

Secretory production of recombinant proteins is the most important application of the methylotrophic yeast Pichia pastoris, a species that permits mass production of heterologous proteins. To date, the genetic engineering of P. pastoris has relied largely on integrative vectors due to the lack of user-friendly tools. Autonomously replicating Pichia plasmids are expected to facilitate genetic manipulation; however, the existing systems, which use autonomously replicating sequences (ARSs) such as the P. pastoris-specific ARS (PARS1), are known to be inherently unstable for plasmid replication and distribution. Recently, the centromeric DNA sequences of P. pastoris were identified in back-to-back studies published by several groups; therefore, a new episomal plasmid vector with centromere DNA as a tool for genetic manipulation of P. pastoris is ready to be developed.
KEYWORDS: Pichia pastoris, centromere, autonomously replicating plasmid, inverted repeat, plasmid retention
ABSTRACT
The methylotrophic yeast Pichia pastoris is widely used to produce recombinant proteins, taking advantage of this species' high-density cell growth and strong ability to secrete proteins. Circular plasmids containing the P. pastoris-specific autonomously replicating sequence (PARS1) permit transformation of P. pastoris with higher efficiency than obtained following chromosomal integration by linearized DNA. Unfortunately, however, existing autonomously replicating plasmids are known to be inherently unstable. In this study, we used transcriptome sequencing (RNA-seq) data and genome sequence information to independently identify, on each of the four chromosomes, centromeric DNA sequences consisting of long inverted repeat sequences. By examining the chromosome 2 centromeric DNA sequence (Cen2) in detail, we demonstrate that an ∼111-bp region located at one end of the putative centromeric sequence had autonomous replication activity. In addition, the full-length Cen2 sequence, which contains two long inverted repeat sequences and a nonrepetitive central core region, is needed for the accurate replication and distribution of plasmids in P. pastoris. Thus, we constructed a new, stable, autonomously replicating plasmid vector that harbors the entire Cen2 sequence; this episome facilitates genetic manipulation in P. pastoris, providing high transformation efficiency and plasmid stability.
IMPORTANCE Secretory production of recombinant proteins is the most important application of the methylotrophic yeast Pichia pastoris, a species that permits mass production of heterologous proteins. To date, the genetic engineering of P. pastoris has relied largely on integrative vectors due to the lack of user-friendly tools. Autonomously replicating Pichia plasmids are expected to facilitate genetic manipulation; however, the existing systems, which use autonomously replicating sequences (ARSs) such as the P. pastoris-specific ARS (PARS1), are known to be inherently unstable for plasmid replication and distribution. Recently, the centromeric DNA sequences of P. pastoris were identified in back-to-back studies published by several groups; therefore, a new episomal plasmid vector with centromere DNA as a tool for genetic manipulation of P. pastoris is ready to be developed.
INTRODUCTION
The methylotrophic yeast Pichia pastoris (Komagataella spp.) is one of the most widely used host organisms for recombinant protein production; this species exhibits excellent capacity for the secretion of heterologous proteins (1–3). As a eukaryotic organism, P. pastoris is capable of producing many human- and mammalian-derived proteins with carbohydrate modifications (3–5). In addition, this species has several other advantages, such as rapid, high-density cell growth (permitting a doubling time of approximately 2 h in yeast extract-peptone-glucose [YPD] medium) (6), the achievement of an optical density (OD) of ∼500 at 600 nm in a jar fermentor (3), and potent, inducible expression (under the control of methanol-inducible alcohol oxidase 1 [AOX1] promoter) (3). Thus, this organism is suited for production of proteins as well as for studies of protein expression machinery such as protein folding and secretion (7–12). A number of enzymes and therapeutic proteins have been produced in P. pastoris (13–15). Several of these proteins have been commercially available for many years, and some have been approved by the U.S. Food and Drug Administration (FDA) for clinical use (16–19).
To generate recombinant strains for the expression of heterologous proteins in P. pastoris, genomic integration vector systems (not circular plasmids) have been commonly utilized (2, 3, 21, 22). Although genomic integration systems are very profitable to generate a strain that does not require the use of selective medium during industrial large-scale processes, they are comparatively burdensome, requiring linearization of the plasmid DNA and screening of the resulting transformants for those in which the donor DNA has correctly integrated into the target chromosomal locus. In this context, the linearized DNA generally does not transform the cells as efficiently as circular DNA and is inserted into random chromosomal locations in a certain proportion of the transformants (23).
Autonomously replicating sequences (ARSs), which serve as the origins of DNA replication during mitosis and are often utilized to replicate the plasmids in host cells, were first identified in the genome of budding yeast, Saccharomyces cerevisiae (24, 25). A P. pastoris-specific ARS, designated PARS1, was identified over 30 years ago (26). Similar to the S. cerevisiae-specific ARS, PARS1 enabled the high-efficiency transformation of P. pastoris by circular plasmids. Recently, a 452-bp ARS element (designated panARS) was identified in Kluyveromyces lactis and shown to facilitate transformation in a wide range of yeast species, including P. pastoris (27). More recently, a mitochondrial DNA (mtDNA) fragment was discovered to function as a novel ARS in P. pastoris (28). However, plasmids bearing these ARS sequences are unstable or poorly stable for replication and distribution in P. pastoris, a species for which centromeric DNA sequences remained (until recently) unknown (29–33).
The centromeres of the budding yeast S. cerevisiae have been extensively characterized, and a short 125-bp consensus region of the centromeric DNA sequences is known to be sufficient for full function during mitosis and meiosis in this yeast (31, 34–36). Incorporation of the centromere sequences and the specific ARSs confers replicative stability to the plasmids and the yeast artificial chromosomes in S. cerevisiae (37–39). Therefore, it would also be of value to identify centromeric sequences for P. pastoris. The genome sequences of this species have been determined in the following three P. pastoris strains: P. pastoris (Komagataella phaffii) strain GS115 (40), P. pastoris (Komagataella pastoris) strain CBS704 (DSMZ 70382) (41), and P. pastoris (K. phaffii) strain CBS7435 (NRRL Y-11430) (42). However, given that centromere sequences can be quite variable among yeast species and are known to diverge rapidly during evolution (43, 44), approaches based on homology searches have not been successful in identifying putative centromeres within P. pastoris genome sequences.
Most recently, Sturmberger et al. published a refined reference genome for the CBS7435 strain and also proposed candidate P. pastoris centromeres (45). Coughlan et al. structurally defined the centromeres of P. pastoris by transcriptome sequencing (RNA-seq) and chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) using binding by the histone H3-like centromere protein Cse4 (46). Through these studies, these researchers found that each of the putative centromeres on the four P. pastoris chromosomes contained inverted repeat structures. However, no laboratory has (to our knowledge) constructed plasmids containing the P. pastoris-specific centromeres, meaning that these centromeres remain unavailable for functional application (e.g., in cloning) or for more detailed characterization.
In the initial phase of the present study, we performed the RNA-seq transcriptome mapping of P. pastoris CBS7435 using next-generation sequencing (NGS), permitting us to independently identify putative centromeric DNA sequences, which are transcriptionally silent. Close examination of the chromosome 2 (Chr 2) centromere DNA sequences (Cen2) permitted the successful identification of a very short autonomously replicating sequence. Moreover, fluorescent reporter gene expression experiments using flow cytometry (FCM) revealed that the entire Cen2 region was needed to achieve high-stability plasmid retention. By using this full-length P. pastoris centromeric DNA sequence, we created a new, stable, autonomously replicating plasmid vector for use in this yeast.
RESULTS AND DISCUSSION
RNA-seq transcriptome analysis for identification of putative centromeric DNA sequences.
