

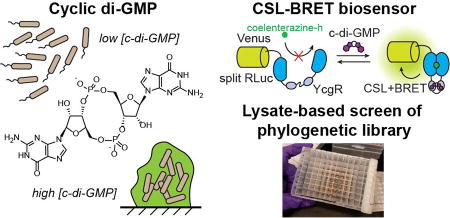
Abstract
Bacteria colonize highly diverse and complex environments, from gastrointestinal tracts, to soil and plant surfaces. This colonization process is controlled in part by the intracellular signal cyclic di-GMP, which regulates bacterial motility and biofilm formation. To interrogate cyclic di-GMP signaling networks, a variety of fluorescent biosensors for live cell imaging of cyclic di-GMP have been developed. However, the need for external illumination precludes the use of these tools for imaging bacteria in their natural environments, including in deep tissues of whole organisms and in samples that are highly autofluorescent or photosensitive. The need for genetic encoding also complicates the analysis of clinical isolates and environmental samples. Toward expanding the study of bacterial signaling to these systems, we have developed the first chemiluminescent biosensors for cyclic di-GMP. The biosensor design combines the complementation of split luciferase (CSL) and bioluminescence resonance energy transfer (BRET) approaches. Furthermore, we developed a lysate-based assay for biosensor activity that enabled reliable high-throughput screening of a phylogenetic library of 92 biosensor variants. The screen identified biosensors with very large signal changes (~40 and 90-fold) as well as biosensors with high affinities for cyclic di-GMP (KD <50 nM). These chemiluminescent biosensors then were applied to measure cyclic di-GMP levels in E. coli. The cellular experiments revealed an unexpected challenge for chemiluminescent imaging in Gram negative bacteria, but showed promising application in lysates. Taken together, this work establishes the first chemiluminescent biosensors for studying cyclic di-GMP signaling, and provides a foundation for using these biosensors in more complex systems.
Graphical abstract
INTRODUCTION
The second messenger 3’,5’-cyclic di-guanosine monophosphate (c-di-GMP) is the first discovered cyclic dinucleotide signaling molecule and has been found to be nearly ubiquitous in bacteria.1,2 Within cells, c-di-GMP levels are controlled by complex networks of diguanylate cyclases (DGCs) and c-di-GMP phosphodiesterases (PDEs) that work to tightly regulate the cellular concentration of this second messenger, often in response to extracellular signals.3,4 These signaling networks coordinate a multitude of fundamental cellular functions, including the transition from sessile to motile states, virulence, and cell cycle control. To interrogate the dynamics of c-di-GMP within these complex signaling networks, we and others have developed biosensors for live cell imaging of c-di-GMP. These include protein-based FRET biosensors that have been used in E. coli,5 C. crescentus, S. typhimurium, and P. aeruginosa,6,7 as well as RNA-based fluorescent biosensors that have been used in E. coli and allow for the first time the ability to image c-di-GMP under anaerobic conditions.8
One long-term goal in the field is to analyze c-di-GMP signaling in more complex settings, including bacteria colonizing the gut, and in clinical isolates, mixed cultures, or environmental samples.9,10 None of these applications have been shown with the fluorescent biosensor tools currently available. Due to its reliance on external illumination, fluorescent imaging is often incompatible with imaging in deep tissues of animals and in long-term experiments due to phototoxicity. Additionally, fluorescent biosensors are difficult to apply to clinical isolates, mixed cultures, and environmental samples, because they haven’t been shown to work in complex lysates that exhibit increased autofluorescence and decreased signal due to sample dilution. One alternative technique that does not require external illumination to produce a signal is chemiluminescence imaging.11 Thus, to expand the study of c-di-GMP signaling networks to these more complex systems, we sought to develop the first chemiluminescent biosensors for c-di-GMP.
