

Lactobacillus plantarum is a bacterial species frequently found in the fermentation of vegetables where hydroxycinnamic acids are present. The bacterial metabolism on these compounds during fermentation plays a fundamental role in the biological activity of hydroxycinnamates. L. plantarum strains exhibit an as yet unknown reducing activity, transforming hydroxycinnamates to substituted phenylpropionic acids, which possess higher antioxidant activity than their precursors. The protein machinery involved in hydroxycinnamate reduction, HcrAB, was genetically identified and characterized. The heterodimeric NADH-dependent coumarate reductase HcrAB described in this work provides new insights on the L. plantarum metabolic response to counteract the stressful conditions generated by food phenolics.
KEYWORDS: lactic acid bacteria, hydroxycinnamates, phenolic compounds, reductases, vegetable foods
ABSTRACT
Lactobacillus plantarum is the lactic acid bacterial species most frequently found in plant-food fermentations where hydroxycinnamic acids are abundant. L. plantarum efficiently decarboxylates these compounds and also reduces them, yielding substituted phenylpropionic acids. Although the reduction step is known to be induced by a hydroxycinnamic acid, the enzymatic machinery responsible for this reduction pathway has not been yet identified and characterized. A previous study on the transcriptomic response of L. plantarum to p-coumaric acid revealed a marked induction of two contiguous genes, lp_1424 and lp_1425, encoding putative reductases. In this work, the disruption of these genes abolished the hydroxycinnamate reductase activity of L. plantarum, supporting their involvement in such chemical activity. Functional in vitro studies revealed that Lp_1425 (HcrB) exhibits hydroxycinnamate reductase activity but was unstable in solution. In contrast, Lp_1424 (HcrA) was inactive but showed high stability. When the hcrAB genes were co-overexpressed, the formation of an active heterodimer (HcrAB) was observed. Since L. plantarum reductase activity was only observed on hydroxycinnamic acids (o-coumaric, m-coumaric, p-coumaric, caffeic, ferulic, and sinapic acids), the presence of a hydroxyl group substituent on the benzene ring appears to be required for activity. In addition, hydroxycinnamate reductase activity was not widely present among lactic acid bacteria, and it was associated with the presence of hcrAB genes. This study revealed that L. plantarum hydroxycinnamate reductase is a heterodimeric NADH-dependent coumarate reductase acting on a carbon-carbon double bond.
IMPORTANCE Lactobacillus plantarum is a bacterial species frequently found in the fermentation of vegetables where hydroxycinnamic acids are present. The bacterial metabolism on these compounds during fermentation plays a fundamental role in the biological activity of hydroxycinnamates. L. plantarum strains exhibit an as yet unknown reducing activity, transforming hydroxycinnamates to substituted phenylpropionic acids, which possess higher antioxidant activity than their precursors. The protein machinery involved in hydroxycinnamate reduction, HcrAB, was genetically identified and characterized. The heterodimeric NADH-dependent coumarate reductase HcrAB described in this work provides new insights on the L. plantarum metabolic response to counteract the stressful conditions generated by food phenolics.
INTRODUCTION
Phenolic compounds in foods have attracted great interest due to growing evidence of their beneficial effect on human health. Phenolic acids account for almost one-third of the dietary phenols and are associated with organoleptic, nutritional, and antioxidant properties of foods. Hydroxycinnamic acids, such as p-coumaric, caffeic, ferulic, and sinapic acids, are the most abundant class of phenolic acids in fruits, vegetables, and cereal grains (1). These hydroxycinnamic acids could be transformed during the fermentation of plant-based foods. The fermentative microbiota plays an important role in the biological activity of these acids; therefore, the elucidation of their bacterial metabolism will provide important clues on their beneficial effects.
Lactic acid bacteria are widely present in food fermentations, and Lactobacillus plantarum is the species most frequently found in plant fermentations where phenolic compounds are abundant (2). Previous studies on L. plantarum demonstrated the existence of two metabolic pathways for the transformation of hydroxycinnamates. The most efficient pathway entails the successive action of a phenolic acid decarboxylase (PDC) and a reductase (3). In this pathway, p-coumaric, caffeic, and ferulic acids are decarboxylated by the PDC action to their corresponding vinyl derivatives (vinylphenol, vinylcatechol, and vinylguaiacol, respectively) (4, 5), which are subsequently reduced to the resultant ethyl derivatives (ethylphenol, ethylcatechol, and ethylguaiacol).
The second metabolic pathway for the transformation of hydroxycinnamates in L. plantarum implies the reduction of these acids (p-coumaric, m-coumaric, caffeic, and ferulic acids) to their corresponding phenylpropionic substituted acids [phloretic, 3-(3-hydroxyphenyl)propionic acid (3-HPPA), dihydrocaffeic, and dihydroferulic acids] (6). Nonetheless, the reductase activity appeared to be approximately 100-fold lower than the decarboxylase activity (3). The reduction pathway was more evident when a L. plantarum mutant strain deficient in PDC activity was constructed (3). Thus, when PDC mutant (Δlp_3665) cells were induced with p-coumaric acid, only phloretic acid was detected (3) instead of 4-ethylphenol, which would result from a decarboxylation reaction. These results demonstrated that the enzyme(s) involved in the reduction of hydroxycinnamates (such as p-coumaric acid) was synthesized after its induction by the hydroxycinnamic acid (6). Within this context, a previous study on the global transcriptomic response of L. plantarum exposed to 1.5 mM p-coumaric acid stress (7) revealed that the most upregulated gene by far was pdc (lp_3665), which was induced 112-fold compared to the growth in the absence of this hydroxycinnamate. Interestingly, a marked induction of some other genes coding for putative reductases was also detected. The oxidoreductase responsible for this second pathway remains biochemically and genetically uncharacterized.
Presently, many oxidoreductases of the class EC 1.3.1 acting on carbon-carbon double bonds in the α,β position to a carbonyl group have been described; however, few of them have been reported to work on hydroxycinnamic acids (8). The 2-coumarate reductase subgroup (EC 1.3.1.11) only includes an NADH-dependent reductase partially purified from Arthrobacter extracts which reduces o-coumaric acid to melilotic acid (9). This enzyme was highly specific for o-coumarate, as the activities against cinnamic acid and several substituted cinnamates (including p-coumaric, caffeic, ferulic, and sinapic acids) were very low (10). The subgroup of 2-enoate reductases (EC 1.3.1.31) also included enzymes for the hydrogenation of the C=C bond on hydroxycinnamates. Enoate reductases belong to the rare class of flavoenzymes in which the involvement of iron-sulfur centers in electron transfer has been well established (11). Enoate reductases are characterized by their high stereospecificity, strict regioselectivity, and a rather broad substrate specificity (12). These enzymes, which have been found in numerous Clostridia and other strict anaerobes, have been scarcely studied, most probably due to their high sensitivity to oxygen (13). The substrate specificity of clostridial enoate reductases is very variable, reducing either a broad range of enoates (Clostridium kluyveri and Clostridium tyrobutyricum) or almost exclusively cinnamic acid (Clostridium sporogenes) (14). In the latter case, whereas p-coumaric acid resulted in a relatively good substrate for the enoate reductase from C. sporogenes, the enzyme was almost inactive on caffeic acid (15). Other hydroxycinnamic acids were also substrates for clostridial enoate reductases: p-coumaric acid was reduced by Clostridium acetobutylicum enoate reductase (8) and o-coumaric acid by Clostridium sp. La 1 protein (13). Despite the fact that clostridia are phylogenetically related to lactobacilli, nowadays, the oxidoreductase type acting on the hydroxycinnamate carbon-carbon double bond in L. plantarum remains largely unknown.
In this work, the genes involved in L. plantarum hydroxycinnamate reduction have been identified. In addition, in this study, a hydroxycinnamate reductase belonging to the 2-coumarate reductase subgroup (EC 1.3.1.11) has been genetically identified and characterized.
RESULTS AND DISCUSSION
Identification of the enzyme responsible for hydroxycinnamate reduction in L. plantarum WCFS1.
Previous studies on the global transcriptomic response of L. plantarum exposed to p-coumaric acid revealed a marked induction of genes coding for putative reductases (7). Thus, the third most overexpressed transcript (19.7-fold induction) was lp_1425, which would encode a fumarate reductase/succinate dehydrogenase, flavin adenine dinucleotide (FAD)-binding flavoprotein, NADPH-dependent flavin mononucleotide (FMN) reductase. In addition, two other genes clustered with lp_1425, namely, lp_1424 (putative NADPH-dependent FMN reductase family protein) and lp_1426 (protein of unknown function), were also strongly upregulated (15.3- and 7.2-fold, respectively). To determine if one (or both) of these genes encoding the reductase(s) was involved in hydroxycinnamate reduction, lp_1424 and lp_1425 knockout mutants were constructed by an insertion-duplication strategy. The correct insertion of the donor plasmids into the L. plantarum WCFS1 chromosome was verified by PCR (see Materials and Methods). The wild-type and mutant strains were grown in MRS medium supplemented with 1.5 mM m-coumaric acid. After 10 days of incubation, samples were collected, and the phenolic compounds present in the supernatants were extracted and analyzed by high-pressure liquid chromatography (HPLC) (Fig. 1). The results indicated that these two mutants were unable to reduce m-coumaric acid, most probably because both proteins are required by L. plantarum to reduce hydroxycinnamates. In this assay, m-coumaric acid was chosen as a model compound of hydroxycinnamic acid, since L. plantarum is unable to decarboxylate it, as was reported previously (5), although it reduces this compound, yielding 3-HPPA (6). These results suggested that the two consecutive genes lp_1424 (hcrA, hydroxycinnamate reductase) and lp_1425 (hcrB) are needed for reducing m-coumaric acid.
