

Abstract
The extracellular matrix (ECM) constitutes a viscoelastic environment for cells. A growing body of evidence suggests that the behavior of cells cultured in naturally-derived or synthetic ECM mimics is influenced by the viscoelastic properties of these substrates. Adaptable crosslinking strategies provide a means to capture the viscoelasticity found in native soft tissues. In this work, we present a covalent adaptable hydrogel based on thioester exchange as a biomaterial for the in vitro culture of human mesenchymal stem cells. Through control of pH, gel stoichiometry, and crosslinker structure, viscoelastic properties in these crosslinked networks can be modulated across several orders of magnitude. We also propose a strategy to alter these properties in existing networks by the photo-uncaging of the catalyst 4-mercaptophenylacetic acid. Mesenchymal stem cells encapsulated in thioester hydrogels are able to elongate in 3D and display increased proliferation relative to those in static networks.
1. Introduction
Cells in the body reside in contact with an extracellular matrix (ECM), either in the form of a basement membrane or interstitial matrix. The ECM is a dynamic and interactive substrate; cells are able to remodel their microenvironments, while ECM signals impact cell behavior in development, injury, and disease [1–3]. Seminal studies using in vitro models of the ECM have revealed that matrix mechanics can influence the phenotype of cultured cells in a process known as mechanotransduction. For example, matrix stiffness can influence the differentiation of multipotent stem cells[4] and promote the invasive phenotype of cancer cells [5,6].
In addition to matrix stiffness, recent work has implicated the importance of viscoelastic properties of matrices in influencing cellular behavior. In polymer networks with reversible or associating crosslinks, these bonds are able to rearrange in response to an imposed deformation (stress relaxation) or flow in response to an imposed force (creep). These time-dependent properties have been shown to affect cell spreading in both two and three dimensions [7,8], cell fate decisions[9], and the regenerative ability of encapsulated cells [10]. Motivating these studies is the fact that soft tissues exhibit viscoelastic properties, so model systems with reversible crosslinks may better represent the mechanical properties of these native environments. As cells exert traction forces on adaptable networks, they are able to remodel their surroundings, allowing them to adopt physiologically relevant spread morphologies in three dimensions. Beyond capturing the viscoelastic properties of native tissue, hydrogels with reversible crosslinks have potential uses in other areas of regenerative medicine, such as controlled drug delivery and bioprinting [11,12].
A number of strategies have been put forth to create networks with viscoelastic behavior, including crosslinks formed through interactions among hydrogen bonding [13], guest-host [14], ionic [15], and covalent adaptable functionalities [16]. Covalent adaptable networks are based on reversible covalent crosslinks, such as the Diels-Alder, imine, and disulfide-forming reactions. In this work, we present thioester exchange as a strategy to afford dynamic crosslinks at physiological conditions. The exchange reaction involves the nucleophilic attack of a thioester by a thiolate anion, generating a new thioester and released thiolate. Thioesters (such as those of coenzyme A) are widely used in biosynthesis, and the transthioesterification reaction has been exploited in the context of native chemical ligation. The rate of thioester exchange in aqueous solutions is a function of the nucleophilicity and protonation state of the thiolated species, and has been shown for alkyl thioesters to greatly outpace the competing hydrolysis reaction at physiological pH[17]. Hydrogel networks incorporating thioesters have recently been reported as a dissolvable wound sealant[18], but to date, this chemistry has not been explored as an adaptable network scaffold for tissue engineering and regenerative biology applications. In this contribution, thiol-ene photopolymerization was chosen for network formation to leverage the ability of thiols to participate in both the thiol-ene and thioester exchange reations (Scheme 1). Furthermore, the photopolymerization provides spatial and temporal control over network formation, opening the possibility for in-situ and/or transdermal polymerizations.
Scheme 1. Strategy for a photopolymerized thioester exchange hydrogel.

When exposed to light in the presence of a photoinitiator, a multifunctional thiol and bifunctional alkene species react through a thiol-ene photoclick reaction. Excess thiols present in the network react with backbone thioesters in a thioester exchange reaction.
