

Abstract
Sphingolipids such as ceramide are important constituents of cell membranes. The ceramide transfer protein (CERT) moves ceramide from the endoplasmic reticulum to the Golgi apparatus in a nonvesicular manner. Hyperphosphorylation of the serine-repeat motif (SRM) adjacent to the pleckstrin homology (PH) domain of CERT down-regulates the inter-organelle ceramide transport function of CERT. However, the mechanistic details of this down-regulation remain elusive. Using solution NMR and binding assays, we herein show that a hyperphosphorylation-mimetic CERT variant in which 10 serine/threonine residues of SRM had been replaced with glutamate residues (the 10E variant) displays an intramolecular interaction between SRM and positively charged regions of the PH domain, which are involved in the binding of this domain to phosphatidylinositol 4-monophosphate (PI4P). Of note, the binding of the PH domain to PI4P-embedded membranes was attenuated by the SRM 10E substitutions in cell-free assays. Moreover, the 10E substitutions reduced the Golgi-targeting activity of the PH-SRM construct in living cells. These results indicate that hyperphosphorylated SRM directly interacts with the surface of the PH domain in an intramolecular manner, thereby decreasing the PI4P-binding activity of the PH domain. In light of these findings, we propose that the hyperphosphorylation of SRM may trigger the dissociation of CERT from the Golgi apparatus, resulting in a functionally less active conformation of CERT.
Keywords: ceramide, lipid transport, phosphorylation, lipid-protein interaction, nuclear magnetic resonance (NMR), CERT, intrinsically disordered, pleckstrin homology domain
Introduction
Lipids are the major constituents of all cell membranes and play dynamic roles in organelle structures and functions. In eukaryotic cells, many lipid types are newly synthesized in the endoplasmic reticulum (ER)4 and then delivered to other organelles. Whereas the transport of proteins from the ER to the Golgi apparatus predominantly occurs via membrane vesicles, lipid ceramide is transported from the ER to the Golgi apparatus by the ceramide transport protein CERT in a nonvesicular manner (1). Recent studies have shown that various lipid transfer proteins mediate the inter-organelle transport of lipids in a wide range of organisms: from the single cell eukaryote yeast to vertebrates and plants (2–7). Thus, regulating the function of each lipid transfer protein is crucial for the homeostasis of cellular lipidomes. Nevertheless, the mechanisms by which the functions of lipid transfer proteins are regulated currently remain unclear.
CERT is composed of several functional modules that enable this protein to mediate the intermembrane transfer of ceramide at ER-Golgi membrane contact sites (1, 8); its N-terminal pleckstrin homology (PH) domain binds to phosphatidylinositol 4-monophosphate (PI4P) and serves as a Golgi-targeting module, whereas the C-terminal lipid transfer START domain catalyzes the intermembrane transfer of ceramide. The two phenylalanines in an acidic tract (FFAT) motif, which is located in a potentially intrinsically disordered region between the two domains, binds to the ER-resident scaffold protein VAP.
When CERT is expressed in mammalian cells, multiple (up to 10) amino acid residues in a serine-repeated motif (SRM; 132SMVSLVSGASGYSATSTSS150; serine/threonine residues in the motif are underlined) of the protein are phosphorylated (9). When these 10 serine/threonine residues of SRM were replaced with glutamic acid residues (the so-called 10E mutation; see also Fig. 1B), the resultant CERT 10E mutant reduced the PI4P-binding activity and ceramide transfer activity (9). The function of CERT is also repressed when the casein kinase 1γ2 isoform, which acts as a kinase to phosphorylate the serine/threonine residues sequentially in SRM, is overexpressed (10). Therefore, the 10E mutant appears to functionally mimic the endogenous multiphosphorylated SRM form of CERT; however, glutamic acid and aspartic acid are chemically different from phospho-serine/threonine (11). The repression of PI4P-binding activity by the 10E mutation was cancelled by removing the START domain, and vice versa, and the repression of ceramide transfer activity was rescued by removing the PH domain, suggesting an inhibitory mechanism by conformational changes to mask the PH and START domains with each other (9). SRM is conserved well among the CERT orthologues of various animals (8), suggesting that the regulatory role of SRM for the function of CERT is crucial for various organisms.
Figure 1.

NMR experiments revealed that CERT SRM(10E) interacts with CERT PH in an intramolecular manner. A, superposition of the 2D 1H-15N HSQC spectra of the isolated CERT PH domain (black), PH-SRM(WT) (blue), and PH-SRM(10E) (red). The region of SRM signals, indicated by the gray dotted lined box, is enlarged and shown in Fig. S1. B, the specific region of 2D 1H-15N HSQC spectra shown in A, indicated by the dashed lined box, is enlarged without superimposition. The positions of the signals in the isolated CERT PH domain are indicated by dotted lines. The bottom panel indicates schematic representations of isolated PH, PH-SRM(WT), and PH-SRM(10E) analyzed in the present study. C, normalized chemical shift differences (Δδ) in individual residues between CERT PH-SRM(WT) and PH-SRM(10E). Dashed orange and red lines indicate 0.03 and 0.06 ppm of normalized chemical shift changes, respectively. As denoted at the top of the graph, 1.22 and 1.75 indicate the Δδ values (ppm) of Glu-138 and Glu-146 residues, respectively. The numbers on the horizontal axis indicate the residue numbers of CERT PH-SRM (amino acids 24–152). The signals, which are visible only in PH-SRM(10E), are indicated by obelisks, whereas those invisible in both WT and 10E are denoted with double daggers. The asterisks indicate that 1H-15N correlation signals were severely degenerated. P, proline residues (Pro-78 and Pro-102). D, mapping of amino acid residues that showed significant chemical shift perturbations upon the hyperphosphorylation-mimetic 10E substitution on SRM. Orange and red, residues that showed chemical shift changes of 0.03–0.06 and >0.06 ppm, respectively. Dark gray, residues that were not detected in the NMR spectra, which are identical to the residues denoted by the obelisks in C. N and C, positions of the N and C termini of the CERT PH domain, respectively. SRM is adjacent to the C terminus of the CERT PH domain. A surface representation of mapping on the solution structure of the CERT PH domain (PDB code 2RSG) was prepared using PyMOL (Schroedinger LLC, New York).
A large-scale human genetic study recently revealed that a mental retardation disorder with dominant inheritance is caused by a missense mutation in the human CERT (or COL4A3BP) gene, which replaces Ser-132 with leucine (12). Because a priming phosphorylation at Ser-132 by protein kinase D is a prerequisite for the casein kinase 1–catalyzed sequential phosphorylation of the following serine/threonine array in the SRM of CERT (9, 10, 13), the alanine replacement of Ser-132 is predicted to cause the loss of SRM phosphorylation, thereby rendering the CERT mutant constitutively active (9, 13). These previous biochemical findings have provided a mechanical explanation for how the S132L substitution in CERT causes the genetically dominant phenotype; the substitution destroys the repressive regulatory mechanism mediated by the hyperphosphorylated SRM on CERT, and the resultant constitutively active CERT is detrimental to the development of the central neuronal network system. If this explanation is correct, the SRM-dependent regulation of CERT is crucial to human health. Nevertheless, the SRM-dependent regulatory mechanism of CERT has not yet been elucidated at the atomic level.