To develop a useful tool for more easily making recombinant strains, we aimed to create a new plasmid vector that would permit highly stable, autonomous replication in P. pastoris. This process required identifying the putative centromeric DNA sequences and ARSs present in the P. pastoris genome. We therefore performed RNA-seq experiments for transcriptome analyses of P. pastoris CBS7435 during culturing in buffered glycerol complex (BMGY) and buffered methanol complex (BMMY) media. In the course of mapping the obtained RNA-seq reads to the P. pastoris reference genome sequence (42), we detected the presence, on each of the four chromosomes (Chr 1 to 4), of one large nontranscribed region; these domains were present under both of the tested culture conditions (Fig. 1). This result provided independent confirmation of a similar observation in two earlier reports (45, 46). We found that these nontranscribed regions ranged from 8 to 11 kb in length, each containing a nearly complete inverted repeat sequence (2 to 2.7 kb in length) flanking a nonrepetitive central core region (0.9 to 1.4 kb) (Fig. 2A; see also Table S1 in the supplemental material). Based on these observations, we predicted that these distinctive regions corresponded to centromeric DNA sequences (47). Two other groups had very recently conducted identification of centromeric regions and reported earlier; our results and interpretations were well consistent with these two earlier reports (45, 46).
FIG 1.

Putative location of P. pastoris centromeres indicated by RNA-seq reads mapped to the reference genome sequence. The putative centromere regions are large nontranscribed regions. Each dark triangle corresponds to the location of the putative centromere on each chromosome.
FIG 2.

The transformation efficiencies of the plasmids constructed with putative centromere regions and various lengths of the Cen2 region. (A) Schematic diagram of putative centromere and truncated versions used for identification of autonomously replicating sequences. Cen2, full-length centromere region of chromosome 2; CC2, central core region of Cen2; LOR2, left outer repeat region of Cen2; ROR2, right outer repeat region of Cen2; Cen1, full-length centromere region of chromosome 1; Cen3, full-length centromere region of chromosome 3; Cen4, full-length centromere region of chromosome 4; green, inverted repeat region; magenta, central core region. (B) Representative images of colonies formed by yeast transformants growing on selective solid medium. P. pastoris strain CBS7435 was transformed with pUC19-Cen2 (left) or pUC19-LOR2(1–111) (right) and spread on YPD solid medium containing 100 μg/ml Zeocin. (C) The number of CFU per microgram of DNA obtained by transformation of yeast cells. The standard deviation is calculated according to the number of CFU in triplicates (n = 3).
Investigation of plasmid replication ability with centromeric DNA sequences (Cen1 to Cen4).
To facilitate further investigation and characterization of these putative centromeric DNA sequences of chromosome 1 to 4 (Cen1 to Cen4), we constructed the plasmids, each harboring a Zeocin resistance-encoding gene along with full-length Cen1 to Cen4 (Fig. 2A and Table 1). Each plasmid was used to transform CBS7435, and transformation efficiencies were determined by normalizing the numbers of Zeor colonies (numbers of CFU per microgram of DNA). Interestingly, the largest plasmid, which contained the full-length Cen2 (6,655 bp), yielded a transformation efficiency of 1.4 × 104 ± 0.7 × 104 CFU/μg (Fig. 2C). Similarly, plasmids carrying either the full-length Cen1 or Cen3 yielded transformation efficiencies of 1.2 × 103 ± 0.3 × 103 to 9.5 × 103 ± 0.6 × 103 CFU/μg. In contrast, for unknown reasons, the plasmid containing Cen4 yielded few transformants (Fig. 2C). However, a previous report in which the mapping data for K. phaffii replication origins (48) were reanalyzed suggested that all four centromeres were located at the early replicating regions and more likely contained ARSs (46). Among the four centromere sequences, Cen2, which exhibited the highest transformation efficiency, was the longest (6,655 bp) and was located close to the center of the 2.4-Mbp chromosome 2 (Fig. 1 and Table S1). To further investigate the origins of replication, we focused on the Cen2 centromeric DNA sequence.
TABLE 1.
Yeast strains and plasmids used in this study
| Strain or plasmid | Specific features | Source |
|---|---|---|
| Strains | ||
| CBS7435 | Pichia pastoris wild-type strain (NRRL Y-11430 or ATCC 76273) | ATCC |
| T38473-EGFP | CBS7435 harboring a reporter cassette (GFP under the control of GAP promoter) chromosomally integrated at the T38473 locus) | This study |
| P01 | CBS7435 harboring Zeor marker chromosomally integrated at the PpPaox1 locus | This study |
| BY5242 | Hansenula polymorpha 8V (leu2Δ) strain (NRRL Y-5445 or CBS4732) | NBRP |
| Plasmids | ||
| pUC19 | Cloning vector | TaKaRa Bio |
| pUC19_HpPgap_HpPgap_HpTmox_Zeo | HpPgap-HpPgap-HpTmox-Zeo in pUC19 | This study |
| pUC19-Cen2 | Cen2 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2CC2 | LOR2-CC2 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2 | LOR2 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-CC2 | CC2 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(1–2409) | LOR2(1–2409) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(1–1800) | LOR2(1–1800) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(1–1200) | LOR2(1–1200) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(1–600) | LOR2(1–600) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(1–111) | LOR2(1–111) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(84–213) | LOR2(84–213) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(191–291) | LOR2(191–291) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-LOR2(84–2699) | LOR2(84–2699) in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-Cen1 | Cen1 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-Cen3 | Cen3 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-Cen4 | Cen4 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-PpPgap-EGFP_Pp-PpTgap1 | PpPgap-EGFP_Pp-PpTgap1 in pUC19_HpPgap_HpPgap_HpTmox_Zeo | This study |
| pUC19-Cen2-EGFP | PpPgap-EGFP_Pp-PpTgap1 in pUC19-Cen2 | This study |
| pUC19-LOR2CC2-EGFP | PpPgap-EGFP_Pp-PpTgap1 in pUC19-LOR2CC2 | This study |
| pUC19-LOR2-EGFP | PpPgap-EGFP_Pp-PpTgap1 in pUC19-LOR2 | This study |
| pUC19-LOR2(1–111)-EGFP | PpPgap-EGFP_Pp-PpTgap1 in pUC19-LOR2(1–111) | This study |
| pUC19-PARS1-EGFP | PpPgap-EGFP_Pp-PpTgap1, PARS1 and Zeor marker in pUC19 | This study |
| pUC19-T38473-EGFP | PpPgap-EGFP_Pp-PpTgap1-T38473 and G418 resistance marker in pUC19 | This study |
| pUC19_PpPaox1_PpPgap-PpTaox1 | PpPaox1_PpPgap-PpTaox1 and Zeor marker in pUC19 | This study |
| pUC19-Cen2-EGFP-ACT1 | PpPgap-EGFP_Pp-ACT1-PpTgap1 in pUC19-Cen2 | This study |
Characterization of the necessary region for plasmid replication and distribution in Cen2 sequence.
Figure 2C summarizes the transformation efficiencies of the plasmids constructed with various lengths of the Cen2 region. For the purposes of describing this analysis, we have abbreviated Cen2 as LOR2-CC2-ROR2, where LOR2 and ROR2 correspond to (respectively) the left and right outer repeat (OR) regions of Cen2, and CC2 corresponds to the central core (CC) region of Cen2. Plasmids carrying either LOR2 alone (2,699 bp) or LOR2 plus CC2 (LOR2-CC2; 3,956 bp) yielded transformation efficiencies of 1.7 × 104 ± 0.1 × 104 to 1.6 × 104 ± 0.3 × 104 CFU/μg. In contrast, a plasmid containing CC2 alone (1,257 bp) yielded few transformants (Fig. 2C). Since the sequence homology between LOR2 and ROR2 was 99.9% (Table S2), we did not analyze the plasmid carrying ROR2 alone (2,699 bp).
Among the plasmids harboring various truncated versions of LOR2, truncations containing an outer end of Cen2, i.e., the CC2 distal edge of LOR2, extending anywhere from bp 1 to 111 of LOR2 [LOR2(1–111)] through bp 1 to 2409 of LOR2 [LOR2(1–2409)] yielded transformants (Fig. 2C). The LOR2 truncation constructs carrying lengths of LOR2 ranging from 600 to 2,409 bp showed higher transformation efficiencies (1.9 × 104 ± 0.3 × 104 to 2.6 × 104 ± 0.3 × 104 CFU/μg) than the plasmid carrying the 111-bp fragment (3.1 × 103 ± 1.2 × 103 CFU/μg). In contrast, plasmids containing LOR2 truncated from the outer (CC2 distal) end [LOR2(84–213), LOR2(191–291), and LOR2(84–2699)] yielded transformants at very low efficiencies (67 ± 64 to 2.1 × 102 ± 0.2 × 102 CFU/μg) (Fig. 2C). These results demonstrated that sequences at the outer (CC2 distal) end of LOR2 were required for obtaining high transformation efficiencies.
