

The high-risk human papillomaviruses (HPVs) are the etiological cause of cervical cancer and the most common sexually transmitted infection. While the majority of infections may be asymptomatic or cause only benign lesions, persistent infection with the oncogenic high-risk HPV types may lead to serious diseases, such as cervical cancer, anogenital carcinoma, or head and neck oropharyngeal squamous cell carcinoma. The identification of virus-host protein interactions provides insights into the mechanisms of viral DNA persistence, viral genome replication, and cellular transformation. Elucidating the mechanism of early events in the virus replication cycle as well as of integration of viral DNA into host chromatin may present novel antiviral strategies and targets for counteracting persistent infection. The E2 protein is an important viral regulatory protein whose functions are mediated through interactions with host cell proteins. Here we explore the interaction of E2 with SMC5/6 and the functional consequences.
KEYWORDS: host virus interaction, mass spectrometry, papillomavirus, viral replication
ABSTRACT
The papillomavirus E2 protein executes numerous essential functions related to viral transcription, replication of viral DNA, and viral genome maintenance. Because E2 lacks enzymatic activity, many of these functions are mediated by interactions with host cellular proteins. Unbiased proteomics approaches have successfully identified a number of E2-host protein interactions. We have extended such studies and have identified and validated the cellular proteins structural maintenance of chromosome 5 (SMC5) and SMC6 as interactors of the viral E2 protein. These two proteins make up the core components of the SMC5/6 complex. The SMC5/6 complex is a member of the conserved structural maintenance of chromosomes (SMC) family of proteins, which are essential for genome maintenance. We have examined the role of SMC5/6 in various E2 functions. Our data suggest that SMC6 is not required for E2-mediated transcriptional activation, E1/E2-mediated transient replication, or differentiation-dependent amplification of viral DNA. Our data, however, suggest a role for SMC5/6 in viral genome maintenance.
IMPORTANCE The high-risk human papillomaviruses (HPVs) are the etiological cause of cervical cancer and the most common sexually transmitted infection. While the majority of infections may be asymptomatic or cause only benign lesions, persistent infection with the oncogenic high-risk HPV types may lead to serious diseases, such as cervical cancer, anogenital carcinoma, or head and neck oropharyngeal squamous cell carcinoma. The identification of virus-host protein interactions provides insights into the mechanisms of viral DNA persistence, viral genome replication, and cellular transformation. Elucidating the mechanism of early events in the virus replication cycle as well as of integration of viral DNA into host chromatin may present novel antiviral strategies and targets for counteracting persistent infection. The E2 protein is an important viral regulatory protein whose functions are mediated through interactions with host cell proteins. Here we explore the interaction of E2 with SMC5/6 and the functional consequences.
INTRODUCTION
Papillomaviruses (PVs) infect cells in the basal layers of stratified squamous epithelia, and the papillomavirus replication cycle progresses as the host cell differentiates. PVs have a small double-stranded DNA genome which encodes a small number of early proteins (E1 to E7) and two late proteins (L1 and L2) that are translated from multicistronic transcripts driven by an early promoter and a late promoter, respectively. In the case of high-risk human papillomaviruses (HPVs), the viral E6 and E7 oncoproteins induce the alterations that can lead to precancerous transformations in host cells (1). E6- and E7-induced dysregulation of the cell cycle and inhibition of apoptosis normally support virus replication in squamous epithelial cells that have otherwise exited the cell cycle. One of several activities of the viral E2 protein is the repression of the HPV early promoter and transcription of the E6 and E7 oncogenes (2–5). Integration of viral DNA into host chromatin and disruption of the E2 open reading frame (ORF) can lead to unregulated expression of E6 and E7, an event that can promote progression to cervical cancer (6–8).
In addition to the transcriptional repression of E6 and E7, E2 can also function as a potent transcriptional activator. It has an auxiliary role in viral DNA replication and is essential for the maintenance of the viral genomes in dividing cells (9). The viral E1 and E2 proteins are DNA replication proteins required for the initial establishment of the 8-kb viral genome as an extrachromosomal episome within the host nucleus, where E2 loads the E1 helicase onto the viral origin to initiate replication to a low copy number (10). Interactions of the viral E2 protein with the viral genomes and specific host proteins facilitate the tethering of viral episomal genomes to host mitotic chromosomes and allow genomes to persist in the nucleus as host cells divide (9). As the host cell differentiates, the viral DNA is amplified in a differentiation-dependent process in which E1 and E2 increase the copy number of viral DNA for packaging into virions (11). A more thorough understanding of the host cellular proteins that the E2 proteins engage will provide insight into how this viral protein executes its various functions.
Structural maintenance of chromosome (SMC) proteins are conserved in eukaryotes and have several roles in the establishment and maintenance of chromosome structure. The SMC protein family encompasses three major complexes: SMC1/3 (cohesin), SMC2/4 (condensin), and SMC5/6. They share a similar architecture, where the individual SMC components are long polypeptides of 1,000 to 1,300 amino acids that are folded at a central hinge domain. SMC5/6 is activated by the DNA damage response (DDR) and is essential for DNA double-strand break repair through homologous recombination (HR) and the completion of DNA replication (12–16). SMC5/6 is also required for the segregation of repetitive chromosome regions and the rescue of collapsed replication forks (12, 15, 16). It recruits HR factors, such as SMC1/3, which is thought to promote HR by maintaining the close proximity of sister chromatids at double-strand breaks (17).
We have used an unbiased proteomic approach to confirm that SMC5/6 is an interactor of the PV E2 protein. In this study, we investigated possible roles of the SMC5/6 complex in the PV life cycle and found that it associates with DNA replication foci and may play a role in the maintenance of the HPV DNA genomes.
RESULTS
Papillomavirus E2 interacts with SMC5/6.
Previous studies determined that SMC5 and SMC6, the core components of the SMC5/6 complex, interact with E2 proteins from different papillomavirus types (18, 19). The SMC5 and SMC6 proteins have long coiled-coil domains bent at a hinge region and globular, DNA-binding domains at the N and C termini. The two SMC heterodimers bind through their hinge domains, while the globular domains are connected by a kleisin subunit (Fig. 1A). The other components that make up the SMC complexes are referred to as non-SMC elements (NSE). In humans these are termed NSE1, NSE2, NSE3, and NSE4. Human NSE1 contains a RING-like domain that is required for the formation of the NSE1-NSE3-NSE4 subcomplex. The C-terminal domain of NSE3 interacts with NSE4, and together, the NSE1-NSE3-NSE4 subcomplex bridges the SMC5 and SMC6 head domains (Fig. 1A). NSE2, also referred to as MMS21, binds directly to SMC5 and is an E3 SUMO ligase that is required for DNA damage repair but is not required for stability of the complex (20).
FIG 1.

E2 interacts with SMC6. (A) The SMC5/6 complex. Colors represent the NWD score of each coimmunoprecipitated component detected by mass spectrometry in 293T cells. An NWD of ≥1 signifies a high-confidence interaction with BPV1 E2. (B) C33A cells were transiently transfected with HPV6b, -18, -5, or -8 or BPV1 E2 or with HPV16 E6. E2-protein complexes were immunoprecipitated from cell lysates overnight and visualized by Western blotting. (C) C33A cells were transiently transfected with HPV18, HPV5, or BPV1 E2 or with HPV16 E1. HA-tagged proteins were immunoprecipitated, and bound proteins were visualized by Western blotting. Blotting for actin was used to control for loading. (D) C33A cells were transfected with HPV16 or HPV31 E2 inducibly expressed from a pMEP4 plasmid. Cells were treated with 2 μM cadmium chloride for 5 h to induce E2 expression before immunoprecipitation and Western blotting.