Results
Hyperphosphorylation-mimetic CERT SRM interacts with the CERT PH domain in an intramolecular manner
The three-dimensional structure of the PH domain of CERT has been resolved by NMR and X-ray diffraction analyses, and these structural biological studies have identified the subregions and amino acid residues that are crucial for PI4P-binding activity in the CERT PH domain (14, 15). We measured the 2D 1H-15N heteronuclear single quantum correlation (HSQC) spectra of uniformly 15N-labeled CERT derivatives (Fig. 1, A and B): (i) the isolated PH domain (CERT PH domain only), (ii) the nonphosphorylated WT PH-SRM fragment derived from CERT (referred to as PH-SRM(WT)), and (iii) the hyperphosphorylation-mimetic PH-SRM fragment derived from CERT 10E (referred to as PH-SRM(10E)). As a result, more prominent chemical shift changes were observed in several amino acids of the PH domain of the PH-SRM(10E) fragment than in those of the PH-SRM(WT) fragment, when the signal pattern of the isolated PH domain was used as the reference (Fig. 1, A and B). Chemical shift differences between PH-SRM(WT) and PH-SRM(10E) (Fig. 1C) were then mapped on the tertiary structure of the PH domain to show the distribution of the amino acid residues with marked chemical shift alterations by the hyperphosphorylation-mimetic modification to SRM (Fig. 1D). The residues with marked chemical shift alterations were localized in a limited region on the PH domain (Fig. 1, C and D), which suggests that SRM(10E) interacts with the CERT PH domain. The SRM(10E)-interacted region on the CERT PH domain (Fig. 1C) partially overlapped with the previously identified region to recognize the headgroup of PI4P (14). These results suggest that hyperphosphorylated SRM in CERT, at least partially, masks the PI4P-binding site in the CERT PH domain.
Protein phosphorylation preferentially occurs within intrinsically disordered protein regions (16, 17). A bioinformatics analysis previously predicted that SRM is intrinsically disordered (8). A comparison of the 2D NMR spectrum between PH and PH-SRM (WT and 10E) revealed that 13 signals (Glu-118, Ser-126, Arg-128, His-130, Gly-131, Met-133, Val-134, Ser-141, Tyr-143, Ser-144, Ser-147, Thr-148, and Ser-149) derived from the non-PH region (corresponding to amino acids 118–152 in PH-SRM) were invisible in PH-SRM(WT) (Fig. S1), suggesting that nonphosphorylated SRM structurally fluctuates in the intermediate time scale of NMR spectroscopy. In contrast, eight of their counterpart signals (Glu-118, Ser-126, Arg-128, Met-133, Val-134, Glu-141, Tyr-143, and Glu-144) were clearly visible in PH-SRM(10E) (Fig. S1). In addition, the line widths of signals derived from the non-PH region were sharper in PH-SRM(10E), and, as a consequence, the intensities of these signals (e.g. Ser-123, Glu-124, Ser-125, Ala-140, and Ala-145) improved, whereas the signal line width and intensity derived from the PH domain were independent of the 10E mutation in the adjacent SRM moiety (Fig. S1). This indicates that structural fluctuation in a micro- to millisecond time scale at the SRM region, which induces the line broadening of the SRM moiety of the WT, was suppressed in the 10E form. Along with the additional chemical shift differences from the CERT PH domain, these results suggest that the 10E mutation in PH-SRM enables the SRM region to interact with the PH domain, thereby changing the nature of the structural fluctuation in this region to make some signals visible. It is also important to note that the sharpness of the NMR signals of all three proteins implies their monomer behavior in the solution.
The hyperphosphorylation-mimetic SRM attenuates the degree of the chemical shift change in the CERT PH domain upon the addition of diC4-PI4P
To verify whether the CERT PH domain–PI4P interaction is modulated by the CERT PH domain-SRM(10E) interaction, we performed chemical shift perturbation NMR experiments to titrate diC4-PI4P, a soluble form of PI4P with a short acyl-chain, into uniformly 15N-labeled CERT PH, PH-SRM(WT), or PH-SRM(10E) proteins. Lys-32, Trp-33, Arg-66, and Trp-95 of CERT typically constitute the PI4P-binding pocket of the PH domain and are crucial for the recognition of the PI4P headgroup (14). Upon the addition of an identical concentration of diC4-PI4P, chemical shift changes in these amino acid residues were less prominent in PH-SRM(10E) than in the isolated PH domain (Fig. 2 and Fig. S2), whereas chemical shift changes in PH-SRM(WT) led by the diC4-PI4P titration were similar to those in the isolated PH domain (Fig. 2 and Fig. S2). This result indicates that hyperphosphorylated SRM, but not its nonphosphorylated form, physically competes with PI4P binding to the CERT PH domain.
Figure 2.

The hyperphosphorylation-mimetic SRM attenuates the degree of chemical shift changes in the CERT PH domain upon the addition of diC4-PI4P. The specific regions of the spectra, indicated by the dashed line boxes with the notations of B, C, and D in Fig. S2 are enlarged and shown in A, B, and C, respectively. The protein concentration of these NMR samples was 0.1 mm, in the presence or absence of 1.6 mm diC4-PI4P. A–C, 2D 1H-15N HSQC spectra of the isolated PH (left NMR spectrum), PH-SRM(WT) (center NMR spectrum), and PH-SRM(10E) (right NMR spectrum); the protein concentration of these NMR samples was 0.1 mm in the presence or absence of 1.6 mm diC4-PI4P. The plots of chemical shift changes upon interaction with diC4-PI4P are shown (right panels). The asterisk shown in the 2D 1H-15N HSQC spectrum of PH-SRM(WT) in C indicates unknown signals.
The hyperphosphorylation-mimetic SRM attenuates binding of the CERT PH domain to PI4P-embedded and -free phospholipid membranes
To test whether SRM(10E) affects the function of the adjacent PH domain, the binding activity of PH-SRM for the phospholipid membranes, “liposomes,” was assayed in conventional co-precipitation experiments. In this assay, we employed two types of phospholipid compositions: type A, consisting of 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoethanolamine (POPE), and 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoserine (POPS), and type B, consisting of POPC and POPE. Although the Golgi membrane lipid composition was closer to type A than type B (18), the potential electrostatically repulsive effect of the SRM(10E) mutation on anionic liposomes may be prevented on the type-B matrix. When assayed with type-A liposomes, the isolated PH domain, PH-SRM(WT), and PH-SRM(10E) bound to liposomes in a PI4P dose-dependent manner. Among the PI4P concentrations examined (0, 0.5, and 5 mol %), PH-SRM(10E) was slightly less active for binding to type-A liposomes than PH-SRM(WT) and the isolated PH domain, and this difference was the most prominent for binding to 0.5% PI4P-containing liposomes (Fig. 3A). When assayed with type-B liposomes, all three proteins exhibited clear enhancements in binding activity in the presence of 5% PI4P, but not 0.5% PI4P, over their binding activity to the PI4P-free control (Fig. 3B). PH-SRM(10E) was slightly less active than PH-SRM(WT) and the isolated PH domain with type-B liposomes containing 5%, but not 0.5%, PI4P (Fig. 3B). In addition, attenuation levels at the given PI4P concentrations appeared to be affected by co-existing PS.
Figure 3.