Chemiluminescent biosensors generally utilize luciferases, enzymes which catalyze the oxidation of small molecule luciferin substrates to produce light.12 Luciferase-based biosensors typically fall into one of two categories: complementation of split luciferase (CSL) or bioluminescence resonance energy transfer (BRET)-based biosensors. In CSL systems, a sensor domain is used to split the luciferase protein into two non-functional halves, such that analyte binding results in a conformational change that reconstitutes the luciferase to produce a change in signal intensity.13–17 In BRET systems, the sensor domain is inserted between a fluorescent protein acceptor on one side and an intact luciferase donor on the other side, and analyte binding leads to a change in BRET efficiency between donor and acceptor.18–21 CSL biosensors generally produce large signal changes, however their signal intensity is often low due to poor reconstitution of luciferase activity. BRET biosensors generally have high signal intensity and are ratiometric, however their signal change upon analyte binding is typically small. To overcome these drawbacks, the Nagai lab combined the CSL and BRET approaches and used Nano-lantern (NL) or enhanced Nano-lantern (eNL) scaffolds to produce chemiluminescent biosensors with large signal changes (up to 4-fold with original NL and 6-fold with eNL) and good signal intensity for imaging.22–24 They used these biosensors to perform single-cell imaging of Ca2+, cyclic AMP, and ATP in HeLa cells, D. discoideum cells, and Arabidopsis plant leaves, respectively.22
In this work, we applied the CSL-BRET strategy to develop the first chemiluminescent biosensors for c-di-GMP. First, an insertion site for the E. coli c-di-GMP binding protein YcgR (EcYcgR) within the NL scaffold was found that generated a functional biosensor. Next, we mined sequence databases for diverse YcgR homologs and selected a set of 92 proteins for functional characterization within the biosensor scaffold. These phylogenetic variants were synthesized as codon optimized sequences for cloning into the NL scaffold. The resulting phylogenetic biosensor library was expressed and rapidly screened in a lysate-based assay for response to c-di-GMP. A large number of biosensors were identified with improved affinity, signal change, and stability compared to the initial EcYcgR biosensor. This new collection of chemiluminescent biosensors includes ones with the highest affinity for a protein-based c-di-GMP biosensor (<50 nM) and others with the largest signal change for a CSL-BRET biosensor (up to 90-fold). These biosensors were applied to measure c-di-GMP levels in live E. coli, and revealed challenges for chemiluminescence imaging in Gram-negative bacteria. Subsequently, we developed a bacterial lysate assay for diguanylate cyclase activity using the biosensors. This work provides the first chemiluminescent biosensors for measuring c-di-GMP levels in bacteria and lays the groundwork for live cell imaging of c-di-GMP dynamics without external illumination.
RESULTS AND DISCUSSION
Design of chemiluminescent biosensor for cyclic di-GMP
PilZ domain-containing proteins bind c-di-GMP and have previously been used to generate genetically encoded FRET biosensors for c-di-GMP.5,6,25 For the initial design, the PilZ domain-containing protein YcgR from E. coli (EcYcgR) was chosen as the sensor domain to insert into the yellow Nano-lantern (YNL) scaffold, which is an NL scaffold that uses Venus as the fluorescent acceptor (Figure 1a). EcYcgR has previously been shown to bind to c-di-GMP with an affinity of ~800 nM and undergo a large conformational change upon binding.26
Fig. 1.

Four biosensors were designed that differed in either the split site of RLuc8 (91/92 or 228/229) or the deletion/retention of a flexible N-terminal region of RLuc8 (1.0 or 1.1 scaffold) (Figure 1b). These designs were chosen based on previously developed NL sensors.22 The four biosensors were constructed, expressed, and purified from E. coli, then tested for response to c-di-GMP. While all of the biosensors produced a chemiluminescent signal, only biosensors with EcYcgR inserted at the 91/92 site of RLuc8 produced a c-di-GMP-dependent change in signal (Figure 1c). For further characterization, the biosensors were expressed and purified from E. coli co-expressing the c-di-GMP-specific phosphodiesterase PdeH (also called YhjH) to ensure that minimal to no c-di-GMP remain bound. The EcYcgR-91 biosensor has a dissociation constant (KD) for c-di-GMP of 615 ± 115 nM and gives a 2.6-fold change in chemiluminescent signal intensity upon binding (Figure 1d). The measured affinity for the biosensor is slightly improved compared to previously reported affinity values for EcYcgR alone,26 and the signal change and intensity are comparable to previously developed NL biosensors.22 Additional in vitro binding tests with related compounds showed that the biosensor is highly specific for c-di-GMP (Figure 1d).
Lysate-based assay of biosensor performance
One of the benefits of chemiluminescent biosensors compared to fluorescent or FRET-based biosensors is that because external illumination is not required, output signal is not compromised by solution autofluorescence. Accordingly, we find that biosensor activity can be reliably assayed in lysates of cells grown in complex media with no further purification. Growth and assay procedures were optimized to permit the use of small culture volumes and to produce consistent growth between wells, which led to the development of a 96-well plate format lysate activity assay for biosensor constructs (Figure S1). Importantly, by measuring in lysates rather than whole cells, new biosensor designs can be rapidly and reliably screened with any analyte of interest at a set concentration, including non-cell permeable compounds like c-di-GMP.
Using this assay workflow, various mutants of EcYcgR were tested (Figure S2a). These mutations have been characterized in other YcgR proteins, and are predicted to alter the binding stoichiometry to c-di-GMP (M1; R113L mutant),27 abolish binding to c-di-GMP (M2, R118D), or improve affinity for c-di-GMP (M3; S147A mutant).26 A double mutant combination of M1 and M3 mutations (M4; R113L, S147A) was also tested. While M2 severely reduced binding affinity as expected, the other mutations also led to decreased affinity or signal change (Figure S2b, c; Supplemental Notes). In addition, a small library of biosensors with different linkers flanking EcYcgR was analyzed using the lysate-based screen. It is well known that linker length and composition can produce large changes in the affinity and/or signal change of both CSL and BRET-type biosensors,14,28 but to our knowledge, this had not yet been tested in NL biosensors. Six different linker designs were tested spanning a length of 2–5 residues on either side of EcYcgR that were designed to be either flexible or rigid (Figure S2a). However, none of the new linker designs produced signal changes better than the original linkers used (Figure S2d; Supplemental Notes).