Additionally, we observed that the sizes of the emerging colonies (growing on the Zeocin-containing plates after transformation) varied depending on the length of the Cen2 domain. Whereas the plasmid containing full-length Cen2 yielded normal-sized colonies, the LOR2(1–111) plasmid yielded only very small colonies (Fig. 2B). Since the transformation efficiency of the plasmid containing full-length Cen2 was higher than that of the plasmid containing LOR2(1–111), this result implied that full-length Cen2 provided more complete centromere activity for P. pastoris, presumably permitting more-stable plasmid replication and distribution. Together with the above observations, these data again indicated that a centromere-associated ARS is located within the outer (CC2 distal) end of LOR2 (or ROR2) within the Cen2 region, such that the LOR2 region of bp 1 to 111 is sufficient for plasmid replication.
Evaluation of autonomous replication activity and plasmid retention of the newly constructed vectors with Cen2 sequences.
To assess the autonomous replication activity of our constructs in P. pastoris, we analyzed the proportion of cells carrying the plasmids. Previous work has shown that the proportion of cells expressing a green fluorescent protein-encoding reporter gene (GFP) corresponds to the plasmid retention rate (49). We therefore used FCM to detect single-cell fluorescence levels and determined the proportions of strongly fluorescent and weakly fluorescent cells in populations of cells transformed with plasmids carrying the GFP reporter gene expression cassette in combination with various lengths of the Cen2 centromere sequences.
For comparison, we used a control plasmid harboring the conventional PARS1 (164 bp), a sequence that is also derived from chromosome 2 (nucleotides 1980709 to 1980872) (26). Enhanced GFP (EGFP) expression plasmids containing Cen2, LOR2-CC2, LOR2, LOR2(1–111), or PARS1 sequence were constructed and used to transform the CBS7435 strain (Table 1 and Fig. 3A). As an additional control, we included a strain carrying a chromosomally integrated EGFP expression cassette. The transformants were cultivated in BMGY medium in the presence or absence of antibiotic (Zeocin) selection (the CBS7435 control and the integrant control were grown only in the absence of selection) for 24 h, and the proportions of fluorescent cells were measured with a flow cytometer.
FIG 3.

Evaluation of autonomously replicating activity and plasmid retention in P. pastoris. (A) EGFP expression plasmids containing Cen2 (pUC19-Cen2-EGFP), LOR2-CC2 (pUC19-LOR2CC2-EGFP), LOR2 (pUC19-LOR2-EGFP), LOR2(1–111) [pUC19-LOR2(1–111)-EGFP], or PARS1 (pUC19-PARS1-EGFP). (B) CBS7435 harboring pUC19-Cen2-EGFP (Cen2), pUC19-LOR2CC2-EGFP (LOR2-CC2), pUC19-LOR2-EGFP (LOR2), pUC19-LOR2(1–111)-EGFP [LOR2(1–111)], and pUC19-PARS1-EGFP (PARS1) was grown in BMGY medium with or without 200 μg/ml Zeocin for 24 h. The strains CBS7435 (control) and T38473-EGFP (genomic integration) were grown in BMGY medium without Zeocin for 24 h. The GFP expression level of each cell was measured by FCM, and the histogram data from 10,000 cells per strain are shown. For each strain, three different transformants were analyzed, and one typical histogram is displayed.
In the case of the strain harboring the integrated EGFP expression cassette, most of the cells exhibited a narrow range of GFP fluorescence (Fig. 3B, genomic integration). On the other hand, when the transformant harboring the PARS1 plasmid was grown in the presence of Zeocin, 55% of the cells displayed GFP fluorescence, with fluorescent signal exhibiting a broad distribution (Fig. 3B, PARS1). This result indicated that while PARS1 provides autonomous replication activity, plasmid replication or partitioning was weak, such that almost half (45%) of the cells lost the plasmid during culturing. We compared this pattern to that obtained with plasmids carrying full-length or truncated Cen2 sequences. When the plasmids with LOR2 or LOR2-CC2 were used, 43 to 45% of the cells displayed GFP fluorescence, a proportion similar to that seen with the PARS1 plasmid (Fig. 3B, LOR2 and LOR2-CC2). On the other hand, only 27% of the cells harboring the LOR2(1–111)-containing plasmid exhibited GFP fluorescence [Fig. 3B, LOR2(1–111)]. Since the LOR2(1–111) construct yielded only very small colonies (Fig. 2B), these results showed that the 111-bp Cen2 sequence permits replication but suffers from severe instability due to the functional centromere sequence.
In contrast, when the full-length Cen2-containing plasmid was used, 84% of the cells displayed GFP fluorescence; notably, the shape of the GFP histogram consisted of a single peak with a narrow distribution of fluorescence (Fig. 3B, Cen2). When the cells transformed with the circular plasmids were grown under nonselective conditions (i.e., without Zeocin), almost no GFP fluorescence was observed (Fig. 3B). This observation implied that the introduced circular plasmids existed as autonomous (nonintegrated) DNA, as evidenced by the cells' ability to be cured of the plasmids during cultivation in the absence of selective pressure. Furthermore, the cells with the Cen2 plasmid showed a single peak with narrow fluorescence distribution until 100 h of cultivation (Fig. S1).
Together, these results indicated that the centromere sequence containing an ARS region needs to build the expression vector to direct the replication and segregation of the episomal DNA. A comparison of Cen2 with PARS1 showed that there is no sequence similarity (Table S2), and this difference may affect plasmid replication and plasmid distribution. Underscoring anew the results, the entire Cen2 region (containing both inverted repeat sequences) is required for the accurate replication and distribution of a plasmid during cell division, thereby permitting the stable, autonomous replication of an episome in P. pastoris.
To further evaluate the stability of plasmid retention in the cells during repeated subculturing, we performed FCM analysis of the cells harboring the full-length Cen2 plasmid for GFP expression, examining the populations of the fluorescent cells. The transformant was cultivated and transferred into fresh selective medium at 24-h intervals. As shown in Fig. 4, the cells with the Cen2 plasmid showed a single peak with narrow fluorescence distribution until the third cycle of subculturing (corresponding to 22 generations total). This pattern indicated that the Cen2 plasmid was stably maintained in P. pastoris cells for at least 20 generations.
FIG 4.
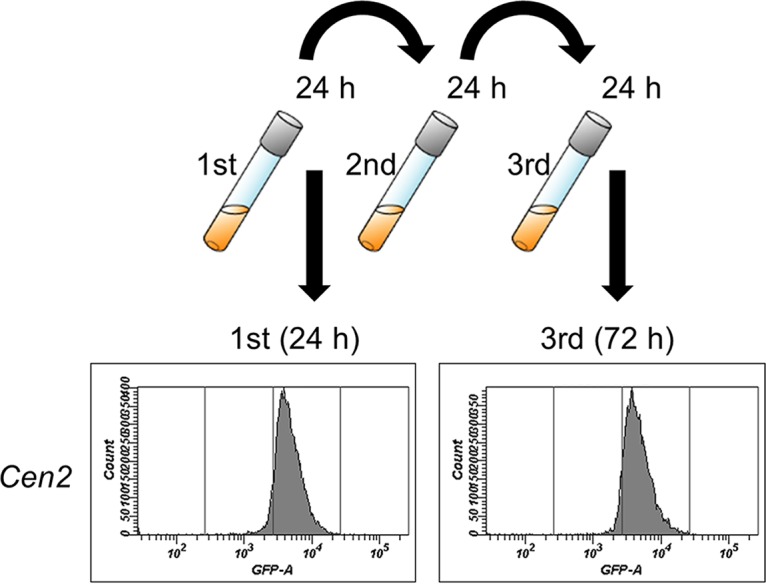
Evaluation of autonomously replicating activity and plasmid retention through repeated subculture in P. pastoris. CBS7435 harboring pUC19-Cen2-EGFP (Cen2) was grown for 24 h (starting from an initial OD660 of 0.1) in BMGY medium supplemented with 200 μg/ml Zeocin. The cells then were subcultured (initial OD660 of 0.1) to fresh selective medium every 24 h. GFP expression in individual cells was measured by flow cytometry (FCM) at 24-h intervals, and the histogram data from a total of 10,000 cells at each time point are shown. Three different transformants were analyzed, and one typical histogram is displayed.