The hyperphosphorylation-mimetic SRM attenuates the binding of the CERT PH domain to PI4P-embedded and -free phospholipid membranes. The binding of purified recombinant proteins to liposomes consisting of POPC, POPE, and POPS (14.3:4.8:0.8, mol/mol/mol) with various mol % PI4P (A) or of POPC and POPE (16:4, mol/mol) with various mol % PI4P (B) was assayed using a co-precipitation method as described under “Experimental procedures.” Error bars, S.D. calculated from the average value of three experiments. Statistical analyses were performed using an unpaired one-tailed Student's t test, and p values are shown. Shown is an FCS analysis of the binding of the CERT PH domain to PI4P-embedded (C) and -free (D) phospholipid membranes. C, an FCS analysis of the binding of isolated PH (black), PH-SRM(WT) (blue), and PH-SRM(10E) (red) to PI4P-embedded liposomes was performed using a single-molecule fluorescence technique. D, the translational diffusion time was measured by FCS with various concentrations of PI4P-free liposomes in the same manner as described in C. Error bars of the vertical axis of C and D, S.D. calculated from the average values of five experiments.
The co-precipitation of PH-SRM proteins with liposomes was slightly attenuated by the 10E mutation, even with PI4P-free POPC/POPE liposomes, which did not contain acidic phospholipids (Fig. 3B). Hence, the inhibitory effect of SRM(10E) on binding to PI4P-free liposomes did not appear to be simply due to electrostatic repulsion between the negatively charged SRM(10E) moiety and the surface of phospholipid bilayers.
We also examined the avidity of PH-SRM for phospholipid membranes in fluorescence correlation spectroscopy (FCS) experiments. When titrated with PI4P-embedded liposomes, the diffusion of PH-SRM(10E) was significantly faster than that of PH-SRM(WT) and the isolated PH domain in the middle concentration range (33–330 μm) of PI4P-embedded liposomes, whereas no significant difference was observed in the higher concentration range (Fig. 3C), which is consistent with the results of the co-precipitation assay (Fig. 3, A and B). When titrated with PI4P-free liposomes, the diffusion of PH-SRM(10E) was significantly faster than that of PH-SRM(WT) and the isolated PH domain in the higher concentration range (>330 μm) of liposomes (Fig. 3D), although the signals obtained fluctuated, presumably due to low affinity for PI4P-free phospholipid membranes (14). The results of co-precipitation and FCS analyses indicated that the hyperphosphorylation-mimetic SRM attenuates the binding of the CERT PH domain to not only PI4P-embedded, but also PI4P-free phospholipid membranes.
The hyperphosphorylation-mimetic SRM attenuates the Golgi localization of the CERT PH-SRM construct
We next examined whether the attenuated PI4P-binding activity detected in cell-free systems reflected the behavior of the PH-SRM(10E) construct expressed in cells. Because it had not yet been established whether PH-SRM(WT) remained in a nonphosphorylated form when expressed in cells, we employed an additional construct referred to as PH-SRM(S132A) with a replacement of the 132nd amino acid residue from serine to alanine; this construct was expected not to receive the priming phosphorylation at serine 132, which is essential for multiple phosphorylation in SRM (9). Thus, PH-SRM(S132A) was expected to serve as an ideal nonphosphorylated counterpart of PH-SRM(10E). When the PH domain only and three PH-SRM constructs (i.e. WT, 10E, and Ser-132) with an N-terminal HA tag were ectopically expressed in HeLa cells, Western blot analysis with an anti-HA antibody showed that the expression levels of the three PH-SRM constructs were similar, whereas that of the PH domain only was markedly lower (Fig. 4A). Thus, we did not use the PH domain construct in subsequent analyses.
Figure 4.

Attenuation of Golgi targeting by the 10E mutation in the PH-SRM construct. The colocalization of the HA-tagged CERT PH-domain or CERT PH-SRMs with the Golgi marker GM130 was analyzed. A, HeLa cells were transfected with an expression plasmid encoding the HA-CERT PH domain, HA-PH-SRM(WT), HA-PH-SRM(S132A), HA-PH-SRM(10E), or an empty vector and cultured for 24 h before harvesting. The lysate of harvested cells was prepared as described previously (31) and subjected to SDS-PAGE followed by Western blotting (WB) with the anti-HA antibody (α-HA) and anti-glyceraldehyde-3-phosphate dehydrogenase antibody (α-GAPDH). The mobilities of molecular mass standards (Bio-Rad Precision Plus protein standards) with the indicated mass values are shown at the right of the panel. B, HeLa cells transfected with expression plasmids for HA-PH-SRM(WT), HA-PH-SRM (S132A), and HA-PH-SRM(10E) were grown on coverslips, subjected to indirect immunocytochemistry doubly labeled with the anti-HA antibody (green) and anti-GM130 antibody (magenta), and observed by confocal microscopy. Typical staining patterns are shown. Bar, 10 μm. C, the percentage of colocalization of HA-PH-SRM(WT), HA-PH-SRM(S132A), and HA-PH-SRM(10E) with GM130 was calculated using ZEN software (Carl Zeiss). Means ± S.E. were calculated from the data of 23–36 cells for each group. Statistical analyses were performed using an unpaired one-tailed Student's t test, and p values are shown. For details of the calculation, see also the legend to Fig. S3.
When the intracellular distribution of these ectopically expressed proteins was examined by indirect immunostaining, the PH-SRM(WT) and PH-SRM(S132A) constructs both clearly localized to the perinuclear structure, which was co-stained with the Golgi marker GM130, whereas PH-SRM(10E) was dispersed throughout the cytoplasm (Fig. 4B). Although CERT has been suggested to target the medial/trans Golgi regions (1), to which SMS1 is localized (19), we were unable to obtain antibodies that are useful for immunocytochemistry of an endogenous trans Golgi region marker protein. Thus, we herein employed GM130, which is predominantly distributed to the cis Golgi region as a Golgi marker. It is important to note that all of the HA-tagged proteins used were strongly distributed to the nucleus (Fig. 4B), possibly due to nonspecific permeation of these proteins through nuclear pores.
To quantitatively compare HA-tagged proteins in their colocalization with GM130, we performed a pixel-based analysis on images (Figs. S3 and S4). The HA-tagged CERT START domain, which has no Golgi-targeting or ER-targeting motifs, was distributed throughout the cytoplasm without accumulating in specific organelles (except for the nucleus) (Fig. S3A), thereby allowing us to rationally set a cross-hair bar at the x axis to represent the border between the Golgi- and non-Golgi cytoplasmic areas in the 2D map (Fig. S3B). The ratio of the total intensities of the pixels of each HA-tagged protein colocalized with GM130 to the total intensities of the pixels of the protein in a cell was assessed in 23–36 cells, and its average value was used to reflect the activity of the protein to target the Golgi apparatus (Figs. S3B and S4). The quantitative analysis confirmed that Golgi targeting was weaker with PH-SRM(10E) than with PH-SRM(WT) and PH-SRM(S132A) (Fig. 4C and Fig. S4). These results demonstrated that the attenuated PI4P-binding activity of PH-SRM(10E) affects its Golgi-targeting activity in living cells.
