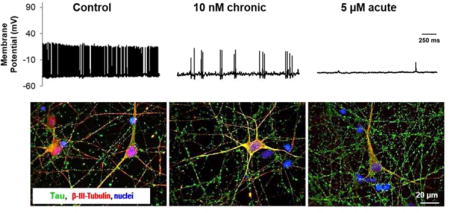
Patch-clamp recordings were taken between 28–30 DIV in control (column A), 10 nM chronic (column B), and 5 μM acute (column C) treated cortical neurons. Recordings from cells acutely treated with 10 nM Aβ were also taken but there were no significant difference from the control (p > 0.05). (Top row) Voltage-clamp recording of inward sodium and outward potassium currents (−70 mV holding potential, 10 mV steps). (Second row) Neurons fired repeatedly in response to depolarizing current injections at a −70 mV holding potential. (Third row). A single AP was generated using a 3 ms depolarizing current pulse of 1–2 nA. (Bottom row) Gap free recordings were performed for up to 5 m to measure spontaneous activity of patched cells. (Inset) Phase contrast image of patched neuron. Scale = 20 μM.
Figure 3. Changes in electrophysiological characteristics of hiPSC-derived neurons treated with either acute or chronic Aβ.
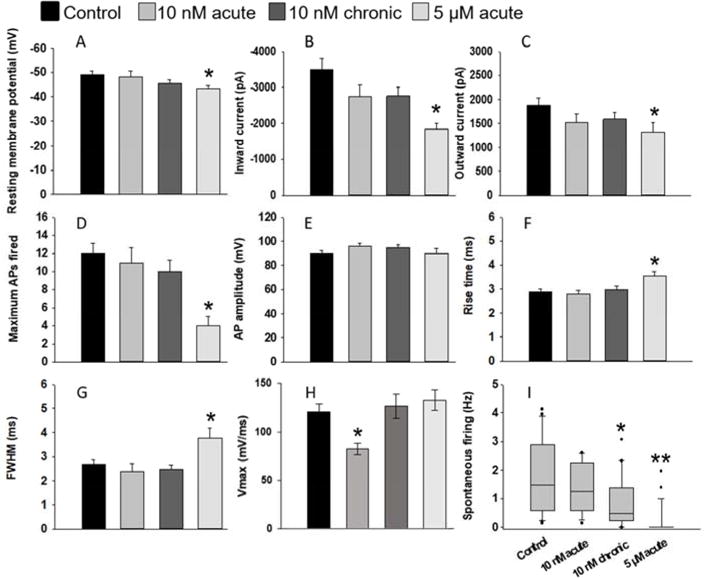
A) Resting membrane potential, B) peak inward current, C) peak outward current, D) maximum APs fired in response to depolarizing current injections at −70 mV, E) and peak action AP are represented at 28 DIV. Whereas the resting membrane potential, peak inward and outward currents, and maximum number of APs were maintained across control and 10 nM conditions, these parameters were significantly reduced in acute, 5 μM treated cultures (p < 0.05). Treatment with 5 μM of Aβ resulted in an even more significant drop in spontaneous neural firing activity (p < 0.01). Time to peak AP and AP duration, and represented by rise time (F) and FWHM (G) respectively was significantly increased in 5 μM treatment of Aβ (p < 0.05). Inversely, the maximum rate of rise (Vmax; H) was significantly reduced in 5 μM treatment of Aβ but was unaffected in other conditions (p < 0.03). 10 nM Aβ acute treatment did not have a significant effect on spontaneous APs fired during gap-free recordings (I) but chronic 10 nM Aβ resulted in a significant drop in the spontaneous activity of these cultures (p = 0.02). Error bars represent SEM.
The maximum number of action potentials (APs) fired during stimulation while holding the membrane potential at −70 mV was calculated for each condition. No significant differences were observed between control and 10 nM treated cultures (p > 0.05); however, 5 μM Aβ-treated cultures were significantly lower than all other conditions (p < 0.007, Figure 3D). Despite this difference, AP amplitudes were not significantly different between any condition tested (p = 0.410, Figure 3E). Membrane resistance was also not significantly different between any condition (data not shown, p = 0.239). However, while the membrane resistance and AP amplitude was unaffected, AP waveforms were altered in 5 μM Aβ-treated cultures. Specifically, AP rise time (Figure 3F), calculated full width at half maximum (FWHM) (Figure 3G), and the maximum rate of rise (Vmax) (Figure 3H) were significantly different in 5 μM Aβ-treated cultures compared with control and 10 nM treated cells (p < 0.05), indicating that supraphysiolgical doses of Aβ trigger a widening of APs in cultured neurons. Decay slope and decay time were not significantly different between conditions (p < 0.05; Supplemental Table 1).