Examination of cell growth and average plasmid copy number and plasmid extraction.
The cell growth was measured by monitoring the optical density of cultures at 660 nm (OD660) every 30 min over a period of 48 h with shaking at 30°C. The growth curve of yeast cells harboring the Cen2 plasmid was similar to that of the strain integrating the Zeocin resistance-encoding selective marker on the chromosome (P01 strain) (Fig. 5A and Fig. S2). In contrast, the growth of cells harboring the LOR2(1–111)-containing plasmid was slow compared with that of the strain P01 (Fig. 5A and Fig. S2). These results indicated that the Cen2 sequence has no negative effects on cell growth. In addition, there was no difference in maximum achievable ODs between the strain harboring the Cen2 plasmid and other comparative strains (Fig. 5B).
FIG 5.
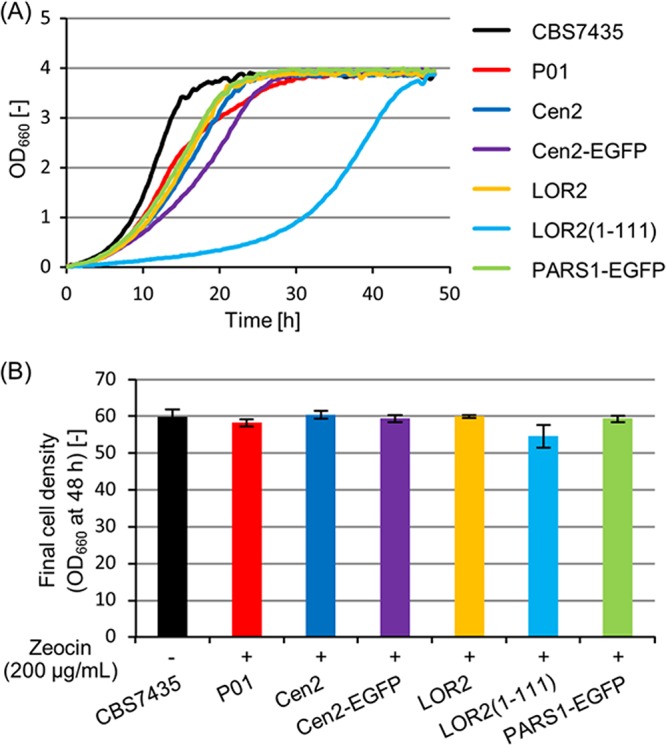
Measurement of cell growth. The strain CBS7435 was grown in BMGY medium without Zeocin for 48 h. The strains P01 and CBS7435 harboring pUC19-Cen2 (Cen2), pUC19-Cen2-EGFP (Cen2-EGFP), pUC19-LOR2 (LOR2), pUC19-LOR2(1–111) [LOR2(1–111)], and pUC19-PARS1-EGFP (PARS1-EGFP) were grown in BMGY medium with 200 μg/ml Zeocin for 48 h. (A) The growth curve of yeast cells. Cell growth was automatically monitored in L-shaped test tubes by using a bio-photorecorder. (B) The final cell density (OD660) after 48 h of cultivation. OD660 was measured by a UV-VIS spectrophotometer. Error bars represent the standard deviations from three independent transformants (n = 3).
In addition, average plasmid copy number was measured by quantitative PCR (qPCR). The EGFP expression plasmid containing Cen2 or PARS1 sequence was used to transform the CBS7435 strain (Table 1 and Fig. 3A). As a control, we included a strain chromosomally integrating a single-copy EGFP expression cassette on the genome. The transformants were cultivated in BMGY medium with antibiotic (Zeocin) selection (the integrant control was grown without selection). In the case of the strain integrating the EGFP expression cassette, approximately a single copy of the EGFP gene was detected per cell (Fig. 6). When the Cen2 plasmid was used, almost a single copy of the EGFP gene per cell was also detected (Fig. 6). On the other hand, the PARS1 plasmid strain had a slightly lower average copy number than that of the Cen2 strain (P < 0.05, t test). This result may be attributed to the loss of plasmid in the cells in concurrence with the observation on FCM analysis. These results indicated that the full-length Cen2 sequence provided a stable single-copy plasmid.
FIG 6.
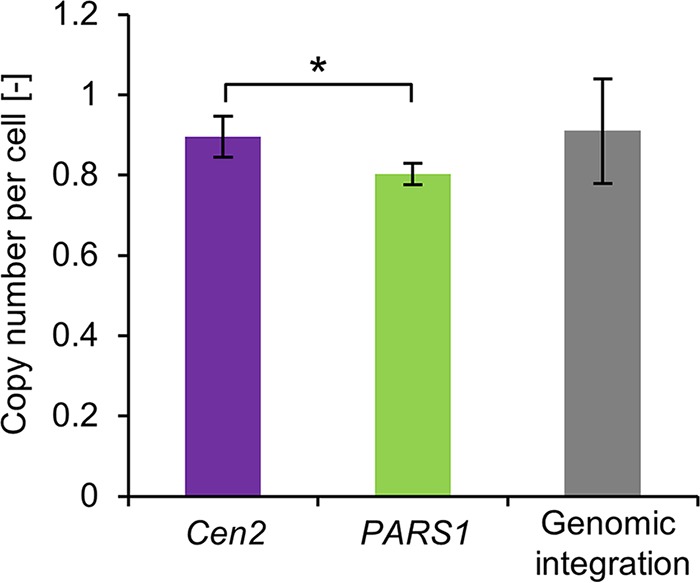
Average plasmid copy number per cell. CBS7435 harboring pUC19-Cen2-EGFP (Cen2) and pUC19-PARS1-EGFP (PARS1) was grown in BMGY medium with 200 μg/ml Zeocin for 24 h. The strain T38473-EGFP (genomic integration) was grown in BMGY medium without Zeocin for 24 h. Copy number was determined by qPCR. Error bars represent the standard deviations from three independent transformants (n = 3). Statistical significance was assessed by a t test (*, P < 0.05).
Finally, we assessed whether it was possible to reisolate the Cen2 plasmids from the transformed yeast cells. Notably, for screening applications, it would be advantageous to recover episomal DNA from the transformed cells, for instance, to permit sequencing of inserts or repetition of transformation. Indeed, this critical attribute was previously demonstrated for the PARS1-containing plasmid (26). In the present work, given the low copy number expected for a centromere-containing episome, the Cen2 plasmid DNA was recovered from the yeast cells and then amplified by transformation into Escherichia coli. After extraction and purification of the Cen2 plasmid DNA from transformed E. coli cells, restriction enzyme digestion and DNA sequencing of the plasmid were performed to confirm the plasmid's identity. Critically, the structure of the reisolated plasmid was consistent with that of the original plasmid; no deletions and mutations were observed (data not shown). Based on these results, we concluded that the Cen2 plasmid, harboring the full-length Cen2 sequence consisting of a nearly complete inverted repeat sequence flanking a nonrepetitive central core region (LOR2-CC2-ROR2), is available as a new autonomously replicating plasmid vector, exhibiting the centromere/autonomously replicating (CEN/ARS) activity in P. pastoris.
Conclusions.
In this study, we used RNA-seq data and genome sequence information to independently identify P. pastoris centromeric DNA sequences. These sequences, present on each of the four chromosomes, consist of a long, inverted repeat flanking a nonrepetitive central core (abbreviated as LOR2-CC2-ROR2). By focusing on the chromosome 2 centromere, we demonstrated that the ARS is located on the outer (CC2 distal) end of LOR2 (or ROR2) of the Cen2 region. We further demonstrated that the full-length Cen2 is needed for the stable replication and distribution of the plasmid in P. pastoris. Thus, we successfully constructed a new, stable, autonomously replicating P. pastoris plasmid vector containing the full-length Cen2 sequence. This new plasmid vector exhibits high stability for plasmid retention, facilitating genetic manipulation in this yeast. This vector is therefore expected to serve as a powerful and useful tool for research application in Pichia. Furthermore, this new plasmid vector has the potential to expedite cloning and high-throughput screening in P. pastoris, accelerating metabolic and genome engineering and high-level protein production in this organism.