PH-SRM(WT) and PH-SRM(S132A) exhibited similar mobilities in SDS-PAGE, suggesting that the SRM region in PH-SRM(WT) was not phosphorylated in HeLa cells under the conditions used (Fig. 4A). Nevertheless, PH-SRM(WT) was significantly less active in Golgi targeting than PH-SRM(S132A) (Fig. 4C). Although the reason for this discrepancy remains unclear, PH-SRM(WT) may be phosphorylated at a few sites in SRM, which may not change its molecular weight to a discernible level in an SDS-PAGE analysis.
Discussion
We previously demonstrated that the phosphomimetic 10E mutation in the SRM of CERT simultaneously down-regulated the functions of the PH and STAR domains and that the repressed activity of the PH domain in CERT 10E was reversed by the removal of the START domain, whereas that of the START domain was achieved by the removal of the PH domain (9). These findings led to the model of the hyperphosphorylation of SRM inducing a conformational change in the overall structure of CERT to mutually mask the functional regions of the N-terminal PH domain and C-terminal START domain (9). This model was recently supported by a structural study by Prashek et al. (20), who showed that isolated PH and START domains of CERT interacted with each other and that specific regions on the surface of the PH domain, which are involved in the interaction between the two domains, are partially overlapped with the PI4P-binding site. In addition, Prashek et al. (20) demonstrated that amino acid substitutions in the PH-START–interacting surface abrogated the repressive effects of the CERT 10E mutation.
In the present study, we revealed that the hyperphosphorylation-mimetic mutation influenced the function of the PH domain, which was evident even in the absence of the START domain (Figs. 1 and 2). Moreover, the attenuated PI4P-binding activity of the PH-SRM(10E) construct affected its Golgi-targeting activity in HeLa cells. Thus, in this study, we elucidated that hyperphosphorylation of the SRM directly participates in the functional regulation of CERT, and we will add new findings to the previously reported model that phosphorylated SRM may also partially attenuate the function of the PH domain by directly interacting with the domain (Fig. 5); the phosphorylation of CERT SRM on the Golgi apparatus and the interaction between phosphorylated CERT SRM and the CERT PH domain will be a trigger for the dissociation of CERT from the Golgi apparatus. It is still not clear whether the hyperphosphorylated SRM and START domain can bind simultaneously to the PH domain or if these two would compete for the binding on the PH domain. However, it is evident that both of the interactions are inhibitory to the PI4P binding. In addition, because these interactions are highly dynamic, both interactions would most likely be interexchanged with each other in solution. It might be reasonable to speculate that the hyperphosphorylated SRM can interact first with the PH domain because the SRM region is sterically closer to the PH domain than the START domain. Moreover, the SRM-PH interaction might draw the START domain near to the PH domain. In this sense, the interaction between the PH domain and phosphorylated SRM can be an “intermediate” form toward a more drastic conformational rearrangement of CERT and will lead the PH-START interdomain interaction for mutually and functionally silencing conformations. The START domain is required for the repression of the PI4P-binding activity of the CERT PH domain under these conditions (9, 20). The inactive conformer is mainly distributed in the cytosol as an inactive pool of CERT, whereas inactive CERT is dephosphorylated in response to specific stimuli (e.g. the loss of cellular SM) (9). Protein phosphatase 2Cϵ (PP2Cϵ), which may associate with VAP on the ER, may be responsible for the dephosphorylation of the SRM of CERT (21). After dephosphorylation at SRM, CERT changes to its active form, in which Golgi localization and ceramide transfer activities are restored.
Figure 5.
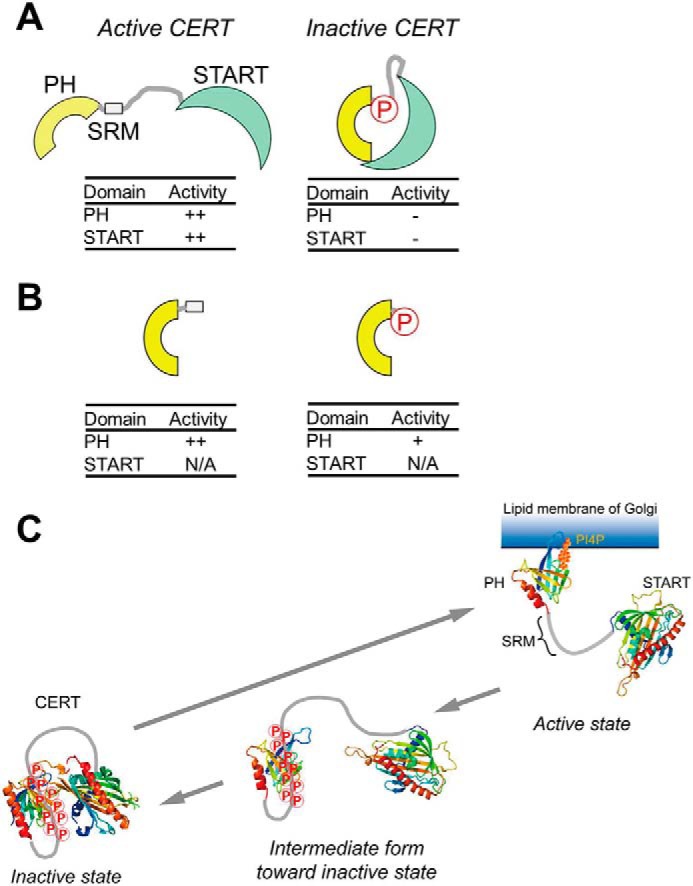
A schematic model of the phosphoregulation of the structure and function of CERT. The hyperphosphorylation of SRM induces a conformational change in CERT to mutually mask the N-terminal PH domain and C-terminal START domain (A), as described in a previous study (9). On the other hand, as new findings in the present study, we demonstrate that phosphorylated SRM may also partially attenuate the function of the PH domain by directly interacting with the PH domain (B). N/A, not applicable. C, a functional diagram of the structural and functional regulation of CERT based on previously reported insights (A) and the results of the present study (B). On the Golgi membrane, the SRM of CERT is hyperphosphorylated by the Golgi-resident protein kinase PKD or CKIγ (9). The interaction between hyperphosphorylated SRM and the CERT PH domain (PDB code 2RSG) may facilitate the dissociation of CERT from the PI4P-embedded Golgi membrane. After dissociation from the Golgi membrane, CERT with hyperphosphorylated SRM may undergo a more drastic conformational rearrangement toward a state in which PH-START interdomain mutual functional silencing may be attained (9). The interaction between hyperphosphorylated SRM and the CERT PH domain identified in this study would be a first step of the conformational rearrangement of the CERT to lead to functional regulation. Notably, whereas the interaction between the PH domain and phosphorylated SRM occurs in an intramolecular manner, the interaction between the PH and START domains (PDB code 5JJD) may occur between different monomers in a CERT homooligomer (also see “Discussion”). Nevertheless, the schematic model tentatively illustrates only the intramolecular interaction between the two domains in a CERT monomer for simplicity.
PH-SRM(WT) and PH-SRM(10E) both behave as a monomer in our solution NMR assays. However, full-size CERT (or GPBPΔ26) has been suggested to form a homooligomer (22, 23). Thus, although the interaction between the PH domain and phosphorylated SRM occurs in an intramolecular manner, that between the PH and START domains in the inactive conformation may occur between different monomers in a CERT homooligomer.
