

Abstract
Fast photochemical oxidation of proteins (FPOP) is a footprinting technique used in mass spectrometry-based structural proteomics. It has been applied to solve a variety of problems in different areas of biology. A FPOP platform requires a laser, optics, and sample flow path properly assembled to enable fast footprinting. Sample preparation, buffer conditions, and reagent concentrations are essential to obtain reasonable oxidations on proteins. FPOP samples can be analyzed by LC-MS methods to measure the modification extent, which is a function of the solvent-accessible surface area of the protein. The platform can be expanded to accommodate several new approaches, including dose responsible studies, new footprinting reagents, and two-laser pump-probe experiments. Here, we briefly review FPOP applications and in a detailed manner describe the procedures to set up an FPOP protein footprinting platform.
1. Introduction
Mass spectrometry (MS) is a rapidly developing tool in structural biology research. Compared to X-ray crystallography (X-ray), cryogenic electron microscopy (cryo-EM), and nuclear magnetic resonance (NMR), MS approaches require lower sample amounts and assure faster analysis turnaround. MS studies can provide structural information of proteins in solution, although the spatial resolution is low and does not yet give atomic coordinates like those from cryo-EM, NMR, and X-ray crystallography. MS analysis provides m/z measurements that inform on protein modifications (peptide and amino-acid readouts) that can be related to structure, changes in structure, and dynamics.
Protein footprinting is a MS-based analysis that studies protein structure and interactions within macromolecular assembly. The chemical reactions used in footprinting include limited cleavage, hydrogen deuterium exchange (HDX), and modifications of amino acid side chains by irreversible covalent labeling. Limited cleavage (proteolysis) of proteins and nucleic acids is an early footprinting strategy to study interactions between proteins and DNAs [1]. With improvements in MS resolving power, accurate mass, and speed, HDX and other covalent labeling have seen increased use, now providing more structure resolution, depth, and accuracy of footprinting [2]. Compared to HDX, the other labeling approaches generate irreversible modifications on protein surfaces that permit careful and stringent sample cleanup and analysis by, for example, bottom up proteomics methods. Those modifications provide information related to solvent accessibility, structure dynamics, and distance constraints that reflect protein native structure.
Hydroxyl radical footprinting (HRF) is the most widely used irreversible covalent labeling method to characterize protein structure and dynamics [3]. It utilizes hydroxyl radicals (·OH) to oxidize amino acid side chains on the protein surface. Hydroxyl radicals can be generated in aqueous solution by Fenton reactions [4], UV photolysis of hydrogen peroxide [5,6], and radiolysis of water by synchrotron X-ray [7], in-source electrical discharge [8], 137Cs γ-irradiation [9], and plasma irradiation [10]. Although the experimental methods, reaction products, and mechanisms vary among those methods, a common goal is to limit the number of exposure and minimize reaction time to avoid misleading modification-induced unfolding. Long-time footprinting of protein can give misleading results because the early stages of modification can cause denaturation, allowing in later stages additional labeling that does not reflect the original protein structure. Because the purpose of this method paper is to review fast photochemical oxidation of proteins (FPOP), we will not present any further review of the other approaches [2,11,12].
FPOP is an approach to footprint proteins by radicals generated by laser photolysis in a flow system. FPOP limits the exposure of proteins with reactive species to a sub-millisecond time scale by incorporating two design features. First, radicals are generated in a flow system where a protein solution passes through a window irradiated by a pulsed laser beam. Each sample “plug” or region within the window is exposed once to the pulsed laser beam for a short time (ns). Second, the lifetimes of the primary ·OH radicals are kept short (sub-ms) by scavenging them in the solution to reduce their lifetimes. Therefore, FPOP footprinting is sufficiently fast that it can capture rapid structure changes, as is mentioned in the next section.
We will first briefly review example problems in biochemistry/biophysics that can be investigated using FPOP. We will offer guidance for platform setup, sample preparation, data acquisition and result analysis. Besides the generic FPOP setup for hydroxyl radical footprinting, we will also describe several expansions of the platform for kinetics studies, new reagents, and protein dynamics studies. We will also draw from our experience some troubleshooting tips for addressing problems in primary FPOP experiments. We also supply in supplemental information a parts list used to set up an FPOP platform.
2. FPOP applications
FPOP offers opportunities to answer a variety of structural biology questions as we did in a recent review [13]. The following brief list of applications serve as specific examples of general applications.
Locate protein-protein and protein-ligand binding regions. For example FPOP can report accurately the heme-binding region in myoglobin [14] and the sites of calcium/peptide binding in calmodulin [15]. FPOP can also map the binding regions of antibody-antigen complexes [16–20], and thereby provide insights into structural issues of therapeutic proteins.
Serve as a fast footprinting method. The approach can be used uniquely to capture protein conformational changes (folding) on sub-millisecond time scale [6,17,18], even shorter than most protein folding processes. Indeed, by using a two-laser approach, temperature-induced folding of barstar protein can be followed at the global protein level down to peptide and even amino-acid residue levels [22,23]. As another example, FPOP is able to reveal fast loop motions in TEM β-lactamase variants, motions that are invisible to slower methods [24].
Provide a platform to study protein aggregation by probing structural difference in oligomeric states of ApoE isoforms [25]. It can specifically characterize the solvent accessibility changes during various stages of amyloid protein aggregation, affording peptide-level and sometimes amino-acid-residue resolution [26].
Study membrane proteins. So far, FPOP can delineate the soluble regions of membrane proteins and provide comparative data for their interactions with surfactants and detergents [27,28]. These applications describe the first steps in using footprinting to map the interactions of membrane proteins with ligands and with the lipids that constitute a cell wall.
Investigate solvent-exposed proteins in live cells [29]. The fast turnaround of the labeling can maintain cell viability while probing protein-solvent accessibility in a native environment [30]. By adopting a microflow system with sheath buffer, the performance of in-cell footprinting has been greatly increased [31].
Despite the successful application mentioned above, there are limitations of hydroxyl radical footprinting by FPOP as there are for any method. Some proteins are oxidized simply by exposure to hydrogen peroxide. This can occur either prior to or following laser irradiation. Measures can be taken to minimize and/or control for this problem. Another problem occurs in studies of trans-membrane proteins. Although residues in contact with water can be footprinted, regions within the membrane present challenges. Other problems and possible updates to FPOP platform will be discussed in later sections.
3. General Platform setup
Having established some classes of FPOP applications, we now describe its setup, methods for data processing, and opportunities to expand functionality of the FPOP platform. The emphasis will be on experimental details and on the requirements for setting up an FPOP apparatus for those readers who are interested in adopting this approach in structural proteomics. We will next consider the generic FPOP platform for hydroxyl radical footprinting.