This 96-well format lysate-based activity assay enables biosensor constructs to be rapidly assessed without individually purifying each protein. A small library of EcYcgR mutants and linker variants was tested, but none showed improved biosensor performance. While a larger randomized library-based approach could lead to improved affinity or signal change, the biosensor would remain limited by the characteristics of the sensor domain, EcYcgR. Thus, we instead decided to focus on altering the sensor domain itself in the next round of optimization.
Phylogenetic screen for improved c-di-GMP binding domains
Our lab has previously used phylogenetic libraries in the development of RNA-based fluorescent biosensors, and we have found that such an approach can lead to rapid improvements in biosensor characteristics.8,29 A small number of YcgR proteins have been tested in FRET-based biosensors for c-di-GMP, with the best design exhibiting a –60.6% signal change and 195 nM affinity.6,7 For a wider screen, all PilZ-containing proteins with domain architectures similar to EcYcgR (YcgR-PilZ and YcgR_2-PilZ) were obtained from the Pfam database. From this set of 840 sequences resulting from the bioinformatic query, 92 sequences were synthesized and cloned into the NL scaffold (Table S2) using the design found to work best with the EcYcgR sensor domain (91/92 insertion site, 1.0 scaffold). This set included 34 sequences chosen from the genomes of candidate thermophilic bacteria, as we hypothesized that these would produce more stably folded biosensors. Additionally, c-di-GMP binding to PilZ domains has been found to be largely entropically driven,30 so we expected that sensor domains from thermophiles may bind with higher affinity to c-di-GMP than those from mesophiles. Of the remaining 58 sequences, at least three were previously characterized YcgR proteins27,30,31 and the remainder were chosen from a large variety of different bacterial genomes.
All sequences were codon optimized for E. coli expression to aid screening. In addition, an mCherry fluorescent tag32 was introduced at the C-terminus to measure the relative folding stability of each biosensor variant. During purification of EcYcgR biosensors, it was found that truncated proteins were produced at appreciable levels during expression, suggesting the degradation of improperly folded biosensors (Figure S3a). In contrast, the purified YNL had little-to-no truncated products. Thus, the ratio of mCherry fluorescence (C-terminal) to Venus fluorescence (N-terminal) provides a proxy for the relative amount of full-length biosensor in cells and can be determined in a high-throughput manner via flow cytometry. This ratio for each phylogenetic variant was normalized to the ratio for the EcYcgR biosensor (mCh/Venus = 1). Library members with mCherry/Venus fluorescence ratios above 1 were considered more well-folded than EcYcgR, while library members with fluorescence ratios lower than 1 were considered less well-folded (Figure S3b). The mCherry-tagged version of the EcYcgR biosensor was tested in the lysate-based assay and showed that the mCherry tag had little-to-no effect on biosensor performance (Figure S3c).
To determine signal change and relative affinity for c-di-GMP, all candidate biosensors were tested in the lysate-based assay with two different concentrations of c-di-GMP (Figure 2, S4a). Less than 10% (8 sequences) were inactive and produced low chemilumescent signal, likely due to poor expression and/or misfolding. Of the sequences that were active, over 40% (34 sequences) showed signal changes better than EcYcgR in response to 20 μM c-di-GMP (~1.86-fold). Two constructs showed signal changes of ~17-fold and ~28-fold, which are larger than signal changes for previously developed NL biosensors.22,24 Interestingly, while the large majority of these variants all showed signal changes in the positive direction, 3 sequences showed signal changes in the negative direction, suggesting that they may undergo a different sort of structural rearrangement upon binding to c-di-GMP. Of all tested constructs, over 60% (56 sequences) showed relative folding stability better than the EcYcgR biosensor, and 6 showed greater stability than the YNL scaffold itself. Out of the 31 thermophilic variants that were active, 25 showed better folding stability than EcYcgR, supporting the hypothesis that thermophilic sensor domains produce more well-folded biosensors in the NL scaffold. Taken together, this lysate-based screen of a large phylogenetic library of YcgR sensor domains resulted in multiple biosensors that exhibit the largest signal changes of any NL biosensors to date, as well as many biosensors that are more well-folded and have larger signal change than the original EcYcgR biosensor.
Fig. 2.