The CERT PH domain has two distinct but overlapping regions that are crucial for high-affinity binding to PI4P-embedded phospholipid membranes; one is a pocket that accommodates the negatively charged headgroup of PI4P, and the other is a basic groove that weakly associates with acidic phospholipid membranes (14). Seven amino acid residues (Lys-32, Trp-33, Thr-34, Asn-35, Trp-40, Gln-41, and Tyr-96) of the CERT PH domain are shared in both regions (15). Some of the shared residues (Trp-33, Thr-34, Trp-40, and Ser-93) were mapped as residues perturbed by the 10E mutation in SRM (Fig. 1C) (14). Because Trp-33 and Trp-40 play pivotal roles in the interaction between the PH domain and phospholipid bilayers (14), these results suggest that interference in these residues with the adjacent SRM(10E) attenuates not only the recognition of PI4P, but also the phospholipid association of the CERT PH domain.
The state of PI4P in the Golgi membrane is another key factor in the regulation of the Golgi targeting of CERT (24–28). Although the actual concentration of PI4P in the Golgi apparatus remains unknown, a previous study estimated that phosphatidylinositol (PI) accounted for 5–7% of all phospholipids in the Golgi apparatus in two mammalian cell lines (29). If 10% of PI is converted to PI4P in the steady state, the phospholipid matrix of the Golgi apparatus is calculated to contain 0.5–0.7% PI4P (if all Golgi PI4Ps are oriented to the cytosol side, the cytosolic leaflet of the Golgi phospholipid bilayers may have 1.0–1.4% PI4P). When POPC/POPE/POPS liposomes with 5 mol % PI4P were used, the percentage of PH-SRM(WT) co-precipitated with liposomes was 87.9 ± 3.8% (n = 3), and that of PH-SRM (10E) was 54.8 ± 13.3% (n = 3). On the other hand, when POPC/POPE/POPS liposomes with 0.5 mol % PI4P were used, the percentage of PH-SRM(WT) co-precipitated was 14.3 ± 1.4% (n = 3), and that of PH-SRM(10E) was 4.0 ± 1.5% (n = 3) (Fig. 3A). The ratio of co-precipitation percentage values of PH-SRM(10E) to PH-SRM(WT) was lower for 0.5 mol % PI4P-containing liposomes (4.0/14.3 ∼0.28) than for 5 mol % PI4P-containing liposomes (54.8/87.9 ∼0.62), meaning that the SRM(10E)-dependent attenuation of the binding of PH-SRM to POPC/POPE/POPS liposomes was more prominent when 0.5% PI4P co-existed rather than 5% PI4P (Fig. 3, A and B). The SRM(10E)-dependent attenuation of the binding of PH-SRM to the Golgi apparatus was also clearly observed in living cells (Fig. 4). Thus, these results may be reflective of the average concentration of PI4P at the Golgi region, being ∼0.5%.
It would be an interesting question to ask how many Ser/Thr residues in the SRM have to be phosphorylated to execute the inhibitory effect on the PH domain. However, it is always difficult to know how well Glu/Asp mutations mimic phospho-Ser/phospho-Thr, although Glu (or Asp) may qualitatively mimic phosphorylated Ser/Thr. Thus, it is inappropriate to extrapolate the mimicry to quantitative evaluation. Indeed, we previously demonstrated that triple replacement of amino acid residues at 130HGS132 with Asp (H130D/G131D/S132D) resulted in bypassing the priming phosphorylation by protein kinase D to trigger the sequential phosphorylation events downstream of Ser-135 in the SRM, whereas neither single (S132D) nor double (G131D/S132D) replacement allowed triggering of a casein kinase 1 cascade unless the kinase was overproduced (10). More studies will be needed to address the question described above.
In addition to SRM, CERT has another phosphorylation site (Ser-315 adjacent to the FFAT motif for binding to the endoplasmic reticulum-resident scaffold protein VAP) (30), and the interplay between these phosphorylation sites results in a fine regulatory system for the ceramide trafficking function of CERT (31). In various proteins, a phosphorylated region binds to its adjacent domain, and the intramolecular regulation of the function of the adjacent domain occurs, which is exemplified by the autoinhibition of the membrane-binding activity of the C2 domain of the PTEN protein (32), DNA binding by the transcription factor Ets-1 (33), and the intra-/intermolecular interactions of various domains in Src family kinases (34). CERT is one of the most extensively investigated lipid transfer proteins from the aspect of post-translational regulation, and the model described above may serve as a typical framework for the regulatory mechanisms of lipid transfer proteins.
Experimental procedures
Expression and purification of CERT PH, PH-SRM(WT), and PH-SRM(10E) recombinant proteins
The cDNA fragments encoding the CERT PH, PH-SRM(WT), and PH-SRM(10E) polypeptides were chemically synthesized (Invitrogen/Thermo Fisher Scientific, Yokohama, Japan) (synthesized sequences are listed in Table 1) and cloned into the EcoRI/XhoI site on the pET28a(+) plasmid (Novagen) using the In-Fusion Cloning Technology (TaKaRa Bio, Shiga, Japan). Escherichia coli BL21(DE3) cells (Stratagene/Agilent Technologies, Santa Clara, CA) were transfected with these plasmids, and the overexpressed CERT PH, PH-SRM(WT), and PH-SRM(10E) recombinant proteins with an N-terminal His6tag were uniformly 15N- and 13C/15N-labeled in bacteria as described previously (14). After lysis of the bacteria, the recombinant proteins were purified to ∼90% homogeneity using polyhistidine-affinity chromatography described previously (1). The purified His6-tagged proteins were used in the phospholipid membrane-binding assay. In NMR and FCS analyses, the N-terminal His6 tag of the purified proteins was removed by proteolytic digestion with the tobacco etch virus protease, and tag-free recombinant proteins were purified to homogeneity by ion-exchange chromatography as described previously (14).
Table 1.
DNA sequences
Capital letters, sequences derived from human CERT or CERT 10E cDNA; underlined letters, EcoRI and XhoI sites; double-underlined letters, TAG stop codon; gray box, region coding the TEV protease-cleavable site.

NMR spectroscopy
All NMR experiments were performed on a Bruker Avance 600- or 800-MHz spectrometer equipped with a TXI cryogenic probe. All NMR spectra were recorded at 25 °C. Recombinant proteins were dissolved at a concentration of 0.1 mm in 10 mm HEPES-NaOH buffer (pH 7.2) containing 100 mm NaCl, 5 mm tris(2-carboxyethyl)phosphine hydrochloride, and 7% (v/v) D2O. The main-chain resonance assignments of PH-SRM(WT) and PH-SRM(10E) were established by performing sets of multidimensional triple resonance experiments, 3D HNCACB, 3D CBCA(CO)NH, 3D HNCO, and 3D HN(CA)CO (14). All NMR spectra were processed by NMRPipe (35) and analyzed with Sparky (Goddard and Kneller, SPARKY 3-NMR Assignment and Integration Software, University of California, San Francisco, CA). In the chemical shift perturbation studies, the averaged chemical shift changes (Δδ) in the 2D 1H-15N HSQC spectra were calculated with the equation, Δδ = ((ΔδH)2 + (ΔδN/5)2)½, where ΔδH and ΔδN are the chemical shift changes of the amide proton and 15N nucleus in ppm.