3.1 Laser, optics and sample flow path
The FPOP platform consists of three components: laser, optics, and sample flow path (Figure 1). Proper configuration of all component is essential for successful footprinting experiments. Here, we demonstrate the setup of each part and discuss the principles behind those designs.
Figure 1.

(A) Scheme for a general FPOP setup. (B) A real photo showing the setup from (A). (C) Laser-burn mark on a plastic sticker after 20 pulses with laser intensity of ~20 mJ/pulse. (D) Transparent window on a piece of silica tubing after removal of coating with flame. (A)(B) are adapted from K.S. Li et al, Acc. Chem. Res. 51 (2018) 736–744. doi:10.1021/acs.accounts.7b00593 with permission.
Laser
Krypton fluoride (KrF) excimer laser is the commonly used laser source for FPOP experiments, and there are a few vendors. The laser provides ns pulses of UV laser at 248 nm. Although the absorbance of H2O2 is not maximum at this wavelength, the quantum yield is sufficient to generate adequate amount of hydroxyl radicals [32]. Light of this wavelength is minimally absorbed by proteins in aqueous solution. The pulse width of KrF laser is generally tens of nanoseconds, much shorter than ·OH lifetime and timescale of protein higher order structural motions.
The number of ·OH generated is positively affected by laser pulse intensity when the optics are fixed. The laser intensity also gradually decreases with its usage, requiring periodic refilling of the krypton fluorine gas mixture. Therefore, it is recommended that laser intensity be measured at the beginning of an FPOP experiment. The laser pulse intensity (usually greater than 15 mJ/pulse but dependent on the optics) can be measured by a pyroelectric energy sensor placed at the exit of the KrF laser or other positions in the laser path. The pyroelectric sensor also measures the laser shot rate (f, in Hz), which is used to determine the sample flow rate, as discussed later.
Optics
The optics focus the laser beam onto sample flow path (Figure 1). Laser focusing is critical for high photon density and, thus, for higher yield of hydroxyl radicals. A spherical and a cylindrical plano-convex lenses are used for focusing in the setup (Figure 1). The spherical lens focuses the beam to a tiny spot of ~ 2 mm x 2 mm. The cylindrical lens further compresses the beam in one dimension to form a ~ 2 mm x 1 mm spot with photo density doubled (Figure 1d). A height of 1 mm is sufficient to cover the sample capillary, which usually has an 100–400 μm outer diameter (O.D., we use 350 μm O.D.). An iris with adjustable aperture can be used to reduce the spot width when a smaller spot is desired (e.g., in two-laser experiments). A laser stop is placed behind the capillary to terminate the laser beam for safety.
Laser spot shape can be recorded on heat-sensitive paper, a piece of plastic or paper sticker with color printing that will be bleached by UV laser. The sticker is placed behind the capillary with color printing facing the laser beam. After a few laser shots, the laser-spot shape is imprinted on the sticker. When the laser beam and sample capillary are properly aligned, the capillary will leave its shadow on the imprint after firing just a few laser shots. For example, the imprint (Figure 1D) from 20 shots at ~ 20 mJ/pulse is readily detected. The burn imprint allows measurement of laser spot width (l, in mm). The spot width will be used to calculate exclusion volume (E.V.) to determine sample flow rate (see later).
Sample flow path
Radical generation and footprinting reaction take place at the intersection of laser and sample flow path. A convenient carrier for the sample solution can be made from fused silica tubing. The coating on the tubing is removed by an ethanol burner to form a transparent window for the UV laser (Figure 1). A syringe pump delivers a constant sample flow rate (Q, in μL/min) through the silica tubing. The tubing holder is mounted on a 3-dimensional adjustable platform for careful alignment with the laser beam.
During an experiment, the sample solution passing through the lens region is irradiated segment-by-segment. Ideally, each segment is only irradiated once, assuming it were plug flow (Figure 2). The Exclusion volume (E.V.) is defined as fraction of solution that is not exposed to laser if the plug flow is ideal. It is calculated from inclusion volume by Equation 1, where d is inner diameter (I.D.) of the tubing, l is laser spot width, f is laser shot rate and Q is sample flow rate.
Figure 2.
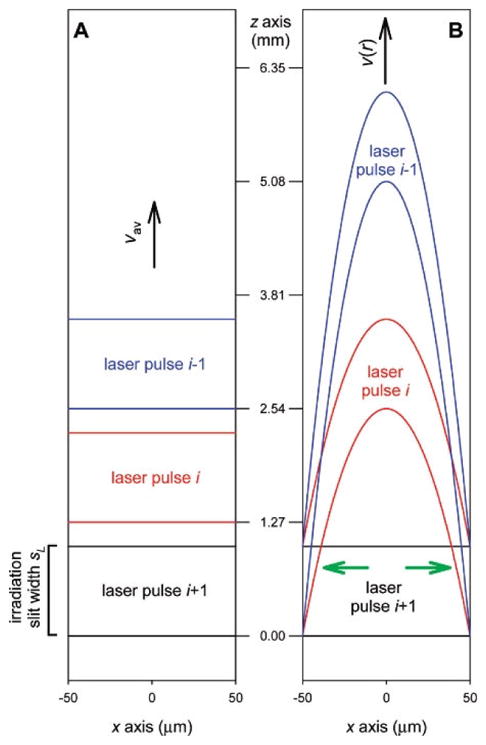
Simulated models for sample flow in a capillary (A) Plug flow (B). laminar flow. Capillary I.D. 100 μm, irradiation slit width 1 mm, flow rate 60 μL·min−1. Adapted from L. Konermann et al, Anal. Chem. 2010, 82, 6667–6674 doi:10.1021/ac101326f with permission.
| (Equation 1, SI unit) |
Plug flow is a simplified model for calculation of exclusion volume. In fact, flow profile in the capillary is determined by capillary I.D., flow velocity, dynamic viscosity, and density, affording a Reynolds number. In practice, the flow velocity is low, resulting in a laminar flow [33]. Owing to the parabolic profile of laminar flow, part of the solution flowing closest to the tubing wall is moving slowest and is most likely to be exposed to the laser more than once (Figure 2). To minimize this exposure, at least 25% E.V. is recommended in practice. When E.V. = 30%, the solution subject to multiple exposure is less than 10% of solution irradiated by laser [33]. With known tubing I.D. from the manufacture and laser-spot width measured above, laser shot rate can be calculated from target flow rate or vice versa, allowing one to configure either the laser shot rate on the KrF laser or the sample flow rate on the syringe pump to ensure a specified E.V. For example, for a capillary of 150 μm I.D., when the spot width is 2 mm and laser shot rate is 7 Hz, the flow rate is calculated to be 21.2 μL/min for 30% E.V.