In vitro characterization of phylogenetic biosensor variants
From the screening panel, a subset of biosensors that displayed either increased affinity, large signal change, or rare negative signal change were chosen for re-screening and further analysis. For in vitro characterization, all phylogenetic biosensor variants were recloned without the mCherry tag and expressed, then purified from cells co-expressing PdeH to ensure that minimal to no c-di-GMP remain bound. Biosensors that showed maximal signal in response to 1 µM c-di-GMP in the original screen were predicted to have increased affinity; we further selected only biosensors that displayed positive signal change of at least 2.5-fold. Out of the 31 sequences with positive signal changes greater than EcYcgR, 9 met these criteria. Re-screening in the lysate-based assay with lower concentrations of c-di-GMP confirmed that all 9 sequences had affinities better than or equal to EcYcgR, while 7 of the sequences appeared to have affinities <200 nM (Figure S4b). As purified proteins, these 7 sequences span a range of affinities, all <200 nM, with 5 of the sequences exhibiting affinities <50 nM (Table 1). Interestingly, the CbYcgR biosensor showed a decrease in signal intensity at high concentrations of c-di-GMP, suggesting a secondary binding site (Figure S5a).
Table 1.
Characteristics of Selected Sensor Variants
| sequence | KDa (nM) | Δsignalb | Hill coefficienta | mCh/Venusc |
|---|---|---|---|---|
| EcYcgRd | 354 ± 18 | 3 | 2.3 ± 0.23 | 1 |
| Selected for High Affinity | ||||
| TmYcgR | <50 | 6.1 | 1.5 ± 0.04 | 1.7 |
| TuYcgR | <50 | 6.3 | 1.4 ± 0.04 | 2.7 |
| TbYcgR | <50 | 7.9 | 1.6 ± 0.1 | 1.4 |
| CpYcgR | <50 | 6.3 | 1.9 ± 0.1 | 1.7 |
| CbYcgR | <50 | 18.8 | 1.2 ± 0.04 | 0.73 |
| TcYcgR | 59 ± 2 | 4.6 | 1.7 ± 0.1 | 1.6 |
| BvYcgR | 105 ± 2 | 4.9 | 1.9 ± 0.05 | 2.7 |
| Selected for High Signal Change | ||||
| DnYcgR | 303 ± 15 | 39.3 | 1.8 ± 0.1 | 0.61 |
| ToYcgR | 379 ± 23 | 24.5 | 2.2 ± 0.2 | 1.1 |
| Selected for Negative Signal Change | ||||
| PtYcgR | <50 | −7.7 | −1.8 ± 0.1 | 1.6 |
| BtYcgR | 286 ± 17 | −89 | −2 ± 0.2 | 0.52 |
The two biosensor variants that showed large signal changes in the lysate-based screen, DnYcgr and ToYcgR, demonstrated signal changes of ~40-fold and ~25-fold as purified proteins, respectively (Table 1; Figure S5a). These signal changes are about 10× higher than those seen in any previously developed NL biosensors. To our knowledge, the largest signal change reported for any rationally designed, single chain CSL-type biosensor was a 70-fold change for a cAMP biosensor.14 Interestingly, the largest signal change we characterized was for BtYcgR, which exhibited negative signal change and showed ~90-fold maximal decrease in chemiluminesence activity in response to c-di-GMP (Figure S5a). BtYcgR was among the most poorly folded variants as measured by the mCh/Venus fluorescence ratio, which may contribute to its large signal change. Of the other 2 biosensor sequences with negative signal changes, PtYcgR recapitulated the lysate-based results, while BtYcgR did not. The biosensors with negative signal changes could be useful for measuring decreases in c-di-GMP, for example, due to phosphodiesterase activity.
The ligand selectivities for three of the high affinity biosensors were tested, and all retained high selectivity for c-di-GMP versus (3’3’)-cAG and pGpG (Figure 3a). Both CpYcgR and TbYcgR biosensors showed >100-fold selectivity against (3’3’)-cAG and no appreciable binding to pGpG. TuYcgR also showed >100-fold selectivity against (3’3’)-cAG, but was slightly less selective against pGpG than the other biosensors were. Interestingly, our lab has shown that riboswitch-based biosensors for c-di-GMP have exquisitely selectivity against other cyclic di-nucleotides, with no appreciable binding, but ~2000-fold selectivity against pGpG,8 which is the opposite trend to what is observed for these protein-based biosensors.
Fig. 3.

Another key characteristic of these biosensors is signal intensity. CSL-type biosensors rely on the complementation of a split enzyme to produce signal, so their signal intensity is often only a fraction of the brightness of the non-split protein. In previously developed NL biosensors, for example, the brightest design achieved 35% of the intact YNL signal and was sufficient for single cell imaging experiments.22 Two of our highest affinity biosensors variants, TuYcgR and CpYcgR, gave maximal signal intensities of ~40% and 30%, respectively, of the intact YNL signal (Figure 3b). However, while total protein concentration was the same for each sample based on Venus domain absorbance,33 the biosensor samples contained visible amounts of a truncated product including Venus, whereas the YNL sample was a single protein band (Figure S5b). Thus, the relative chemiluminescence intensities we measured likely underestimate the actual brightness of the biosensors.