FIG 1.
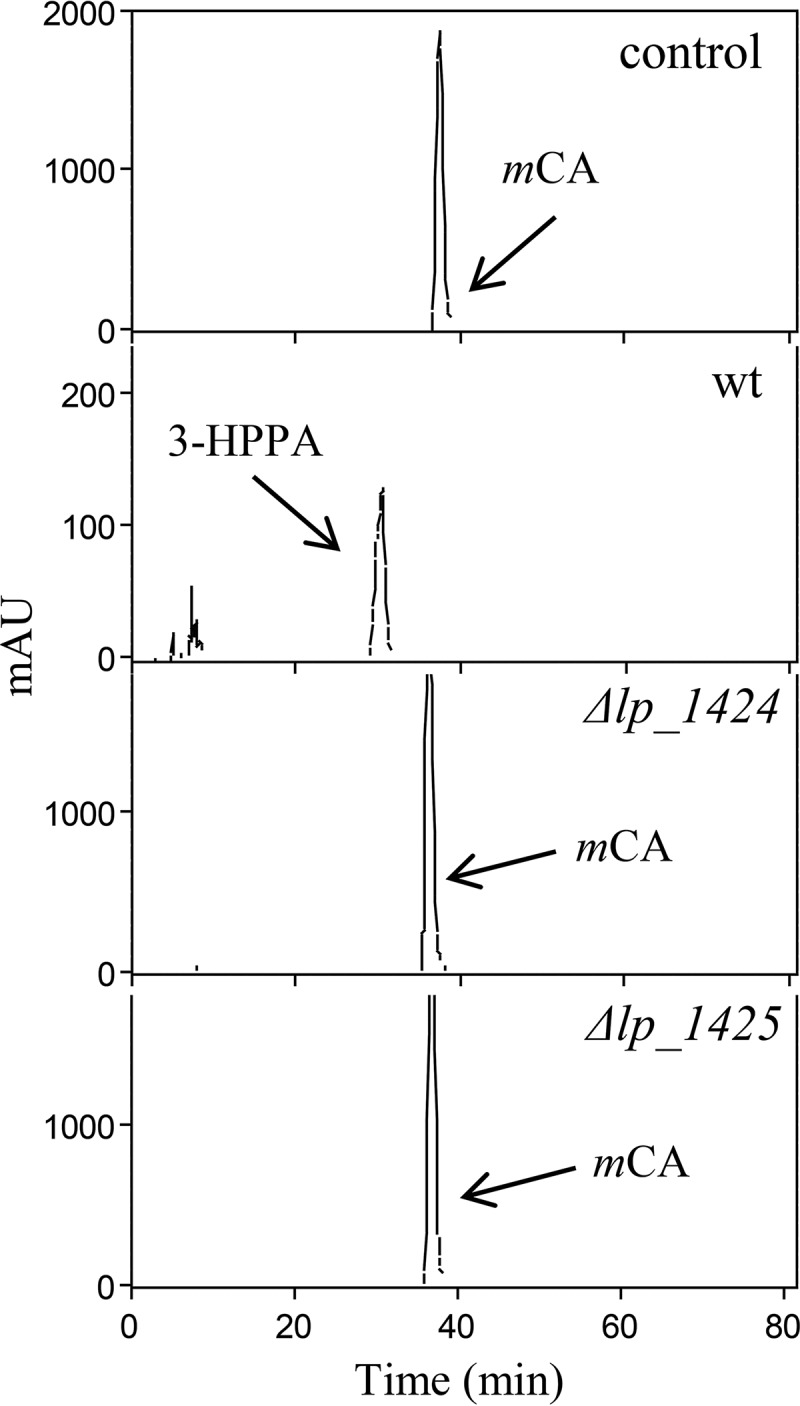
Effect of disruption of lp_1424 (hcrA) and lp_1425 (hcrB) on hydroxycinnamate reduction in L. plantarum WCFS1. HPLC chromatograms of L. plantarum cultures incubated in 1.5 mM m-coumaric acid are shown for L. plantarum WCFS1 (wild type [wt]), L. plantarum WCFS1(pUCE191-hcrA) (Δlp_1424 mutant), and L. plantarum WCFS1(pUCE191-hcrB) (Δlp_1425 mutant). Results for uninoculated medium are also shown (control). The m-coumaric acid (mCA) and 3-(3-hydroxyphenyl) propionic acid (3-HPPA) detected are indicated. Chromatograms were recorded at 280 nm.
Genetic organization and expression profile of the hcr locus of L. plantarum WCFS1.
To characterize in deeper detail the roles of hcrA and hcrB in the context of hydroxycinnamate reduction, their genetic organization and expression profile in L. plantarum WCFS1 were analyzed (Fig. 2A). To determine the transcriptional profile of the lp_1420 to lp_1427 region, total RNA from L. plantarum WCFS1 cells grown in MRS medium supplemented with p-coumaric acid (1.5 mM) was isolated and analyzed by reverse transcriptase PCR (RT-PCR) (Table 1, Fig. 2B). To investigate the potential involvement of the lp_1422 and hcrC genes in the reduction of hydroxycinnamates, their corresponding L. plantarum WCFS1 knockout mutants were constructed. Whereas the detection of reductase activity in the hcrC mutant grown in the presence of m-coumaric acid indicated that HcrC plays no role (or an ancillary one) in the reduction of hydroxycinnamates, the lack of activity in the knockout lp_1422 mutant (see Fig. S1 in the supplemental material) supports the participation of HcrR, an LysR-type transcriptional regulator, in the reduction process.
FIG 2.