MATERIALS AND METHODS
Yeast strains and media.
P. pastoris strain CBS7435 (NRRL Y-11430 or ATCC 76273) was obtained from the ATCC (American Type Culture Collection, Manassas, VA, USA). Hansenula polymorpha 8V (leu2Δ) strain BY5242 (NRRL Y-5445 or CBS4732) was obtained from the National BioResource Project (NBRP). YPD medium contained 10 g/liter yeast extract (Nacalai Tesque, Kyoto, Japan), 20 g/liter Bacto peptone (BD Biosciences, San Jose, CA, USA), and 20 g/liter glucose. YPG medium contained 10 g/liter Bacto yeast extract (BD Biosciences), 20 g/liter hipolypepton (Nihon Pharmaceutical, Tokyo, Japan), and 20 g/liter glycerol. BMGY medium (buffered glycerol complex medium) contained 10 g/liter Bacto yeast extract, 20 g/liter hipolypepton, 13.4 g/liter yeast nitrogen base without amino acids (YNB) (BD Biosciences), 0.4 mg/liter biotin (Nacalai Tesque), 100 mM potassium phosphate buffer (pH 6.0), and 20 g/liter glycerol. BMMY medium (buffered methanol complex medium) contained 10 g/liter Bacto yeast extract, 20 g/liter hipolypepton, 13.4 g/liter YNB, 0.4 mg/liter biotin, 100 mM potassium phosphate buffer (pH 6.0), and 20 g/liter methanol. For solid medium, agar was added at 20 g/liter.
Strain cultivation and RNA extraction for RNA-seq.
P. pastoris strain CBS7435 was inoculated into 30 ml of YPG medium and cultured with shaking at 30°C for 24 h to obtain a preculture. The obtained preculture (1 ml) was inoculated into 100 ml of BMGY or BMMY medium and cultured with shaking at 30°C until the optical density at 600 nm (OD600) reached 2 to 3. The culture was centrifuged at 4,000 × g and 4°C for 10 min, the supernatant was decanted, and the harvested cells were immediately flash-frozen in liquid nitrogen. The cells were fragmented in liquid nitrogen with a muddler and vortexed in 5 ml of Isogen II (Nippon Gene, Tokyo, Japan). RNase-free water (2 ml) was added; the resulting suspension was vortexed for 15 s and then allowed to stand at 25°C for 10 min. The suspension was centrifuged at 16,000 × g and 25°C for 20 min. An aliquot (25 μl) of p-bromoanisole (Wako Pure Chemical Industries, Osaka, Japan) was added to the resulting supernatant, and the mixture was vortexed for 15 s and allowed to stand at 25°C for 5 min. The resulting solution was centrifuged at 16,000 × g and 25°C for 10 min. An aliquot (5 ml) of isopropanol was added to the resulting supernatant, and the mixture was mixed and allowed to stand at 25°C for 10 min. The mixture then was centrifuged at 16,000 × g and 25°C for 10 min. The resulting pellet was washed with 2 ml of ethanol and centrifuged at 16,000 × g and 25°C for 3 min, and the supernatant was decanted. The resulting pellet was air dried before being dissolved in 200 μl of RNase-free water; the resulting solution was purified using an RNeasy minikit (Qiagen, Hilden, Germany), and the product was designated the purified total RNA sample.
Sequencing and transcriptome mapping.
Sequence libraries were constructed using a TruSeq Stranded mRNA LT sample prep kit (Illumina, San Diego, CA, USA) and sequenced on a HiSeq 2000 instrument (Illumina). Mapping of the reads to the reference genome sequence of CBS7435 was performed using CLC Genomics Workbench software (Qiagen).
Plasmid vector construction.
All plasmids used in this study are listed in Table 1. All primers used for plasmid construction are listed in Table 2.
TABLE 2.
Oligonucleotide primers used in this study

The P. pastoris backbone plasmids used in this study were constructed by analogy to the plasmid vector of the methylotrophic yeast Hansenula polymorpha (Ogataea polymorpha) protein expression system (50). The plasmid containing the Zeocin resistance-encoding selective marker was constructed as follows. A DNA fragment containing the H. polymorpha MOX terminator (HpTmox) was PCR amplified from BY5242 genomic DNA using primers 1 and 2. The amplified fragment was digested with HindIII and EcoRI and ligated into HindIII- and EcoRI-cleaved pUC19 vector (TaKaRa Bio, Shiga, Japan). The resultant plasmid was named pUC19_HpTmox. A DNA fragment containing the H. polymorpha GAP promoter (HpPgap) was PCR amplified from BY5242 genomic DNA using primers 3 and 4. The amplified fragment was digested with EcoRI and ligated into MunI-cleaved pUC19_HpTmox. The resultant plasmid was named pUC19_HpPgap_HpTmox. A DNA fragment containing the H. polymorpha GAP promoter (HpPgap) was PCR amplified from BY5242 genomic DNA using primers 5 and 6. The amplified fragment was digested with BamHI and ligated into BamHI-cleaved pUC19_HpPgap_HpTmox. The resultant plasmid was named pUC19_HpPgap_HpPgap_HpTmox. A DNA fragment containing the Zeocin resistance-encoding selective marker was ordered from GeneArt Gene Synthesis (Thermo Fisher Scientific, Waltham, MA, USA) and PCR amplified using primers 7 and 8. The amplified fragments were digested with EcoRI and ligated into the EcoRI-cleaved pUC19_HpPgap_HpPgap_HpTmox. The resultant plasmid was named pUC19_HpPgap_HpPgap_HpTmox_Zeo.
The plasmids used for investigating the characteristics of the putative centromeric DNA sequences were constructed as follows. A DNA fragment containing one half of Cen2 (LOR2-CC2) was PCR amplified from CBS7435 genomic DNA using primers 9 and 10, and a DNA fragment containing the other half of Cen2 (CC2-ROR2) was PCR amplified from CBS7435 genomic DNA using primers 11 and 12; the amplified fragments were digested with NotI and PstI and with PstI and XbaI, respectively, and ligated together into NotI- and SpeI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmid was named pUC19-Cen2. The DNA fragments containing LOR2-CC2, LOR2, and CC2 were PCR amplified from CBS7435 genomic DNA using primer pairs 9 and 13, 9 and 14, and 15 and 13, respectively. Each amplified fragment was digested with NotI and ligated into the NotI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmids were named pUC19-LOR2CC2, pUC19-LOR2, and pUC19-CC2, respectively. The DNA fragments containing LOR2(1–2409), LOR2(1–1800), LOR2(1–1200), LOR2(1–600), LOR2(1–111), LOR2(84–213), and LOR2(191–291) were PCR amplified from CBS7435 genomic DNA using primer pairs 9 and 16, 9 and 17, 9 and 18, 9 and 19, 9 and 20, 21 and 22, and 23 and 24, respectively. Each amplified fragment was digested with NotI and XbaI and ligated into NotI- and SpeI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmids were named pUC19-LOR2(1–2409), pUC19-LOR2(1–1800), pUC19-LOR2(1–1200), pUC19-LOR2(1–600), pUC19-LOR2(1–111), pUC19-LOR2(84–213), and pUC19-LOR2(191–291), respectively. A DNA fragment containing LOR2(84–2699) was PCR amplified from CBS7435 genomic DNA using primers 21 and 14. The amplified fragment was digested with NotI and ligated into the NotI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmid was named pUC19-LOR2(84–2699).