3.2 Sample preparation, FPOP process and sample collection
Generating chemical modification on proteins while in its native conformation in solution is critical for successful footprinting experiments. The overall modification level can affect data quality and confidence in the results. Too low an oxidation extent may challenge the detection limit of the mass spectrometer and its ability to produce suitable product-ion spectra to locate the modification. Excessive oxidation may cause saturation of the modification level and cause loss of control in the quantification of the unmodified peptides. Sample preparation, FPOP process and sample collection steps are essential for successful FPOP experiments.
3.2.1 Sample preparation
A typical sample for FPOP contains protein and ligands of interest, H2O2, and a chemical radical scavenger in appropriate buffer. Owing to the high sensitivity of modern mass spectrometers, the amount of protein in each FPOP sample can be as low as a few micrograms (low nanomoles). However, protein and ligand concentrations should be biologically relevant (e.g., consistent with good binding as determined by their binding constants). Protein conformation and function should be in a native state prior to footprinting.
Buffer components
Buffer components affect both protein function and oxidation level generated by FPOP. Although protein function in certain buffers can be assayed by variety of biological methods, it is not subject to discussion here. The acid base pair and other chemicals in a buffer solution can scavenge the free radicals and diminish oxidation induced by FPOP. A study shows that different buffers have different effects on oxidation strength in synchrotron radiolysis of water, and small molecule additives such as ATP and EDTA also affect the outcome [7,11]. An FPOP dosimetry experiment shows that tris-HCl concentrations decrease the hydroxyl radicals that are available to react with the dosimeter [34]. Phosphate buffer saline (PBS) has been used in many FPOP experiments because it is compatible with many biological systems, and the buffer components do not react significantly with ·OH. Reducing substances (e.g., dithiothreitol (DTT) and solubilizing components, dimethyl sulfoxide (DMSO)) have strong quenching effects on hydroxyl radicals. Those components should be avoided or kept at very low concentration. If such chemicals are critical for protein solubility or activity, one should consider using other footprinting reagents developed for FPOP. If the buffer components vary among sample groups, a non-interfering reporter peptide can be added to the sample to even out the oxidative strength in later data processing. Those expansions for FPOP setup will be described in later sections.
H2O2 and scavenger
H2O2 and scavenger concentrations are major determinants of the protein oxidation level. There are a few radical dosimeters that can be used to determine the ·OH concentrations produced in either a flow system [34–36] or with synchrotron radiation [7]. Notably, oxidation of angiotensin, a model peptide, has a positive correlation with H2O2 concentration and a negative correlation with tris-HCl buffer [34]. In practice, we find 15–20 mM H2O2 with 20–30 mJ/pulse laser energy generates appropriate oxidation levels on calmodulin and other proteins of this MW (Figure 4). Dosimetry experiments show that approximately 1 mM ·OH is produced from 15 mM H2O2 under our experimental conditions with no scavenger added [35]. Although this is a remarkable number of radicals, allowing good yields of modified proteins and peptides, the radical lifetime needs to be controlled.
Figure 4.
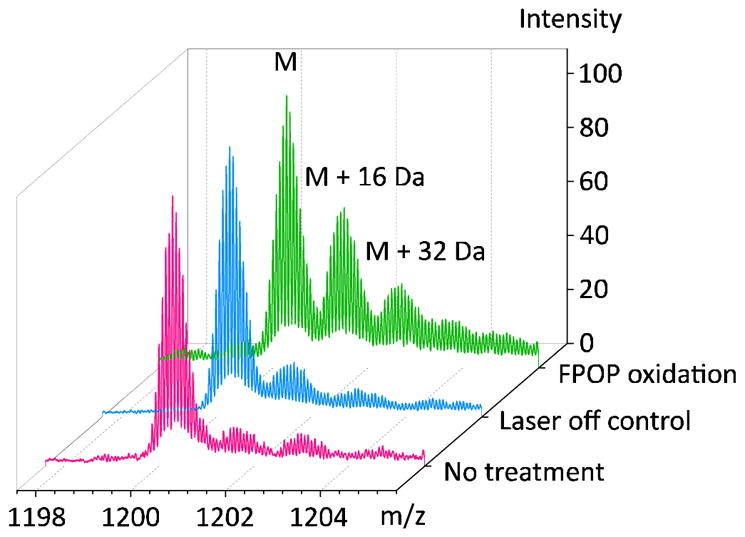
Comparison of oxidation extent on calmodulin without any treatment (pink), with H2O2 and laser off (blue), with H2O2 and laser on (green). Charge state +14 is shown. FPOP experiment parameters: calmodulin 2 μM, histidine 2 μM, H2O2 20 mM, laser energy 24 mJ/pulse, spot width 2.2 mm, shot rate 7.2 Hz, capillary I.D. 150 μm, flow rate calculated and set as 22.4 μL/min, E.V. 25%.
To control the life time of ·OH in solution, a chemical scavenger is added to consume some of the ·OH in competition with the protein. Kinetic modeling shows that with the scavenger added, the primary ·OH lifetime can be controlled to be in the microsecond timescale [6]. Some of the amino acids are good candidates for scavengers, because their reaction rate constants with ·OH are well known and large (nearly diffusion-controlled) [11]. Furthermore, some amino acids are used to stabilize therapeutic antibodies, and their inclusion in an experiment assures compatibility with the protein handling in biotechnology [37]. Glutamine, histidine or methionine are commonly used as scavengers, although there are questions about the effectiveness of glutamine [36].
3.2.2 FPOP process
Before running FPOP samples, the laser intensity should be recorded and the laser spot width measured. Furthermore, the sample flow rate or laser shot rate should be configured as described in section 3.1. To perform an experiment, proteins and ligands are prepared as stock solutions. Solutions of H2O2 and the scavenger are added, and the sample is loaded into a syringe on a syringe pump. The laser pulsing and the syringe pump are started immediately. The sample “plugs” are irradiated for hundreds of laser shots (the whole sequence is complete in minutes). The sample is collected at the end of the capillary in an Eppendorf tube containing quenching solution, which is described below in 3.2.3.
The H2O2 solution should be added to the sample immediately before starting the run to minimize oxidation of the test protein prior to laser treatment. The time between adding H2O2 and injection can be controlled among samples by using a timer. If there is concern or evidence that too much oxidation occurs to the protein from exposure to the hydrogen peroxide, a micro-tee mixer device can be used to introduce H2O2 to the sample flow [38]. A coaxial laminar flow mixer can also be used, although its design was originally for protein-folding studies [39]. The mixing device is located just before the laser spot to reduce the mixing time from minutes to seconds (Figure 3). Additionally, the data precision increases when using the mixing device [38].