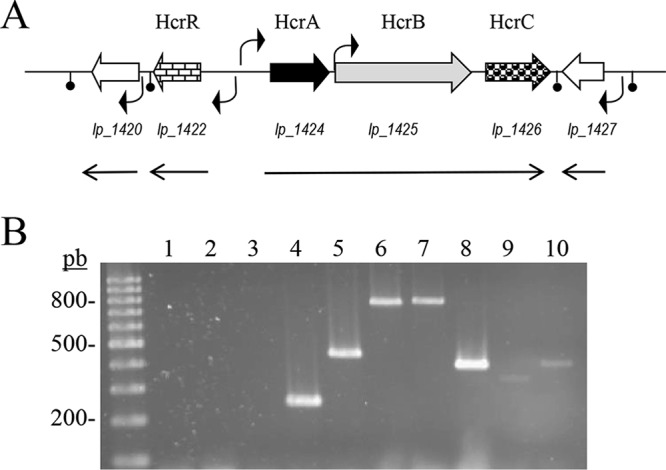
Genetic organization of the L. plantarum WCFS1 chromosomal region containing the hydroxycinnamate reductase encoding genes. (A) NCBI accession NC_004567, positions 1299251 to 1309330; arrows indicate genes. The shaded genes encode proteins putatively involved in hydroxycinnamate reduction (hcr). The locations of putative promoters and transcription terminators are also indicated. The size and direction of the transcripts revealed by reverse transcription are also shown. (B) Transcriptional analysis by RT-PCR of the L. plantarum WCFS1 genome in hydroxycinnamate reductase locus. RT-PCR amplification with primers designed to amplify internal gene regions or intergenic regions: lane 2, hcrR (primers 1328 and 1329, 628 bp); lane 3, hcrR-hcrA (1327 and 1229, 861 bp); lane 4, hcrA (1228 and 1229, 241 bp); lane 5, hcrA-hcrB (1222 and 1330, 432 bp); lane 6, hcrB (877 and 878, 758 bp); lane 7, hcrB-hcrC (1385 and 1052, 778 bp); lane 8, hcrC (1051 and 1952, 384 bp); lane 9, hcrC-lp_1427 (1226 and 1331, 323 bp); lane 10, lp_1427 (1332 and 1333, 395 bp). The control without cDNA is also shown (lane 1). Left lane, 100-bp molecular size ladder. Numbers indicate some of the molecular sizes.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′)a | Amplified fragment and/or cloning strategy |
|---|---|---|
| 877878 | GGGGTACCTGCTGTCAAAGAAGTGACGGCTCTAGAAGCTAAAATCACCGCATCAGC | The 758-bp PCR hcrB internal fragment was double-digested with KpnI/XbaI and cloned into KpnI/XbaI double-digested pUCE191 plasmid generating pUCE191-hcrB Amplification of a 758-bp hcrB internal region |
| 889 890 | TAACTTTAAGAAGGAGATATACATATGAAATTTGTTGGAATTGTTGGAA GCTATTAATGATGATGATGATGATGGGCCGTCACCGGATCAACGTGCTC | The 2.4-kb hcrB gene amplified with 889 and 890 primers was introduced into pURI3-Cter vector by using an LIC strategy The 3.1-kb hcrA and hcrB genes amplified with 1631 and 890 primers were introduced into pURI3-Cter vector by using an LIC strategy |
| 918 919 | AACCGCGACAATGTTTTGATTTTGTGAACGGCAGTTTCAGTGT | Amplification of a 62-bp lp_2057 (ldhD) fragment for RT-qPCR |
| 976 | ATGAAATTTGTTGGGATT | Amplification with oligo 1233 to corroborate the correct insertion of pUCE191-hcrA into the L. plantarum WCFS1 chromosome |
| 1051 1052 | GGGGTACCAACCAGCTTATATGA GCTCTAGATGAAAGTCGGTGATCCAA | The 384-bp PCR hcrC internal fragment was double-digested with KpnI/XbaI and cloned into KpnI/XbaI double-digested pUCE191 plasmid generating pUCE191-hcrC Amplification of a 384-bp hcrC internal fragment Amplification with 1385 and 1052 primers for a 778-bp hcrB-hcrC intergenic region |
| 1222 1223 | CACTGGTGATGCCAAATATTGAA GCCCTGATCATCAAAAGCTTGT | Amplification of a 65-bp hcrA fragment for RT-qPCR Amplification with 1222 and 1330 primers for a 432-bp hcrA-hcrB intergenic region |
| 1224 1225 | CGGCAGCCCTGACCAA GCCGGCATCAACGTAACG | Amplification of a 198-bp hcrB fragment for RT-qPCR |
| 1226 1227 | TGACATCGACTGGCCCAAT TGCCCTTTGTCAATGCTTCA | Amplification of a 56-bp hcrC fragment for RT-qPCR Amplification with 1226 and 1331 primers for a 323-bp hcrC-lp_1427 intergenic region |
| 1228 1229 | GGGGTACCGAACAGCACCAGTAGC GCTCTAGAATCTGGCGTAAGTGC | The 241-bp PCR hcrA internal fragment was double-digested with KpnI/XbaI and cloned into KpnI/XbaI double-digested pUCE191 plasmid generating pUCE191-hcrAAmplification of a 241-bp hcrA internal region Amplification with 1327 and 1229 primers for an 861-bp hcrR-hcrA intergenic region |
| 1233 | AGCGGATAACAATTTCACACAGGA | 24-mer reverse sequencing primer (−48) for pUCE19/pUCE191 |
| 1242 | ATGAAATTTGTTGGAATTGTTG | Amplification with primer 1233 to corroborate the correct insertion of pUCE191-hcrB into the L. plantarum WCFS1 chromosome |
| 1243 | ATGCAAAAACTAATGCCAGTC | Amplification with primer 1233 to corroborate the correct insertion of pUCE191-hcrC into the L. plantarum WCFS1 chromosome |
| 1327 | CTTTTTCAACATTGCGGATGC | Amplification with primer 1229 for an 861-bp hcrR-hcrA intergenic region |
| 1328 1329 | CATTAACGCTTGGGGCAAAGG GACTTATTATCAATATTTGACG | Amplification for a 628-bp hcrR internal fragment |
| 1330 | CGTCAGCCGTCGCAATTTTAGTGG | Amplification with primer 1222 for a 432-bp hcrA-hcrB intergenic region |
| 1331 | GACTTGCCCATCATCGCCTTGC | Amplification with primer 1226 for a 323-bp hcrC-lp_1427 intergenic region |
| 1332 1333 | CGATAAAGTCGCCTTTAGGTAATTG CAGTCCCGAACAAGTGACGCG | Amplification for a 395-bp lp_1427 internal fragment |
| 1385 | TGCCGTGGCACCATTTTAC | Amplification with primer 1052 for a 778-bp hcrB-hcrC intergenic region |
| 1627 1628 | TCGGCTGGTACCTATTGCCCCTA AACTTGGCGTCGTCTAGATGAC | The 335-bp PCR hcrR internal fragment was double-digested with KpnI/XbaI and cloned into KpnI/XbaI double-digested pUCE191 plasmid generating pUCE191-hcrR |
| 1630 | ATGCCAAAATCAGATTCCAGCGA | Amplification with primer 1233 to corroborate the correct insertion of pUCE191-hcrR into the L. plantarum WCFS1 chromosome |
| 1631 1632 | TAACTTTAAGAAGGAGATATACATATGAAATTTGTTGGGATTGTTGGGA GCTATTAATGATGATGATGATGATGTTCCTCCCCAGTCACTTGCCGCCA | The 0.6-kb hcrA gene amplified with primers 1631 and 1632 was introduced into pURI3-Cter vector by using an LIC strategy The 3.1-kb hcrA and hcrB genes amplified with primers 1631 and 890 were introduced into pURI3-Cter vector by using an LIC strategy |
| 1655 1656 | CARATHYTNGAYGCNCCNGGNGT CATCATYTGNGWRAANCCCATNCC | Degenerate primers coding for the HcrB conserved motifs QILDAPGV (1655) and GMGF(S/T)QMM (1656). They are used to amplify a 1.4-kb internal fragment of hcrB in lactic acid bacteria |
M = A or C; S = G or T; R = G or A; Y = C or T; D = A, G or T; H = A, C, or T, and N = G, A, C, or T. Engineered restriction sites are underlined. The nucleotides pairing the vector sequence are in italics.
Moreover, since the transcriptional analysis showed cotranscription of the hcrA, hcrB, and hcrC genes (Fig. 2), it could be possible that the disruption of hcrA may prevent the induction of the other downstream cotranscribed genes. This, in turn, could be the basis of reductase activity abolition. Quantitative RT-PCR experiments demonstrated the presence of polar effects on the hcrB transcription due to the disruption of the upstream hcrA gene (Fig. 3).
FIG 3.

Relative expression levels of hcrAB genes in hcr knockout mutants in response to 1.5 mM m-coumaric acid. Expression levels were calculated with the 7500 Fast system relative quantification software using L. plantarum ldh as the endogenous gene and growth in the absence of m-coumaric acid as a growth condition calibrator. Expression levels of hcrA and hcrB genes are represented by black and gray bars, respectively. The experiments were done in triplicate. The mean values and the standard errors are shown. *, P < 0.1.
Therefore, as a whole, it could be concluded that the presence of a functional HcrB protein is a minimum requirement for hydroxycinnamate reductase activity in L. plantarum, since the hcrB knockout mutant, despite producing HcrA protein, lacks reductase activity.
Hydroxycinnamate reductase activity of HcrA and HcrB proteins.
Once the necessity of the HcrB protein was determined for the reducing activity of L. plantarum, it was determined whether this protein is sufficient for this bacterium to exhibit such enzymatic activity in vitro. With this aim, the hcrA and hcrB genes were cloned either individually or jointly, as they appeared on the L. plantarum WCFS1 chromosome. Details about the cloning strategy can be found in Table 1. The correct sequences of the recombinant plasmids pURI3-Cter-HcrA, pURI3-Cter-HcrB, and pURI3-Cter-HcrAB were verified by DNA sequencing.
Cell extracts were prepared from Escherichia coli BL21 cells harboring the recombinant plasmids. The pURI3-Cter-HcrA cell extract showed one major protein band of approximately 24 kDa by SDS-PAGE, which was absent in control cells containing the pURI3-Cter vector plasmid alone (see Fig. S2A). The molecular mass of this overproduced protein is in good agreement with the one deduced from its amino acid sequence as derived from the hcrA gene. These extracts were incubated in the presence of 1.5 mM m-coumaric acid, and the reaction products were extracted with ethyl acetate and analyzed by HPLC (Fig. S2B). Similar to the extracts from control cells, those with the HcrA protein contained m-coumaric acid but not the expected reduced product 3-HPPA.
The HcrA protein was purified by immobilized-metal affinity chromatography (IMAC). The purified protein showed the characteristic yellow color indicative of the binding of an oxidized flavin cofactor. However, similar to in cell extracts, when this pure HcrA protein was added to a reaction mixture containing FMN (7.5 mM) and NADH or NADPH (15 mM), no reduction of m-coumaric acid was detected (Fig. S2B).