2. Materials and Methods
2.1 Synthesis of thioester di(vinyl ether) (TEDVE)
Thioester di-isocyanate from ref. [19] (500 mg, 2.5 mmol, 1.0 equiv.), di(ethylene glycol) vinyl ether (661 mg, 5.0 mmol, 2.0 equiv), and dibutyltin dilaurate (61 mg, 0.1 mmol, 0.04 equiv) were added to a 20 ml round bottom flask equipped with a magnetic stir bar and dissolved in 3 ml anhydrous ethyl acetate. The mixture was brought to 60 °C and refluxed overnight under argon. The product was purified via flash chromatography (20–80% EtOAc in hexane), and concentrated to yield 1.14 g white solid (98% yield).
TEDVE: 98% yield, white solid, Rf=0.49 (80:20 EtOAc:Hexane), 1H NMR (400 MHz, Chloroform-d) δ 6.56–6.50 (m, 2H), 4.29 – 4.21 (m, 6H), 4.08–4.04 (m, 2H), 3.90–3.84 (m, 4H), 3.80 – 3.70 (m, 9H), 3.67 – 3.63 (m, 1H), 3.50 (q, 2H), 3.39 (q, 2H), 3.06 (t, 2H), 2.83 (t, 2H). HRMS ESI+: theor. [M+H]+ 465.1907, found 465.1905 (Δ = −0.4 ppm)
2.2 Synthesis of caged thioester catalyst (NPPOC-MPAA)
To a stirred solution of 2-(2-nitrophenyl)propyl chloroformate (500 mg, 2.05 mmol, 1 equiv.) in 10 ml anhydrous dichloromethane was added 4-dimethylaminopyridine (25.7 mg, 0.21 mmol, 0.1 equiv) and 4-mercaptophenylacetic acid (345.2 mg, 2.05 mmol, 1 equiv). The reaction was stirred for 24h at room temperature under argon. The reaction mixture was then concentrated and purified by flash chromatography (5–50% EtOAc in hexanes with 1% acetic acid) to yield 275 mg (36% yield).
NPPOC-MPAA: 36% yield, white solid, Rf = 0.5 (75:25:1 EtOAc:hexanes:acetic acid), 1H NMR: (400 MHz, Chloroform-d) δ 10.79 (s, 1H), 7.79 (dd, 1H), 7.57 (td, 1H), 7.46 – 7.42 (m, 2H), 7.42 – 7.38 (m, 2H), 7.33 – 7.29 (m, 2H), 4.41 (dd, 2H), 3.69–3.65 (m, 1H), 3.67 (s, 2H), 1.35 (d, 3H). HRMS ESI-: theor.[M-H]− 374.0698, found 374.0704 (Δ = 1.6 ppm)
2.3 Norbornene RGD
The peptide Gly-Arg-Gly-Asp-Ser (GRGDS) was synthesized on rink-amide resin via Fmoc solid phase peptide synthesis (Protein Technologies Tribute peptide synthesizer) and HATU activation. 5-norbornene-2-carboxylic acid was coupled to the free N-terminus by HATU coupling. The peptide was cleaved by treatment with 95:2.5:2.5 (trifluoroacetic acid:triisopropylsilane:water) for 90 min, followed by precipitation and two washes in cold diethyl ether. Crude peptide was purified by reverse-phase HPLC (5–30% acetonitrile in water with 0.01% trifluoroacetic acid) and lyophilized. MALDI-TOF: theor. [M+H]+ 610.30, found: 610.40.