Figure 3.
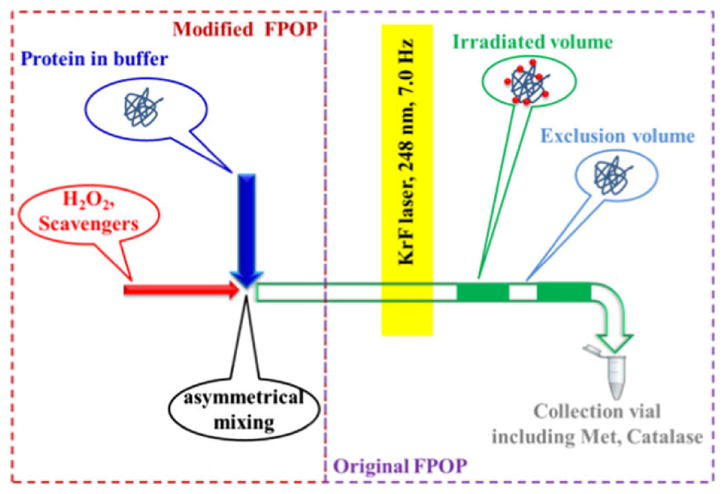
H2O2 and scavenger solution introduced by a rapid mixing device to reduce H2O2 oxidation and improve precision. Adapted from. Y. Zhang et al, J. Am. Soc. Mass Spectrom. 26 (2014) 526–529. doi:10.1007/s13361-014-1055-0 with permission.
Laser-off controls should be done in the same manner as for test samples, except that the laser is off during the flow. The laser-off control allows measurement of oxidation that is a post-translational modification or occurs during sample handling and storage. It is also recommended that laser-off control be done to accompany study of each protein state. Another control is to submit the sample to laser irradiation without H2O2, which is usually of lower concern than oxidation by H2O2 during sample handling.
3.2.3 Sample collection and storage
In the FPOP setup, samples can be collected by placing an Eppendorf tube with quenching solution at the end of sample capillary. Once labeled, changes in protein structure during sample handling and storage do not alter the modification pattern. Nevertheless, the remaining H2O2 and other oxidative species in the solution must be removed to avoid slow oxidation during sample storage. Even when the protein solution is frozen, H2O2 can still oxidize protein in a matter of hours, and the total oxidation may no longer be a reporter of solvent exposure [40]. To prevent post-irradiation oxidation, a quench solution containing methionine and catalase is added to the collector/storage tube. Catalase is an enzyme that decomposes remaining hydrogen peroxide to water and oxygen. Methionine is a good reducing agent for species that are weak oxidizing agents generated in the H2O2 decomposition. After completing the experiment and sample collection, the sample and quenching solution are rapidly mixed in the collection tube. A mixing device located after laser window can also be introduced for that purpose. At this point, the sample can be stored for later processing and analysis.
3.3 MS Data collection and analysis
Protein footprinting by hydroxyl radicals produces both backbone cleavage and side-chain oxidation [11]. Backbone cleavage is nonspecific, difficult to identify or quantify, and is not prominent for FPOP data analysis. Oxidation extent, however, can be measured at the intact protein and peptide levels, or even at the amino-acid residue levels using LC-MS proteomics methods.
As a hydroxyl radical footprinting method, FPOP can produce various changes on amino acid side chains. The most abundant product is oxidation whereby an H is replaced by an OH, causing a mass addition of +16 Da (+15.9949 Da to be accurate). A series of +16 Da peaks can be observed in the mass spectra of labeled proteins (Figure 4). Other products include di-oxidation (+32, 2O), tri-oxidation (+48, 3O), oxidation to carbonyl (+14, O -2H), oxidation and dehydration (−2, +O -H2O), decarboxylation (−30, O -2H -CO2), etc. [3]. Those mass shifts, along with that from single oxidation, are used to identify oxidized peptides by database searching for proteolytic peptides.
Intact protein measurement
Intact protein masses can be measured by MS to evaluate the footprinting yield provided the intact protein is well-behaved by MS. After desalting by solid-phase extraction or on-line HPLC, the protein can be analyzed by electrospray ionization (ESI). Reversed-phase columns and acidic aqueous/organic solvents can be used for this LC-MS analysis, leading to protein denaturation and extensive charging in the spectrometer. A representative spectrum (Figure 4) illustrates the oxidation of a calmodulin sample as measured on a high-performance Q-TOF instrument. One alternative is native LC/MS and another is to accompany native MS with ion mobility [41].
Location of modifications at the peptide level
Oxidation extent is a measure of solvent accessibility, and changes in labeling extent reflect changes in protein binding or conformation. The bottom-up proteomic strategy with MS is a suitable and powerful approach to obtain peptide and residue level oxidation information. FPOP generates covalent modifications that are stable, enabling optimization of protein digestion and LC-MS/MS methods to provide nearly complete sequence coverage of the proteins. Protease (e.g., trypsin, chymotrypsin, Asp-N, or their combinations) are used to digest FPOP samples. Chromatography (e.g., reversed-phase, size-exclusion) separations can be used to separate the peptides formed by proteolysis. Peptides and modifications are identified by MS/MS with collisional activated dissociation (CAD) or ETD. Many software packages can use suitable protein databases to identify the peptides and their modifications. Examples are Protein Metrics [40], Mascot [41], PEAKS Studio, and Proteome Discoverer. The mass changes produced by oxidative footprinting are included in the searching parameters to identify peptides with oxidation.
Quantification of oxidative extents
Relative quantification of modification extent can be achieved by extracted ion chromatogram with accurate masses of wild type and modified peptides from reversed-phase chromatography (Figure 5A) [13,14,42]. Notably, the elution time of oxidized peptide is often different than that of the wild-type peptide, allowing one to evaluate the interference from peptide oxidation during electrospray process [43]. Modification extents at the amino acid residue level can be calculated when a peptide with different modification sites can be separated by chromatography (Figure 5A). However, that is not always the case because the separation capability is limited or there is ambiguity in product-ion assignment. The data processing is now expedited with the software provided by Protein Metrics.
Figure 5.

(A) Flow chart of FPOP data processing by bottom-up proteomics and reversed phase LC-MS/MS. Oxidation extent on peptide is calculated as modified / (modified + unmodified) using their peak area on extracted ion chromatogram. Adapted from K.S. Li et al, Acc. Chem. Res. 51 (2018) 736–744. doi:10.1021/acs.accounts.7b00593 with permission. (B) “FPOP histogram” and differential plot identify key residues in epitope regions of an interleukin-23/Fab complex. Adapted from J. Li et al, Anal. Chem. 89 (2017) 2250–2258. doi:10.1021/acs.analchem.6b03058 with permission.