On the other hand, SDS-PAGE analysis of pURI3-Cter-HcrB cell extracts showed an intense 90-kDa protein band which was absent from control cells (Fig. 4A). The molecular mass of this protein agrees well with that expected for HcrB (88 kDa). When these extracts were incubated with 1.5 mM m-coumaric acid, it was observed that this hydroxycinnamic acid was fully reduced to 3-HPPA. As was the case for HcrA, the purified HcrB protein also showed a yellow color indicative of a complex between the protein and an oxidized form of flavin cofactors. When this protein was incubated with FMN (7.5 mM) and NADH (15 mM), it was able to reduce the m-coumaric acid present in the in vitro reaction (Fig. 4B).
FIG 4.

Purification and enzymatic activity of E. coli extracts expressing L. plantarum HcrB and recombinant HcrB protein. (A) SDS-PAGE analysis of the expression and purification of the His6-tagged HcrB. Results of analysis of soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3-Cter) (lane 1) or E. coli BL21(DE3)(pURI3-Cter-HcrB) (lane 2), flowthrough from the affinity resin (lane 3), and fractions eluted from His affinity resin (lanes 4 to 8). The 8% gel was stained with Coomassie blue. Molecular mass markers are located on the left (SDS-PAGE standards; Bio-Rad). (B) HPLC chromatograms showing hydroxycinnamate reductase activity of soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3-Cter) (control) or E. coli BL21(DE3)(pURI3-Cter-HcrB) (HcrB) incubated in 1.5 mM m-coumaric acid. HPLC chromatograms also showed the reductase activity of purified His6-HcrB protein (500 μg) (HcrB) or without protein (control). The m-coumaric acid (mCA) and 3-(3-hydroxyphenyl)propionic acid (3-HPPA) detected are indicated. Chromatograms were recorded at 280 nm.
Regarding the dependence of the reducing activity of m-coumaric acid by HcrB on the cofactors used, control experiments revealed, as expected, a lack of activity in the absence of FMN or NADH (data not shown). On the other hand, no activity was observed upon the replacement of NADH with NADPH, indicating that the HcrB activity is NADH dependent. These results indicate that HcrB utilizes FMN and NADH as cofactors to reduce m-coumaric acid to 3-HPPA and that HcrA lacks this in vitro enzymatic activity.
m-Coumaric acid was reduced to 3-HPPA when pure HcrB protein was assayed, although a major peak for m-coumaric acid remained (Fig. 4B). However, protein extracts from E. coli producing recombinant HcrB were able to reduce all the m-coumaric acid present in the reaction mixture (Fig. 4B). Therefore, these cell extracts were used to assay reductase activity against five additional hydroxycinnamic acids (p-coumaric, o-coumaric, ferulic, caffeic, and sinapic acids) and cinnamic acid (Fig. 5). A schematic representation of these acids is shown in Fig. S3. All the hydroxycinnamic acids assayed were reduced to their corresponding phenylpropionic acids, such as p-coumaric acid to phloretic acid, m-coumaric acid to 3-HPPA, o-coumaric acid to melilotic acid or 3-(2-hydroxyphenyl)propionic acid (2-HPPA), ferulic acid to hydroferulic acid, caffeic acid to hydrocaffeic acid, and sinapic acid to hydrosinapic acid. The identification of these compounds was carried out by comparing the retention times and spectral data of each peak with those of standards from commercial suppliers. The study revealed that HcrB needs a hydroxyl group substituent on the benzene ring for activity, as reduction was only observed on hydroxycinnamates (o-coumaric, m-coumaric, p-coumaric, caffeic, ferulic, and sinapic acids).
FIG 5.

Reductase activity of HcrB on several hydroxycinnamic acids. HPLC chromatograms of E. coli BL21(DE3)(pURI3-Cter) (control) or E. coli BL21(DE3)(pURI3-Cter-hcrB) (HcrB) cell extracts incubated at 37°C for 16 h. The hydroxycinnamic acids assayed were p-coumaric (pCA), m-coumaric (mCA), o-coumaric (oCA), ferulic acid (FA), caffeic acid (CA), and sinapic acid (SA). A nonhydroxy-derived cinnamic acid was used as the control (cinnamic acid [CiA]). The detected corresponding reduced acids, such as phloretic acid (PhlA) and 3-(3-hydroxyphenyl)propionic (3-HPPA), 3-(2-hydroxyphenyl)propionic (2-HPPA), melilotic, hydroferulic (HFA), hydrocaffeic (HCA), and hydrosinapic (HAS) acids, are indicated. Chromatograms were recorded at 280 nm.
The acids reduced by HcrB are in agreement with previous results when the metabolism of hydroxycinnamic acids was studied in L. plantarum cell cultures (6), except for the o-coumaric acid metabolism, which was not observed. The reason for the absence of o-coumarate reductase activity in L. plantarum cultures is currently unknown. Searching for a possible explanation for this difference, we studied the relative expression of all the described hcr genes (hcrR, hcrA, hcrB, and hcrC) upon exposure to a hydroxycinnamic acid (Fig. 6). The responses to the presence of cinnamic acid and gallic acid (a hydroxybenzoic acid) were also included in the study. Unexpectedly, the expression results did not explain the difference observed, as hcrABC genes were induced in the presence of o-coumaric as well as in response to other hydroxycinnamic acids. Despite the fact that cinnamic acid is not a substrate for the reductase, the hcrABC genes were also induced, possibly due to its structural similarity. However, the studied genes were not induced in response to the presence of a hydroxybenzoic acid (gallic acid).
FIG 6.

Relative transcriptional expression of hcr genes in L. plantarum WCFS1 in response to the presence of several hydroxycinnamic acids. L. plantarum cultures were exposed for 10 min to 1.5 mM hydroxycinnamic acid (p-coumaric, m-coumaric, o-coumaric, ferulic, caffeic, or sinapic acid). As controls, a nonhydroxy-derived cinnamic acid (cinnamic acid) and a hydroxybenzoic acid (gallic acid) were assayed. Expression levels were calculated with the 7500 Fast system relative quantification software using L. plantarum ldh as the endogenous gene and growth in the absence of acid as a growth condition calibrator. Expression levels of hcrR, hcrA, hcrB, and hcrC genes are represented by brick-like shading, black color, gray color, and dotted-like shading bars, respectively. The experiments were done in triplicate. The mean values and the standard errors are shown. *, P < 0.1.
Role of HcrA in reductase activity.
The results obtained above support that HcrB reduces hydroxycinnamates and that HcrA lacks this enzymatic activity. Purified HcrB protein in the presence of appropriate cofactors (FMN and NADH) reduces in vitro, only partially, m-coumaric acid. To elucidate a possible role of HcrA in hydroxycinnamate reductase activity, the hcrA and hcrB genes were also jointly cloned, as they appeared on the L. plantarum WCFS1 chromosome. When cell extracts were prepared from E. coli BL21 harboring pURI3-Cter-HcrAB recombinant plasmid, the two expected bands (22 and 87 kDa) were observed on SDS-PAGE, which were absent from control cells containing the pURI3-Cter vector plasmid alone (Fig. 7A). Since only the HcrB protein has a His6 tag (at its C-terminal end) according to the cloning strategy of hcrAB in the pURI3-Cter plasmid, it was surprising to observe both proteins coeluting from the IMAC step (Fig. 7B), since it reveals that both proteins form a stable complex.
FIG 7.
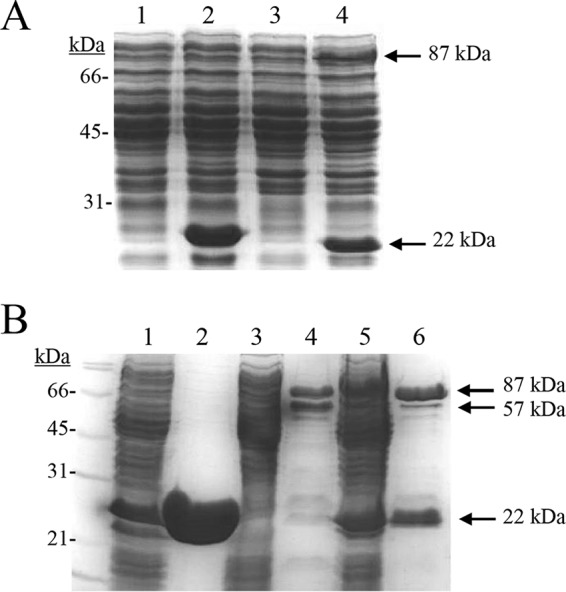
SDS-PAGE analysis of the production and purification of HcrA, HcrB, and HcrAB recombinant proteins. (A) Soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3-Cter) (lane 1), E. coli BL21(DE3)(pURI3-Cter-HcrA) (lane 2), E. coli BL21(DE3)(pURI3-Cter-HcrB) (lane 3), and E. coli BL21(DE3)(pURI3-Cter-HcrAB) (lane 4). (B) Purification of HcrA (lane 2), HcrB (lane 4), and HcrAB (lane 6) recombinant proteins from the corresponding E. coli BL21 cell extracts (lane 1, pURI3-Cter-HcrA; lane 3, pURI3-Cter-HcrB; lane 5, pURI3-Cter-HcrAB). HcrA protein, 22 kDa; HcrB protein, 87 kDa; hydrolyzed HcrB, 57 kDa.