2.4 Hydrogel fabrication
Poly(ethylene glycol) (PEG) hydrogel networks were synthesized from an 8-arm-poly(ethylene glycol) thiol (PEG-8SH) macromer (20 kDa, JenKem Technology) and the thioester-containing divinyl crosslinker (TEDVE) using a photoinitiated, thiol-ene step polymerization. To create hydrogels lacking the thioester functionality, tri(ethylene glycol) divinyl ether (Sigma) was substituted for TEDVE. Stoichiometry was then manipulated at a constant 5%wt polymer to synthesize TEDVE crosslinked hydrogels with either a 20% excess of thiol groups or a 5% excess of vinyl groups. A tri(ethylene glycol) divinyl ether crosslinked hydrogel with 5% excess vinyl group was used as a control. LAP (synthesized as previously described)[20] was used as a photoinitiator at a final concentration of 1 mM and initiated with 365nm UV light. For mechanical testing, collimated UV light was applied with a mercury arc lamp (EXFO Omnicure) equipped with a 365 nm bandpass filter and set to an intensity of 10 mW cm−2. The amount of irradiation varied slightly with composition; excess thiol, excess ene, and control formulations were exposed for 100s, 200s, and 100s respectively. Increased LAP concentrations of 5mM, 18mM, and 5mM, were chosen for excess thiol, excess ene, and control formulations at high pH conditions (i.e., pH 8.0, 8.5, and 9.0). For cell encapsulations, 1 mM LAP, a lower light intensity (4 mW cm−2) and longer duration (7 min) were employed. Norbornene-functionalized RGD was incorporated at 1 mM to facilitate cellular adhesion.
2.5 Mechanical characterization of hydrogels
Hydrogels were photopolymerized in situ on a rheometer (TA Instruments HR3); storage (G′) and loss (G″) moduli were measured using an oscillatory strain input with an amplitude of 1% and a frequency of 1 Hz. To vary the solution pH, buffered solutions (200 mM) were created using MES (pH 6 & 6.5), MOPS (pH 7, 7.4 and 8) and Tris (pH 8.5 and 9). After photopolymerization, a 10% strain was applied over the course of 5 s and the total generated stress was monitored for 20 min. Stress relaxation curves were normalized to their initial value and fit to a stretched exponential function:
| (eq. 1) |
Where σ/σ0 is the normalized stress, t is the experiment time, τk is the fitted time constant, and β (0<β <1) is the stretching exponent. Fitting was performed in Matlab using the fmincon function with the condition (0<β<1).
2.6 Mesenchymal stem cell isolation, culture, and encapsulation
Primary human mesenchymal stem cells (hMSCs) were isolated from bone marrow samples by differential attachment as described previously[21] and used at the third passage for all studies. hMSCs were cultured in low-glucose DMEM supplemented with 10% fetal bovine serum, 50 U ml−1 each penicillin and streptomycin, and 50 ng ml−1 recombinant human fibroblast growth factor (FGF-2, Peprotech). For encapsulation, cells were removed from polystyrene using a trypsin/EDTA solution, resuspended in medium lacking phenol red and mixed with hydrogel precursor solution at a final concentration of 2×106 cells ml−1 prior to light exposure. To facilitate handling, hydrogel drops were formed adhered to 12mm thiol-functionalized glass coverslips.
2.7 Live/Dead analysis
Cell viability was determined using a membrane integrity assay, the calcein AM/ethidium homodimer-1 assay (Live/Dead, ThermoFisher). After incubation for 1 h, cell-laden hydrogels were imaged with a confocal microscope (LSM 710 – Zeiss). At least three fields of view were acquired for each of three gels at all time points. Three dimensional stacks were analyzed using Imaris image analysis software (Bitplane).
2.8 Proliferation assay
The fraction of proliferating cells was determined by incorporation of 5-ethynyl-2′-deoxyuridine (EdU) over an 18 h pulse from the second to third day of culture. EdU was visualized by staining with AlexaFluor 594 Azide using the Click-iT EdU kit (ThermoFisher) according to the manufacturer’s protocol. Cell nuclei were counterstained with Hoechst before confocal imaging. At least three fields of view were analyzed for each of three gels in each condition. The fraction of proliferating cells was reported as the fraction of total nuclei staining positive for EdU.