A DNA fragment containing one half of Cen1 (LOR1-CC1) was PCR amplified from CBS7435 genomic DNA using primers 25 and 26, and a DNA fragment containing the other half of Cen1 (CC1-ROR1) was PCR amplified from CBS7435 genomic DNA using primers 27 and 28; the amplified fragments were digested with NotI and SacI and with SacI and BamHI, respectively, and ligated together into NotI- and BamHI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmid was named pUC19-Cen1. A DNA fragment containing one half of Cen3 (LOR3-CC3) was PCR amplified from CBS7435 genomic DNA using primers 29 and 30, and a DNA fragment containing the other half of Cen3 (CC3-ROR3) was PCR amplified from CBS7435 genomic DNA using primers 31 and 32; the amplified fragments were digested with NotI and AgeI and with AgeI and BamHI, respectively, and ligated together into NotI- and BamHI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmid was named pUC19-Cen3. A DNA fragment containing one half of Cen4 (LOR4-CC4) was PCR amplified from CBS7435 genomic DNA using primers 33 and 34, and a DNA fragment containing the other half of Cen4 (CC4-ROR4) was PCR amplified from CBS7435 genomic DNA using primers 35 and 36; the amplified fragments were digested with NotI and SpeI and with SpeI and BamHI, respectively, and ligated together into NotI- and BamHI-cleaved pUC19_HpPgap_HpPgap_HpTmox_Zeo vector. The resultant plasmid was named pUC19-Cen4.
The plasmids containing the GFP reporter gene were constructed as follows. To construct the PpPgap-EGFP_Pp-PpTgap1 cassette, a DNA fragment containing the P. pastoris GAP promoter (PpPgap) was PCR amplified from CBS7435 genomic DNA using primers 37 and 38. A DNA fragment containing the codon-optimized enhanced green fluorescent protein (EGFP) gene sequence for P. pastoris (EGFPPp), ordered from GeneArt Gene Synthesis (Thermo Fisher Scientific), was PCR amplified using primers 39 and 40. The DNA fragment containing the P. pastoris GAP1 terminator (PpTgap1) was PCR amplified from CBS7435 genomic DNA using primers 41 and 42. These three fragments (PpPgap, EGFPPp, and PpTgap1) were simultaneously linked and inserted into the BamHI- and BglII-digested pUC19_HpPgap_HpPgap_HpTmox_Zeo vector using a Clontech In-fusion HD Cloning kit (TaKaRa Bio), yielding pUC19-PpPgap-EGFP_Pp-PpTgap1.
The plasmids used for investigating plasmid retention were constructed as follows. The DNA fragment containing the PpPgap-EGFP_Pp-PpTgap1 cassette was PCR amplified from pUC19-PpPgap-EGFP_Pp-PpTgap1 using primer pair 43 and 44 or pair 45 and 44. The amplified fragments were inserted into the XbaI-digested pUC19-Cen2 and pUC19-LOR2(1–111) plasmids, yielding pUC19-Cen2-EGFP and pUC19-LOR2(1–111)-EGFP, respectively. The DNA fragment containing the PpPgap-EGFP_Pp-PpTgap1 cassette was PCR amplified from pUC19-PpPgap-EGFP_Pp-PpTgap1 using primers 46 and 44. The amplified fragment was inserted into XbaI- and NotI (partial)-digested pUC19-LOR2CC2, yielding pUC19-LOR2CC2-EGFP. A DNA fragment containing LOR2 was released from pUC19-LOR2 by digestion with NotI and then inserted into NotI-digested pUC19-LOR2CC2-EGFP, yielding pUC19-LOR2-EGFP. A DNA fragment containing PARS1 was PCR amplified from CBS7435 genomic DNA using primers 47 and 48. The amplified fragment was digested with NotI and ligated into NotI-digested pUC19-LOR2CC2-EGFP, yielding pUC19-PARS1-EGFP.
To construct the integrative plasmids for expressing the GFP reporter gene, a plasmid containing the G418 resistance-encoding selective marker was constructed as follows. A DNA fragment containing the H. polymorpha GAP promoter (HpPgap) and the G418 resistance-encoding selective marker was ordered from GeneArt Gene Synthesis and PCR amplified using primers 49 and 50. The resulting amplicon was digested with HindIII and EcoRI and then ligated into HindIII- and EcoRI-digested pUC19, yielding pUC19_G418. The plasmid used for single-crossover type integration into the CCA38473.1 terminator locus was constructed as follows. A DNA fragment containing the CCA38473.1 terminator (T38473; 600 bp) was PCR amplified from CBS7435 genomic DNA using primers 51 and 52. The amplified fragment was inserted into XbaI-cleaved pUC19_G418 using an In-fusion HD Cloning kit, yielding pUC19_T38473-G418. The DNA fragment containing the PpPgap-EGFP_Pp-PpTgap1 cassette was PCR amplified from pUC19-PpPgap-EGFP_Pp-PpTgap1 using primers 37 and 53. The amplified fragment was inserted into BamHI- and BglII-digested pUC19_T38473-G418, yielding pUC19-T38473-EGFP.
The integrative plasmid containing the Zeocin resistance-encoding selective marker was constructed as follows. A DNA fragment containing the Zeocin resistance-encoding selective marker was prepared by digesting pUC19_HpPgap_HpPgap_HpTmox_Zeo with EcoRI and then ligated into EcoRI-digested pUC19, yielding pUC19_Zeo. The plasmid used for single-crossover type integration into the P. pastoris AOX1 promoter locus was constructed as follows. A DNA fragment containing the P. pastoris AOX1 promoter (PpPaox1) was PCR amplified from CBS7435 genomic DNA using primers 54 and 55. The amplified fragment was inserted into PstI- and HindIII-cleaved pUC19_Zeo using an In-fusion HD Cloning kit, yielding pUC19_PpPaox1_Zeo. A DNA fragment containing the P. pastoris GAP promoter (PpPgap) was PCR amplified from CBS7435 genomic DNA using primers 56 and 57, and a DNA fragment containing the P. pastoris AOX1 terminator (PpTaox1) was PCR amplified from CBS7435 genomic DNA using primers 58 and 59. The amplified fragments were inserted into BamHI-cleaved pUC19_PpPaox1_Zeo using an In-fusion HD Cloning kit, yielding pUC19_PpPaox1_PpPgap-PpTaox1.
To construct a standard plasmid for qPCR, a plasmid containing both the EGFP gene and the ACT1 gene encoding actin was constructed as follows. A DNA fragment containing the P. pastoris ACT1 gene was PCR amplified from CBS7435 genomic DNA using primers 60 and 61. The amplified fragments were inserted into XhoI-cleaved pUC19-Cen2-EGFP using an In-fusion HD Cloning kit, yielding pUC19-Cen2-EGFP-ACT1.
Transformation.
P. pastoris strain CBS7435 was transformed by electroporation as previously described (51). In short, competent cells (60 μl) were mixed with plasmid in a 0.2-cm electroporation cuvette, incubated on ice for 5 min, and electroporated in a Bio-Rad Gene Pulser (Bio-Rad Laboratories, Hercules, CA, USA) with settings of 1,500 V, 25 μF, and 200 Ω. After a pulsing step, 1 ml of YPD medium was added immediately to the cuvette, and the cells were transferred to a sterile tube. The tube was incubated at 30°C without shaking for 1 h. Thereafter, transformants were harvested by centrifugation at 3,000 × g at 25°C for 5 min, and 960 μl of supernatant was discarded. The transformants were resuspended in the remaining YPD supernatant, plated to YPD selection agar plates containing 100 μg/ml Zeocin (phleomycin D1) (InvivoGen, San Diego, CA, USA), and grown at 30°C.
The number of CFU per microgram of DNA was determined from the numbers of colonies generated on the YPD selectable solid medium. Data are presented as means ± standard deviations for three replicates (representing three transformations).
To construct yeast strain T38473-EGFP, which has a single-copy chromosomally integrated GFP gene, CBS7435 was transformed by electroporation with EcoRV-linearized pUC19-T38473-EGFP plasmid. Transformants were grown on YPD selection agar plates containing 500 μg/ml G418 (Nacalai Tesque), and integration of the plasmid was confirmed by colony PCR.
To construct yeast strain P01, CBS7435 was transformed by electroporation with NsiI-linearized pUC19_PpPaox1_PpPgap-PpTaox1 plasmid. Transformants were grown on YPD selection agar plates containing 100 μg/ml Zeocin, and integration of the plasmid was confirmed by colony PCR.
Evaluation of plasmid retention.