Finally, we measured the response time for one of the best-performing biosensors, CpYcgR. To test the kinetics of biosensor activation, c-di-GMP was co-injected with chemiluminescent substrate at time 0 into solutions containing the biosensor. To provide comparison to prebound or unbound controls, the CpYcgR biosensor was pre-incubated with or without c-di-GMP before substrate was injected at time 0. The t1/2 value for the co-injected sample was 9–9.5 sec compared to the t1/2 value of 0.2 sec for prebound biosensor (Figure 3c). This experiment shows that the CSL-BRET biosensor undergoes a c-di-GMP-dependent conformational change that activates chemiluminescence in less than one minute, which is similar to the results for YcgR-based FRET biosensors used for live cell imaging.6
Taken together, in vitro characterization of phylogenetic variants revealed a set of biosensors with good signal changes that span a range of affinities for c-di-GMP, including several incorporating novel YcgR proteins that have <50 nM KD values. One biosensor showed positive signal changes of ~40-fold and another showed negative signal changes of ~90-fold, which are among the largest signal changes observed for any CSL-type biosensors. Finally, the biosensors showed strong signal intensities and fast activation in response to c-di-GMP.
Measuring cellular levels of c-di-GMP with chemiluminescent biosensors
As an initial test of our new NL biosensors for measuring c-di-GMP in live cells, we co-expressed a subset of the mCherry-tagged biosensors in E. coli along with a diguanylate cyclase (WspR-D70E) or a phosphodiesterase (PdeH) to generate high or low levels of c-di-GMP, respectively. After overnight growth in autoinduction media, cells were pelleted and washed with PBS, then chemiluminescent substrate was added and total signal was measured in a plate reader. The mCherry fluorescence signal of the cells was also measured so that chemiluminescent signal intensity could be normalized to the amount of full-length biosensor (LUM/mCherry FL). However, to our surprise, the majority of biosensors did not show the expected signal increase with WspR-D70E (Figure 4a). Instead, several of them showed decreases in total signal when comparing WspR-D70E versus PdeH overexpressing cells, which was also observed for YNL alone.
Fig. 4.

The result for YNL suggested that WspR-D70E overexpression could be decreasing substrate diffusion into cells due to increased biofilm formation, which is a phenotypic consequence of high c-di-GMP levels. Thus, we repeated the experiment but lysed the cells using the same protocol as the lysate-based assay before adding the chemiluminescent substrate. With no barriers to substrate diffusion, all the biosensors except for TuYcgR showed signal increases with WspR-D70E lysates versus PdeH lysates, whereas YNL showed no significant difference (Figure 4b). In addition, the normalized signal intensities were also much larger in cell lysates compared to live cells, which is consistent with substrate diffusion affecting the signal in intact cells.
Thus, we revised the plan to develop a rapid assay for measuring c-di-GMP levels in cell lysates instead. In this assay, the purified CpYcgR biosensor is added directly to lysed cells and signal is measured after addition of chemiluminescent substrate in a plate reader (Figure 4c). Due to E. coli having low endogenous levels of c-di-GMP,4 we could not detect differences in c-di-GMP lysate levels between cells expressing inactive enzyme (WspR-G249A, i.e. endogenous levels) or expressing a c-di-GMP specific phosphodiesterase (PdeH). However, from these data we were able to observe clear differences in c-di-GMP lysate levels between cells expressing active diguanylate cyclases (WspR-WT, WspR-D70E, and PleD) and control cells containing empty vector. Notably, the signal strength and fold-change of the CpYcgR biosensor was almost identical in cell lysates and in buffer alone.
Conclusion
This work represents, to our knowledge, the development of the first chemiluminescent biosensors for c-di-GMP. After initial validation of the NL biosensor design, a phylogenetic library of YcgR-like proteins was rapidly screened in a lysate-based assay to identify variants with improved biosensor characteristics. The two key benefits of this lysate-based assay are that the biosensors can be screened without purification and for binding to any ligand of interest. We envision that a similar approach can be used to screen YcgR mutants in a high-throughput manner to generate chemiluminescent biosensors for other cyclic dinucleotides, such as c-di-AMP, (2’,3’)-cGAMP, and (3’,3’)-cGAMP.34,35
The screen identified a panel of biosensor variants with improved affinity, signal change, folding stability, and signal intensity compared to the initial EcYcgR biosensor and other protein-based biosensors for c-di-GMP. While Nano-lantern-based biosensors have been used to make live cell measurements in eukaryotic systems, to our knowledge, they had not been applied in bacterial systems prior to this study. Our results suggest that changes in diffusion of the chemiluminescent substrate, coelenterazine-h, due to increased biofilm formation may be an issue for live cell experiments in bacteria, which can be overcome in part by performing measurements with the biosensors in cell lysates. Thus, we developed a rapid plate reader-based assay for measuring diguanylate cyclase activity in cell lysates, which could be applied to other types of complex samples, such as clinical isolates or microbial co-cultures. Future work will focus on developing ratiometric, rather than intensity-based, chemiluminescent biosensors using the panel of sensor domains discovered here to overcome limitations in bioavailability of substrate. An encouraging precedent is a ratiometric BRET system that has been employed to monitor chemotaxis receptor kinase activity in live E. coli.36 Ultimately we aim to study c-di-GMP dynamics in biological systems for which fluorescent biosensors are less useful, such as in plant-pathogen interactions or intestinal bacteria in the gut.