2.9 Cell morphology analysis
hMSCs were fixed in a 4% paraformaldehyde solution for 30 min at room temperature, washed with PBS, followed by permeabilization with 0.5% Tween-20 in PBS for 1 h. Non-specific adsorption sites were blocked by incubation overnight at 4°C in a solution of 10% horse serum, 1% BSA, and 0.01% Tween-20 in PBS. Hydrogels were then incubated with rhodamine phalloidin and DAPI. Images of at least 10 individual cells were captured for each of the three gels at all time points, then analyzed using Imaris image analysis software. Specifically, images were subjected to a universal threshold to obtain 3D renderings representative of the observed cell morphology. The sphericity of each rendering was then calculated as the ratio of the surface area of a perfect sphere with the same volume as the cell to the measured surface area of the cell.
2.10 Statistical analysis
All experiments were performed with three replicates, and statistical significance was determined using Student’s t-test. Statistical significance was determined by a P-value of 0.05 or less. Statistical analyses were performed using Prism (GraphPad) software.
3. Results and discussion
3.1 Network formation
Hydrogels were formed by photocrosslinking eight-arm PEG thiol macromers with the thioester di(vinyl ether) (TEDVE) crosslinker. The thiol-ene photoclick reaction is known to result in rapid, homogeneous, and biocompatible polymerization at ambient conditions.[22] Furthermore, the thiol-ene reaction is compatible with a wide range of functional groups, including the thioester moiety [23,24]. Consistent with these results, upon light illumination (365 nm, 4 mW cm−2) with 0.03 wt% (1 mM) photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), the gel point is reached within 60 s, and the final shear storage modulus of 1.69 ± 0.09 kPa within 7 min (Figure 1b). For comparison, hydrogels lacking the thioester group were synthesized using tri(ethylene glycol) divinyl ether, and these hydrogel formulations displayed nearly identical gelation kinetics and achieved a final storage modulus similar to the thioester networks (Figure 1b–c).
Figure 1. Thioester hydrogels formed through thiol-ene chemistry.

a) 8-arm PEG star macromers end-functionalized with thiols are crosslinked by divinyl molecules with and without thioesters to yield adaptable and static networks, respectively. b) Representative rheological traces of hydrogel formation in thioester and control hydrogels. Storage (G′) and loss (G″) moduli are recorded during photopolymerization (365 nm, 4 mW cm−2, 1 mM LAP) in cell culture medium (DMEM with 10% serum). Inset shows greater detail of the crossover point. c) Crosslinking with thioester and control divinyl monomers results in similar final storage moduli.
3.2 Stress relaxation in crosslinked thioester networks
After confirming the ability to form networks with TEDVE, we characterized the viscoelastic properties of the thioester networks. Free thiols are required for the exchange reaction; therefore, we hypothesized that network dynamics could be controlled by tuning the concentration of thiols in the final network. To test this, we compared the stress relaxation rates of otherwise identical networks where the free thiol concentration was varied by changing the stoichiometry of the network precursors, PEG-8SH and TEDVE. In a polymerization with 20% excess thiols, these remain as pendant functionalities after the thiol-ene reaction and are able to participate in the thioester exchange reaction. A seen in adaptable hydrazone[8]- and ionically[9]- crosslinked hydrogels, the thioester networks do not fit a simple Maxwell model for stress relaxation (exponential decay), so we therefore chose to fit the normalized stress (σ/σ0) using the empirical stretched exponential function:
| (eq. 1) |
where τk represents the time constant for stress relaxation and the stretching exponent β (0<β<1) represents the distribution of relaxation times. The mean relaxation time (<τ>) can then be calculated as the area under the curve using the gamma function (Γ) [25] to compare rates among samples:
| (eq. 2) |
Thioester hydrogels exhibit stress relaxation at pH 7.4, with a mean relaxation time of 11,000 s (Figure 2a). This rate is slower than that reported for native soft tissues[9], but in line with other adaptable hydrogel chemistries, such as the hydrazone reaction, that have been put forth for tissue engineering purposes [26,27]. In contrast, for hydrogels polymerized with a slight excess (5%) of vinyl functional groups, nearly all the thiols are consumed during the polymerization, and the time constant for stress relaxation becomes 650,000 s, nearly two orders of magnitude slower. The small amount of stress relaxation seen in the latter networks may be due to a small concentration of unreacted thiols. In control networks lacking the thioester functionality, stress relaxation plateaus at a smaller value (ca. 2%) over the 20 min experiment. The storage modulus of each of these networks remains unchanged over the time course of the experiment. A slight increase in stress relaxation was observed for the same networks formed in cell culture medium with 10% serum, as used for cell encapsulations (Supplementary Figure S5). Thiols from the medium participate in the thiol-ene reaction, altering the network stoichiometry and structure.