A clever alternative quantification strategy for site specific modification is MS/MS by electron transfer dissociation (ETD). Isomers of modified peptides are coeluted from size-exclusion chromatography, and modification sites are quantified by product ion intensity [44].
The oxidation extents on the whole peptide or amino-acid residue levels are analyzed to obtain information on solvent accessibility changes among different sample groups (e.g., bound vs. unbound, wild type vs. mutant). Data can be plotted as “FPOP histograms” that report differences between states (Figure 5B). Student’s t-test can be applied to determine the significance of oxidation-extent differences between sample groups. Normalized Protection Factors (NPF), the radical response rate of a residue normalized against the intrinsic reactivity of that free amino acid, can also report solvent accessible surface areas from hydroxyl radical footprinting data [45,46]. However, more effort is still needed to provide appropriate statistical methods for FPOP.
4. Expansion of the FPOP platform
We discussed the strengths of hydroxyl radical footprinting by FPOP in a variety of biological studies and some limitations earlier in this article. Despite its strengths, the general FPOP setup is not a one-solves-all technique, and more development is needed. For example, ligands or cofactors in a protein sample can scavenge radicals and cause systematic errors in measuring oxidation extent. The intrinsic rate constants for hydroxyl radicals reacting with amino acids are over a large range, and this challenges the ability of FPOP to map interfaces comprised of both low and highly reactive residues. Therefore, the FPOP platform needs to evolve to accommodate these and other problems that must be faced in biological applications. Indeed, the platform has a flexible design that provides opportunities for customization and upgrades. In this section, we describe several expansions for the general FPOP setup. These modifications allow inclusion of FPOP kinetics in comparison studies, new footprinting reagents, and studies of protein folding.
4.1 Reporter peptide
In the FPOP experiment, the laser irradiation generates a large amount of ·OH (~ 0.5 – 1 mM) compared to the concentration of the protein (2 – 10 μM) but comparable to the “effective concentration” of reactive sites. Although small changes in protein concentration usually will not affect oxidation, large variations in ·OH may occur from sample to sample when the solution contains adventitious scavengers (e.g., TCEP, ATP, enzyme cofactors, lipids, DMSO, unexpected proteins). One approach to this problem is to add a short peptide (e.g., leu-enkephalin, YGGFL) as a reporter to calibrate oxidative potential (Figure 6A) [47]. The reporter peptide must have exposed reactive residues but should not bind to the protein. Its oxidation extent is determined by integrating the chromatographic peak areas of the unmodified and the +16 species. Incorporation of the reporter peptide also enables dose-response studies where the dose is a function of time and time is controlled by varying the scavenger concentration. Each protein state is footprinted for various ·OH life times (·OH lifetime is determined by scavenger concentration). Longer times of reaction give higher levels of reporter oxidation. Plotting extents of protein modification vs. those of the reporter gives a time-dependent, dose-response curve that clearly pinpoints when differences in solvent accessibility occur in protein states (Figure 6B) [47]. The reporter peptide approach achieves spatial resolution at the peptide and sometimes the residue levels. The dose-response plots have more statistical weight than a single time point and afford an HDX-like kinetic display that is familiar to investigators.
Figure 6.

(A) Reduced oxidation extents on a reporter peptide show the effect of adventitious scavenger in SOD1 samples. (B) Dose-dependent curves show regions that are differentially protected on WT SOD1 vs A4V mutation. Adapted from B. Niu et al, J. Am. Soc. Mass Spectrom. 28 (2017) 389–392. doi:10.1007/s13361-016-1552-4 with permission.
4.2 New reagents
New footprinting reagents can offer broader reactivity to enable greater coverage and higher resolution for the determination of critical binding residues (e.g., epitopes). Instead of broader coverage, some applications pose more focused questions on targeted residues where less-reactive, more specific reagents can play a role. For these applications, more facile data processing will also lead to higher experimental efficiency and faster turnaround rate.
Several types of reagents have been developed on the FPOP platform, including iodination [48], carbenes [49], SO4−• [50], and trifluoromethylation [51] for protein footprinting. Each of the new reagents has unique properties and reactivity with the 20 amino acids, expanding the library of footprinters.
Trifluoromethyl radicals (·CF3) radicals are generated by reaction of sodium trifluoromethanesulfinate (NaSO2CF3) with ·OH generated from H2O2 by the KrF excimer laser [51]. One ·CF3 substitution creates an unusually large mass shift of +68 (+ CF3 -H) compared to ·OH, enabling resolution from natural post-translational modifications and adducts. These new footprinting radicals can modify nearly all 20 amino acids except for cysteine and methionine, thereby increasing the coverage of footprinting. This method has been implemented on the FPOP platform without any hardware changes.
Carbenes are highly reactive diradicals that undergo insertion into X-H bonds, especially when X is a heteroatom. Carbenes can be produced on the FPOP platform by photolysis of diazirines at 355 nm (using a Nd-YAG laser with a third harmonic generator) [49]. No scavenger is needed because the reactive species have ns lifetimes, and they are readily quenched by water. Furthermore, the hydrophobicity and reactivity of carbenes can be tailored by their structure, offering versatility in controlling the regions where they react (e.g., soluble portions vs. membranes) [54].
A more specific reagent is the iodide radical, which can be generated on the FPOP platform by photolysis of iodobenzoic acid at 248 nm to give ·I and presumably ·C6H4COOH [48]. It may be that protein radicals are produced by ·C6H4COOH and then quenched by the more selective ·I These latter radicals only target histidine and tyrosine, yielding a characteristic mass shift of +126 (+ I -H). As one testimony to its footprinting ability, the iodide reactions showed large differences between myoglobin and its apo form, notably in the region of the F helix that exists only in the holo state.
4.3 Two-laser experiments
A second laser can be incorporated for time-resolved experiments to follow fast protein folding [22,23]. In this design, one laser (the “pump”) induces protein conformational change by a temperature jump, and the other laser (“probe”) photolyzes a suitable precursor to give reactive species that report time-dependent changes in solvent accessibility. Controlling the time between the laser pulses allows the protein folding to be followed from μs and up to ms.