Spontaneous APs were recorded from patch-clamped cells with a gap-free protocol where current was held between −35 and −40 mV (Figure 3H). Again, 5 μM Aβ-treated cultures were significantly hindered in function (p < 0.001). Similarly, chronic 10 nM treatment was significantly lower than control and 10 nM acute conditions (p = 0.02), suggesting impaired functional performance in both 5 μM acute and 10 nM chronic conditions. This reduction in spontaneous activity in both conditions was suggested as we have previously shown similar findings in rat cultures treated with 100 nM Aβ over just 24 hrs40.
Immunocytochemical analysis of tau expression levels
Immunocytochemical analysis by confocal microscopy highlighted the presence of tau protein in cultured neurons under all examined conditions (Figure 4A). Control and Aβ treated cultures were stained for the presence of total tau protein as early-stage tauopathies may include not only hyperphosphorylated tau but other toxic oligomers45,46. The anti-tau antibody used detected both phosphorylated and nonphosphorylated tau. Analysis of positive staining intensity revealed increases in tau accumulation in response to Aβ treatment, mimicking previously-reported Aβ-tau protein interactions47–49. Calculation of statistical differences suggested an upregulation of tau expression in 5 μM treated cultures with a greater than 90% confidence interval (Figure 4B, p = 0.057). The slight increase observed in 10 nM treated cultures was more modest, and insufficient to suggest any significant changes in tau expression over the time-frame investigated.
Figure 4. Tau protein is upregulated in hiPSC-derived neurons treated with higher-than-physiologic concentrations of Aβ.
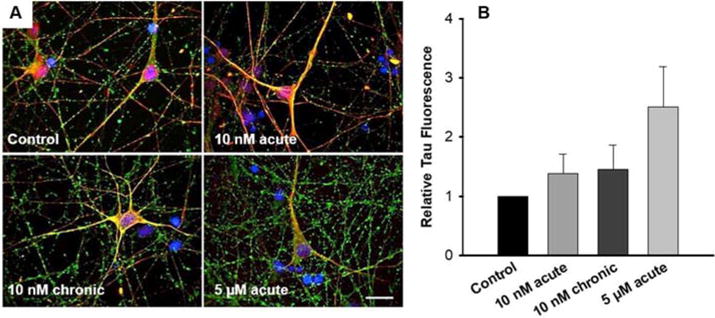
(A) Cells were fixed at culture endpoints (28–30 DIV) and stained for beta-ill tubulin (red), tau (green), and DAPI (blue). Scale bar = 20 μm. (B) Image analysis of tau protein intensity was analyzed relative to control tau positive staining while accounting for differences in cell viability (p = 0.057).
Discussion
A human-based neuronal culture system incorporating an accurate AD pathophysiological phenotype would be of considerable value to drug development and disease modeling applications. While patient derived iPSC models of AD are available13,50,51, cell line variability may inhibit the widespread applicability of conclusions drawn from specific lines. Commercially available iPSC-derived human cells offer an extremely reproducible alternative for general screening purposes. Study of Aβ-mediated phenotypic responses in cultured wild type neurons offers a widely available platform for generalized assessment of neurodegeneration in human tissues. Therapeutic screens and/or mechanistic studies of AD progression, once tested in a generalized model, could then be advanced to specific patient iPSC lines for continued personalized evaluation. Many previous studies have used acute, high doses of Aβ to model AD effects on cells in vitro. However, our results indicate that although such treatments induce severe pathological phenotypes rapidly in cultured human neurons, this method may not best recapitulate the AD disease state as it occurs in vivo.
AD patients undergo a slow neurodegenerative process which takes years to manifest in a detectable phenotype52,53. In the early-stages of AD pathology, patients exhibit synaptic dysfunction, amyloid beta accumulation in the brain but low concentrations of Aβ1–42 in the cerebral spinal fluid (CSF), a high tau/p-tau ratio in the CSF, and beginning stages of cortical/hippocampal diminishment54–56. This alteration in functional performance precedes significant cell death, which usually only occurs in later stages of AD progression37,38 Acute high doses of Aβ in vitro are more closely related to late-stage AD modeling and do little to accurately recapitulate the slow, progressive nature of the condition. A model system that more closely mimics the mild cognitive impairment stage of the disease would therefore be more suitable for studies on early AD pathophysiology. Furthermore, such a system would be beneficial for screening compounds for their capacity to prevent disease progression before late-stage cell death occurs.