Figure 2. Thiol-thioester exchange in thioester-containing hydrogels.

a) Stress relaxation at pH 7.4 for non-thioester gels (gray), on-stoichiometry thioester networks (red) and thioester networks with excess thiol (blue). b) Frequency sweep of excess thiol thioester (blue) and non-thioester (gray) hydrogels. All values are normalized to the storage modulus at 1 rad s−1. c) Dissolution of thioester hydrogel networks using 1 M cysteine bath at pH 7.4. d) Stress relaxation of thioester hydrogels with excess network thiols at various pH. Dotted lines represent fitted stretched exponentials. e) Stress relaxation in stoichiometric thioester networks (no free network thiols) at various pH. Dotted lines represent fitted stretched exponentials. f) Fitted time constants from equation 2 for stress relaxation in thioester networks as a function of pH and stoichiometry.
A frequency sweep was also performed to evaluate the time-dependence of the storage and loss moduli (Figure 2b). At frequencies below 0.01 rad s−1, the loss modulus in thioester networks with excess thiols increases relative to that of the control hydrogels, in line with the stress relaxation experiments. As further confirmation of the thioester exchange mechanism, a thioester hydrogel was placed in an aqueous solution of cysteine (1 M). Exchange with monofunctional thiols in solution leads to decrease in the concentration of elastically effective crosslinks and a reduction in the network connectivity. Over time, the exchange eventually results in reverse gelation of the hydrogel at ~30 min. (Figure 2c). This rate is similar to that seen in thioester networks used as dissolvable wound sealants [18]. In the context of regenerative medicine, this result indicates that cells cultured in thioester networks can be collected (e.g., for passaging, analyses such as flow cytometry, or transplantation) simply by placing the cell-laden gels in a thiol-rich medium.
The thioester exchange reaction is also predicted to be a function of the concentration of free thiolate, and therefore, solution pH for a given concentration of free network thiol. To confirm this behavior, stress relaxation tests were performed on hydrogels formed from precursor solutions buffered to pH values ranging from 6.0 to 9.0. Interestingly, the thiol-ene polymerization was strongly affected by the solution pH, and polymerization was greatly retarded in solutions with pH above 8.0 (Supplementary Table 1). This is likely due to the reaction between a thiyl radical and deprotonated thiol – the concentration of which increases with increasing pH – to form a radical disulfide anion, sequestering the reactive radical species [28]. Work is currently underway to further characterize this reaction. Therefore, an elevated concentration of LAP (5 mM) was used to form these networks for rheological characterization, and the [LAP] was further increased to stoichiometric levels (18 mM) for excess alkene hydrogel formulations to ensure full conversion of the thiol species. As expected, the stress relaxation rate increased dramatically with increasing pH (Figure 2d). For networks formed with a slight excess of alkenes, stress relaxation was not noticeable over the 20 min experiment below pH 8.5 (Figure 2e). At elevated pH (≥ 8.5), it is likely that thioester hydrolysis begins to play a role [17]. The effect of thioester bond hydrolysis is two-fold: while the hydrolysis itself will lead to stress relaxation in the form of decreased network connectivity, a free thiol is also released, which catalyzes network reorganization. Therefore, a small amount of hydrolysis can convert the elastic network to a viscoelastic network.