We have used FPOP with a two-laser setup to study temperature-induced folding of the barstar protein [22,23]. The “pump” was the 1900 nm, generated by Raman shifting the fundamental 1065 nm of a Nd-YAG laser. The 1900 nm light couples to the bending motion of the solvent water molecule to give a temperature jump in the flow system. The KrF excimer laser then generates hydroxyl radicals to capture conformation changes of barstar by the time-dependent differences in oxidation. The observation of decreasing oxidation on barstar with increasing time delay shows that folding of the protein is occurring, and LC-MS analysis locates the regions that are folding. Changes of oxidation extent on peptide and residue level are consistent with the concept that protein folding occurs around a hydrophobic nucleus.
5. Troubleshooting tips
Oxidation level is low
Footprinting by FPOP can fail if the oxidative modifications are too sparse or low in amount. Low oxidation levels may result from flaws in instrument setup or sample preparation. Instrument setup can be checked by using small model peptides or proteins (e.g., calmodulin, ubiquitin, or leu-enkephalin in phosphate buffer saline) as tests. Having a simple platform to measure the extent of oxidation of the intact peptide or protein (e.g., ESI or Native MS) expedites the check.
If no oxidation is apparent, the instrument setup (e.g., laser energy and laser-capillary alignment) should be examined. The transparent window on the silica capillary should be checked for laser damage. Laser shot rate or sample flow rate should be recalibrated to guarantee the desired exclusion volume. The sample flow rate should be configured properly on the syringe pump, and even more basically, one should insure that the correct syringe is used.
To troubleshoot sample preparation, one should examine if there are any reducing chemicals in protein samples. H2O2 concentration can decompose after longer term storage. Concentration of H2O2 stock solution can be measured spectrophotometrically at 240 nm using a molar extinction coefficient of 43.6 M−1cm−1 [55]. Slowly forming bubbles in the sample-collection tube is an indication for the presence of H2O2 in the sample. Better sample purification, if possible, or another footprinting reagent should be considered if the buffer contains reducing chemicals, or the protein contains many residues that are not reactive with hydroxyl radicals.
Oxidation level is too high
Over-oxidation can also result from many factors. Once again, a check on the intact protein (prior to digestion) can be done by a simple MS or LC-MS method, as described above. The scavenger concentration should also be checked. Too slow flow rates can result in multiple shots on each sample fraction, causing over oxidation. Sample flow rate and laser shot rate should be recalibrated and reconfigured if necessary. Leakage in the flow system, including around tube fittings and syringe seals can also slow down the sample flow past the laser and cause over oxidation.
Low protein coverage
Many reasons can cause low coverage on proteins after database search, such as low digestion efficiency, poor chromatography, improper MS setting and inaccurate searching parameters. Here we only list a few possibilities. Proper enzymes or their combinations should be selected based on the proteolytic sites in protein sequence. Digestion conditions are to be optimized, especially for rigid proteins or large protein complexes with multiple disulfide bonds. Chromatography type, gradient and flow rate should be checked. Development in LC techniques provides opportunities to improve retention and separation of peptides. MS configurations should be examined. Data acquisition methods such as data independent scan can be considered. During database search, the searching parameters such as mass accuracy, protein database, modifications sites and masses and FDR cutoff should be examined. Peptides with unexpected or diversified post translational modifications are hard to identify during database search, especially for glycosylated proteins. Those problems, however requires efforts and will likely be solved with progresses in LC, MS and software algorisms.
6. Conclusion and prospects
FPOP was developed as a hydroxyl radical footprinting approach with advantages of fast reaction and limited exposure [6]. Its basic platform consists of a laser, optics and flow system. LC-MS, bottom-up proteomics and database searching software are needed for the analysis, and such analyses are mature thanks to the demands of proteomics. New software developments permit faster data processing. Good chromatography resolution enables measuring solvent accessibility on the residue level. Solvent accessibility information from protein footprinting has been used alone or combined with data from other physical methods to evaluate or generate structural models [45,56,57]. With rapidly growing computer algorithms and even artificial intelligence, obtaining coarse-grained structural models may be possible by using footprinting data as constraints, much like that done in NMR with chemical shifts. With multiple possibilities of platform expansions and upgrades, we expect that more applications will employ FPOP and other footprinting approaches to investigate structural biology questions in the future.
Supplementary Material
Figure 7.
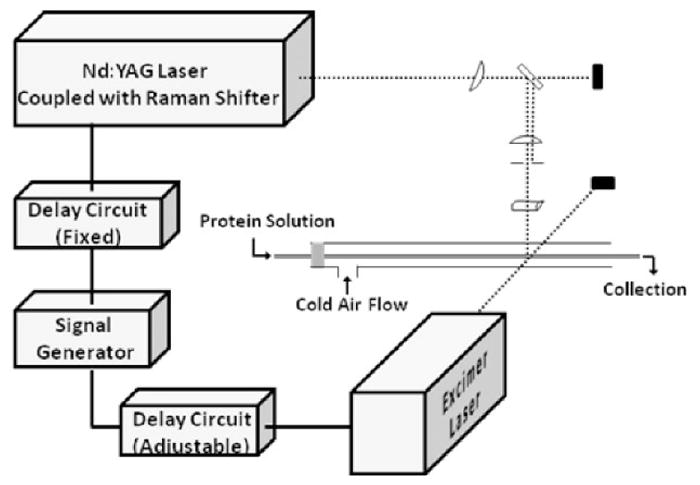
Scheme for the FPOP setup with two lasers to study protein folding. Adapted from J. Chen et al, Journal of the American Chemical Society. 132 (2010) 15502–15504. doi:10.1021/ja106518d.
Table 1.
Reagents developed for protein footprinting by FPOP
| Reagent | Radical | Wavelength | Mass shift | Target amino acid | Reference |
|---|---|---|---|---|---|
| H2O2 | ·OH | 248 nm | +16 Da | C, M, W, Y, F, H, L, I,R, K, V, S, T, P, N… | [6,52] |
| Na2S2O8 | SO4−• | 248 nm | +16 Da | M, Y, W, F, E, H, S, P, D, T, K, Q… | [50] |
| CF3SO2Na | ·CF3 | 248 nm | +68 Da | R, H, K, D, E, S, T, N, Q, G, P, A, V, I, L, F, Y, W | [51] |
| Iodobenzoic acid | ·I | 248 nm | +126 Da | Y, H | [48] |
| Diazirine | Carbene | 355 nm | Various | Y, E, D, H, K, C, W, R, Q, M… | [49,53] |
Highlights.