Using a mass spectrometry-based proteomics approach, we determined the cellular interacting proteins that interact with bovine papillomavirus 1 (BPV1) E2 and confirmed its binding to SMC5 and SMC6. A similar proteomics pipeline was previously used to define the interaction proteome of HPV E6 and E7 proteins (21, 22). For the E2 analysis, 293T cell lines stably expressing hemagglutinin (HA)-tagged BPV1 E2, E2TR, or E8^E2C proteins were used as the starting material in immunoprecipitation (IP) experiments. HA immunoprecipitations recovered the E2 bait proteins and any associated cellular proteins. The E2-protein complexes were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and subsequent analysis with the Comparative Proteomics Analysis Software Suite (CompPASS). CompPASS assigns a normalized weighted D (NWD) score to each protein detected by LC-MS/MS. The NWD score is higher when an interacting protein is more abundant, when it is detected more frequently, or when the interaction is more reproducible in an experiment of interest than in a library of other experiments. High-confidence interacting proteins (HCIPs) are those for which the NWD score is greater than or equal to 1 (23). SMC6 and SMC5 were identified as HCIPs of BPV1 E2, with NWD scores of 3.9 and 1.84, respectively. Neither protein was detected in complex with BPV1 E2TR or with E8^E2C (Fig. 1A; see also Tables S1 to S3 in the supplemental material). Other components of the complex identified in these proteomics experiments were MMS21 and NSE1. Their NWD scores were less than 1, suggesting that the interaction was detected but did not meet the threshold for significance (Fig. 1A; Table S1).
To validate these interactions, confirm them in a second cell type, and extend our observation to PV E2 proteins from additional virus types, C33A cell lines were transiently transfected with plasmids encoding HA-tagged E2 proteins. Anti-HA immunoprecipitation experiments recovered E2 and its associated proteins and validated the interaction of BPV1 E2 with SMC6. These also demonstrated that HPV6b, -18, -5, and -8 E2 proteins bound to SMC6 but that SMC6 did not coprecipitate with HPV16 E6 or E1 proteins (Fig. 1B and C). SMC6 levels did not appear to be influenced by the presence of E2 (Fig. 1C). Unlike other E2 proteins, which were expressed in a pOZN vector, expression of HPV16 E2 and HPV31 E2 could be visualized by Western blotting only when expressed from the cadmium-inducible pMEP4 vector. Immunoprecipitation of these proteins indicated that HPV16 and -31 could pull down both SMC6 and the previously confirmed interactor, Brd4, as a positive control (Fig. 1D). Together with data from previous studies, these data suggest that the SMC5/6-E2 interaction is conserved among several different papillomaviruses.
Of several components of the SMC5/6 complex identified in the mass spectrometry experiments, SMC6 consistently had the highest NWD scores (Table S1). This suggests that an interaction of E2 with this complex may be mediated by SMC6. We next sought to validate the interaction with SMC5 and determine whether SMC6 is required for the E2-SMC5 interaction. To test this, SMC6 was depleted by small interfering RNA (siRNA) transfection of C33A cells stably expressing FLAG-tagged HPV8 or HPV18 E2. Anti-FLAG immunoprecipitation revealed that the E2 interactor bromodomain protein 4 (Brd4) was recovered even when SMC6 levels were reduced, whereas the SMC5-E2 interaction occurred only in the presence of SMC6 (Fig. 2). However, because SMC5 levels were reduced with the loss of SMC6, we cannot conclude that SMC6 is required for the interaction of E2 with SMC5. A reduction in SMC5 levels with an siRNA targeting SMC6 has been reported in previous studies, in which SMC6 depletion destabilized the entire SMC5/6 complex (24, 25). Thus, experiments employing targeted depletion of SMC6 may represent a condition in which the entire SMC5/6 complex is lost from cells. However, pulldown of SMC5 by HPV8 and -18 E2 confirmed the E2 interaction with SMC5 (Fig. 2).
FIG 2.
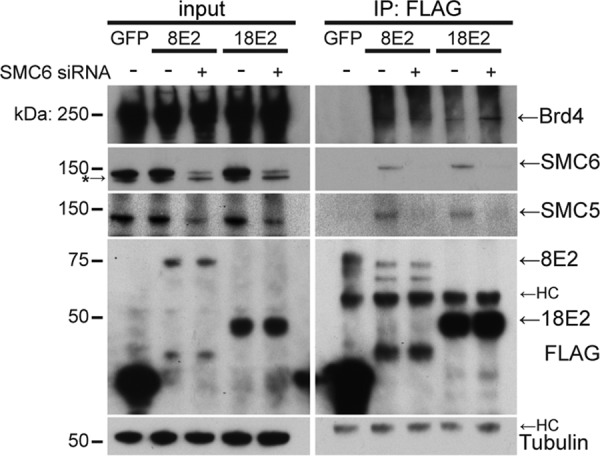
Analysis of the E2 interaction with SMC5/6. Western blotting for E2-protein complexes was performed in the presence of SMC6 siRNA or a control siRNA. C33A cells stably expressing FLAG-tagged HPV E2 or GFP as a negative control were transfected with SMC6 siRNA or control siRNA. At 3 days postransduction, cells were lysed and subject to immunoprecipitation of FLAG-tagged proteins. siRNA-mediated knockdown and coimmunoprecipitation of Brd4, SMC6, and SMC5 were assessed by immunoblotting. HC, heavy chain. A lower nonspecific band was detected by the SMC6 antibody used in this experiment and is denoted by an asterisk.
SMC5/6 is not required for E2 transcription functions.
The papillomavirus E2 protein can function both as a transcriptional repressor and as a transcriptional activator. A genome-wide siRNA screen previously conducted by the Howley laboratory that included siRNAs to SMC5 and SMC6 did not identify either one to be an effector of its repression function of the HPV early E6/E7 promoter (26). To examine whether SMC6 contributes to E2-dependent transcriptional activation, we tested whether its depletion could affect E2-dependent transcriptional activation. C33A cells were reverse transfected with either control, Brd4, or SMC6 siRNA (Fig. 3A). Seventy-two hours later these cells were transfected with an E2-responsive firefly luciferase reporter plasmid (p2x2xE2BS-Luc), which contains four E2 binding sites, the E2 expression plasmid, and a Renilla luciferase reporter gene (27). BPV1 E2 alone enhanced the luciferase expression from the reporter plasmid (Fig. 3B). The E2 transactivation function is mediated by Brd4, and consistent with previous results, depletion of Brd4 by siRNA inhibited the transcriptional activation function of E2 (28). In contrast, depletion of SMC6 had no significant effect on the luciferase activity of the E2-dependent reporter plasmid, suggesting that SMC6 is not required for E2-mediated transactivation of a reporter gene (Fig. 3B). We have found similar results in experiments with HPV16 E2-mediated transactivation.
FIG 3.
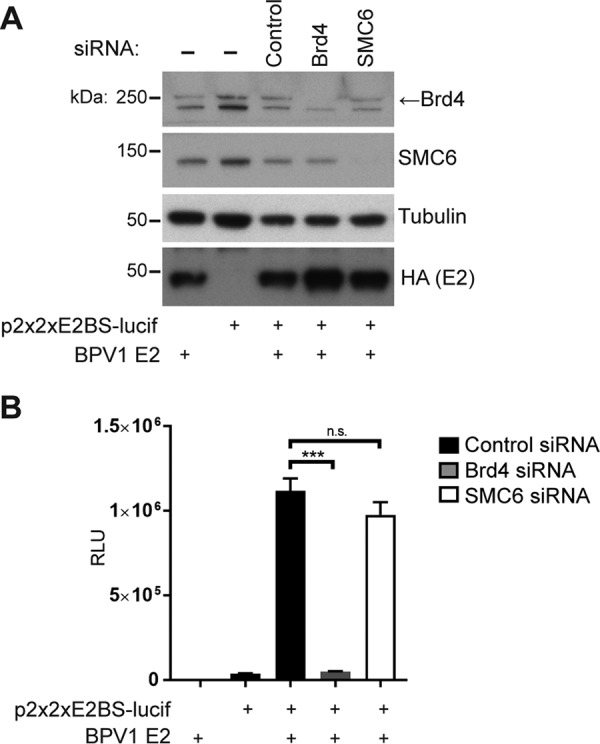
SMC5/6 does not influence E2-mediated transactivation of a reporter gene. (A) Western blot of lysates from C33A cells after transfection of siRNAs targeting Brd4 and SMC6. (B) Firefly luciferase activity of an E2 reporter construct containing E2 binding sites normalized to that of Renilla luciferase. The data represent the averages from three experiments. Statistical significance was measured using a standard Student's t test. ***, P < 0.0001; n.s., not significant. RLU, relative light units.