METHODS
Chemiluminescence measurements
All chemiluminescence measurements were performed in opaque white, 96-well LUMITRAC 600 plates (Grenier). Briefly, proteins and ligands were added to assay buffer [50 mM HEPES (pH 7.2), 100 mM KCl, 10 mM DTT, 0.1% BSA] to the given final concentrations in 100 µL reaction volume, then incubated at 28 °C for at least 10 min to reach binding equilibrium. Unless otherwise noted, all affinity and kinetics measurements were made using 50 nM protein, and all signal change, brightness, and selectivity measurements were made using 100 nM protein. Chemiluminescent substrate was prepared by diluting coelenterazine-h to 60 µM in reagent buffer [50 mM HEPES (pH 7.2), 100 mM KCl, 300 mM ascorbate], and allowing the solution to equilibrate to RT for at least 30 min. For equilibrated biosensor measurements, chemiluminescence at 28 °C was measured on a SpectraMax i3× platereader (Molecular Devices) by injecting 20 µL of chemiluminescent substrate, then integrating total chemiluminescent signal for 10 sec after a 3 sec delay. For biosensor kinetics measurements, chemiluminescence at 28 °C was measured over 3 min in 200 ms intervals with 100 ms integration times starting at substrate injection. For c-di-GMP binding kinetics, c-di-GMP was co-injected with substrate for a final concentration of 2 µM c-di-GMP.
Lysate-based assay for biosensor activity
Single colonies of BL21(DE3) star E. coli cells transformed with pET24-biosensor-mCherry plasmids were resuspended in 500 μL of P-0.5G non-inducing media [0.5% glucose, 25 mM (NH4)2SO4, 50 mM KH2PO4, 50 mM Na2HPO4, 1 mM MgSO4]37 supplemented with 100 µg/mL kanamycin in 2.2 mL 96-well deep well plates (VWR), then grown at 37 °C, 325 rpm, for 24 h to generate pre-cultures. A 5 µL aliquot of each pre-culture was used to inoculate 500 µL of ZYP-5052 auto-induction media [25 mM (NH4)2SO4, 50 mM KH2PO4, 50 mM Na2HPO4, 1 mM MgSO4, 0.5% (v/v) glycerol, 0.05% glucose, 0.2% α-lactose, 1% tryptone, and 0.5% yeast extract]37 supplemented with 100 µg/mL kanamycin in deep well plates. Two identical plates of auto-induction media were inoculated to permit aeration with shaking and to yield 1 mL of combined culture for each sample. Plates were grown at 37 °C, 325 rpm, for 20 h for biosensor expression. After 20 h of growth, equivalent cultures were combined into a single plate.
For flow cytometry analysis of biosensor expression and stability, 2 µL of each culture was added to 200 µL of phosphate-buffered saline (PBS) [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4]. The mean fluorescence intensities were analyzed on an Attune NxT flow cytometer (Life Technologies). Venus fluorescence was measured with a 488 nm laser for excitiation and a 530/30 filter for emission. mCherry fluorescence was measured with a 561 nm laser for excitation and a 620/15 filter for emission.
For chemiluminescence measurements, the remaining culture was harvested in the deep-well plate by centrifugation at 4700 rpm for 10 min at 4 °C. Supernatant media was removed and each cell pellet was resuspended in 360 µL screening buffer [50 mM Tris (pH 7.5), 100 mM KCl, 5% glycerol, 2 mM EDTA, 300 µg/mL lysozyme, 1 mM PMSF]. Cells were gently lysed for 1 h at 4 °C and lysates were clarified by centrifugation at 4700 rpm for 40 min at 4 °C. To obtain three samples, 90 µL aliquots of clarified lysates were pipetted into three wells of opaque white, 96-well LUMITRAC 600 plates (Grenier), being careful not to disturb the pelleted cell debris. Each aliquot was mixed with 10 µL of either buffer, 10 µM c-di-GMP, or 200 µM c-di-GMP [in 50 mM HEPES (pH 7.2), 100 mM KCl] to produce final concentrations of 0, 1 µM or 20 µM c-di-GMP. Mixtures were incubated at 28 °C for at least 10 min to allow binding to occur, then chemiluminescence was measured as described above. Signal fold-changes were calculated for at least two biological replicates by dividing the total chemiluminescence signal with added c-di-GMP by the total signal without added c-di-GMP.