Previous studies have used human cell sources to investigate the effect of high (nonphysiologic), acute doses on Aβ-induced cytotoxicity and cell cycle induction57 and low (more physiologic) concentrations of Aβ over acute periods58. However, assessment of the functional response of these cells to physiologically representative levels of Aβ over extended culture periods has not been performed in human cells. We therefore investigated treatment of hiPSC-derived neurons with low-doses of Aβ over extended time-courses to evaluate the ability for these cells to mimic the in vivo pathophysiology of early AD progression more closely.
Our results more likely closely mirror the in vivo effects of Aβ in human neural tissue at early stages of AD progression. The collected data highlight that chronic treatment of Aβ at a physiologic dose results in impaired neuronal firing capacities without significant cell death. Acute, high dose A(3 treatment produced a more pronounced reduction in spontaneous activity, but this was coupled with a significant reduction in cell viability, indicating a more rapid progress toward cell death than is typically seen in AD patients. No significant differences were seen in AP amplitudes when under current clamp, indicating treated cells were capable of firing APs as compared to healthy control cells, but only when membrane potentials were artificially maintained. AP waveforms were unaltered by chronic or acute 10 nM treatment, but AP rise time and FWHM were significantly increased when acutely treated with a high dose of Aβ. These results are supported by previous studies which demonstrate how different Aβ doses result in significant and conflicting electrophysiological responses in cultured rodent neurons40, 59. Furthermore, changes in AP rise time, Vmax and FWHM in acute high dose Aβ treated cells may indicate a change in sodium conductance and rectifying potassium channel activity in this condition60. Such drastic functional changes are poorly representative of the early AD phenotype. A recent in vivo study examining cortical and thalamic electrophysiology in a rat model of AD demonstrated that the disease state produced a significant reduction in neural activity without alterations in AP waveform metrics61. Our presented data correlate with the findings of this study and support the hypothesis that chronic low dose Aβ treatment produces functional phenotypes in cultured human neurons that more closely recapitulate AD pathology. Furthermore, these findings confirm that acute high dose Aβ treatment induces functional phenotypic changes in cultured neurons that are not observed in in vivo models of AD.
Tau is a major structural protein in neurons responsible for stabilizing axonal microtubules, and has been shown to be constitutively expressed in iCell neurons specifically46. Neurodegenerative tauopathies, concomitant with AD, include a reduced association of tau with microtubules, the accumulation of intracellular bundles of highly insoluble, straight or paired helical tau filaments, and the hyperphosphorylation of tau and the subsequent formation of neurofibrillary tangles (NFTs)45. Moreover, in stages leading up to (and during) the appearance of NFTs, neurotoxic tau oligomers are present and have been shown to contribute to neurodegeneration in drosophila and transgenic mice models of AD41,62–64. As such, the increased presence of tau aggregates in neurons is an established hall mark of neuronal degeneration in AD and associated tauopathies.
In this study, immunocytochemical analysis of tau protein in cultured cells indicated a significant increase in tau accumulation in response to acute high doses of Aβ. Conversely, neither acute, nor chronic 10 nM Aβ treatments produced significant increases of tau protein. This result was consistent with clinical observations as Aβ-mediated impairment of electrophysiological function is known to precede accumulation of tau in the brain during early AD progression65. These data provide further evidence to support the hypothesis that chronic low-dose Aβ is more appropriate for modeling early AD stages in vitro. Previous studies have demonstrated that the increased presence of Aβ does not upregulate tau gene expression, but rather promotes the formation of larger tau aggregates through hyperphosphorylation of specific sites47,66–69. Given this, the observed increase from tau immunocytochemical analysis detected in these experiments is believed to result from increased aggregation of the protein within cultured neurons, thereby increasing the detection of the positive fluorescence signal above background levels.
Taken together these results suggest that acute, non-physiological doses of Aβ poorly represent early AD phenotypes, in terms of modeling electrophysiological function, cell death, and tau aggregation. Chronic, physiological levels of Aβ more accurately recreate the mild functional impairment of neurons likely present during initial neurodegenerative stages of AD. The stability of these commercial hiPSC-derived neurons over extended time courses, and the more accurate representation of disease phenotype, demonstrate the utility of the described system for modeling early AD progression in vitro. As such, this system is highly amenable to studies designed to further our understanding of AD progression and provide a valuable potential tool for therapeutic drug screening.