An alignment of the amino acid sequences of HcrA and HcrB revealed that the latter protein has an N-terminal region highly similar to HcrA; the first N-terminal 200-amino-acid sequence region is approximately 55% identical to that of HcrA (Fig. S4A). Moreover, both genes are expressed in a single transcript with a 2-bp intergenic region. Given these observations, the coexpression and purification of HcrA plus HcrB-His6 could result in the purification of the active holoenzyme.
Size exclusion chromatography was used to determine the oligomeric state of the individually purified HcrA, HcrB, and HcrAB protein forms. The elution profile of HcrA displayed a single symmetric peak that is consistent with a molecular species with an average mass in solution of 44 kDa. Since the molecular mass expected for HcrA is 22.9 kDa, this result indicates that the most probable species in solution for HcrA is a dimer. Unfortunately, the results obtained with HcrB or HcrAB samples were not conclusive, since numerous peaks corresponding to high-molecular-mass species were observed, indicating protein aggregation. Nonetheless, the smallest species observed with freshly prepared samples were close to a homodimeric complex for HcrB (170 kDa) or a heterodimeric complex for HcrAB (110 kDa) (data not shown).
In addition to the HcrB aggregation in solution observed during size exclusion chromatography, it was observed that this protein became partially hydrolyzed after its purification, with the end product of this partial hydrolysis being a well-defined species with an approximate molecular mass of 57 kDa according to SDS-PAGE (Fig. 7B). It was noted that when HcrB was coproduced with HcrA, this proteolysis was significantly avoided. To identify the 57-kDa molecular species, the corresponding gel band was subjected to fingerprint mass spectrometry. This analysis revealed that this core and stable fragment corresponded to the C-terminal 523 amino acid residues of HcrB (56,906-Da molecular mass); therefore, the hydrolysis occurred between the K289 and Q290 residues. Hence, QVAQPAV is the N-terminal sequence of this fragment.
A domain analysis of the HcrB protein at the NCBI site revealed the presence of three conserved domains (Fig. S4B). From the N to C termini of the protein sequence, we found COG0431 [amino acid residues 1 to 175, NAD(P)H-dependent FMN reductase (SsuE)], COG3976 (amino acid residues 179 to 275, FMN-binding domain [FMN_bind]), and COG1053 (amino acid residues 321 to 797, succinate dehydrogenase/fumarate reductase, flavoprotein subunit [SdhA]) domains. As determined by the proteomic analysis, the N-terminal end of the 57-kDa fragment would be located between the domains involved in FMN reductase activity (COG0431 and COG3976) and the FAD-binding domain (COG1053). The proposed model for the modular tridimensional structure of HcrB protein bound to FMN and FAD is shown in Fig. S4C.
The protein domain analysis of the HcrA protein showed the presence of only the COG0431 domain. Therefore, the COG0431 domain is present in the two contiguous proteins, HcrA and HcrB. The presence of two similar genes predicted to encode two 20-kDa proteins each containing flavin mononucleotide (FMN) reductase conserved domains has been previously described in Lactobacillus johnsonii and other lactobacilli (16, 17). Similar to L. plantarum HcrAB reductase, in the L. johnsonii reductase, the coexpression of both reductase proteins facilitated the solubility of the active heterodimer. The genes encoding the subunits of this reductase are conserved in all other members of the Lactobacillus acidophilus group (L. gasseri, L. acidophilus, and L. delbrueckii). Homology searches in more distant species revealed that genes homologous to those from L. plantarum and L. johnsonii are encountered as consecutive genes in several species belonging to the Streptococcus, Enterococcus, and Pediococcus genera. In all these cases, the first upstream gene is similar in sequence and size to HcrA (or lj_0548 and lj_0549 from L. johnsonii) and is followed by a second larger gene, of which the N-terminal 200-amino-acid residue domain is homologous to the HcrB N-terminal domain. This type of arrangement is also present in a more distant species from the Actinobacteria phylum, Atopobium parvulum (16). In all the aforementioned species, the proteins encoded by these genes are annotated as fumarate reductases, NADH dehydrogenases, or flavin reductases. Apart from the L. johnsonii reductase, there are no experimental data supporting these annotations. The only proteins described so far for hydroxycinnamate reductase activity are enoate reductase; however, this work on the HcrA and HcrB proteins in L. plantarum describes a new type of heterodimeric enzyme involved in the reduction of the carbon-carbon double bond present in hydroxycinnamic acids.
Hydroxycinnamate reduction in lactic acid bacteria.
Once the involvement of L. plantarum hcr genes in hydroxycinnamate reductase activity was demonstrated, it seemed probable that other species of lactic acid bacteria could also reduce these phenolic acids. Database searches revealed the existence of similar hcr genes in other lactic acid bacterial species (Fig. 8). The conservation of the same hcrAB genetic organization in these bacteria supports the involvement of HcrA protein in hydroxycinnamate reduction. The amino acid sequences of HcrA and HcrB homologues from 11 lactic acid bacterial species were aligned (see Fig. S5 and S6, respectively). The degree of identity exhibited among these HcrA-like proteins ranged from 50% (Enterococcus columbae and Streptococcus mutans proteins) to 99% (Enterococcus casseliflavus and Enterococcus flavescens proteins). The identity among HcrB proteins was slightly higher, ranging from 63% (Lactobacillus pentosus and Clostridium beijerinckii) to 99% (E. casseliflavus and E. flavescens). These alignments enabled the identification of conserved amino acid domains. Taking into account the high identity found among HcrA and HcrB proteins (50%), only domains from HcrB were chosen to design degenerate oligonucleotides to detect its presence by PCR. Oligonucleotides 1655 and 1656 amplified a 1.4-kb internal fragment of hcrB in lactic acid bacteria.
FIG 8.

Genetic organization of hydroxycinnamate reductases from several lactic acid bacteria, such as Lactobacillus plantarum WCFS1 (NC_004567.2), Lactobacillus paraplantarum DSM 10667 (NZ_CP013130.1), Lactobacillus pentosus DSM 20314 (GCA_001433755.1), Lactobacillus fabifermentans DSM 21115 (NZ_AYGX00000000.2), Lactobacillus alimentarius DSM 20249 (NZ_AZDQ00000000.1), Lactobacillus paralimentarius DSM 13961 (NZ_AZDH00000000.1), Lactobacillus dextrinicus DSM 20335 (NZ_AYYK00000000.1), Enterococcus phoeniculicola ATCC BAA-412 (NZ_ASWE00000000.1), Enterococcus columbae DSM 7374 (NZ_AHYW00000000.1), Enterococcus flavescens ATCC 49996 (NC_020995.1), Enterococcus casseliflavus ATCC 12755 (GCA_000191365.1), Enterococcus gilvus ATCC BAA-350 (NZ_ASWH00000000.1), Streptococcus parasanguinis ATCC 15912 (NC_015678.1), Streptococcus uberis ATCC BAA-854 (NC_012004.1), Streptococcus equinus ATCC 9812 (NZ_JNLO00000000.1), Streptococcus gallolyticus ATCC 43143 (NZ_CP018822.1), Streptococcus mutans ATCC 700610 (NC_004350.2), Clostridium beijerinckii ATCC 51743 (NZ_CP010086.2), and Clostridium saccharobutylicum DSM 13864 (NC_022571.1). Arrows indicate genes. Genes having putative identical functions are represented by identical shading. The genes having brick-like shading encode putative transcriptional regulators. Genes coding for putative reductases similar to HcrA and HcrB are represented by black and gray arrows, respectively.
To associate the presence of the hcrB gene and the ability to reduce hydroxycinnamic acids, selected strains of lactic acid bacteria were grown in culture medium containing m-coumaric acid, and their supernatants were analyzed for the production of 3-HPPA (Fig. 9). In addition, their DNAs were used as the templates in PCR experiments using oligonucleotides 1655 and 1656 to detect the presence of the hcrB gene (Fig. 9A). The strains belonging to Lactobacillus brevis, Lactobacillus sakei, Lactobacillus fructivorans, Leuconostoc citreum, Enterococcus durans, Enterococcus faecalis, Enterococcus faecium, and Enterococcus hirae did not amplify the expected fragment. Contrarily, PCR experiments gave positive amplicons in all the strains belonging to the L. plantarum group analyzed (L. plantarum, L. pentosus, and L. paraplantarum) and in E. casseliflavus DSM 20680, Enterococcus gallinarum DSM 24841, and Streptococcus gallolyticus UCN34 strains. These results indicated that hcrB is a gene conserved in all the strains from the L. plantarum group. Database searches of complete genomes from these strains confirmed this result. Surprisingly, differences in the presence of hcrB were found among the enterococcal strains. HcrB-like proteins were found in E. casseliflavus and E. gallinarum species. However, despite the fact that numerous E. faecium and E. faecalis strains have been sequenced, proteins similar to HcrB were only found in E. faecalis 06-MB-DW-09 (accession number S4D3PU_ENTFL) and two E. faecium strains (accession numbers T2NMF6_ENTFC and A0A24J1P6_ENTFC). Strain variability and the possibility of an erroneous taxonomic identification of these strains could not be ruled out (18).