Phospholipid membrane-binding assay: A co-precipitation method
POPC, POPE, and POPS were from Avanti Polar Lipids (Alabaster, AL). l-α-Phosphatidylinositol-4-monophosphate was from Cayman Chemicals (Ann Arbor, MI). The anti-His tag antibody was from Medical and Biological Laboratories Co., Ltd. (Nagoya, Japan). The binding activity of purified recombinant proteins to phospholipid membranes was assayed by a co-precipitation method. In brief, purified recombinant protein was precentrifuged at 100,000 × g at 4 °C for 30 min on the day of the assay, and the supernatant was used for the assay. Liposomes with a composition of POPC/POPE/POPS (14.3:4.8:0.8, mol/mol/mol) or POPC/POPE (16:4, mol/mol) with various amounts of PI4P (0%, 0.5%, and 5 mol %) were formed in buffer C (25 mm HEPES-NaOH (pH 7.4) containing 100 mm NaCl and 1 mm EDTA) by bath-type sonication. Liposomes (20 nmol of phospholipids/20 μl of the suspension/assay) and 20 μl of binding buffer (25 mm sodium phosphate buffer (pH 7.4) containing 150 mm NaCl and 0.5 mg/ml BSA) were mixed, and purified protein (2 pmol of protein/10 μl/assay) was added. The final buffer contained 0.3 mg/ml BSA. The mixture was incubated at 4 °C for 30 min. After being incubated, liposomes were precipitated by centrifugation at 100,000 × g at 4 °C for 30 min. The amounts of the purified recombinant protein in the supernatant and pellet fractions were assessed by Western blotting with the anti-His tag antibody, and the ratios of the precipitated His6-tagged proteins to the sum of the His6-tagged proteins in the pellet and supernatant fractions were calculated.
FCS experiments
Purified recombinant proteins were dialyzed against a fluorescence-labeling buffer (10 mm HEPES-NaOH buffer (pH 7.5) containing 1 mm tris(2-carboxyethyl)phosphine hydrochloride) and then concentrated to 0.1 mm (liquid volume, ∼500 μl) by ultrafiltration with the Amicon Ultra set (Merck-Millipore, Burlington, MA). The concentrated proteins were conjugated with maleimide-ATTO633, a fluorescent dye (ATTO-TEC, Siegen, Germany), according to the guidance for protein fluorescent labeling released by Invitrogen/Thermo Fisher Scientific. The maleimide-ATTO633 fluorescent dye was covalently attached to the sulfhydryl groups of the side chains of four cysteine residues (Cys-27, Cys-65, Cys-70, and Cys-84) of the PH domain. There were no cysteine residues in the non-PH region of the PH-SRM constructs used in the present study. After the elimination of unconjugated fluorescent dye by size-exclusion Superdex 200 chromatography (GE Healthcare), 10 nm ATTO633-labeled proteins was subjected to measurements of translational diffusion times with various concentrations of PI4P-embedded liposomes in FCS experiments using the single-molecule fluorescent detection system FluoroPoint View (Olympus, Tokyo, Japan) at 25 °C as described previously (36). PI4P-embedded and PI4P-free small unilamellar liposomes were prepared with a Mini-Extruder (Avanti Polar Lipids, Inc., Alabaster, AL), and the concentrations of these liposomes, representing apparent concentrations of input phospholipids, were estimated as described previously (14).
Expression of HA-tagged proteins in HeLa cells
In the CERT 10E construct we made previously (9), an EcoRI site exists just downstream of the sequence encoding the SRM region. This EcoRI site was removed by PCR-mediated mutagenesis without alterations in the amino acid sequence of the region as follows: a mixture of the template plasmid pBS/nHA-hCERT 10E and a set of primers (5′-GAAGAGGAGGAAGAGTTCAAGAAAGGCC-3′ and 5′-GGCCTTTCTTGAACTCTTCCTCCTCTTC-3′) were subjected to PCR using DNA polymerase KOD PLUS (Toyobo) (thermal cycle program: 94 °C for 2 min; 12 cycles of 98 °C for 20 s, 55 °C for 60 s, and 68 °C, for 8 min; and 68 °C for 2 min). After chloroform/phenol extraction and ethanol precipitation of the reacted sample, purified DNA was treated with DpnI to digest the residual template plasmid and then introduced into the bacteria. The plasmid retrieved from the bacteria was named pBS/nHA-hCERT 10E(ΔEcoRI). We confirmed that the new CERT 10E version had the desired point mutation without any undesired mutations by Sanger sequencing. The plasmid was digested with EcoRI and XhoI, and the resulting ∼1.8-kb fragment encoding nHA-hCERT 10E(ΔEcoRI) was subcloned into the EcoRI/XhoI sites of pcDNA3.1(+)Neo to make pcDNA3.1/nHA-CERT 10E(ΔEcoRI). To ensure that the recombinant PH domain and PH-SRM proteins were composed of 23–117 residues and 23–152 residues, respectively, of full-size CERT, mammalian expression constructs were prepared as follows: a cDNA fragment encoding the PH domain only was amplified by PCR with a template (pcDNA3.1/nHA-CERT WT) and a primer set of primer 1 (5′-GACTACGCCGAAGCTTCTGTGGAGCGCTGCGGGGTC-3′) and primer 2 (5′-CGCGCCGGCCCTCGAGCTAAGTCTTGTGCTGTTCAATGG-3′); a cDNA fragment encoding PH-SRM(WT) was amplified with a template (pcDNA3.1/nHA-CERT WT) and a primer set of primer 1 and primer 3 (5′-CGCGCCGGCCCTCGAGCTACTTGAATGAAGAGGTGGATG-3′); a cDNA fragment encoding PH-SRM(10E) was amplified with a template (pcDNA3.1/nHA-CERT 10E(ΔEcoRI)) and a primer set of primer 1 and primer 4 (5′-CGCGCCGGCCCTCGAGCTACTTGAACTCTTCCTCCTCTTC-3′). The DNA sequence encoding an N-terminal HA epitope was added to these constructs by PCR with respective DNA fragments obtained above as templates and various primer sets: primer 5 (5′-GGTGGTACGGGAATTCACCATGTACCCATATGACGTCCCGGACTACGCCGAAGCT-3′) and primer 2 for HA-tagged PH domain only; primer 5 and primer 3 for HA-tagged PH-SRM(WT); primer 5 and primer 4 for HA-tagged PH-SRM(10E). The amplified cDNA fragments were subcloned into the EcoRI/XhoI sites of pcDNA3.1(+)Neo to make pcDNA3.1/nHA-CERT PH, pcDNA3.1/nHA-PH-SRM(WT), and pcDNA3.1/nHA-PH-SRM(10E(ΔEcoRI)). pcDNA5/FT nHAcFL-hCERT ST, an expression plasmid for the N-terminal HA- and C-terminal FLAG-tagged START domain of CERT (amino acid residues 370–598 of human CERT), was constructed as follows: a ∼0.7-kb fragment was amplified by PCR with pBS/hCERT as the template and a set of primers (5′-CACAAGCTTCGAAGGTTGAAGAGATGGTGCAG-3′ and 5′-GGCTCGAGCTAAGATCTGAACAAAATAGGCTTTCCTGC-3′) (thermal cycle program: 94 °C for 2 min; 5 cycles of 98 °C for 20 s, 58 °C for 30 s, and 70 °C for 1 min; and 16 cycles of 98 °C for 20 s, and 70 °C for 60 s with 3-s increments per cycle; and 70 °C for 2 min). After digestion with HindIII and XhoI, the fragment was cloned between the HindIII and XhoI sites of the pET28a vector to produce the plasmid pET/hCERT ST. After the digestion of pET/hCERT ST with HindIII and BglII, the resultant ∼0.7-kb fragment was cloned between the HindIII and BglII sites of the pBS-nHAcFL vector (30) to produce pBS/nHAcFL-hCERT ST. After the digestion of pBS/nHAcFL-CERT ST with ApaI and NotI, the resultant ∼0.7-kb fragment was cloned between the ApaI and NotI sites of the pcDNA5/FRT/TO vector (Life Technologies, Inc.) to produce pcDNA5/FT nHAcFL-hCERT ST.