Materials and Methods
Cell culture
Human iPSC-derived neurons (Cellular Dynamics International, iCell Neurons NRC-100-010-001) were plated at 700 cells per mm2 according to manufacturer’s instructions. Cells were plated in maintenance medium (NRM-100-121-001 with NRM-100-031-001) and incubated overnight at 37°C 5% C02. Plated cells were changed to Hybrid Systems Laboratory Medium 1 (HSL1, see Supplemental Table 2) without phenol red as phenol red may interact with amyloidogenic compounds70. HSL1 was specifically developed to facilitate the long-term survival and maturation of hippocampal neurons71,72 Medium was changed every 3–4 days and maintained over 28–30 days in vitro (DIV). Chronic treatment of 10 nM Aβ, a dose close to physiological Aβ concentrations41–43, began at 7 DIV to give cellular networks enough time to establish robust connections71. Medium was changed completely with freshly prepared oligomers to eliminate any possibility of buildup of higher-weight oligomers. Acute, 48 hr treatments of 10 nM and 5 μM Aβ were performed 2 days prior to patch clamp electrophysiology (26–28 DIV).
Surface modification
Cells were plated onto laminin-coated (Sigma, L2020, 3.3 μg/mL) coverslips which had been treated with the self-assembled monolayer, N-1(3-[trimethoxysilyl]propyl)-diethylenetriamine (DETA, United Chemical Technologies Inc., Bristol, PA, T2910KG). Briefly, plasma-cleaned glass coverslips (Thomas Scientific 1217N81, 18 mm, no. 1.5) were silanized with a 0.1% (v/v) mixture of DETA in freshly distilled toluene (Fisher T2904) as described previously73. The DETA-treated glass was heated to just below the boiling point of toluene (70°C), rinsed with toluene, reheated to 70°C, and then dried in an oven overnight at (100–105°C). Surfaces were validated via contact angle measurement and X-ray photoelectron spectroscopy as described previously74. Laminin was added to DETA-treated glass overnight at 4°C. DETA has previously been used for long-term culture of multiple cell types, including neurons72,74–77 and enhances the adsorption of laminin onto the culture surfaces (see Supplemental Figure 1).
Amyloid-β oligomer preparation
Amyloid-β oligomers (Bachem, H-1368) were prepared as described previously78. Briefly, Aβ peptide was monomerized using HFIP (Sigma-Aldrich, 105228) and dried overnight. Peptide films were further dehydrated with dry nitrogen and stored under desiccant at −80°C until use. Peptide films were resuspended in dimethylsulphoxide (DMSO) and diluted to 100 μM in Neurobasal A medium without phenol red (Invitrogen, 12349-015). Peptide suspensions were allowed to oligomerize for 24 hrs at 5°C before use. Preparations of Aβ1–42 were centrifuged at 14,000 rpm to remove any large fibrils and the supernatant was then diluted in phenol red-free medium for immediate use. The final concentration of DMSO was ≤ 0.1%.
Oligomer characterization
Aβ protein was prepared using a protocol known to produce low-weight oligomers78. To confirm the success and quality of our oligomerization protocol, gel electrophoresis of pure Aβ protein was performed, followed by a coomassie blue stain. Mini-PROTEAN® 10–20% Tris-Tricine Precast Gels (Bio-Rad, 456–3113) were loaded with the purified Aβ peptide suspended in Tricine Sample Buffer (Bio-Rad, #161–0739), and electrophoresed for 1–2 hrs at 120 V in Tris-Tricine SDS buffer (Sigma-Aldrich, T1165). Gels were then stained with Coomassie Brilliant Blue (Bio-Rad, 161-0786), and destained (Bio-Rad, Coomassie Brilliant Blue R-250 Destaining Solution 161-0438161-0438) to evaluate low-weight oligomer formation (Supplemental Figure 2).
MTT assay
MTT solution (Thiazolyl Blue Tetrazolium Bromide (Sigma, M5655) at 5 mg/mL in PBS) was added to cultures at a 1:5 ratio and incubated for 2–3 hrs. The MTT solution/culture medium was then removed and fresh lysis buffer (1% SDS and 0.57% glacial acetic acid in DMSO) was added to wells, triturated, and transferred to a 96-well plate (Falcon, 3-353070). Cell viability was then assessed using a plate reader (Bio-Tek, KC4 Synergy FIT) to detect emission levels at 570 nm and thereby quantify purple formazan byproduct levels in each sample. Emission levels were normalized to background levels achieved when measuring MTT/culture medium solution from wells with no cells plated.