Previous studies have indicated that SMC5/6 restricts hepatitis B virus (HBV) infection by repressing the transcription of viral genes (25, 29). We therefore next examined whether SMC5/6 had an effect on the transcription of papillomavirus genes in CIN612-9E cells. CIN612-9E cells are a cervical squamous intraepithelial neoplasia grade 1 (CIN1) cell line that harbors episomal copies of HPV31 genomes and that differentiates in response to increased calcium levels (30). This differentiation results in the increased expression of differentiation markers, such as involucrin, which are amplified by cellular differentiation independently of viral gene expression (Fig. 4A). These cells were transduced with pLKO.1 carrying an SMC6 short hairpin RNA (shRNA) (shSMC6 #1) or pLKO.1 carrying green fluorescent protein (GFP) shRNA (shGFP), and the levels of SMC6 RNA (Fig. 4A) and various viral gene RNAs were examined by quantitative real-time PCR (qRT-PCR) in both undifferentiated and calcium-differentiated CIN612-9E cells. Although shRNA-mediated depletion of SMC6 RNA was achieved in the cells transduced with the shSMC6-expressing lentivirus, this depletion of SMC6 had no significant effect on the basal transcription of HPV genes or on their differentiation-dependent induction (Fig. 4B).
FIG 4.

SMC5/6 does not influence transcription of viral genes. (A) (Left) SMC6 knockdown in undifferentiated and differentiated CIN612-9E cells was measured by qRT-PCR. (Right) qRT-PCR for involucrin RNA was used as a marker of differentiation in CIN612-9E cells. (B) Viral early (E2, E6, E7), intermediate (E1^E4), and late (L1) genes in undifferentiated cells (3 days postinfection) and differentiated cells (7 days postinfection) were measured by qRT-PCR. The data represent the averages from three experiments. All data are normalized to those for cyclophilin A. *, P < 0.05.
SMC5/6 is not required for E1/E2-mediated transient DNA replication.
The E2 protein initiates replication of viral DNA by loading the E1 helicase onto the replication origin (10). Transient transfection of E1- and E2-encoding plasmids can be used to promote an E1/E2-mediated plasmid DNA replication that may represent the initial genome amplification that occurs immediately after PV infection (11). To determine whether SMC5/6 has a role in this function of E2, stable C33A cell lines expressing SMC6 shRNA or GFP shRNA, as a negative control, were generated by lentiviral transduction. Of the 11 shRNAs tested, those labeled numbers 1 and 2 produced the best knockdown of SMC6 and were used for subsequent experiments (Fig. 5A). The C33A-GFP shRNA and C33A-SMC6 shRNA #1 stable cell lines were used in the assays whose results are presented in Fig. 5B and C. These cells were cotransfected with plasmids encoding HPV16 E1 and HPV16 E2 and a plasmid containing the HPV16 origin of replication (ori). SMC6 protein levels were assessed by Western blotting at the time of the experiment (Fig. 5B). Replicated DNA was linearized, and input DNA was digested by DpnI. DNA was then detected by Southern blotting with a probe targeting the HPV16 ori. The level of replicated DNA was not reduced in the SMC6 knockdown cells, indicating that SMC6 is not required for E1/E2-mediated transient viral DNA replication (Fig. 5C). Indeed, the higher levels of replicated DNA observed might suggest that SMC6 could function as a replication restriction factor, although further experiments would be needed to explore that possibility.
FIG 5.
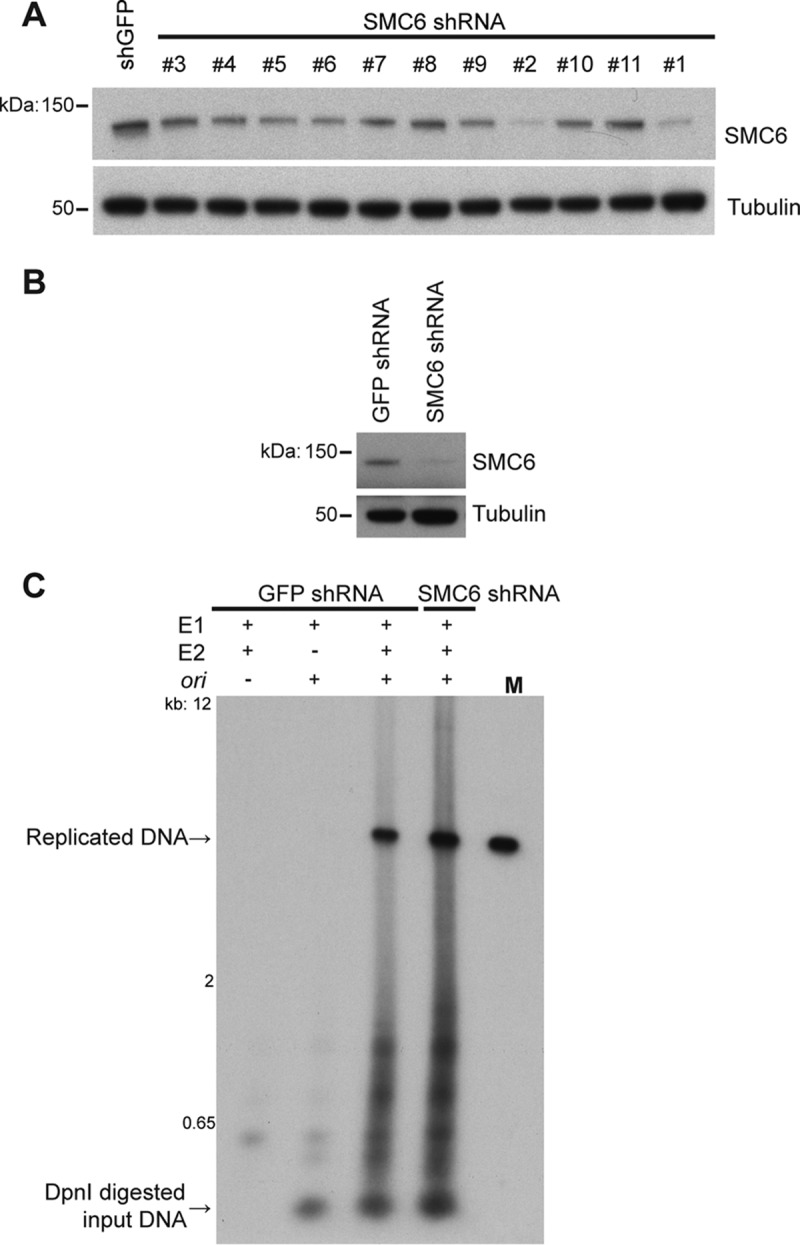
SMC5/6 is not required for optimal E1/E2-mediated DNA replication. (A) SMC6 levels in C33A cells at 4 days postransduction with the indicated shRNAs were detected by Western blotting. (B) Western blot showing SMC6 knockdown in C33A cells for the experiment whose results are shown in panel C. (C) Southern blotting using a probe for the HPV16 ori detected replicated and input DNA from C33A shGFP or shSMC6 #1 cell lines transfected with HPV16 E1 and E2 and the HPV16 ori-containing plasmid. Lane M, marker (HPV16 genome excised from the vector by digestion with the enzyme BamHI).