FIG 9.

Hydroxycinnamate reductase activity in lactic acid bacteria. (A) PCR amplification of the HcrB gene. Chromosomal DNA from several lactic acid bacteria was used for PCR amplification with oligonucleotides 1655 and 1656 to amplify 1.4 kb of the hcrB gene. PCR products were subjected to gel electrophoresis and stained with Gel Red. Left lane, λ/EcoT14I (TaKaRa) molecular size marker. Numbers indicate some of the molecular sizes. (B) HPLC chromatograms of supernatants from lactic acid bacteria grown for 10 days at 30°C in MRS medium supplemented with 1.5 mM m-coumaric acid. The m-coumaric acid (mCA) and 3-(3-hydroxyphenyl)propionic acid (3-HPPA) detected are indicated. Chromatograms were recorded at 280 nm. The strains assayed were Enterococcus casseliflavus DSM 20680 (1), E. durans DSM 20633 (2), E. faecalis DSM 20478 (3), E. faecium CECT 4102 (4), E. gallinarum DSM 24841 (5), E. hirae DSM 20160 (6), Lactobacillus brevis CECT 5354 (7), L. fermentum CECT 4007 (8), L. fructivorans CECT 4785 (9), L. paraplantarum DSM 10641 (10), L. plantarum subsp. argentoratensis DSM 16365 (11), L. plantarum subsp. plantarum ATCC 14917 (CECT 748) (12), L. plantarum DSM 10492 (13), L. pentosus DSM 16366 (14), L. sakei subsp. carnosus DSM 15831 (15), Leuconostoc citreum CECT 4025 (16), and Streptococcus gallolyticus UCN34 (17).
An HPLC analysis of the supernatants from cultures of these bacteria in the presence of m-coumaric acid indicated that only those possessing the hcrB gene were able to reduce this hydroxycinnamic acid to 3-HPPA (Fig. 9B). Only strains from the L. plantarum group as well as E. casseliflavus DSM 20680, E. gallinarum DSM 24841, and S. gallolyticus UCN34 strains produced 3-HPPA from m-coumaric acid. However, the strains analyzed from the L. brevis, L. sakei, L. fructivorans, Leuconostoc citreum, E. durans, E. faecalis, E. faecium, and E. hirae species did not reduce the hydroxycinnamic acid present in the culture medium. From these bacterial species, the metabolism of hydroxycinnamates has only been studied in L. brevis (19); L. brevis strains were not able to reduce the hydroxycinnamic acids assayed.
The results obtained suggest that the ability to reduce hydroxycinnamates is not widely extended among lactic acid bacterial strains. Moreover, the ability to reduce these acids is related to the presence of the hcrB gene found in these bacteria. Hydroxycinnamate reductase activity has not been widely studied in lactic acid bacteria. In addition to L. plantarum, the ability to reduce some hydroxycinnamic acids has been described only in Lactobacillus paracollinoides (formerly L. pastorianus var. quinicus) (20, 21), Lactobacillus fermentum (22, 23), Lactobacillus curvatus (24), Lactobacillus reuteri (23), Lactobacillus rossiae (24), Weissella cibaria (24), Clostridium aerotolerans (25), and Clostridium xylanolyticum (25) strains. As the two clostridial species, in addition to hydroxycinnamates, also reduced cinnamic acid and other cinnamates (o-, m-, and p-methoxycinnamic acid, p-methylcinnamic, and 3,4,5-trimethoxycinnamic acid), the presence of an enoate reductase in these strains could be envisaged.
In this work, the description of the L. plantarum heterodimeric NADH-dependent hydroxycinnamate reductase identifies the alternative metabolism to decarboxylation of hydroxycinnamates in lactic acid bacteria. Barthelmebs et al. (3) demonstrated that L. plantarum has three inducible enzymatic activities for the degradation of hydroxycinnamates involved in the stress response induced by phenolic acids, particularly in acidic media, which are the natural habitats of lactic acid bacteria (Fig. 10). PDC decarboxylase production appears as the most efficient and rapid cellular response to convert these acids into less toxic derivatives (3). The physiological significance of hydroxycinnamate metabolism has been poorly investigated, but its degradation is a mechanism to detoxify such compounds. The reduction of hydroxycinnamic acids involves a hydrogen donor and the reoxidation of the reduced cofactor NADH, thus providing a metabolic advantage through NAD+ regeneration. It has been suggested that strictly heterofermentative lactic acid bacteria mainly use hydroxycinnamic acids as external acceptors of electrons and gain additional metabolic energy to counteract the stressful conditions generated by phenolics (24).
FIG 10.

Schematic representation of hydroxycinnamic acid metabolism in L. plantarum WCFS1. For R1 (OH) and R2 (H), the represented compounds are p-coumaric acid (A), vinyl phenol (B), and phloretic acid (C). For R1 (OH) and R2 (OH), the compounds are caffeic acid (A), vinyl catechol (B), and hydrocaffeic acid (C). For R1 (OH) and R2 (OCH3), the compounds are ferulic acid (A), vinylguaiacol (B), and hydroferulic acid (C). For R1 (H) and R2 (OH), only the reduction reaction is carried out and the represented compounds are m-coumaric acid (A) and 3-(3-hydroxyphenyl)propionic acid (C).
So far, only enoate reductases have been described for the reduction of cinnamates/hydroxycinnamates. However, this work identifies the L. plantarum hydroxycinnamate reductase and provides a genetic description of a 2-coumarate reductase (subgroup EC 1.3.1.11) from the oxidoreductases of the class EC 1.3.1 acting on carbon-carbon double bonds.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
L. plantarum WCFS1 used through this study was kindly provided by Michiel Kleerebezem (NIZO Food Research, The Netherlands). This strain is a colony isolate of L. plantarum NCIMB 8826, which was isolated from human saliva. In this study, an additional 25 L. plantarum strains were analyzed. L. plantarum NC8 was kindly provided by L. Axelsson (Norwegian Institute of Food, Fisheries and Aquaculture Research). Seven strains were purchased from the Spanish Type Culture Collection (CECT): L. plantarum CECT 220 (ATCC 8014), CECT 221 (ATCC 14431), CECT 223, CECT 224, CECT 749 (ATCC 10241), CECT 4645 (NCFB 1193), and the type strain L. plantarum subsp. plantarum CECT 748 (ATCC 14917, DSMZ 20174). Six strains were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ): L. plantarum DSM 1055, DSM 2648, DSM 10492, DSM 13273, DSM 20246, and the type strain L. plantarum subsp. argentoratensis DSM 16365. Ten strains were isolated from grape must or wine of different wine-producing areas of Spain over the period from 1998 to 2001: L. plantarum RM28, RM31, RM35, RM38, RM39, RM40, RM41, RM71, RM72, and RM73 (26). In addition, two Lactobacillus paraplantarum and three Lactobacillus pentosus strains purchased from the DSMZ, DSM 10641 (ATCC 10776) and DSM 10667T, and DSM 16366, DSM 20314, and DSM 20199, respectively, were also analyzed. Strains from other lactic acid species were also purchased from the CECT or DSMZ collections, such as Enterococcus casseliflavus (DSM 20680), E. durans (DSM 20633), E. faecalis (DSM 20478), E. faecium (CECT 4102 and DSM 20477), E. gallinarum (DSM 24841), E. hirae (DSM 20160), Lactobacillus brevis (CECT 5354, CECT 216, and CECT 4121), L. fermentum (CECT 4007), L. fructivorans (CECT 4785), L. sakei (DSM 15831), and Leuconostoc citreum (CECT 4025 and CECT 4700). Streptococcus gallolyticus subsp. gallolyticus UCN34 (CIP 110142) was kindly provided by Philipe Glaser (Institut Pasteur, France).
Lactic acid bacteria were routinely grown in MRS broth. When hydroxycinnamate reductase activity was assayed, MRS medium was supplemented with filter-sterilized hydroxycinnamic acids at a 1.5 mM final concentration. Where appropriate, erythromycin was added to the culture medium at 10 μg/ml and lincomycin at 100 μg/ml. The inoculated media were incubated at 30°C in the dark, without shaking, for 7 days. Incubated media with cells and without hydroxycinnamic acid were used as the controls. The phenolic compounds present in the supernatants were extracted using a standard protocol involving two extraction steps with one-third of the reaction mixture volume of ethyl acetate.
Construction of L. plantarum hcr knockout mutants.