HeLa cells (ATCC CCL2) were cultured as described previously. HeLa cells were seeded at 30,000 cells/well in a 24-well plate (for immunocytochemistry, a coverslip (12-mm diameter) was preplaced on the bottom of each well) and cultured in 1 ml of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum for 24 h. After transfection with expression plasmids with Lipofectamine reagent (Thermo Fisher Scientific) in Opti-MEM (Thermo Fisher Scientific), a serum-free medium, for 4 h, transfected cells were cultured in normal medium for 20 h and then subjected to Western blotting or immunocytochemistry. Western blotting to detect HA-tagged proteins was performed using cell lysates from transfected cells as described previously (31). The indirect immunostaining of HeLa cells grown on coverslips was performed as described previously.
Immunocytochemistry
The indirect immunostaining of HeLa cells grown on coverslips was performed as described previously (31). In brief, cells were fixed with 4% neutralized formaldehyde/PBS, permeabilized with 0.1% Triton X-100/PBS, blocked with 3% BSA/PBS, and incubated with a 250-fold diluted solution of a rat monoclonal anti-HA antibody (Roche Applied Science) or monoclonal mouse anti-GM130 antibody (BD Biosciences) as primary antibodies, followed by an incubation with the secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 594 (Thermo Fisher Scientific). The coverslips were mounted on FluoromountTM (Diagnostic BioSystems), and images were captured using a confocal laser-scanning microscope (Axio Observer Z1, Carl Zeiss) equipped with an LSM 700 system (Carl Zeiss Microimaging, LLC) using excitation at 488 and 555 nm and an α Plan-Apochromat ×100/1.46 numerical aperture Oil DIC M27 oil immersion objective lens. Images were prepared using ZEN software (Carl Zeiss). The nucleus was stained with 4′,6-diamidino-2-phenylindole.
A quantitative pixel-based colocalization analysis was performed with ZEN software (Carl Zeiss), according to the manufacturer's manual. Images of cells with HA intensity signals greater than 250 units were not selected for the quantitative colocalization analysis because they were out of the dynamic range of the analysis. In the 2D map constructed, the x and y axes represent the intensity (as an arbitrary unit) of HA and GM130 signals, respectively, whereas the colors represent the frequency of occurrence. The CERT START domain having no Golgi-targeting or ER-targeting modules was distributed throughout the cytoplasm without accumulating in specific organelles (except for the nucleus) (Fig. S3A). The distribution pattern of the CERT START domain allowed us to rationally set a cross-hair bar at the x axis to represent the border between Golgi and non-Golgi areas in the 2D map (Fig. S3A). All of the HA-tagged proteins used were prominently distributed to the nucleus (Fig. 4B), possibly due to the nonspecific permeation of these proteins through nuclear pores. Because nucleus-distributed HA proteins were characterized by high-intensity HA signals (∼100–250 units) with low-intensity GM130 signals (<50 units) in the 2D map, a cross-hair bar was set at the y axis to represent the border between the nucleus and nonnucleus areas in the map (Fig. S3A). Pixels of HA-tagged proteins distributed to the area excluding the nucleus and non-Golgi cytoplasmic areas were regarded as those of GM130-colocalizing HA-tagged proteins, and pixel numbers were counted in images of 23–36 transfected cells for each construct.
Author contributions
T. S. performed NMR experiments, analyses of NMR data, and manuscript preparation. D. E. and K. K. performed biochemical and cell biological experiments, analyses of biochemical and cell biological data, and manuscript preparation. C. K., T. F., K. T., and I. S. conducted and supported this project and prepared the manuscript. K. H. and H. T. directed this project and prepared the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Momoko Yoneyama (Institute for Protein Research, Osaka University, Japan) for technical assistance with the preparation of recombinant CERT proteins. We also thank Drs. Yukiko Miura and Miyuki Kawano (Department of Biochemistry and Cell Biology, National Institute of Infectious Diseases, Japan) for the construction of the plasmid pcDNA5/FT nHAcFL-hCERT ST and its precursors.
This work was primarily supported by METI and NEDO, MEXT Scientific Research on Innovative Areas Grant 17H06417 (to K. H.) and in part by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED and AMED-CREST from AMED under Grant JP17gm0910005j0003 of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- ER
- endoplasmic reticulum
- SRM
- serine-repeat motif
- PH
- pleckstrin homology
- PI
- phosphatidylinositol
- PI4P
- phosphatidylinositol 4-monophosphate
- FFAT
- two phenylalanines in an acidic tract
- HSQC
- heteronuclear single-quantum correlation
- FCS
- fluorescence correlation spectroscopy
- 2D and 3D
- two- and three-dimensional, respectively
- CERT
- ceramide transfer protein
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoserine
- HA
- hemagglutinin
- PDB
- Protein Data Bank.