SMC5/6 influences maintenance of viral episomal DNA.
To determine whether the SMC5/6 complex has a role in the maintenance of viral DNA, CIN612-9E cells were transduced with GFP or SMC6 shRNA #1 lentiviruses and total DNA was harvested at several time points postinfection. Southern blotting suggested that SMC6 knockdown resulted in the loss of predominantly the monomeric episomal forms of viral DNA by 14 days (two passages) postransduction (Fig. 6A). The levels of HPV31 total DNA were also assessed by quantitative PCR (qPCR), revealing an approximately 50% reduction of viral DNA 3 days after transduction in four experiments (Fig. 6B). Despite the loss of viral episomes, cells depleted of SMC6 did not display a growth deficiency (Fig. 6C). Linearization of viral DNA through digestion with XbaI reduced concatemeric, integrated, nicked, and episomal forms of viral DNA into one band that ran at 8 kbp. Southern blotting for linearized DNA indicated that viral DNA was still present in these cells in SMC6 knockdown cells (Fig. 6D). We propose that this remaining DNA may be integrated in the mammalian genome and that the loss of SMC5/6 over the course of several days may lead to selection for cells that can proliferate in the absence of viral episomes.
FIG 6.

SMC6 influences the maintenance of the episomal form of HPV31 DNA. (A) Total DNA was extracted from CIN612-9E cells at 3 and 14 days after lentiviral shRNA transduction. (Top) A probe targeting the HPV31 URR was used to identify viral DNA by Southern blotting; (bottom) ethidium bromide staining of the Southern blot for DNA. (B) Quantification of total viral DNA extracted from CIN612-9E cell lines at 3 days posttransfection. DNA was measured by qPCR, and the results were normalized to those for beta-actin (n = 4). (C) Number of cells counted by use of a hemocytometer 7 days after plating 100,000 cells per plate. (D) Southern blotting with a probe targeting HPV31 detects the XbaI-digested linear form at approximately 8 kbp. The bottom half was probed for tissue plasminogen activator gene (tPa) to control for loading (1 to 2 kbp). Lane M, marker for linear HPV31 DNA (1 ng HPV31 genome excised from the pLit vector).
SMC5/6 is not required for differentiation-dependent amplification of viral DNA.
SMC1 is required for differentiation-dependent amplification of HPV31 DNA in CIN612-9E cells, and SMC5/6 has been shown to recruit SMC1 to damaged cellular replication forks for their repair (17, 31). We therefore next examined whether SMC5/6 also had a role in the PV replication cycle. CIN612-9E cells were transduced with control (GFP) shRNA or SMC6 shRNA #1 or #2. Undifferentiated cells were collected at 3 days postransduction, and a subset was grown to confluence in high-calcium medium to induce differentiation. Cellular DNA was extracted and digested with BamHI, transferred to nitrocellulose filters, and probed for HPV31 DNA. BamHI cleaves the mammalian DNA but not HPV31 DNA. Calcium treatment led to an increase in involucrin mRNA in these cells (Fig. 7A), and the SMC6 shRNAs reduced the levels of SMC6 compared to those in control cells (Fig. 7B). Southern blotting was used to assess the differentiation-dependent amplification of HPV31 DNA in these cells. Increased levels of episomal DNA were observed in all CIN612-9E cell lines after calcium-mediated differentiation (Fig. 7C). This suggests that SMC5/6 is not required for differentiation-dependent amplification of viral DNA, although we cannot rule out the possibility that it is required and that the reduced levels of SMC5/6 remaining after shRNA depletion might be sufficient to support this level of DNA replication. However, a marked decrease in viral DNA, particularly for the monomeric episomal form, occurred in the undifferentiated cells 3 days after shRNA transduction, suggesting a role for SMC6 in episome maintenance (Fig. 7C). However, the increase in episomal DNA with calcium-induced differentiation suggests that, although markedly reduced, some of the episomal form is still present in these cells (Fig. 7C).
FIG 7.

SMC6 is not required for differentiation-dependent amplification of viral DNA. CIN612-9E cells were lentivirally transduced with control or SMC6 shRNA on day 0. Uninfected cells were collected at day 0. At 3 days postinfection, undifferentiated cells were harvested for analysis by qRT-PCR and Southern blotting or underwent 96 h of differentiation in high-calcium medium. (A) qRT-PCR measured involucrin levels as a marker of keratinocyte differentiation. (B) SMC6 mRNA levels were measured by qRT-PCR in CIN612-9E cells lentivirally transduced with SMC6 shRNA or GFP shRNA. All qRT-PCR data are normalized to those for cyclophilin A (n = 4). (C) (Left) Southern blotting detected HPV31 DNA from CIN612-9E cells. (Top) Total DNA was digested with BamHI targeting only mammalian DNA for visualization of the various forms of viral DNA. (Middle) Alternatively, DNA was digested with XbaI to linearize viral DNA (8 kbp). (Bottom) The Southern blot containing XbaI-digested DNA was probed for the human tPa gene as a loading control (1 to 2 kbp). (Right) Densitometric analysis of the episomal form of DNA as visualized by Southern blotting. P values were calculated using Student's t test. ***, P < 0.0001; **, P < 0.001; *, P < 0.05.
SMC5/6 is present at HPV31 replication foci.
E2 is required for the formation of viral DNA replication foci in differentiated cells, where various cellular DNA damage response and replication proteins are present and where viral DNA is replicated (32). Because SMC5/6 is a core component of the DNA damage response and may play a role in viral DNA maintenance, we sought to determine whether it is present at viral DNA replication centers. Replication protein A (RPA) is among the numerous components of the DDR that have been previously shown to be recruited to viral replication foci (32–35). It is required for the repair of double-strand breaks, where it binds to single-stranded DNA and facilitates homologous recombination (36, 37). We sought to determine whether the localization of SMC5/6 is altered in HPV-positive cells, using RPA as a marker for viral replication centers. Immunofluorescence was used to examine the localization of SMC5/6 components SMC5, SMC6, and NSE4 in differentiated CIN612-9E cells. Here, SMC6, SMC5, and NSE4 colocalized with RPA in differentiated CIN612 cells; however, this colocalization was not observed in differentiated HPV-negative NIKS cells, a spontaneously immortalized human keratinocyte cell line (38) (Fig. 8A to C). However, because SMC5/6 is required for the loading of RPA onto stalled replication forks in host DNA, it is plausible that the observed RPA-SMC5/6 colocalization is unrelated to PV infection of these cells (12). Therefore, to determine whether SMC5/6 is present at viral replication centers, fluorescent in situ hybridization (FISH) with a probe targeting HPV31 was used to visualize viral DNA in CIN612 cells. Here, SMC5 and SMC6 both colocalized with viral DNA, which was most notably observed with the larger foci (Fig. 8D and E). Further studies will need to address the functional significance of this association with the larger replication foci. Three-dimensional rendering of stacks of images throughout the nuclei confirmed that SMC6 is present within viral DNA foci (Fig. 8E). These data suggest that localization of the SMC5/6 complex is altered in HPV-positive cells and that it is present at viral replication centers, supporting its role in some aspect of papillomavirus DNA replication or episome maintenance.
FIG 8.