Brief review of applications of fast photochemical oxidation of proteins (FPOP)
Introduction to the principles of FPOP
Instructions on platform assembly
Details on experimental protocols
Expansions of the FPOP platform
Acknowledgments
The preparation of this review was support by the NIH (Grant no. 2P42GM103422). We thank Protein Metrics for developing and sharing software for FPOP data processing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Chance MR. Protein Footprinting Comes of Age: Mass Spectrometry for Biophysical Structure Assessment. Mol Cell Proteomics. 2017;16:706–716. doi: 10.1074/mcp.O116.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Chance MR. Structural Mass Spectrometry of Proteins Using Hydroxyl Radical Based Protein Footprinting. Anal Chem. 2011;83:7234–7241. doi: 10.1021/ac200567u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shcherbakova I, Mitra S, Beer RH, Brenowitz M. Fast Fenton footprinting: a laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucleic Acids Res. 2006;34:e48–e48. doi: 10.1093/nar/gkl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharp JS, Becker JM, Hettich RL. Analysis of Protein Solvent Accessible Surfaces by Photochemical Oxidation and Mass Spectrometry. Anal Chem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- 6.Hambly DM, Gross ML. Laser Flash Photolysis of Hydrogen Peroxide to Oxidize Protein Solvent-Accessible Residues on the Microsecond Timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Gupta S, Sullivan M, Toomey J, Kiselar J, Chance MR. The Beamline X28C of the Center for Synchrotron Biosciences: a National Resource for Biomolecular Structure and Dynamics Experiments Using Synchrotron Footprinting. J Synchrotron Radiat. 2007;14:233–243. doi: 10.1107/S0909049507013118. [DOI] [PubMed] [Google Scholar]
- 8.Maleknia SD, Chance MR, Downard KM. Electrospray-assisted modification of proteins: a radical probe of protein structure. Rapid Commun Mass Spectrom. 1999;13:2352–2358. doi: 10.1002/(SICI)1097-0231(19991215)13:23<2352::AID-RCM798>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Smedley JG, Sharp JS, Kuhn JF, Tomer KB. Probing the pH-Dependent Prepore to Pore Transition of Bacillus anthracis Protective Antigen with Differential Oxidative Protein Footprinting. Biochemistry (Mosc) 2008;47:10694–10704. doi: 10.1021/bi800533t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minkoff BB, Blatz JM, Choudhury FA, Benjamin D, Shohet JL, Sussman MR. Plasma-Generated OH Radical Production for Analyzing Three-Dimensional Structure in Protein Therapeutics. Sci Rep. 2017;7:12946. doi: 10.1038/s41598-017-13371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu G, Chance MR. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 12.Maleknia SD, Downard KM. Advances in radical probe mass spectrometry for protein footprinting in chemical biology applications. Chem Soc Rev. 2014;43:3244–3258. doi: 10.1039/C3CS60432B. [DOI] [PubMed] [Google Scholar]
- 13.Li KS, Shi L, Gross ML. Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Acc Chem Res. 2018;51:736–744. doi: 10.1021/acs.accounts.7b00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hambly D, Gross M. Laser flash photochemical oxidation to locate heme binding and conformational changes in myoglobin. Int J Mass Spectrom. 2007;259:124–129. doi: 10.1016/j.ijms.2006.08.018. [DOI] [Google Scholar]
- 15.Zhang H, Gau BC, Jones LM, Vidavsky I, Gross ML. Fast Photochemical Oxidation of Proteins for Comparing Structures of ProteinZLigand Complexes: The CalmodulinZPep[de Model System. Anal Chem. 2011;83:311–318. doi: 10.1021/ac102426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones LM, Sperry JB, Carroll JA, Gross ML. Fast Photochemical Oxidation of Proteins for Epitope Mapping. Anal Chem. 2011;83:7657–7661. doi: 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Y, Chen G, Wei H, Huang RYC, Mo J, Rempel DL, Tymiak AA, Gross ML. Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. J Am Soc Mass Spectrom. 2014;25:2084–2092. doi: 10.1007/s13361-014-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY-C, Julien SH, Lin Z, Moore K, Mueller L, Noriega C, Sejwal P, Sheppard P, Stevens B, Chen G, Tymiak AA, Gross ML, Schneeweis LA. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal Chem. 2017;89:2250–2258. doi: 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li KS, Chen G, Mo J, Huang RYC, Deyanova EG, Beno BR, O’Neil SR, Tymiak AA, Gross ML. Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Anal Chem. 2017;89:7742–7749. doi: 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Wecksler AT, Molina P, Deperalta G, Gross ML. Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry. J Am Soc Mass Spectrom. 2017;28:850–858. doi: 10.1007/s13361-017-1601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gau BC, Sharp JS, Rempel DL, Gross ML. Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Anal Chem. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Rempel DL, Gross ML. Temperature Jump and Fast Photochemical Oxidation Probe Submillisecond Protein Folding. J Am Chem Soc. 2010;132:15502–15504. doi: 10.1021/ja106518d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Rempel DL, Gau BC, Gross ML. Fast Photochemical Oxidation of Proteins and Mass Spectrometry Follow Submillisecond Protein Folding at the Amino-Acid Level. J Am Chem Soc. 2012;134:18724–18731. doi: 10.1021/ja307606f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart KM, Ho CMW, Dutta S, Gross ML, Bowman GR. Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat Commun. 2016;7:12965. doi: 10.1038/ncomms12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gau B, Garai K, Frieden C, Gross ML. Mass Spectrometry-Based Protein Footprinting Characterizes the Structures of Oligomeric Apolipoprotein E2, E3, and E4. Biochemistry (Mosc) 2011;50:8117–8126. doi: 10.1021/bi200911c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li KS, Rempel DL, Gross ML. Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J Am Chem Soc. 2016;138:12090–12098. doi: 10.1021/jacs.6b07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Zhang H, Niedzwiedzki DM, Jiang J, Blankenship RE, Gross ML. Fast Photochemical Oxidation of Proteins Maps the Topology of Intrinsic Membrane Proteins: Light-Harvesting Complex 2 in a Nanodisc. Anal Chem. 2016;88:8827–8834. doi: 10.1021/acs.analchem.6b01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watkinson TG, Calabrese AN, Ault JR, Radford SE, Ashcroft AE. FPOP-LC-MS/MS Suggests Differences in Interaction Sites of Amphipols and Detergents with Outer Membrane Proteins. J Am Soc Mass Spectrom. 2017;28:50–55. doi: 10.1007/s13361-016-1421-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chea EE, Jones LM. Analyzing the structure of macromolecules in their native cellular environment using hydroxyl radical footprinting. Analyst. 2018 doi: 10.1039/C7AN01323J. [DOI] [PubMed] [Google Scholar]