Immunocytochemistry
Cultures were fixed using 4% paraformaldehyde for 20 mins before being rinsed with phosphate-buffered saline (PBS) at 4°C. Cells were then permeabilized for 20 mins with 0.2% Triton X-100 in PBS and incubated at room temperature for 2 hrs in blocking buffer (2.5% donkey serum, 2.5% goat serum, and 0.5% BSA) to prevent non-specific antigen binding. Primary antibodies rabbit monoclonal anti-microtubule associated protein tau (abcam, ab32057, 1:250), and chicken polyclonal anti-beta-ill tubulin (Millipore, ab9354, 1:1,000) were diluted in blocking buffer then added to the permeabilized cultures and maintained overnight at 4°C. Cells were then rinsed four times with PBS and incubated with their respective secondary antibodies diluted in blocking serum: goat anti-chicken Alexa 568 (A11041) and donkey anti-rabbit Alexa 488 (A21206) at 1:200 (Molecular Probes) for 2 hrs at room temperature. After rinsing four times in PBS, cells were mounted with ProLong® antifade reagent (Life Technologies, P36930) onto glass slides. The cells were imaged using a Zeiss LSM 510 confocal microscope. Expression of the respective antibodies was then quantified with ImageJ79 by measuring the integrated density and normalizing to cell number. Results are expressed relative to values recorded for untreated controls. A minimum of 50 fields of view at 40x magnification were quantified per condition.
Patch-clamp electrophysiology
Electrophysiological properties of hiPSC-derived neurons were investigated after 28–30 DIV. Whole-cell patch clamp recordings were performed on the 37°C heated stage of an upright microscope (Axioscope FS2, Carl Zeiss, Göttingen, Germany). Extracellular recordings were performed in the same medium used for cell culture (+10 mM HEPES buffer), including drug treatments. The intracellular recording solution (140 mM K-gluconate, 4 mM NaCI, 0.5 mM CaCI2, 1 mM MgCI2, 1 mM EGTA, 5 mM HEPES acid, 5 mM HEPES base, and 5 mM Na2ATP) was loaded into borosilicate glass patch pipettes (BF150-86-10; Sutter, Novato, CA) produced with a pipette puller (Sutter, P97). Patch pipettes with a resistance in the range of 6–10 MΩ were used for all recordings. Voltage-clamp and current-clamp experiments were performed with a Multiclamp 700B amplifier (Axon Instruments, Foster City, CA, USA). Signals were filtered at 2 kHz and digitized at 20 kHz with an Axon Digidata 1322A interface. Offset potentials were nulled before formation of a gigaΩ seal and fast and slow capacitance was compensated for in all recordings. Membrane potentials were corrected by subtraction of a 15 mV tip potential, calculated using Axon’s pCIamp 10 software (Axon Instrument, Foster City, CA, USA). Recordings with an access resistance of less than 22 MΩ were not included as a high access resistance indicates poor patch quality/insufficient access to the interior of the cell. To generate a single action potential (AP), a 3 ms depolarizing current pulse of sufficient intensity (1–2 nA) was applied. Inward and outward currents were evoked by a series of 250 ms depolarizing steps from −70 to +170 mV with +10 mV increments. Depolarization-evoked repetitive firing was recorded in current clamp mode. Gap-free recordings of spontaneous activity of patched neurons were performed for up to 5 m while holding the cell membrane at optimal threshold (between −45 and −50 mV). Cells which required more than 200 pA of current to maintain voltage clamp were excluded as excessive application of current is indicative of poor patch quality or membrane integrity. All recordings and analysis were performed using the pCIamp 10 software suite. Action potential rise times were calculated as the time taken to reach 90% maximum AP amplitude from 10% of the maximum amplitude. Decay slope and time were calculated from 90% to 10% of the action potential. Full width at half maximum was calculated as the width of the action potential (in ms) at half the maximum amplitude. Action potential rise time and FWHM were calculated during the flattest portion of the AP to allow for easy comparison between conditions.
Statistical analysis
One-way analysis of variance (ANOVA) tests were performed to assess differences between treatments of hiPSC-derived neurons followed by Student-Newman-Keuls Method tests. Post hoc Student-Newman-Keuls Method tests were also performed on data sets that did not have equal variance (resting membrane potential, MTT) but had a normal distribution of data. Data that had equal variance but were not normally distributed were analyzed using ANOVA on ranks, followed by the Dunn’s method for multiple comparisons (inward and outward currents, max rep firing, rise time). Square root transformations were performed on the spontaneous firing and maximum repetitive firing data sets and log transformations were performed on decay slope and decay time data sets to properly condition them for ANOVA analysis. Statistical differences in tau protein staining intensity between each Aβ treatment and untreated controls were independently assessed via student’s t-test. Each experiment included data collected from at least 3 independent cultures. Data is presented as mean ± standard error of the mean.