SMC5/6 localizes to PV replication foci. HPV-negative NIKS cells and cells of the HPV31-positive cell line CIN612-9E were grown on coverslips, fixed, and permeabilized for analysis by immunofluorescence (IF). Cells underwent 5 days of differentiation in 1.5 mM calcium medium. Blue represents nuclear DAPI staining. (A to C) Samples were stained with anti-SMC6 (green) and anti-RPA (red) antibodies (A), anti-NSE4 (green) and anti-RPA (red) antibodies (B), and anti-SMC5 (green) and anti-RPA (red) antibodies (C). (D) Fluorescent in situ hybridization (FISH) was used to detect HPV31 DNA (green) in calcium-differentiated CIN612-9E cells. Anti-SMC5 was used to visualize SMC5 (red) localization. (E) HPV31 DNA (green) was visualized using FISH, and red indicates SMC6 staining. z-stack images of the nucleus were collected and deconvolved, and a 3D reconstruction was generated in Imaris software.
DISCUSSION
The papillomavirus E2 protein is an essential viral regulatory factor with important functions in viral transcription, viral DNA replication, and viral genome maintenance (9). These E2 functions appear to be largely conserved among different PV types, although various E2-host protein interactions may vary among PVs. For example, Brd4 is a cellular interacting partner of E2 proteins of all PV types that have been examined; however, the affinities of Brd4-E2 binding and the functions of this interaction vary among PV species (39–41). While an E2 interaction with Brd4 is required for the transactivation function of all E2 types tested so far, it has been shown to be required for the tethering function of only a subset of papillomaviruses: HPV1, rabbit oral papillomavirus (ROPV), and those of the Deltapapillomavirus genus (including BPV) (41–43). Here, we identified and validated the interactions of the host factors SMC5 and SMC6 with E2. We investigated the conservation of the interaction among PV types and its potential roles in the papillomavirus replication cycle. Our data suggest that the SMC5/6 interaction may be mediated by SMC6 and is conserved among a number of different PV E2 proteins within the Alphapapillomavirus, Betapapillomavirus, and Deltapapillomavirus genera. Colocalization with HPV31 DNA foci suggests that this interaction may also extend to HPV31 E2. While our data suggest that this interaction extends to the E2 proteins of different PV types, it remains to be determined whether or not the role of the interaction is also conserved among PV types.
Because of the interaction of E2 with SMC5/6, we examined whether SMC5/6 might be involved in any activities associated with E2. SMC5/6 has been previously linked to virus-host cell interactions. It acts as a restriction factor for hepatitis B virus (HBV), where it associates with extrachromosomal reporters and the HBV genome to directly inhibit transcription (25). This is counteracted by an interaction with the HBV X protein, which targets SMC6 for degradation (25, 29, 44). We did not find evidence that SMC5/6 affected E2 transcriptional activation or affected transcription of viral genes. SMC6 protein levels were also not influenced by the presence of E2. This suggests different roles of SMC5/6 in the replication cycles of HBV and papillomaviruses.
E2 plays an important role in viral DNA replication, and so we next sought to determine whether SMC5/6 influenced PV DNA replication. It is generally accepted that PVs undergo three phases of DNA replication during their replication cycle: initial DNA replication (genome establishment), maintenance replication, and vegetative (differentiation-dependent) DNA amplification. We found that SMC5/6 was not required for E1/E2-mediated DNA replication, which may be representative of the initial replication that occurs soon after cells are infected, nor was it required for vegetative DNA amplification. However, in cells in which viral episomes are established, SMC6 depletion did influence viral DNA maintenance. The mechanism of the initial transient replication is believed to be conserved among PVs and requires the viral E1 protein (45, 46). During maintenance replication, circular viral episomes are maintained at a relatively constant copy number of 50 to 200 copies per cell throughout several cell divisions and are partitioned into the nuclei of two daughter cells (47). During this stage of the viral replication cycle, SMC5/6 may have a role in the resolution of multimeric viral DNA to monomeric forms or in tethering HPV31 genomes to mitotic chromatin. Another possibility is that SMC5/6 has some other role in PV replication in the maintenance phase. The DDR and HR machinery has been suggested to facilitate the initial and differentiation-dependent amplification of PV episomes, as well as maintenance of episome stability (31, 34, 48–50). Whether and how PVs switch between mechanisms of DNA replication during different phases of the replication cycle and the role of the DDR/HR machinery during these stages are not yet well understood, but it is unlikely that viral DNA replication adheres to conventional rules of chromosomal DNA replication. SMC5/6 has been shown to have a role in chromosome dynamics (51), and our data suggest that it might also have a role in PV genome dynamics, possibly in resolving replicated genomes.
Total viral DNA decreased in the absence of SMC6 in CIN612 cells. This was most prominent for the monomeric episomal form but may have also been in the form of head-to-tail concatemers that migrate at a high molecular weight and that may or may not have been integrated into host chromosomes. It has been suggested that the formation of head-to-tail concatemers of viral DNA occurs through a recombination-dependent mechanism (52). It has also been postulated that the fusion of PV and human DNA occurs through a homology-mediated DNA repair pathway, such as homologous recombination (53, 54). Unlike retroviruses, papillomaviruses do not encode a factor that facilitates viral DNA integration. Papillomavirus replication occurs near fragile sites in host DNA, where integration is believed to be facilitated by double-stranded DNA breaks in host DNA (54–61). Here we show that SMC5/6 was observed to localize at papillomavirus replication foci in differentiated keratinocytes, similar to several other HR and DDR factors (31, 34, 35, 48–50). Disruption of the cellular HR process by depletion of SMC5/6 may affect the resolution of replicated forms of the DNA from multimers to monomers or disrupt a recombination-mediated mechanism of maintenance replication, oligomerization, and/or integration. Thus, depletion of this complex may have led to selection for cells that harbored integrated copies prior to SMC5/6 depletion. Our data indicate that further studies on the role of the HR and DDR machinery in PV genome maintenance are warranted.
MATERIALS AND METHODS
Plasmids.
To generate the pHAGE lentiviral expression vectors used in the proteomics studies, BPV1 E2 and E2TR (P. M. Howley laboratory plasmid numbers p7211 and p7212, respectively) were PCR amplified from a BPV1 genomic plasmid and E8/E2C was PCR amplified from the pE8E2 plasmid and cloned into the lentiviral vector using Gateway cloning (p7214) (Invitrogen) (62, 63). Human papillomavirus E2 ORFs with XhoI and NotI restriction sites at their 5′ and 3′ ends, respectively, were amplified from cloned HPV genomes (22, 64, 65) and subcloned into the pOZ-N-HA-PURO vector (where PURO stands for puromycin resistance) using the XhoI and NotI sites, and the sequence was verified. The E2 proteins used in this study were from HPV6b (p6976), HPV18 (p6869), HPV5 (p6980), HPV8 (p6981), and BPV1 (p6989). E2 and control (GFP) proteins were also recombined into the Phage-CMV-N-FLAG (3×)-HA vector (a gift of J. Wade Harper, Harvard Medical School) by Gateway cloning [phage-CMV-N-FLAG (3×)-HA-HPV8 E2, p7960; phage-CMV-N-FLAG (3×)-HA-HPV18 E2, p7961; phage-CMV-N-FLAG (3×)-HA-GFP, p7959]. The HPV16 E6 and E1 plasmids have been previously described (21, 27). The p2x2xE2BS-Luc and p16ori plasmids have been previously described (27, 66). The pMEP4 HPV16 and HPV31 E2 plasmids have also been previously described (19).
Cell lines.
C33A and 293T cells were maintained according to standard protocols in Dulbecco modified Eagle medium (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (HyClone) and 1% penicillin-streptomycin (Gibco/Invitrogen). Retroviral transduction of 293T cells was used to generate stable cell lines for HA-immunoprecipitation/mass spectrometry (IP/MS) analysis. CIN612-9E cells were cultured in E medium with 3T3-J2 fibroblast feeders according to the protocol previously described by Fehrmann and Laimins (67). Feeders were removed using Versene prior to protein, RNA, or DNA analysis of CIN612-9E cells. For the immunofluorescence and FISH experiments, CIN612-9E and NIKS cells were cultured in F medium with 3T3-J2 fibroblast feeders, as described previously (38).