- 30.Espino JA, Mali VS, Jones LM. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal Chem. 2015;87:7971–7978. doi: 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- 31.Rinas A, Mali VS, Espino JA, Jones LM. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal Chem. 2016;88:10052–10058. doi: 10.1021/acs.analchem.6b02357. [DOI] [PubMed] [Google Scholar]
- 32.Goldstein S, Aschengrau D, Diamant Y, Rabani J. Photolysis of Aqueous H2O2: Quantum Yield and Applications for Polychromatic UV Actinometry in Photoreactors. Environ Sci Technol. 2007;41:7486–7490. doi: 10.1021/es071379t. [DOI] [PubMed] [Google Scholar]
- 33.Konermann L, Stocks BB, Czarny T. Laminar Flow Effects During Laser-Induced Oxidative Labeling for Protein Structural Studies by Mass Spectrometry. Anal Chem. 2010;82:6667–6674. doi: 10.1021/ac101326f. [DOI] [PubMed] [Google Scholar]
- 34.Xie B, Sharp JS. Hydroxyl Radical Dosimetry for High Flux Hydroxyl Radical Protein Footprinting Applications Using a Simple Optical Detection Method. Anal Chem. 2015;87:10719–10723. doi: 10.1021/acs.analchem.5b02865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niu B, Zhang H, Giblin D, Rempel DL, Gross ML. Dosimetry Determines the Initial OH Radical Concentration in Fast Photochemical Oxidation of Proteins (FPOP) J Am Soc Mass Spectrom. 2015;26:843–846. doi: 10.1007/s13361-015-1087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vahidi S, Konermann L. Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds. J Am Soc Mass Spectrom. 2016:1–9. doi: 10.1007/s13361-016-1389-x. [DOI] [PubMed] [Google Scholar]
- 37.Falconer RJ, Chan C, Hughes K, Munro TP. Stabilization of a monoclonal antibody during purification and formulation by addition of basic amino acid excipients. J Chem Technol Biotechnol. 2011;86:942–948. doi: 10.1002/jctb.2657. [DOI] [Google Scholar]
- 38.Zhang Y, Rempel DL, Zhang H, Gross ML. An Improved Fast Photochemical Oxidation of Proteins (FPOP) Platform for Protein Therapeutics. J Am Soc Mass Spectrom. 2014;26:526–529. doi: 10.1007/s13361-014-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L. Submillisecond Protein Folding Events Monitored by Rapid Mixing and Mass Spectrometry-Based Oxidative Labeling. Anal Chem. 2013;85:8618–8625. doi: 10.1021/ac401148z. [DOI] [PubMed] [Google Scholar]
- 40.Hambly DM, Gross ML. Cold Chemical Oxidation of Proteins. Anal Chem. 2009;81:7235–7242. doi: 10.1021/ac900855f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Downard KM, Maleknia SD, Akashi S. Impact of limited oxidation on protein ion mobility and structure of importance to footprinting by radical probe mass spectrometry. Rapid Commun Mass Spectrom. 2012;26:226–230. doi: 10.1002/rcm.5320. [DOI] [PubMed] [Google Scholar]
- 42.Charvatova O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: Application to galectin-1. J Am Soc Mass Spectrom. 2008;19:1692–1705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morand K, Talbo G, Mann M. Oxidation of peptides during electrospray ionization. Rapid Commun Mass Spectrom. 1993;7:738–743. doi: 10.1002/rcm.1290070811. [DOI] [PubMed] [Google Scholar]
- 44.Xie B, Sharp JS. Relative Quantification of Sites of Peptide and Protein Modification Using Size Exclusion Chromatography Coupled with Electron Transfer Dissociation. J Am Soc Mass Spectrom. 2016;27:1322–1327. doi: 10.1007/s13361-016-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie B, Sood A, Woods RJ, Sharp JS. Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection. Sci Rep. 2017;7:4552. doi: 10.1038/s41598-017-04689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang W, Ravikumar KM, Chance MR, Yang S. Quantitative Mapping of Protein Structure by Hydroxyl Radical Footprinting-Mediated Structural Mass Spectrometry: A Protection Factor Analysis. Biophys J. 2015;108:107–115. doi: 10.1016/j.bpj.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niu B, Mackness BC, Rempel DL, Zhang H, Cui W, Matthews CR, Zitzewitz JA, Gross ML. Incorporation of a Reporter Peptide in FPOP Compensates for Adventitious Scavengers and Permits Time-Dependent Measurements. J Am Soc Mass Spectrom. 2017;28:389–392. doi: 10.1007/s13361-016-1552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Cui W, Giblin D, Gross ML. New Protein Footprinting: Fast Photochemical Iodination Combined with Top-Down and Bottom-Up Mass Spectrometry. J Am Soc Mass Spectrom. 2012;23:1306–1318. doi: 10.1007/s13361-012-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang B, Rempel DL, Gross ML. Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. J Am Soc Mass Spectrom. 2015;27:552–555. doi: 10.1007/s13361-015-1313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gau BC, Chen H, Zhang Y, Gross ML. Sulfate Radical Anion as a New Reagent for Fast Photochemical Oxidation of Proteins. Anal Chem. 2010;82:7821–7827. doi: 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng M, Zhang B, Cui W, Gross ML. Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angew Chem. 2017;129:14195–14198. doi: 10.1002/ange.201706697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiselar JG, Chance MR. Future directions of structural mass spectrometry using hydroxyl radical footprinting. J Mass Spectrom. 2010;45:1373–1382. doi: 10.1002/jms.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ziemianowicz DS, Bomgarden R, Etienne C, Schriemer DC. Amino Acid Insertion Frequencies Arising from Photoproducts Generated Using Aliphatic Diazirines. J Am Soc Mass Spectrom. 2017;28:2011–2021. doi: 10.1007/s13361-017-1730-z. [DOI] [PubMed] [Google Scholar]
- 54.Manzi L, Barrow AS, Scott D, Layfield R, Wright TG, Moses JE, Oldham NJ. Carbene footprinting accurately maps binding sites in protein–ligand and protein–protein interactions. Nat Commun. 2016;7:13288. doi: 10.1038/ncomms13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noble RW, Gibson QH. The Reaction of Ferrous Horseradish Peroxidase with Hydrogen Peroxide. J Biol Chem. 1970;245:2409–2413. [PubMed] [Google Scholar]
- 56.Huang W, Ravikumar KM, Parisien M, Yang S. Theoretical modeling of multiprotein complexes by iSPOT: Integration of small-angle X-ray scattering, hydroxyl radical footprinting, and computational docking. J Struct Biol. 2016;196 doi: 10.1016/j.jsb.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Armeev GA, Gorkovets TK, Efimova DA, Shaitan KV, Shaytan AK. Modeling of the structure of protein–DNA complexes using the data from FRET and footprinting experiments. Mosc Univ Biol Sci Bull. 2016;71:29–33. doi: 10.3103/S0096392516010016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.