Supplementary Material
Supplemental Table 1: list of the average values, standard error, and number of values collected for all electrophysiological parameters.
Supplemental Table 2: list of medium components, vendors, and concentrations used to culture neurons.
Supplemental Figure 1: XPS analysis comparing protein on glass and DETA surfaces
Supplemental Figure 2: gel electrophoresis and coomassie blue stain of pure, oligomerized samples of Aβ.
Acknowledgments
Funding: This research was funded by National Institute of Health [grant number R01NS050452] and the Department of the Army [grant number W81XWH1410162].
Footnotes
Author contributions: JJH proposed the experimental basis; BJB, ASTS, COM, JJH designed the experiments; BJB, ASTS, COM performed the experiments; BJB, ASTS, CCM, CJL analyzed the data; BJB, ASTS, CCM, JJH interpreted the results; BJB, ASTS, CJL prepared the figures; BJB and JJH drafted the manuscript; BJB and JJH made edits for the final version of the manuscript.
Competing interests: JJH has a potential competing financial interest, in that a company has been formed that potentially could market services for the type of device described herein in which he has a financial interest.
References
- 1.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 2.Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary Costs of Dementia in the United States. N Engl J Med. 2013;368:1326–1334. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casey DA, Antimisiaris D, O’Brien J. Drugs for Alzheimer’s disease: are they effective. PT. 2010;35:208–211. [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou FM, Hablitz JJ. Layer I neurons of rat neocortex. I. Action potential and repetitive firing properties. Journal of Neurophysiology. 1996;76:651–667. doi: 10.1152/jn.1996.76.2.651. [DOI] [PubMed] [Google Scholar]
- 5.Ding Q, Vaynman S, Akhavan M, Ying Z, Gomez-Pinilla F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience. 2006;140:823–833. doi: 10.1016/j.neuroscience.2006.02.084. [DOI] [PubMed] [Google Scholar]
- 6.Clark P. Cell behaviour on micropatterned surfaces. Biosensors and Bioelectronics. 1994;9:657–661. [Google Scholar]
- 7.Clark P, Britland S, Connolly P. Growth cone guidance and neuron morphology on micropatterned laminin surfaces. Journal of Cell Science. 1993;105:203–212. doi: 10.1242/jcs.105.1.203. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, Xu HE. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38:1205–1235. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Res Dis Primers. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 10.Cavanaugh SE, Pippin JJ, Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. ALTEX. 2014;31:279–302. doi: 10.14573/altex.1310071. [DOI] [PubMed] [Google Scholar]
- 11.Götz J, Götz NN. Animal models for Alzheimer’s disease and frontotemporal dementia: a perspective. ASN Neuro. 2009;1:e00019. doi: 10.1042/AN20090042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- 13.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LSB. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, Yamanaka S. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 15.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 16.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-[beta] protein dinners isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tu S, Okamoto S-I, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bitan G, Teplow DB. Rapid Photochemical Cross-Linking-A New Tool for Studies of Metastable, Amyloidogenic Protein Assemblies. Acc Chem Res. 2004;37:357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 20.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 21.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of A beta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 22.Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. Journal of Neuroscience. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Agnaf OMA, Mahil DS, Patel BP, Austen BM. Oligomerization and toxicity of beta-amyloid-42 implicated in Alzheimer’s disease. Biochemical and Biophysical Research Communications. 2000;273:1003–1007. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- 24.Frid P, Anisimov SV, Popovic N, et al. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev. 2007;53:135–160. doi: 10.1016/j.brainresrev.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Hoozemans JJM, Chafekar SM, Baas F, Eikelenboom P, Scheper W. Always around, never the same: Pathways of amyloid beta induced neurodegeneration throughout the pathogenic cascade of Alzheimer’s disease. Current Medicinal Chemistry. 2006;13:2599–2605. doi: 10.2174/092986706778201585. [DOI] [PubMed] [Google Scholar]
- 26.Huang THJ, Yang DS, Plaskos NP, Go S, Yip CM, Fraser PE, Chakrabartty C. Structural studies of soluble oligomers of the Alzheimer beta-amyloid peptide. Journal of Molecular Biology. 2000;297:73–87. doi: 10.1006/jmbi.2000.3559. [DOI] [PubMed] [Google Scholar]