For calcium-induced differentiation of CIN612-9E cells, feeders were removed using Versene, and cells were rinsed with phosphate-buffered saline (PBS) and cultured for 24 h in keratinocyte basal medium (KBM; Lonza) containing SingleQuots additives for keratinocytes (Lonza). Cells were then rinsed with PBS and cultured for 96 h in KBM with 1.5 mM CaCl2.
Immunoprecipitation and mass spectrometry.
HA-tagged bait proteins stably expressed in 293T cells were grown to 90% confluence in four 15-cm plates for immunoprecipitation/mass spectrometry (IP/MS). Cells were harvested in mammalian cell lysis buffer (MCLB; 150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 0.5% [vol/vol] Nonidet P-40, 1 mM EDTA, protease inhibitors). The lysate was sonicated and clarified by centrifugation prior to overnight incubation with monoclonal anti-HA-agarose (Sigma) at 4°C as described previously (21, 22, 65). After thorough washing, proteins were eluted with 250 μg/ml HA peptide (Sigma), concentrated by trichloroacetic acid precipitation, and washed with acetone.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was carried out using previously described methods, where peptides were identified using Sequest software and analyzed using the Comparative Proteomics Analysis Software Suite (CompPASS) (21, 22, 65).
Immunoprecipitation-Western blotting.
pOZ-N-HA-PURO-E2 or negative-control constructs were transiently transfected in C33A cells in 10-cm plates using the Fugene 6 reagent. Cells were harvested at 72 h posttransfection and lysed in MCLB, and the lysate was sonicated and clarified by centrifugation. Anti-HA magnetic beads (catalog number 88836; Pierce) were used to elute HA-tagged proteins and their binding partners at 4°C, as described above. For Western blotting, co-IPs were performed as described above; however, bound proteins were eluted by boiling in reducing sample buffer for 5 min.
For siRNA-immunoprecipitation experiments, 1.5 × 105 cells of each C33A cell line were reverse transfected with 40 nM siRNA (Dharmacon) using 5 μl of the DharmaFECT 2 reagent according to the manufacturer's instructions. Cells were harvested at 96 h posttransfection and lysed in MCLB. The lysate was then sonicated, clarified by centrifugation, and subject to immunoprecipitation overnight at 4°C with anti-FLAG M2 magnetic beads (catalog number M8823; Sigma) before boiling in reducing sample buffer to elute bound proteins. The following primary antibodies were used for Western blotting: actin-horseradish peroxidase (HRP) (catalog number 49900; Abcam), SMC6 (catalog number AT3956a; Abgent), anti-SMC5 (catalog number 18038; Abcam), alpha-tubulin (catalog number T6199; Sigma), Brd4 (catalog number A301-985A50; Bethyl Laboratories), anti-FLAG (catalog number F3165; Sigma), and anti-HA-HRP (catalog number 12013819001; Roche).
Reporter assays.
C33A cells (1.5 × 105) were reverse transfected with 40 nM siRNA using the DharmaFECT 2 reagent (Dharmacon) according to the manufacturer's protocol. After 24 h, cells were transfected with 0.41 μg p2x2xE2BS-Luc, 0.81 μg E2, and 200 ng Renilla luciferase using the Fugene 6 reagent (Roche) at a 3:1 ratio (microliters per microgram of DNA) according to the manufacturer's protocol. Seventy-two hours later, cells were lysed and luciferase activity was measured using a dual-luciferase reporter assay system (Promega) and a SpectraMax L luminescence microplate reader (Molecular Devices). Firefly luciferase readings were normalized to the respective Renilla luciferase readings. siRNAs were obtained from Dharmacon.
Lentiviral knockdowns.
The SMC5 and SMC6 shRNAs used in this study (MmSH00248729, MmSH00249026) were obtained from the DF/HCC DNA resource Core and were expressed from a pLKO.1 PURO vector (Sigma). Two micrograms of shRNA and 0.5 μg of lentivirus packaging plasmids were transfected into 293T cells to produce lentivirus particles. Twenty-four hours later the medium was changed to that specific for the target cells, and the virus-producing cells were allowed to grow for another 24 h. The virus supernatant was then collected and filtered through a 0.45-μm-pore-size filter and titrated in C33A cells to determine the optimal volume for shRNA-mediated knockdown. Stocks of virus were frozen at −80°C. CIN612-9E cells were lentivirally transduced with control or SMC6 shRNA. Uninfected cells were collected at day 0. At 3 days postinfection, undifferentiated cells were harvested for analysis by qRT-PCR and Southern blotting or underwent 96 h of differentiation in high-calcium medium.
Transient papillomavirus DNA replication.
E1/E2-mediated DNA replication was assessed in C33A cells according to a previously described protocol (28, 45). C33A cells (2 × 105) were infected with 1 ml of lentivirus medium supplemented with 4 ml of medium and 0.8 μg/ml Polybrene for 2 h and selected with 0.75 μg/ml puromycin 24 h later. Approximately 72 h later, 5 × 105 cells for each cell line were cotransfected with the previously described plasmids pCMV 16E1 and pCMV 16E2 using the Fugene 6 reagent (Roche) (28). Low-molecular-weight DNA was collected using the Hirt method (68) and treated with DpnI to digest unreplicated transfected DNA, and the replicated DNA was linearized by AccI digestion. DNA was detected by Southern blotting with a probe targeting the HPV16 ori using a digoxigenin (DIG) DNA labeling and detection kit (Roche). The primer sequences used to generate the probe were CAAACCGTTTTGGGTTACAC (forward) and TGCAGTTCTCTTTTGGTGCAT (reverse). The HPV16 genome was excised from the pUC19 vector by BamHI digestion and has been previously described (69).
Quantitative real-time PCR (qRT-PCR).
Total RNA was extracted using a NucleoSpin RNA II kit. The RNA concentration was determined by use of a NanoDrop spectrophotometer, and equivalent amounts were reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Gene expression levels were determined using primers specific for each gene, an Applied Biosystems ABI 7500 fast sequence detection system, and SYBR Select Mastermix (Thermo Fisher Scientific). Serial dilutions of a reference cDNA were used to generate a standard curve and determine the relative amounts of cDNA in each sample. All data were normalized to those for cyclophilin A, which has been previously indicated to not vary during keratinocyte differentiation (70). Predesigned SYBR green primers for SMC6 (H_SMC6_3), involucrin, (H_IVL_1), and cyclophilin A (H_PPIA_1) were purchased from Sigma. The following gene-specific primers were used to measure viral gene transcripts: for HPV31 E2, sense primer 5′-ATGGCTGATCCAGCAGGAC-3′ and antisense primer 5′-CGTTGAGAAAGAGTCTCCATCG-3′; for HPV31 E6, sense primer 5′-AGATTGAATTGTGTCTACTGCAAAGGTGT-3′ and antisense primer 5′-GCTATGCAACGTCCTGTCCACCT-3′; for HPV31 E7, sense primer 5′-TAGGAGGAAGGTGGACAGGA-3′ and antisense primer 5′-GCTGTCGGGTAATTGCTCAT-3′; for HPV31 E1^E4, sense primer 5′-CTACAATGGCTGATCCAGCAGCA-3′ and antisense primer 5′-CGCCGCACACCTTCACTGG-3′; and for L1, sense primer 5′-ACACTTAAAAGATGTCTCTGTGGC-3′ and antisense primer 5′-TCGTGTTACATATTCATCCGTGC-3′.
Southern blotting.