References
- 1. Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., and Nishijima M. (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426, 803–809 10.1038/nature02188 [DOI] [PubMed] [Google Scholar]
- 2. Holthuis J. C., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57 10.1038/nature13474 [DOI] [PubMed] [Google Scholar]
- 3. Hurlock A. K., Roston R. L., Wang K., and Benning C. (2014) Lipid trafficking in plant cells. Traffic 15, 915–932 10.1111/tra.12187 [DOI] [PubMed] [Google Scholar]
- 4. Midzak A., and Papadopoulos V. (2014) Binding domain-driven intracellular trafficking of sterols for synthesis of steroid hormones, bile acids and oxysterols. Traffic 15, 895–914 10.1111/tra.12177 [DOI] [PubMed] [Google Scholar]
- 5. Tamura Y., Sesaki H., and Endo T. (2014) Phospholipid transport via mitochondria. Traffic 15, 933–945 10.1111/tra.12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tatsuta T., Scharwey M., and Langer T. (2014) Mitochondrial lipid trafficking. Trends Cell Biol. 24, 44–52 10.1016/j.tcb.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 7. Chiapparino A., Maeda K., Turei D., Saez-Rodriguez J., and Gavin A. C. (2016) The orchestra of lipid-transfer proteins at the crossroads between metabolism and signaling. Prog. Lipid Res. 61, 30–39 10.1016/j.plipres.2015.10.004 [DOI] [PubMed] [Google Scholar]
- 8. Hanada K. (2014) Co-evolution of sphingomyelin and the ceramide transport protein CERT. Biochim. Biophys. Acta 1841, 704–719 10.1016/j.bbalip.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 9. Kumagai K., Kawano M., Shinkai-Ouchi F., Nishijima M., and Hanada K. (2007) Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J. Biol. Chem. 282, 17758–17766 10.1074/jbc.M702291200 [DOI] [PubMed] [Google Scholar]
- 10. Tomishige N., Kumagai K., Kusuda J., Nishijima M., and Hanada K. (2009) Casein kinase Iγ2 down-regulates trafficking of ceramide in the synthesis of sphingomyelin. Mol. Biol. Cell 20, 348–357 10.1091/mbc.e08-07-0669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mandell D. J., Chorny I., Groban E. S., Wong S. E., Levine E., Rapp C. S., and Jacobson M. P. (2007) Strengths of hydrogen bonds involving phosphorylated amino acid side chains. J. Am. Chem. Soc. 129, 820–827 10.1021/ja063019w [DOI] [PubMed] [Google Scholar]
- 12. Firth H. V., Wright C. F., and DDD Study (2011) The Deciphering Developmental Disorders (DDD) study. Dev. Med. Child. Neurol. 53, 702–703 10.1111/j.1469-8749.2011.04032.x [DOI] [PubMed] [Google Scholar]
- 13. Fugmann T., Hausser A., Schöffler P., Schmid S., Pfizenmaier K., and Olayioye M. A. (2007) Regulation of secretory transport by protein kinase D-mediated phosphorylation of the ceramide transfer protein. J. Cell Biol. 178, 15–22 10.1083/jcb.200612017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sugiki T., Takeuchi K., Yamaji T., Takano T., Tokunaga Y., Kumagai K., Hanada K., Takahashi H., and Shimada I. (2012) Structural basis for the Golgi association by the pleckstrin homology domain of the ceramide trafficking protein (CERT). J. Biol. Chem. 287, 33706–33718 10.1074/jbc.M112.367730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prashek J., Truong T., and Yao X. (2013) Crystal structure of the pleckstrin homology domain from the ceramide transfer protein: implications for conformational change upon ligand binding. PLoS One 8, e79590 10.1371/journal.pone.0079590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., and Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 10.1093/nar/gkh253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bah A., and Forman-Kay J. D. (2016) Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 291, 6696–6705 10.1074/jbc.R115.695056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cluett E. B., Kuismanen E., and Machamer C. E. (1997) Heterogeneous distribution of the unusual phospholipid semilysobisphosphatidic acid through the Golgi complex. Mol. Biol. Cell 8, 2233–2240 10.1091/mbc.8.11.2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huitema K., van den Dikkenberg J., Brouwers J. F., and Holthuis J. C. (2004) Identification of a family of animal sphingomyelin synthases. EMBO J. 23, 33–44 10.1038/sj.emboj.7600034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prashek J., Bouyain S., Fu M., Li Y., Berkes D., and Yao X. (2017) Interaction between the PH and START domains of ceramide transfer protein competes with phosphatidylinositol 4-phosphate binding by the PH domain. J. Biol. Chem. 292, 14217–14228 10.1074/jbc.M117.780007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saito S., Matsui H., Kawano M., Kumagai K., Tomishige N., Hanada K., Echigo S., Tamura S., and Kobayashi T. (2008) Protein phosphatase 2Cϵ is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J. Biol. Chem. 283, 6584–6593 10.1074/jbc.M707691200 [DOI] [PubMed] [Google Scholar]
- 22. Raya A., Revert F., Navarro S., and Saus J. (1999) Characterization of a novel type of serine/threonine kinase that specifically phosphorylates the human goodpasture antigen. J. Biol. Chem. 274, 12642–12649 10.1074/jbc.274.18.12642 [DOI] [PubMed] [Google Scholar]
- 23. Charruyer A., Bell S. M., Kawano M., Douangpanya S., Yen T. Y., Macher B. A., Kumagai K., Hanada K., Holleran W. M., and Uchida Y. (2008) Decreased ceramide transport protein (CERT) function alters sphingomyelin production following UVB irradiation. J. Biol. Chem. 283, 16682–16692 10.1074/jbc.M800799200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tóth B., Balla A., Ma H., Knight Z. A., Shokat K. M., and Balla T. (2006) Phosphatidylinositol 4-kinase IIIβ regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J. Biol. Chem. 281, 36369–36377 10.1074/jbc.M604935200 [DOI] [PubMed] [Google Scholar]
- 25. Peretti D., Dahan N., Shimoni E., Hirschberg K., and Lev S. (2008) Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell 19, 3871–3884 10.1091/mbc.e08-05-0498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banerji S., Ngo M., Lane C. F., Robinson C. A., Minogue S., and Ridgway N. D. (2010) Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the Golgi apparatus requires phosphatidylinositol 4-kinase IIα. Mol. Biol. Cell 21, 4141–4150 10.1091/mbc.e10-05-0424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goto A., Charman M., and Ridgway N. D. (2016) Oxysterol-binding protein activation at endoplasmic reticulum-Golgi contact sites reorganizes phosphatidylinositol 4-phosphate pools. J. Biol. Chem. 291, 1336–1347 10.1074/jbc.M115.682997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Capasso S., Sticco L., Rizzo R., Pirozzi M., Russo D., Dathan N. A., Campelo F., van Galen J., Hölttä-Vuori M., Turacchio G., Hausser A., Malhotra V., Riezman I., Riezman H., Ikonen E., Luberto C., Parashuraman S., Luini A., and D'Angelo G. (2017) Sphingolipid metabolic flow controls phosphoinositide turnover at the trans-Golgi network. EMBO J. 36, 1736–1754 10.15252/embj.201696048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. (1978) [Mstislav Vsevolodovich Keldysh (1911–1978)]. Kosm. Biol. Aviakosm. Med. 12, 83 [PubMed] [Google Scholar]
- 30. Kawano M., Kumagai K., Nishijima M., and Hanada K. (2006) Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 281, 30279–30288 10.1074/jbc.M605032200 [DOI] [PubMed] [Google Scholar]
- 31. Kumagai K., Kawano-Kawada M., and Hanada K. (2014) Phosphoregulation of the ceramide transport protein CERT at serine 315 in the interaction with VAMP-associated protein (VAP) for inter-organelle trafficking of ceramide in mammalian cells. J. Biol. Chem. 289, 10748–10760 10.1074/jbc.M113.528380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolduc D., Rahdar M., Tu-Sekine B., Sivakumaren S. C., Raben D., Amzel L. M., Devreotes P., Gabelli S. B., and Cole P. (2013) Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2, e00691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Desjardins G., Meeker C. A., Bhachech N., Currie S. L., Okon M., Graves B. J., and McIntosh L. P. (2014) Synergy of aromatic residues and phosphoserines within the intrinsically disordered DNA-binding inhibitory elements of the Ets-1 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 111, 11019–11024 10.1073/pnas.1401891111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amata I., Maffei M., and Pons M. (2014) Phosphorylation of unique domains of Src family kinases. Front. Genet. 5, 181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 36. Sugiki T., Yoshiura C., Kofuku Y., Ueda T., Shimada I., and Takahashi H. (2009) High-throughput screening of optimal solution conditions for structural biological studies by fluorescence correlation spectroscopy. Protein Sci. 18, 1115–1120 10.1002/pro.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.