Cellular c-di-GMP measurements with biosensor addition
Single colonies of BL21(DE3) star E. coli cells transformed with pCOLA plasmids encoding c-di-GMP-related enzymes were resuspended in 500 µL of P-0.5G non-inducing media supplemented with 100 µg/mL kanamycin in 2.2 mL 96-well deep well plates (VWR), then grown at 37 °C, 325 rpm, for 24 h to generate pre-cultures. A 5 µL aliquot of each pre-culture was used to inoculate 500 µL of ZYP-5052 auto-induction media supplemented with 100 µg/mL kanamycin in a deep well plate and grown at 37 °C, 325 rpm, for 20 h to allow for protein expression. Cultures were harvested and clarified lysates were prepared as described for the lysate-based assay, except each culture was resuspended in 120 µL screening buffer. For +lysate samples, a 50 µL aliquot of each clarified lysate was mixed with 50 µL of 100 nM biosensor in 2× assay buffer in an opaque white 96-well plate, and the plate was incubated at 28 °C for 10 min. Chemiluminescent substrate was injected and chemiluminesce was measured as described above. For +buffer samples, 50 µL of screening buffer was added instead of the lysate
Supplementary Material
Acknowledgments
FUNDING SOURCES This work was supported in part by NIH grants DP2 OD008677 and R01 GM124589 (to M.C.H.), NIH training grant T32 GM066698 (for A.B.D.), and UC Berkeley College of Chemistry Summer Research Stipend (to W.A.A.). The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
ASSOCIATED CONTENT
The Supporting Information (additional methods, supplementary figures, tables, notes, and references) is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
References
- 1.Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, Braun S, de Vroom E, van der Marel GA, van Boom JH, Benziman M. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic dyguanylic acid. Nature. 1987;325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 2.Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 4.Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, Kaever V, Hengge R. More than Enzymes That Make or Break Cyclic Di-GMP — Local Signaling in the Interactome of GGDEF/EAL Domain Proteins of Escherichia coli. MBio. 2017;8:e01639–17. doi: 10.1128/mBio.01639-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho CL, Chong KSJ, Oppong JA, Chuah MLC, Tan SM, Liang ZX. Visualizing the perturbation of cellular cyclic di-GMP levels in bacterial cells. J. Am. Chem. Soc. 2013;135:566–569. doi: 10.1021/ja310497x. [DOI] [PubMed] [Google Scholar]
- 6.Christen M, Kulasekara HD, Christen B, Kulasekara BR, Hoffman LR, Miller SI. Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell division. Science. 2010;328:1295–1297. doi: 10.1126/science.1188658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mills E, Petersen E, Kulasekara BR, Miller SI. A direct screen for c-di-GMP modulators reveals a Salmonella Typhimurium periplasmic L -arginine – sensing pathway. Sci. Signal. 2015;8:1–12. doi: 10.1126/scisignal.aaa1796. [DOI] [PubMed] [Google Scholar]
- 8.Wang XC, Wilson SC, Hammond MC. Next-generation fluorescent RNA biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Res. 2016;44:e139. doi: 10.1093/nar/gkw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koestler BJ, Waters CM. Bile acids and bicarbonate inversely regulate intracellular cyclic di-GMP in vibrio cholerae. Infect. Immun. 2014;82:3002–3014. doi: 10.1128/IAI.01664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi E, Cimdins A, Lüthje P, Brauner A, Sjöling Å, Landini P, Römling U. “It’s a gut feeling” – Escherichia coli biofilm formation in the gastrointestinal tract environment. Crit. Rev. Microbiol. 2017;7828:1–30. doi: 10.1080/1040841X.2017.1303660. [DOI] [PubMed] [Google Scholar]
- 11.Prescher JA, Contag CH. Guided by the light: visualizing biomolecular processes in living animals with bioluminescence. Curr. Opin. Chem. Biol. 2010;14:80–89. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Saito K, Nagai T. Recent progress in luminescent proteins development. Curr. Opin. Chem. Biol. 2015;27:46–51. doi: 10.1016/j.cbpa.2015.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Kaihara A, Umezawa Y, Furukawa T. Bioluminescent indicators for Ca2+ based on split Renilla luciferase complementation in living cells. Anal. Sci. 2008;24:1405–8. doi: 10.2116/analsci.24.1405. [DOI] [PubMed] [Google Scholar]
- 14.Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 2008;3:346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Lee KC, Bhojani MS, Khan AP, Shilman A, Holland EC, Ross BD, Rehemtulla A. Molecular imaging of Akt kinase activity. Nat. Med. 2007;13:1114–1119. doi: 10.1038/nm1608. [DOI] [PubMed] [Google Scholar]