The yeast cells were grown overnight at 30°C in BMGY medium with or without Zeocin (200 μg/ml), and the cells then were inoculated into 4 ml or 2 ml of fresh BMGY medium with or without Zeocin (200 μg/ml) at an initial optical density at 660 nm (OD660) of 0.1. The cells were incubated at 30°C with shaking at 180 rpm for up to 24 h or 100 h. For evaluation of plasmid retention through repeated subculturing, the cells were transferred repeatedly to fresh selective medium every 24 h (initial OD660 of 0.1). After incubation, the yeast cell samples were diluted with 1 ml of sheath fluid before fluorescence was analyzed by a flow cytometer. Flow cytometry measurements of green fluorescence followed a previously described procedure (52). In brief, fluorescent cells were detected using a BD FACSCanto II flow cytometer equipped with a 488-nm blue laser (Becton, Dickinson, and Company, Franklin Lakes, NJ, USA); the data were analyzed using BD FACSDiva software (version 5.0; Becton, Dickinson, and Company). The GFP fluorescence signal was collected through a 530/30-nm band pass filter, and the GFP-A mean signal of 10,000 cells was defined as green fluorescence intensity.
Monitoring of cell growth.
Cell growth was monitored in L-shaped test tubes by using a bio-photorecorder apparatus (model TVS062CA; Advantec, Tokyo, Japan). The yeast cells were grown overnight at 30°C in BMGY medium with or without Zeocin (200 μg/ml), and the cells then were inoculated into 5 ml of fresh BMGY medium with or without Zeocin (200 μg/ml) at an initial OD660 of 0.1. The cells were incubated at 30°C with shaking at 70 rpm for up to 48 h. The OD660 of the cell suspension was automatically measured every 30 min. The final cell density (OD660) after 48 h of cultivation was measured by a UV-visible light (VIS) spectrophotometer (UVmini-1240; Shimadzu, Kyoto, Japan).
qPCR for average plasmid copy number analysis.
The average plasmid copy number per cell was determined by quantitative PCR (qPCR). The yeast cells were grown overnight at 30°C in BMGY medium with or without Zeocin (200 μg/ml), and the cells then were inoculated into 2 ml of fresh BMGY medium with or without Zeocin (200 μg/ml) at an initial OD660 of 0.1. The cells were incubated at 30°C with shaking at 180 rpm for up to 24 h. The cultures were harvested by centrifugation, and total cellular nucleic acids were extracted with 0.25% sodium dodecyl sulfate, followed by heat treatment at 98°C for 8 min.
The EGFP gene and the ACT1 gene encoding actin were selected as target and reference (housekeeping) genes, respectively. Both genes are present in a single copy on the plasmid (pUC19-Cen2-EGFP and pUC19-PARS1-EGFP) and the CBS7435 genome, respectively. DNA samples and 10-fold serial dilutions of a purified standard plasmid bearing a single copy of both the EGFP gene and ACT1 gene (pUC19-Cen2-EGFP-ACT1) were then amplified on the Stratagene Mx3000P instrument (Agilent Technologies, Santa Clara, CA, USA) with KOD SYBR qPCR mix (Toyobo, Osaka, Japan). The primers for qPCR were 62 and 63 for the EGFP gene and 64 and 65 for the ACT1 gene. The PCR program was as follows: 2 min at 98°C followed by 40 cycles of 10 s at 98°C, 10 s at 60°C, and 30 s at 68°C. The threshold cycle (CT) values were measured, and copy numbers were calculated by using the calibrator standard curves to determine the quantity of plasmid (EGFP) and genome (ACT1) DNA for a given sample in arbitrary units and then calculating their ratio.
Plasmid extraction from yeast.
Transformants on YPD selection agar plates were replicated and grown on new agar plates at 30°C. Plasmid DNA was extracted from each replica transformant using a Clontech Easy Yeast Plasmid Isolation kit (TaKaRa Bio). The extracted plasmid DNA was used to transform E. coli HST08 premium competent cells (TaKaRa Bio).
Supplementary Material
ACKNOWLEDGMENTS
We thank Taiki Yamaji and Misa Ishigami for technical assistance.
This work was supported by the Project Focused on Developing Key Technology for Discovering and Manufacturing Drugs for Next-Generation Treatment and Diagnosis from the Japan Agency for Medical Research and Development (AMED), Japan.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02882-17.
REFERENCES
- 1.Gasser B, Prielhofer R, Marx H, Maurer M, Nocon J, Steiger M, Puxbaum V, Sauer M, Mattanovich D. 2013. Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol 8:191–208. doi: 10.2217/fmb.12.133. [DOI] [PubMed] [Google Scholar]
- 2.Daly R, Hearn MT. 2005. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit 18:119–138. doi: 10.1002/jmr.687. [DOI] [PubMed] [Google Scholar]
- 3.Cereghino JL, Cregg JM. 2000. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 4.Kim H, Yoo SJ, Kang HA. 2015. Yeast synthetic biology for the production of recombinant therapeutic proteins. FEMS Yeast Res 15:1–16. doi: 10.1093/femsyr/fou003. [DOI] [PubMed] [Google Scholar]
- 5.Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM. 2005. Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- 6.Invitrogen. 2014. Pichia expression kit user guide. Publication MAN0000012. Life Technologies, Carlsbad, CA. [Google Scholar]
- 7.Gasser B, Maurer M, Rautio J, Sauer M, Bhattacharyya A, Saloheimo M, Penttilä M, Mattanovich D. 2007. Monitoring of transcriptional regulation in Pichia pastoris under protein production conditions. BMC Genomics 8:179. doi: 10.1186/1471-2164-8-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graf A, Gasser B, Dragosits M, Sauer M, Leparc GG, Tüchler T, Kreil DP, Mattanovich D. 2008. Novel insights into the unfolded protein response using Pichia pastoris specific DNA microarrays. BMC Genomics 9:390. doi: 10.1186/1471-2164-9-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dragosits M, Stadlmann J, Albiol J, Baumann K, Maurer M, Gasser B, Sauer M, Altmann F, Ferrer P, Mattanovich D. 2009. The effect of temperature on the proteome of recombinant Pichia pastoris. J Proteome Res 8:1380–1392. doi: 10.1021/pr8007623. [DOI] [PubMed] [Google Scholar]
- 10.Baumann K, Carnicer M, Dragosits M, Graf AB, Stadlmann J, Jouhten P, Maaheimo H, Gasser B, Albiol J, Mattanovich D, Ferrer P. 2010. A multi-level study of recombinant Pichia pastoris in different oxygen conditions. BMC Syst Biol 4:141. doi: 10.1186/1752-0509-4-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dragosits M, Stadlmann J, Graf A, Gasser B, Maurer M, Sauer M, Kreil DP, Altmann F, Mattanovich D. 2010. The response to unfolded protein is involved in osmotolerance of Pichia pastoris. BMC Genomics 11:207. doi: 10.1186/1471-2164-11-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puxbaum V, Mattanovich D, Gasser B. 2015. Quo vadis? The challenges of recombinant protein folding and secretion in Pichia pastoris. Appl Microbiol Biotechnol 99:2925–2938. doi: 10.1007/s00253-015-6470-z. [DOI] [PubMed] [Google Scholar]
- 13.Spadiut O, Capone S, Krainer F, Glieder A, Herwig C. 2014. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol 32:54–60. doi: 10.1016/j.tibtech.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cereghino GP, Cereghino JL, Ilgen C, Cregg JM. 2002. Production of recombinant proteins in fermenter cultures of the yeast Pichia pastoris. Curr Opin Biotech 13:329–332. doi: 10.1016/S0958-1669(02)00330-0. [DOI] [PubMed] [Google Scholar]
- 15.Spohner SC, Müller H, Quitmann H, Czermak P. 2015. Expression of enzymes for the usage in food and feed industry with Pichia pastoris. J Biotechnol 202:118–134. doi: 10.1016/j.jbiotec.2015.01.027. [DOI] [PubMed] [Google Scholar]
- 16.Walsh G. 2010. Biopharmaceutical benchmarks 2010. Nat Biotechnol 28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 17.Vogl T, Hartner FS, Glieder A. 2013. New opportunities by synthetic biology for biopharmaceutical production in Pichia pastoris. Curr Opin Biotechnol 24:1094–1101. doi: 10.1016/j.copbio.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson CA. 2010. FDA approves kallikrein inhibitor to treat hereditary angioedema. Am J Health Syst Pharm 67:93. doi: 10.2146/news100005. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad M, Hirz M, Pichler H, Schwab H. 2014. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98:5301–5317. doi: 10.1007/s00253-014-5732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reference deleted.