To ascertain the participation of hcr genes on hydroxycinnamate reduction, insertion-duplication mutagenesis was employed. Internal fragments from the hcrR (lp_1422), hcrA (lp_1424), hcrB (lp_1425), and hcrC (lp_1426) genes were cloned into the suicide pUCE191 vector (27) by using the primers described in Table 1. When pUCE191 and its derivatives were used as donor DNA, L. plantarum transformants were selected by plating with 100 μg/ml lincomycin and 10 μg/ml erythromycin, and E. coli transformants were selected by plating with ampicillin at 100 μg/ml. Derivative pUCE191 plasmids, constructed in E. coli, were used to transform L. plantarum WCFS1 competent cells by electroporation according to the method of Aukrust and Blom (28). Knockout mutants were selected by plating on MRS plates containing erythromycin and lincomycin. The correct insertion of the donor pUCE191-derivative plasmid into the L. plantarum WCFS1 chromosome was checked by PCR analysis using primers flanking the target region combined with vector-specific primers (Table 1).
RNA isolation, reverse transcription-PCR, and quantitative PCR.
L. plantarum WCFS1 MRS cultures were grown to an optical density at 600 nm (OD600) of 0.8 to 0.9 and then supplemented with p-coumaric acid or other cinnamic acids at a 1.5 mM final concentration. After 10 min of incubation, the cultures were immediately processed for RNA extraction as previously described (29). After DNase I treatment, the DNA-free RNA was retrotranscribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions. The cDNA obtained in the presence of p-coumaric acid was used for RT-PCR experiments. The hcrR (lp_1422), hcrA (lp_1424), hcrB (lp_1425), hcrC (lp_1426), and lp_1427 genes, as well as the hcrR-hcrA, hcrA-hcrB, hcrB-hcrC, hcrC-lp_1427 intergenic regions, were analyzed by PCR. PCR amplifications were performed using 1 μl of cDNA template and 0.5 μM (each) corresponding primers (Table 1) for 35 cycles (94°C for 30 s, 55°C for 45 s, and 72°C for 1 min). As a control, PCR of DNase-treated RNA was performed with the same primers to check for DNA contamination.
Quantitative gene expression analyses were performed in an ABI Prism 7500 Fast real-time PCR system. Specific primer pairs were designed with the Primer Express 3.0 program to amplify gene internal regions and the endogenous control gene (ldh) (30) (Table 1). The SYBR green method was used, and each assay was performed in triplicate using SYBR green real-time PCR master mix (Applied Biosystems). Amplification was initiated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Control PCRs were included to confirm the absence of primer dimer formation and to verify that there was no DNA contamination. All quantitative PCR (qPCR) assays amplified a single product, as determined by melting-curve analysis and by electrophoresis. A standard curve was plotted with cycle threshold (CT) values obtained from the amplification of known quantities of cDNA and used to determine the efficiency (E) as follows: E = 10−1/slope. To measure L. plantarum gene expression, the amplification of the endogenous control gene was performed simultaneously, and its relative expression was compared with that of the target gene.
Relative quantitative (RQ) expression levels were calculated with the Applied Biosystems 7500 Fast system relative quantification software using the L. plantarum ldh gene as the endogenous gene (30) and growth in the absence of a cinnamic acid as the growth condition calibrator. The results were analyzed using the comparative CT method (also named the double delta-delta CT [2ΔΔCT] method).
Expression of hcr genes in E. coli and purification of the recombinant proteins.
The hcrA, hcrB, and hcrAB genes from L. plantarum WCFS1 were PCR-amplified by HS Prime Start DNA polymerase (TaKaRa) by using the primer pairs 1631 and 1632 (hcrA), 889 and 890 (hcrB), and 1631 and 890 (hcrAB). The purified PCR products were inserted into the pURI3-Cter vector following the restriction enzyme- and ligation-free cloning strategy described previously (31). The vectors produce recombinant proteins having six-histidine affinity tags in their C termini. E. coli DH10B cells were transformed; recombinant plasmids were isolated and verified by DNA sequencing and then used to transform E. coli BL21(DE3) cells for expression.
E. coli BL21(DE3) cells were transformed with the recombinant pURI3-Cter-hcrA, pURI3-Cter-hcrB, and pURI3-Cter-hcrAB plasmids. E. coli cells were grown in LB containing ampicillin (100 μg/ml) until they reached an optical density at 600 nm of 0.4 and were induced by adding IPTG (isopropyl-β-d-thiogalactopyranoside; 0.4 mM final concentration). After induction, the cells were grown at 22°C for 20 h and collected by centrifugation. The cells were resuspended in phosphate buffer (50 mM, pH 6.5). To prevent inclusion body formation, E. coli cells were grown at 22°C in medium containing 1 M d-sorbitol, 2.5 mM glycine betaine, and 0.25 mM IPTG (32). Crude extracts were prepared by French press lysis of the cell suspension (three times at 1,100 lb/in2). The insoluble fraction of the lysate was removed by centrifugation at 47,000 × g for 30 min at 4°C, and the supernatant was filtered through a 0.2-μm filter. The supernatants of the HcrA, HcrB, and HcrAB proteins were analyzed for hydroxycinnamate reductase by adding 1.5 mM m-coumaric acid and incubating at 37°C for 16 h.
For purification of the recombinant His6-tagged proteins, the filtered supernatants were applied to a TALON Superflow resin (Clontech) equilibrated with the buffer described previously containing 0.3 M NaCl and 10 mM imidazole to improve the interaction specificity in the affinity chromatography step. The bound enzymes were eluted using 150 mM imidazole in the same buffer. The purity of the enzymes was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in Tris-glycine buffer.
Size exclusion chromatography.
Fractions containing the enzymes eluted from the TALON Superflow resin (Clontech) were pooled and loaded onto a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare) preequilibrated in Tris buffer (20 mM Tris-HCl, pH 8.5, with 0.1 M NaCl) using an ÄKTAprime plus chromatographic system (GE Healthcare). Fractions with the protein of interest identified by SDS-PAGE were pooled, and the protein was concentrated and stored at −80°C until its use. Protein concentrations were determined spectrophotometrically by UV absorption using extinction coefficients estimated with the ExPASy server (https://web.expasy.org/protparam/).
Enzyme activity assay.
Fractions containing the His6-tagged proteins were pooled and analyzed for hydroxycinnamate reductase activity. Eluted purified HcrA, HcrB, and HcrAB proteins (500 μg) were incubated at 37°C for 18 h in the presence of 1.5 mM m-coumaric acid and 15 mM NAD(P)H, 7.5 mM FMN, and 7.5 mM FAD. The phenolic compounds present in the reaction mixture were extracted with ethyl acetate and analyzed by HPLC with diode array detection (DAD), as described previously (5).
PCR detection of hydroxycinnamate reductase activity.
Genes encoding HcrB proteins were amplified by PCR using chromosomal DNA from several lactic acid bacterial strains. The amplifications were performed by using degenerate primers 1655 and 1656. The reactions were performed using 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 1 min, and extension at 72°C for 30 s. The expected size of the amplicon was 1.4 kb.
Protein identification via mass spectrometry.
The protein band was excised manually and then digested automatically using a Proteineer DP protein digestion station (Bruker-Daltonics, Bremen, Germany). The protocol described by Shevchenko et al. (33) was used for trypsin digestion. The digestion was analyzed in an Ultraflex time of flight mass spectrometer (Bruker-Daltonics) equipped with a LIFT-tandem mass spectrometry (MS-MS) device. Spectra were acquired in the positive-ion mode at a 50-Hz laser frequency, and 100 to 1,000 individual spectra were averaged. An automated analysis of mass data was performed using flexAnalysis software (Bruker-Daltonics). Matrix-assisted laser desorption ionization MS and MS-MS data were combined through the BioTools program (Bruker-Daltonics) to search nonredundant protein databases (NCBInr and Swiss-Prot) using Mascot software (Matrix Science, London, UK).
Sequence data analyses.
A homology search with finished and unfinished microbial genome databases was performed with the BLAST algorithm on the National Center for Biotechnology Information server (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Multiple alignments were made using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/) on the EBI site, after the retrieval of sequences from the GenBank and Swiss-Prot databases. Computer promoter predictions were carried out on the internet (http://fruitfly.org/seq_tools/promoter.html), and predicted transcription terminators were analyzed at the ARNold site (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php#Results).
Statistical analysis.
The two-tailed Student's t tests were performed using GraphPad InStat version 3.0 (GraphPad Software, San Diego, CA) to determine the differences between means. The data are representative means from at least three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. M. Mancheño for the exclusion size chromatography and critical reading of the manuscript and M. V. Santamaría and J. M. Barcenilla for their assistance.
This work was supported by grant AGL2014-52911-R (AEI/FEDER, UE) (MINEICO). L. Santamaría is a recipient of an FPI Fellowship from the MINEICO.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01123-18.