Total DNA was extracted from CIN612-9E cells using previously described methods (71). DNA was quantified by use of the NanoDrop spectrophotometer, and for each experiment, equal amounts were loaded for Southern blotting and visualized with a probe targeting the HPV31 upstream regulatory region (URR). DNA was digested with BamHI-HF (New England BioLabs [NEB]) overnight at 37°C to cleave only mammalian DNA or with XbaI (NEB) to linearize viral DNA. Mammalian DNA cleaved with XbaI produced an ∼1.8-kbp fragment of the tissue plasminogen activator (tPa; gene identifier, 5327), which was used as a loading control for Southern blotting, where the bottom half of the blot was probed for the tPa gene and the top half was probed for the HPV31 URR (∼8 kbp). Probes were generated using the DIG DNA labeling and detection kit (Roche). The primer sequences used to generate the probe were CTGGCTTGTAGTTTCCTGCC (forward) and AAAGCCAGCACTGCAATCAA (reverse). The probe consisted of 206 nucleotides from positions 7576 to 7782 in the HPV31 URR. The HPV31 genome can be found at http://pave.niaid.nih.gov (72). DNA was run on a 0.8% agarose gel, depurinated, denatured, and neutralized before transfer onto a positively charged nylon membrane (Amersham Hybond-N+) overnight. After UV cross-linking at 120 mJ/cm2, the membrane was prehybridized in ULTRAhyb ultrasensitive hybridization buffer (Thermo Fisher Scientific) at 42°C for 30 min prior to overnight hybridization with a probe targeting the HPV31 URR. Membrane washing and detection were done using the DIG DNA labeling and detection kit (Roche) according to the manufacturer's instructions. HPV31 excised from the pLIT31 vector (a gift of L. Laimins) was used as a marker for the linear form on the Southern blot and has been previously described (73).
qPCR for HPV31 DNA.
DNA was extracted from CIN612-9E cells using previously described methods (71). DNA was measured by use of the NanoDrop spectrophotometer (Thermo Fisher Scientific) and digested overnight at 37°C with BamHI-HF and XbaI (New England BioLabs). Serial dilutions of the reference CIN612 DNA ranging from 0.5 ng to 125 ng were used to generate a standard curve for determining relative DNA amounts. Primers for HPV 31 were used to quantify viral DNA, which was normalized to the qPCR data for beta-actin. The following gene-specific primers were used: for HPV31, CTGACCTCCACTGTTATGAGCAA (forward) and CAGCTGGACTGTCTATGACATCCT (reverse); for beta-actin, TACGTCCACCGCAAATGC (forward) and CGCAAGTTAGGTTTTGTCAAGAAA (reverse). qPCR was performed using SYBR Select Mastermix (Thermo Fisher Scientific) and an Applied Biosystems ABI 7500 fast sequence detection system.
Immunofluorescence and FISH.
CIN612-9E cells were seeded on 18-mm glass coverslips and grown to confluence prior to differentiation in KGM medium (Lonza) with 1.5 mM calcium chloride for 5 days. Cells were fixed, blocked, and permeabilized using previously described methods (74). Coverslips were incubated in primary antibodies at 37 degrees for 1 h. The primary antibodies used were mouse anti-SMC6L1 (1:500; catalog number AT3955a; Abgent), rabbit anti-SMC5 (1:100; catalog number 18038 from Abcam or catalog number A300-236A-T from Bethyl), rabbit anti-SMC6 (1:100; catalog number A300-237-T; Bethyl), rabbit anti-NSE4A (1:100; catalog number AP9909a; Abgent), and rat anti-RPA (1:200; catalog number 2208; Cell Signaling). Coverslips were washed and incubated in the secondary antibodies (1:50) Alexa Fluor 488 and Alexa Fluor 594 at 37°C for 30 min.
A FISH probe for HPV31 was generated using a FISH-tag multicolor kit (Thermo Life Technologies). Following immunofluorescence (described above), coverslips were fixed in methanol-acetic acid (3:1) for 10 min, followed by 2% paraformaldehyde for 1 min. Coverslips were washed and treated with 1× RNace-It cocktail (Stratagene) and washed in PBS and ethanol as described previously (74). After air drying for 1 h, each coverslip was incubated in 67.6 ng of FISH probe with Cot-1 DNA (Invitrogen) and hybridization buffer (Empire Genomics) at 75°C for 5 min followed by 37°C overnight. Cells were washed, stained with DAPI (4′,6-diamidino-2-phenylindole), and mounted using Prolong Gold antifade reagent (Life Technologies). Images were captured with a 63× objective lens using a Leica TCS-SP5 laser scanning confocal microscope. Images for three-dimensional (3D) analysis were deconvolved using the Huygens Essential software package (Scientific Volume Imaging B.V., VB Hilversum, Netherlands), manual background selection, and a signal-to-noise ratio of 25:1. Final images were rendered using Imaris software (v7.7.1; Bitplane AG, Zurich, Switzerland).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R35CA197262 to P.M.H. and F31CA189512 to P.B. and also by the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases (grant 1ZIAAI000713 to A.A.M.).
We thank J. W. Harper and his laboratory in cell biology for assistance with LC-MS/MS experiments and analyses and members of the A. A. McBride laboratory for help with the imaging studies.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00356-18.
REFERENCES
- 1.Munger K, Scheffner M, Huibregtse JM, Howley PM. 1992. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv 12:197–217. [PubMed] [Google Scholar]
- 2.Thierry F, Yaniv M. 1987. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J 6:3391–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thierry F, Howley PM. 1991. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol 3:90–100. [PubMed] [Google Scholar]
- 4.Bernard BA, Bailly C, Lenoir MC, Darmon M, Thierry F, Yaniv M. 1989. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J Virol 63:4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang ES, Riese DJ II, Settleman J, Nilson LA, Honig J, Flynn S, DiMaio D. 1993. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J Virol 67:3720–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon S, Allen-Hoffmann BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 69:2989–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeon S, Lambert PF. 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A 92:1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romanczuk H, Howley PM. 1992. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci U S A 89:3159–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc Natl Acad Sci U S A 90:5086–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360–368. doi: 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 12.Ampatzidou E, Irmisch A, O'Connell MJ, Murray JM. 2006. Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26:9387–9401. doi: 10.1128/MCB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Piccoli G, Cortes-Ledesma F, Ira G, Torres-Rosell J, Uhle S, Farmer S, Hwang JY, Machin F, Ceschia A, McAleenan A, Cordon-Preciado V, Clemente-Blanco A, Vilella-Mitjana F, Ullal P, Jarmuz A, Leitao B, Bressan D, Dotiwala F, Papusha A, Zhao X, Myung K, Haber JE, Aguilera A, Aragon L. 2006. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat Cell Biol 8:1032–1034. doi: 10.1038/ncb1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Piccoli G, Torres-Rosell J, Aragon L. 2009. The unnamed complex: what do we know about Smc5-Smc6? Chromosome Res 17:251–263. doi: 10.1007/s10577-008-9016-8. [DOI] [PubMed] [Google Scholar]
- 15.Lindroos HB, Strom L, Itoh T, Katou Y, Shirahige K, Sjogren C. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol Cell 22:755–767. doi: 10.1016/j.molcel.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Menolfi D, Delamarre A, Lengronne A, Pasero P, Branzei D. 2015. Essential roles of the Smc5/6 complex in replication through natural pausing sites and endogenous DNA damage tolerance. Mol Cell 60:835–846. doi: 10.1016/j.molcel.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potts PR, Porteus MH, Yu H. 2006. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J 25:3377–3388. doi: 10.1038/sj.emboj.7601218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang MK, Anderson DE, van Doorslaer K, McBride AA. 2015. A proteomic approach to discover and compare interacting partners of papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics 15:2038–2050. doi: 10.1002/pmic.201400613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan X, Sarangi P, Liu X, Rangi GK, Zhao X, Ye H. 2009. Structural and functional insights into the roles of the Mms21 subunit of the Smc5/6 complex. Mol Cell 35:657–668. doi: 10.1016/j.molcel.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White EA, Kramer RE, Tan MJ, Hayes SD, Harper JW, Howley PM. 2012. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J Virol 86:13174–13186. doi: 10.1128/JVI.02172-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM. 2012. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci U S A 109:E260–E267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. 