- 21.Papakonstantinou T, Harris S, Hearn MTW. 2009. Expression of GFP using Pichia pastoris vectors with zeocin or G-418 sulphate as the primary selectable marker. Yeast 26:311–321. doi: 10.1002/yea.1666. [DOI] [PubMed] [Google Scholar]
- 22.Lin Cereghino GP, Lin Cereghino J, Sunga AJ, Johnson MA, Lim M, Gleeson MA, Cregg JM. 2001. New selectable marker/auxotrophic host strain combinations for molecular genetic manipulation of Pichia pastoris. Gene 263:159–169. doi: 10.1016/S0378-1119(00)00576-X. [DOI] [PubMed] [Google Scholar]
- 23.Schwarzhans J-P, Wibberg D, Winkler A, Luttermann T, Kalinowski J, Friehs K. 2016. Non-canonical integration events in Pichia pastoris encountered during standard transformation analysed with genome sequencing. Sci Rep 6:38952. doi: 10.1038/srep38952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Struhl K, Stinchcomb DT, Scherer S, Davis RW. 1979. High-frequency transformation of yeast: autonomous replication of hybrid DNA molecules. Proc Natl Acad Sci U S A 76:1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huberman JA, Zhu JG, Davis LR, Newlon CS. 1988. Close association of a DNA replication origin and an ARS element on chromosome III of the yeast, Saccharomyces cerevisiae. Nucleic Acids Res 16:6373–6384. doi: 10.1093/nar/16.14.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cregg JM, Barringer KJ, Hessler AY, Madden KR. 1985. Pichia pastoris as a host system for transformations. Mol Cell Biol 5:3376–3385. doi: 10.1128/MCB.5.12.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liachko I, Dunham MJ. 2014. An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res 14:364–367. doi: 10.1111/1567-1364.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarzhans JP, Luttermann T, Wibberg D, Winkler A, Hübner W, Huser T, Kalinowski J, Friehs K. 2017. A Mitochondrial autonomously replicating sequence from Pichia pastoris for uniform high level recombinant protein production. Front. Microbiol 8:780. doi: 10.3389/fmicb.2017.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bitoun R, Zamir A. 1986. Spontaneous amplification of yeast CEN ARS plasmids. Mol Gen Genet 204:98–102. doi: 10.1007/BF00330194. [DOI] [PubMed] [Google Scholar]
- 30.Murray AW, Szostak JW. 1983. Pedigree analysis of plasmid segregation in yeast. Cell 34:961–970. doi: 10.1016/0092-8674(83)90553-6. [DOI] [PubMed] [Google Scholar]
- 31.Clarke L, Carbon J. 1980. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature 287:504–509. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- 32.Lee CC, Williams TG, Wong DW, Robertson GH. 2005. An episomal expression vector for screening mutant gene libraries in Pichia pastoris. Plasmid 54:80–85. doi: 10.1016/j.plasmid.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Camattari A, Goh A, Yip LY, Tan AHM, Ng SW, Tran A, Liu G, Liachko I, Dunham MJ, Rancati G. 2016. Characterization of a panARS-based episomal vector in the methylotrophic yeast Pichia pastoris for recombinant protein production and synthetic biology applications. Microb Cell Fact 15:139. doi: 10.1186/s12934-016-0540-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke L. 1990. Centromeres of budding and fission yeasts. Trends Genet 6:150–154. doi: 10.1016/0168-9525(90)90149-Z. [DOI] [PubMed] [Google Scholar]
- 35.Hyman AA, Sorger PK. 1995. Structure and function of kinetochores in budding yeast. Annu Rev Cell Dev Biol 11:471–495. doi: 10.1146/annurev.cb.11.110195.002351. [DOI] [PubMed] [Google Scholar]
- 36.Biggins S. 2013. The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194:817–846. doi: 10.1534/genetics.112.145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray AW, Szostak JW. 1983. Construction of artificial chromosomes in yeast. Nature 305:189–193. doi: 10.1038/305189a0. [DOI] [PubMed] [Google Scholar]
- 38.Burke DT, Carle GF, Olson MV. 1987. Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. Science 236:806–812. doi: 10.1126/science.3033825. [DOI] [PubMed] [Google Scholar]
- 39.Noskov VN, Chuang R-Y, Gibson DG, Leem S-H, Larionov V, Kouprina N. 2011. Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat Protoc 6:89–96. doi: 10.1038/nprot.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Schutter K, Lin Y-C, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouzé P, Van de Peer Y, Callewaert N. 2009. Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol 27:561–566. doi: 10.1038/nbt.1544. [DOI] [PubMed] [Google Scholar]
- 41.Mattanovich D, Callewaert N, Rouzé P, Lin Y-C, Graf A, Redl A, Tiels P, Gasser B, De Schutter K. 2009. Open access to sequence: browsing the Pichia pastoris genome. Microb Cell Fact 8:53. doi: 10.1186/1475-2859-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Küberl A, Schneider J, Thallinger GG, Anderl I, Wibberg D, Hajek T, Jaenicke S, Brinkrolf K, Goesmann A, Szczepanowski R, Pühler A, Schwab H, Glieder A, Pichler H. 2011. High-quality genome sequence of Pichia pastoris CBS7435. J Biotechnol 154:312–320. doi: 10.1016/j.jbiotec.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 43.Malik HS, Henikoff S. 2009. Major evolutionary transitions in centromere complexity. Cell 138:1067–1082. doi: 10.1016/j.cell.2009.08.036. [DOI] [PubMed] [Google Scholar]
- 44.Roy B, Sanyal K. 2011. Diversity in requirement of genetic and epigenetic factors for centromere function in fungi. Eukaryot Cell 10:1384–1395. doi: 10.1128/EC.05165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sturmberger L, Chappell T, Geier M, Krainer F, Day KJ, Vide U, Trstenjak S, Schiefer A, Richardson T, Soriaga L, Darnhofer B, Birner-Gruenberger R, Glick BS, Tolstorukov I, Cregg J, Madden K, Glieder A. 2016. Refined Pichia pastoris reference genome sequence. J Biotechnol 235:121–131. doi: 10.1016/j.jbiotec.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coughlan AY, Hanson SJ, Byrne KP, Wolfe KH. 2016. Centromeres of the yeast Komagataella phaffii (Pichia pastoris) have a simple inverted-repeat structure. Genome Biol Evol 8:2482–2492. doi: 10.1093/gbe/evw178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishi T, Watanabe T, Nishiyama T, Okubo Y. 9 June 2016. Vector containing centromere DNA sequence and use thereof. International patent WO2016088824.
- 48.Liachko I, Youngblood RA, Tsui K, Bubb KL, Queitsch C, Raghuraman MK, Nislow C, Brewer BJ, Dunham MJ. 2014. GC-rich DNA elements enable replication origin activity in the methylotrophic yeast Pichia pastoris. PLoS Genet 10:e1004169. doi: 10.1371/journal.pgen.1004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishii J, Izawa K, Matsumura S, Wakamura K, Tanino T, Tanaka T, Ogino C, Fukuda H, Kondo A. 2009. A simple and immediate method for simultaneously evaluating expression level and plasmid maintenance in yeast. J Biochem 145:701–708. doi: 10.1093/jb/mvp028. [DOI] [PubMed] [Google Scholar]
- 50.Nishiyama T, Takami H, Nishi T, Takano M. 21 April 2011. Hansenula polymorpha capable of producing antibody, process for production of antibody utilizing same, and antibody produced from same. International patent WO2011046218.
- 51.Faber KN, Haima P, Harder W, Veenhuis M, Ab G. 1994. Highly efficient electrotransformation of the yeast Hansenula polymorpha. Curr Genet 25:305–310. doi: 10.1007/BF00351482. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura Y, Ishii J, Kondo A. 2013. Bright fluorescence monitoring system utilizing Zoanthus sp. green fluorescent protein (ZsGreen) for human G-protein-coupled receptor signaling in microbial yeast cells. PLoS One 8:e82237. doi: 10.1371/journal.pone.0082237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.