REFERENCES
- 1.Shahidi F, Naczk M. 2003. Phenolics in food and nutraceuticals. CRC Press, London, United Kingdom. [Google Scholar]
- 2.Rodríguez H, Curiel JA, Landete JM, de las Rivas B, López de Felipe F, Gómez-Cordovés C, Mancheño JM, Muñoz R. 2009. Food phenolics and lactic acid bacteria. Int J Food Microbiol 132:79–90. doi: 10.1016/j.ijfoodmicro.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Barthelmebs L, Diviès C, Cavin J-F. 2000. Knockout of the p-coumarate decarboxylase gene from Lactobacillus plantarum reveals the existence of two other inducible enzymatic activities involved in phenolic acid metabolism. Appl Environ Microbiol 66:3368–3375. doi: 10.1128/AEM.66.8.3368-3375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavin JF, Barthelmebs L, Guzzo J, van Breeumen J, Samyn R, Travers JF, Diviès C. 1997. Purification and characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum. FEMS Microbiol Lett 147:291–295. doi: 10.1111/j.1574-6968.1997.tb10256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodríguez H, Landete JM, Curiel JA, de las Rivas B, Mancheño JM, Muñoz R. 2008. Characterization of the p-coumaric acid decarboxylase from Lactobacillus plantarum CECT 748T. J Agric Food Chem 56:3068–3072. doi: 10.1021/jf703779s. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2008. Metabolism of food phenolic acids by Lactobacillus plantarum CECT 748T. Food Chem 107:1393–1398. doi: 10.1016/j.foodchem.2007.09.067. [DOI] [Google Scholar]
- 7.Reverón I, de las Rivas B, Muñoz R, López de Felipe F. 2012. Genome-wide transcriptomic responses of a human isolate of Lactobacillus plantarum exposed to p-coumaric acid stress. Mol Nutr Food Res 56:1848–1859. doi: 10.1002/mnfr.201200384. [DOI] [PubMed] [Google Scholar]
- 8.Sun J, Lin Y, Shen X, Jain R, Sun X, Yuan Q, Yan Y. 2016. Aerobic biosynthesis of hydrocinnamic acids in Escherichia coli with a strictly oxygen-sensitive enoate reductase. Metab Eng 35:75–82. doi: 10.1016/j.ymben.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Levy CC. 1964. Metabolism of coumarin by a microorganism: o-coumaric acid as an intermediate between coumarin and melilotic acid. Nature 204:1059–1061. doi: 10.1038/2041059a0. [DOI] [PubMed] [Google Scholar]
- 10.Levy CC, Weinstein GD. 1964. The metabolism of coumarin by a microorganism. II. The reduction of o-coumaric acid to melilotic acid. Biochemistry 3:1944–1947. doi: 10.1021/bi00900a027. [DOI] [PubMed] [Google Scholar]
- 11.Caldeira J, Feicht R, White H, Teixeira M, Moura JJG, Simon H, Moura I. 1996. EPR and Mössbauer spectroscopic studies on enoate reductases. J Biol Chem 271:18743–18748. doi: 10.1074/jbc.271.31.18743. [DOI] [PubMed] [Google Scholar]
- 12.Rohdich F, Wiese A, Feicht R, Simon H, Bacher A. 2001. Enoate reductases of Clostridia. J Biol Chem 276:5779–5787. doi: 10.1074/jbc.M008656200. [DOI] [PubMed] [Google Scholar]
- 13.Bühler M, Simon H. 1982. On the kinetics and mechanism of enoate reductase. Hoppe Seylers Z Physiol Chem 363:609–625. [DOI] [PubMed] [Google Scholar]
- 14.Bougioukou DJ, Stewart JD. 2012. Chapter 27. Reduction of C=C double bonds, p 1111–1163. In Drauz K, Gr̈oger H, May O (ed), Enzyme catalysis in organic synthesis, 3rd ed Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 15.Mordaka PM, Hall SJ, Minton N, Stephens G. 2018. Recombinant expression and characterisation of the oxygen-sensitive 2-enoate reductase from Clostridium sporogenes. Microbiology 164:122–132. doi: 10.1099/mic.0.000568. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hertzberger R, Arents J, Dekker HL, Pridmore RD, Gysler C, Kleerebezem M, Teixeira de Mattos MJ. 2014. H2O2 production in species of the Lactobacillus acidophilus group: a central role for a novel NADH-dependent flavin reductase. Appl Environ Microbiol 80:2229–2239. doi: 10.1128/AEM.04272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valladares RB, Graves C, Wright K, Gardner CL, Lorca GL, González CF. 2015. H2O2 production rate in Lactobacillus jonhsonii is modulated via the interplay of a heterodimeric flavin oxidoreductase with a soluble 28 kd PAS domain containing protein. Front Microbiol 6:716. doi: 10.3389/fmicb.2015.00716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papadimitriou-Olivgeris M, Filippidou S, Kolonitsiou F, Drougka E, Koutsileou K, Fligou F, Dodou V, Sarrou S, Marangos M, Vantarakis A, Anastassiou ED, Petinaki E, Spiliopoulou I. 2016. Pitfalls in the identification of Enterococcus species and the detection of vanA and vanB genes. Lett Appl Microbiol 63:189–195. doi: 10.1111/lam.12610. [DOI] [PubMed] [Google Scholar]
- 19.Curiel JA, Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2010. Ability of Lactobacillus brevis strains to degrade food phenolic acids. Food Chem 120:225–229. doi: 10.1016/j.foodchem.2009.10.012. [DOI] [Google Scholar]
- 20.Ehrmann MA, Vogel RF. 2005. Taxonomic note “Lactobacillus pastorianus” (Van Laer, 1892) a former synonym for Lactobacillus paracollinoides. Syst Appl Microbiol 28:54–56. doi: 10.1016/j.syapm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Whitting GC, Carr JG. 1959. Metabolism of cinnamic acid and hydroxycinnamic acids by Lactobacillus pastorianus var. quinicus. Nature 184:1427–1428. doi: 10.1038/1841427a0. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez-Maldonado AF, Schieber A, Gänzle MG. 2011. Structure-function relationships of the antibacterial activity of phenolic acids and their metabolism by lactic acid bacteria. J Appl Microbiol 111:1176–1184. doi: 10.1111/j.1365-2672.2011.05141.x. [DOI] [PubMed] [Google Scholar]
- 23.Filannino P, Bai Y, di Cagno R, Gobbetti M, Gänzle MG. 2015. Metabolism of phenolic compounds by Lactobacillus spp. during fermentation of cherry juice and broccoli puree. Food Microbiol 46:272–279. doi: 10.1016/j.fm.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Filannino P, Gobbetti M, de Angelis M, di Cagno R. 2014. Hydroxycinnamic acids used as external acceptors of electrons: an energetic advantage for strictly heterofermentative lactic acid bacteria. Appl Environ Microbiol 80:7574–7582. doi: 10.1128/AEM.02413-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chamkha M, Garcia JL, Labat M. 2001. Metabolism of cinnamic acids by some Clostridiales and emmendation of the descriptions of Clostridium aerotolerans, Clostridium celerecrescens and Clostridium xylanolyticum. Int J Syst Evol Microbiol 51:2105–2111. doi: 10.1099/00207713-51-6-2105. [DOI] [PubMed] [Google Scholar]
- 26.Moreno-Arribas MV, Polo MC, Jorganes F, Muñoz R. 2003. Screening of biogenic amine production by lactic acid bacteria isolated from grape must and wine. Int J Food Microbiol 84:117–123. doi: 10.1016/S0168-1605(02)00391-4. [DOI] [PubMed] [Google Scholar]
- 27.Arrecubieta C, García E, López R. 1995. Sequence and transcriptional analysis of a DNA region involved in the production of capsular polysaccharide in Streptococcus pneumoniae type 3. Gene 167:1–7. doi: 10.1016/0378-1119(95)00657-5. [DOI] [PubMed] [Google Scholar]
- 28.Aukrust T, Blom H. 1992. Transformation of Lactobacillus strains used in meat and vegetable fermentations. Food Res Int 25:253–261. doi: 10.1016/0963-9969(92)90121-K. [DOI] [Google Scholar]
- 29.Saulnier DM, Molenaar D, de Vos WM, Gibson GR, Kolida S. 2007. Identification of prebiotic fructooligosaccharide metabolism in Lactobacillus plantarum WCFS1 through microarrays. Appl Environ Microbiol 73:1753–1765. doi: 10.1128/AEM.01151-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiocco D, Crisetti E, Capozzi V, Spano G. 2008. Validation of an internal control gene to apply reverse transcription quantitative PCR to study heat, cold and ethanol stresses in Lactobacillus plantarum. World J Microbiol Biotechnol 24:899–902. doi: 10.1007/s11274-007-9556-7. [DOI] [Google Scholar]
- 31.Curiel JA, de las Rivas B, Mancheño JM, Muñoz R. 2011. The pURI family of expression vectors: a versatile set of ligation independent cloning plasmids for producing recombinant His-fusion proteins. Protein Expr Purif 76:44–53. doi: 10.1016/j.pep.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Ackerley DF, González CF, Park CH, Blake R, Keyhan M, Martin A. 2004. Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl Environ Microbiol 70:873–882. doi: 10.1128/AEM.70.2.873-882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.