2015. The BioPlex Network: a systematic exploration of the human interactome. Cell 162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor EM, Copsey AC, Hudson JJ, Vidot S, Lehmann AR. 2008. Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex. Mol Cell Biol 28:1197–1206. doi: 10.1128/MCB.00767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O, Strubin M. 2016. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531:386–389. doi: 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 26.Smith JA, White EA, Sowa ME, Powell ML, Ottinger M, Harper JW, Howley PM. 2010. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci U S A 107:3752–3757. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakai H, Yasugi T, Benson JD, Dowhanick JJ, Howley PM. 1996. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J Virol 70:1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweiger MR, You J, Howley PM. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol 80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, Wu Y, Yu Y, Xiong Y, Su L. 2016. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16:2846–2854. doi: 10.1016/j.celrep.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD, Laimins LA. 1991. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J Virol 65:2254–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta K, Gunasekharan V, Satsuka A, Laimins LA. 2015. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog 11:e1004763. doi: 10.1371/journal.ppat.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anacker DC, Moody CA. 2017. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res 231:41–49. doi: 10.1016/j.virusres.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol 86:9520–9526. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Binz SK, Sheehan AM, Wold MS. 2004. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 37.Ruff P, Donnianni RA, Glancy E, Oh J, Symington LS. 2016. RPA stabilization of single-stranded DNA is critical for break-induced replication. Cell Rep 17:3359–3368. doi: 10.1016/j.celrep.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O'Connor SL. 2000. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J Investig Dermatol 114:444–455. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 39.Cardenas-Mora J, Spindler JE, Jang MK, McBride AA. 2008. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J Virol 82:7298–7305. doi: 10.1128/JVI.00772-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol 80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sekhar V, Reed SC, McBride AA. 2010. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J Virol 84:543–557. doi: 10.1128/JVI.01908-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. 2009. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J Virol 83:640–650. doi: 10.1128/JVI.01936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117:349–360. doi: 10.1016/S0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 44.Niu C, Livingston CM, Li L, Beran RK, Daffis S, Ramakrishnan D, Burdette D, Peiser L, Salas E, Ramos H, Yu M, Cheng G, Strubin M, Delaney WI, Fletcher SP. 2017. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS One 12:e0169648. doi: 10.1371/journal.pone.0169648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Del Vecchio AM, Romanczuk H, Howley PM, Baker CC. 1992. Transient replication of human papillomavirus DNAs. J Virol 66:5949–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flores ER, Lambert PF. 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol 71:7167–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maglennon GA, McIntosh P, Doorbar J. 2011. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 414:153–163. doi: 10.1016/j.virol.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. 2013. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J Virol 87:3979–3989. doi: 10.1128/JVI.03473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edwards TG, Vidmar TJ, Koeller K, Bashkin JK, Fisher C. 2013. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PLoS One 8:e75406. doi: 10.1371/journal.pone.0075406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spriggs CC, Laimins LA. 2017. FANCD2 binds human papillomavirus genomes and associates with a distinct set of DNA repair proteins to regulate viral replication. mBio 8:e02340-16. doi: 10.1128/mBio.02340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanno T, Berta DG, Sjogren C. 2015. The Smc5/6 complex is an ATP-dependent intermolecular DNA linker. Cell Rep 12:1471–1482. doi: 10.1016/j.celrep.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 52.Orav M, Henno L, Isok-Paas H, Geimanen J, Ustav M, Ustav E. 2013. Recombination-dependent oligomerization of human papillomavirus genomes upon transient DNA replication. J Virol 87:12051–12068. doi: 10.1128/JVI.01798-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, von Knebel Doeberitz M. 2003. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 22:3977–3984. doi: 10.1038/sj.onc.1206629. [DOI] [PubMed] [Google Scholar]
- 54.Senapati R, Senapati NN, Dwibedi B. 2016. Molecular mechanisms of HPV mediated neoplastic progression. Infect Agents Cancer 11:59. doi: 10.1186/s13027-016-0107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McBride AA. 2017. Playing with fire: consequences of human papillomavirus DNA replication adjacent to genetically unstable regions of host chromatin. Curr Opin Virol 26:63–68. doi: 10.1016/j.coviro.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 56.Jang MK, Shen K, McBride AA. 2014. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog 10:e1004117. doi: 10.1371/journal.ppat.1004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cannizzaro LA, Durst M, Mendez MJ, Hecht BK, Hecht F. 1988. Regional chromosome localization of human papillomavirus integration sites near fragile sites, oncogenes, and cancer chromosome breakpoints. Cancer Genet Cytogenet 33:93–98. doi: 10.1016/0165-4608(88)90054-4. [DOI] [PubMed] [Google Scholar]
- 58.Smith PP, Friedman CL, Bryant EM, McDougall JK. 1992. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 5:150–157. doi: 10.1002/gcc.2870050209. [DOI] [PubMed] [Google Scholar]
- 59.Thorland EC, Myers SL, Persing DH, Sarkar G, McGovern RM, Gostout BS, Smith DI. 2000. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res 60:5916–5921. [PubMed] [Google Scholar]
- 60.Dall KL, Scarpini CG, Roberts I, Winder DM, Stanley MA, Muralidhar B, Herdman MT, Pett MR, Coleman N. 2008. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res 68:8249–8259. doi: 10.1158/0008-5472.CAN-08-1741. [DOI] [PubMed] [Google Scholar]
- 61.Gao G, Johnson SH, Vasmatzis G, Pauley CE, Tombers NM, Kasperbauer JL, Smith DI. 2017. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 56:59–74. doi: 10.1002/gcc.22415. [DOI] [PubMed] [Google Scholar]
- 62.Heilman CA, Engel L, Lowy DR, Howley PM. 1982. Virus-specific transcription in bovine papillomavirus-transformed mouse cells. Virology 119:22–34. doi: 10.1016/0042-6822(82)90061-7. [DOI] [PubMed] [Google Scholar]
- 63.Lambert PF, Monk BC, Howley PM. 1990. Phenotypic analysis of bovine papillomavirus type 1 E2 repressor mutants. J Virol 64:950–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bennett EJ, Rush J, Gygi SP, Harper JW. 2010. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 143:951–965. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovelman R, Bilter GK, Glezer E, Tsou AY, Barbosa MS. 1996. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses. J Virol 70:7549–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fehrmann F, Laimins LA. 2005. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 292:317–330. [DOI] [PubMed] [Google Scholar]
- 68.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 69.Flores ER, Allen-Hoffmann BL, Lee D, Sattler CA, Lambert PF. 1999. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 262:344–354. doi: 10.1006/viro.1999.9868. [DOI] [PubMed] [Google Scholar]
- 70.Steele BK, Meyers C, Ozbun MA. 2002. Variable expression of some “housekeeping” genes during human keratinocyte differentiation. Anal Biochem 307:341–347. doi: 10.1016/S0003-2697(02)00045-3. [DOI] [PubMed] [Google Scholar]
- 71.Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 77:2819–2831. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Doorslaer K, Tan Q, Xirasagar S, Bandaru S, Gopalan V, Mohamoud Y, Huyen Y, McBride AA. 2013. The Papillomavirus Episteme: a central resource for papillomavirus sequence data and analysis. Nucleic Acids Res 41:D571–D578. doi: 10.1093/nar/gks984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8^E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stepp WH, Stamos JD, Khurana S, Warburton A, McBride AA. 2017. Sp100 colocalizes with HPV replication foci and restricts the productive stage of the infectious cycle. PLoS Pathog 13:e1006660. doi: 10.1371/journal.ppat.1006660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.