- 16.Takenouchi O, Kanno A, Takakura H, Hattori M, Ozawa T. Bioluminescent Indicator for Highly Sensitive Analysis of Estrogenic Activity in a Cell-Based Format. Bioconjug. Chem. 2016;27:2689–2694. doi: 10.1021/acs.bioconjchem.6b00466. [DOI] [PubMed] [Google Scholar]
- 17.Hattori M, Haga S, Takakura H, Ozaki M, Ozawa T. Sustained accurate recording of intracellular acidification in living tissues with a photo-controllable bioluminescent protein. Proc. Natl. Acad. Sci. 2013;110:9332–9337. doi: 10.1073/pnas.1304056110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito K, Hatsugai N, Horikawa K, Kobayashi K, Matsu-Ura T, Mikoshiba K, Nagai T. Auto-luminescent genetically-encoded ratiometric indicator for real-time Ca2+ imaging at the single cell level. PLoS One. 2010;5:e9935. doi: 10.1371/journal.pone.0009935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, Sternweis PC. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas KH, Sopory S, Visweswariah SS. The GAF domain of the cGMP-binding, cGMP-specific phosphodiesterase (PDE5) is a sensor and a sink for cGMP. Biochemistry. 2008;47:3534–3543. doi: 10.1021/bi702025w. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Cumberbatch D, Centanni S, Shi S, Winder D, Webb D, Johnson CH. Coupling optogenetic stimulation with NanoLuc-based luminescence (BRET) Ca++ sensing. Nat. Commun. 2016;7:13268. doi: 10.1038/ncomms13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito K, Chang Y-F, Horikawa K, Hatsugai N, Higuchi Y, Hashida M, Yoshida Y, Matsuda T, Arai Y, Nagai T. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat. Commun. 2012;3:1262. doi: 10.1038/ncomms2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takai A, Nakano M, Saito K, Haruno R, Watanabe TM, Ohyanagi T, Jin T, Okada Y, Nagai T. Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc. Natl. Acad. Sci. 2015;112:4352–4356. doi: 10.1073/pnas.1418468112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki K, Kimura T, Shinoda H, Bai G, Daniels MJ, Arai Y, Nakano M, Nagai T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016;7:13718. doi: 10.1038/ncomms13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pultz IS, Christen M, Kulasekara HD, Kennard A, Kulasekara B, Miller SI. The response threshold of Salmonella PilZ domain proteins is determined by their binding affinities for c-di-GMP. Mol. Microbiol. 2012;86:1424–1440. doi: 10.1111/mmi.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryjenkov DA, Simm R, Römling U, Gomelsky M. The PilZ domain is a receptor for the second messenger c-di-GMP: The PilZ domain protein YcgR controls motility in enterobacteria. J. Biol. Chem. 2006;281:30310–30314. doi: 10.1074/jbc.C600179200. [DOI] [PubMed] [Google Scholar]
- 27.Ko J, Ryu KS, Kim H, Shin JS, Lee JO, Cheong C, Choi BS. Structure of PP4397 reveals the molecular basis for different c-di-GMP binding modes by pilz domain proteins. J. Mol. Biol. 2010;398:97–110. doi: 10.1016/j.jmb.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Thestrup T, Litzlbauer J, Bartholomäus I, Mues M, Russo L, Dana H, Kovalchuk Y, Liang Y, Kalamakis G, Laukat Y, Becker S, Witte G, Geiger A, Allen T, Rome LC, Chen T-W, Kim DS, Garaschuk O, Griesinger C, Griesbeck O. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods. 2014;11:175–82. doi: 10.1038/nmeth.2773. [DOI] [PubMed] [Google Scholar]
- 29.Su Y, Hickey SF, Keyser SGL, Hammond MC. In Vitro and in Vivo Enzyme Activity Screening via RNA-Based Fluorescent Biosensors for S-Adenosyl- l - homocysteine (SAH) J. Am. Chem. Soc. 2016;138:7040–7047. doi: 10.1021/jacs.6b01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benach J, Swaminathan SS, Tamayo R, Handelman SK, Folta-Stogniew E, Ramos JE, Forouhar F, Neely H, Seetharaman J, Camilli A, Hunt JF. The structural basis of cyclic diguanylate signal transduction by PilZ domains. EMBO J. 2007;26:5153–66. doi: 10.1038/sj.emboj.7601918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul K, Nieto V, Carlquist WC, Blair DF, Harshey RM. The c-di-GMP Binding Protein YcgR Controls Flagellar Motor Direction and Speed to Affect Chemotaxis by a “Backstop Brake” Mechanism. Mol. Cell. 2010;38:128–139. doi: 10.1016/j.molcel.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 33.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 34.Krasteva PV, Sondermann H. Versatile modes of cellular regulation via cyclic dinucleotides. Nat. Chem. Biol. 2017;13:350–359. doi: 10.1038/nchembio.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–970. doi: 10.1016/j.cell.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimizu TS, Delalez N, Pichler K, Berg HC. Monitoring bacterial chemotaxis by using bioluminescence resonance energy transfer: Absence of feedback from the flagellar motors. Proc. Natl. Acad. Sci. 2006;103:2093–2097. doi: 10.1073/pnas.0510958103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.