- 27.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from A beta(1–42) are potent central nervous system neurotoxins. Proc Nat Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large Soluble Oligomers of Amyloid beta-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J Neurosci. 2017;37:152–163. doi: 10.1523/JNEUROSCI.1698-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison JR, Owen MJ. Alzheimer’s disease: the amyloid hypothesis on trial. Br J Psychiatry. 2016;208 doi: 10.1192/bjp.bp.115.167569. [DOI] [PubMed] [Google Scholar]
- 30.Cummings J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci. 2017 doi: 10.1111/cts.12491. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carriba P, Jimenez S, Navarro V, Moreno-Gonzalez I, Barneda-Zahonero B, Moubarak RS, Lopez-Soriano J, Gutierrez A, Vitorica J, Comella JX. Amyloid-[beta] reduces the expression of neuronal FAIM-L, thereby shifting the inflammatory response mediated by TNF[alpha] from neuronal protection to death. Cell Death Dis. 2015;6:e1639. doi: 10.1038/cddis.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi YJ, Chae S, Kim JH, Barald KF, Park JY, Lee SH. Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci Rep. 2013;3:1921. doi: 10.1038/srep01921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko Tr, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine Hr, Catalano SM. Alzheimer’s Therapeutics Targeting Amyloid Beta 1-42 Oligomers I: Abeta 42 Oligomer Binding to Specific Neuronal Receptors Is Displaced by Drug Candidates That Improve Cognitive Deficits. PLoS ONE. 2014;9:e111898. doi: 10.1371/journal.pone.0111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minter MR, Main BS, Brody KM, Zhang M, Taylor J, Crack PJ. Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response in vitro. J Neuroinflammation. 2015;12:71. doi: 10.1186/s12974-015-0263-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheikh AM, Michikawa M, Kim SU, Nagai A. Lysophosphatidylcholine increases the neurotoxicity of Alzheimer’s amyloid (31–42 peptide: Role of oligomer formation. Neuroscience. 2015;292:159–169. doi: 10.1016/j.neuroscience.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 36.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs. 2017;26:735–739. doi: 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varghese K, Molnar P, Das M, Bhargava N, Lambert S, Kindy MS, Hickman JJ. A New Target for Amyloid Beta Toxicity Validated by Standard and High-Throughput Electrophysiology. PLoS One. 2010;5:e8643. doi: 10.1371/journal.pone.0008643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ida N, Hartmann T, Pantel J, Schröder J, Zerfass R, Förstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 43.Kuo YM, Emmerling MR, Lampert HC, Hempelman SR, Kokjohn TA, Woods AS, Cotter RJ, Roher AE. High Levels of Circulating Aβ42 Are Sequestered by Plasma Proteins in Alzheimer’s Disease. Biochem Biophys Res Commun. 1999;257:787–791. doi: 10.1006/bbrc.1999.0552. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen L, Wright S, Lee M, Ren Z, Sauer JM, Hoffman W, Zago W, Kinney GG, Bova MP. Quantifying Amyloid Beta (Aβ)-Mediated Changes in Neuronal Morphology in Primary Cultures. J Biomol Screen. 2012;17:835–842. doi: 10.1177/1087057112441972. [DOI] [PubMed] [Google Scholar]
- 45.Bloom GS, Ren K, Glabe CG. Cultured cell and transgenic mouse models for tau pathology linked to β-amyloid. Biochim Biophys Acta. 2005;1739:116–124. doi: 10.1016/j.bbadis.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis. 2012;48:356–366. doi: 10.1016/j.nbd.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 47.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301I tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 48.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 49.Selenica ML, Brownlow M, Jimenez JP, Lee DC, Pena G, Dickey CA, Gordon MN, Morgan D. Amyloid oligomers exacerbate tau pathology in a mouse model of tauopathy. Neuro-degenerative diseases. 2013;11:165–181. doi: 10.1159/000337230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hossini AM, Megges M, Prigione A, Lichtner B, Toliat MR, Wruck W, Schröter F, Nuernberg P, Kroll H, Makrantonaki E, Zouboulis CC, Adjaye J. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genomics. 2015;16:84. doi: 10.1186/s12864-015-1262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, Santa-Maria I, Zimmer M, Aubry S, Steele JW, Kahler DJ, Dranovsky A, Arancio O, Crary JF, Gandy S, Noggle SA. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE. 2014;9:e84547. doi: 10.1371/journal.pone.0084547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mufson EJ, Mahady L, Waters D, Counts SE, Perez SE, DeKosky ST, Ginsberg SD, Ikonomovic MD, Scheff SW, Binder LI. Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience. 2015;309:51–67. doi: 10.1016/j.neuroscience.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jack CRJ, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron. 2014;84:608–622. doi: 10.1016/j.neuron.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CRJ, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alvarez A, Toro R, Cáceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- 58.Nieweg K, Andreyeva A, Van Stegen B, Tanriöver G, Gottmann K. Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 2015;6:e1709. doi: 10.1038/cddis.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Zhang G, Zhou H, Barakat A, Querfurth H. Opposite Effects of Low and High Doses of Aβ42 on Electrical Network and Neuronal Excitability in the Rat Prefrontal Cortex. PLoS ONE. 2009;4:e8366. doi: 10.1371/journal.pone.0008366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Picken Bahrey HL, Moody WJ. Early Development of Voltage-Gated Ion Currents and Firing Properties in Neurons of the Mouse Cerebral Cortex. J Neurophysiol. 2003;89:1761–1773. doi: 10.1152/jn.00972.2002. [DOI] [PubMed] [Google Scholar]
- 61.Sanganahalli BG, Herman P, Behar KL, Blumenfeld H, Rothman DL, Hyder F. Functional MRI and neural responses in a rat model of Alzheimer’s disease. Neuroimage. 2013;79:404–411. doi: 10.1016/j.neuroimage.2013.04.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Le Corre S, Klafki HW, Plesnila N, Hübinger G, Obermeier A, Sahagún H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: Neurodegeneration Without Neurofibrillary Tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 65.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 66.Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, LaFerla FM. Soluble Aβ Promotes Wild-Type Tau Pathology In Vivo. J Neurosci. 2012;32:17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 69.Nisbet RM, Polanco JC, Ittner LM, Götz J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015;129:207–220. doi: 10.1007/s00401-014-1371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu C, Lei H, Wang Z, Zhang W, Duan Y. Phenol Red Interacts with the Protofibril-Like Oligomers of an Amyloidogenic Hexapeptide NFGAIL through Both Hydrophobic and Aromatic Contacts. Biophys J. 2006;91:3664–3672. doi: 10.1529/biophysj.106.081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berry BJ, Akanda N, Smith AST, Long CJ, Schnepper MT, Guo X, Hickman JJ. Morphological and Functional Characterization of Human Induced Pluripotent Stem Cell-Derived Neurons (iCell Neurons) in Defined Culture Systems. Biotechnol Prog. 2015;31:1613–1622. doi: 10.1002/btpr.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edwards D, Das M, Molnar P, Hickman JJ. Addition of glutamate to serum free culture improves the electrical properties of adult hippocampal neurons in vitro. J Neurosci Methods. 2010;190:155–163. doi: 10.1016/j.jneumeth.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ravenscroft MS, Bateman KE, Shaffer KM, Schessler HM, Jung DR, Schneider TW, Montgomery CB, Custer TL, Schaffner AE, Liu QY, Li YX, Barker JL, Hickman JJ. Developmental neurobiology implications from fabrication and analysis of hippocampal neuronal networks on patterned silane-modified surfaces. J Am Chem Soc. 1998;120:12169–12177. [Google Scholar]
- 74.Das M, Molnar P, Devaraj H, Poeta M, Hickman J. Electrophysiological and morphological characterization of rat embryonic motoneurons in a defined system. Biotechnol Prog. 2003;19:1756–1761. doi: 10.1021/bp034076l. [DOI] [PubMed] [Google Scholar]
- 75.Das M, Bhargava N, Gregory C, Riedel L, Molnar P, Hickman JJ. Adult rat spinal cord culture on an organosilane surface in a novel serum-free medium. In Vitro Cell Dev Biol Anim. 2005;41:343–348. doi: 10.1007/s11626-005-0006-2. [DOI] [PubMed] [Google Scholar]
- 76.Edwards D, Stancescu M, Molnar P, Hickman JJ. Two cell circuits of oriented adult hippocampal neurons on self-assembled monolayers for use in the study of neuronal communication in a defined system. ACS Chem Neurosci. 2013;4:1174–1182. doi: 10.1021/cn300206k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Natarajan A, DeMarse TB, Molnar P, Hickman JJ. Engineered In Vitro Feed-Forward Networks. Journal of Biotechnology and Biomaterials. 2013;3:153. [Google Scholar]
- 78.Klein WL. A beta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 79.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: list of the average values, standard error, and number of values collected for all electrophysiological parameters.
Supplemental Table 2: list of medium components, vendors, and concentrations used to culture neurons.
Supplemental Figure 1: XPS analysis comparing protein on glass and DETA surfaces
Supplemental Figure 2: gel electrophoresis and coomassie blue stain of pure, oligomerized samples of Aβ.