These results further indicate that the choice of thiol is an important parameter in synthesis of these networks. For use as a three-dimensional cell scaffold, the polymerization and operating pH are constrained to a narrow window of physiological conditions (pH 7.4). In these networks, deprotonated thiols negatively affect the polymerization reaction, but are necessary for the subsequent exchange reaction. The alkyl thiols used in this study (i.e., PEG-thiol, pKa ~9.6 based on mercaptoethanol) constitute an appropriate choice based on these parameters; however, it may be desired to increase the thioester exchange rate once the network has been formed. Inspired by methods that have been developed to optimize the rates of native chemical ligation, we turned to the thioester exchange catalyst 4-mercaptophenylacetic acid (MPAA). This catalyst has been shown to accelerate the rate of thioester exchange several-fold without participating in side reactions [29]. To demonstrate the change in stress relaxation resulting from the addition of this compound, we chose to create a photocaged form of the molecule using the 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) functional group. The caged molecule (NPPOC-MPAA) releases free MPAA with a quantum yield of 0.46 upon exposure to 365 nm light (Supplemental Figure S2). When 50 mM of the caged molecule is diffused into a thioester network, a 30 s exposure (365 nm, 10 mW cm−2) results in an immediate change in the stress relaxation profile (Figure 3). Interestingly, this reaction did not result in a change in storage modulus over the course of the experiment (Figure S2), indicating that the equilibrium is shifted strongly towards the more stable alkyl thioester. While the biocompatibility of MPAA remains to be evaluated, this general strategy may be applied with various thiols to spatiotemporally increase the rate of network reorganization in thioester hydrogels.
Figure 3. Uncaging 4-mercaptophenyl acetic acid (MPAA) increases the rate of stress relaxation in thioester networks.
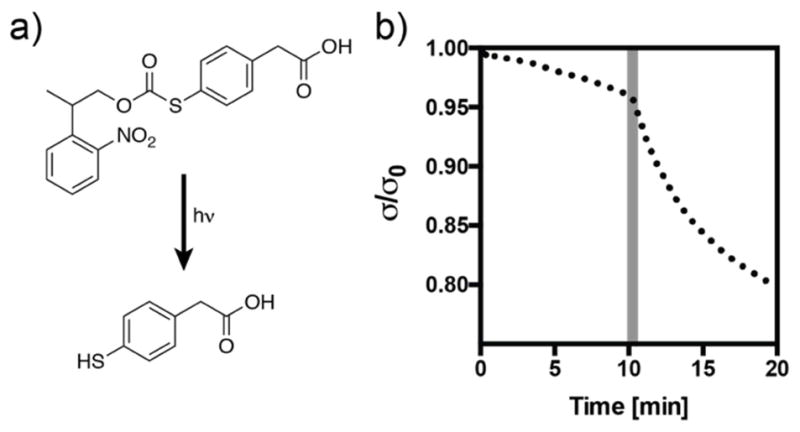
a) Structure of the caged compound NPPOC-MPAA and the product of the uncaging reaction after longwave UV irradiation. b) Stress relaxation increases upon uncaging of MPAA following a 30 s pulse of light (365 nm, 10 mW cm−2) at 10 min (shaded region).
3.3 Cell encapsulation
Thioester hydrogels were next evaluated for their utility as a three-dimensional cell culture platform. With a rapidly increasing body of literature pointing to the effects of dynamic substrates on adherent or encapsulated cells, there is growing need for new biocompatible dynamic chemistries that may be used for experiments with cells or as injectable cell delivery systems. For this work, primary hMSCs were encapsulated in thioester hydrogels at a final concentration of 2 million cells ml−1 by mixing hydrogel precursors with concentrated cell suspensions and inducing gel formation with cytocompatible UV irradiation conditions (1 mM photoinitiator LAP, 4 mW cm−2, 365 nm light, 7 min). As PEG lacks any intrinsic biological activity, norbornene-functionalized RGD peptide (1 mM) was incorporated into these networks to allow hMSC-matrix interactions through integrin binding [30]. To create networks with varied viscoelastic properties, the precursor stoichiometry was varied either with 20% excess thiols or 5% excess alkenes, as in the previous section. As a static control, the divinyl crosslinker lacking the thioester functionality was included in selected formulations.
hMSC viability was determined by incubating cells with Calcein AM and Ethidium homodimer-1 to stain for live and dead cells, respectively. Encapsulated hMSCs had a relatively high level of viability (≥ 84%) under all polymerization conditions and remained viable over the course of the experiment (Figure 4a–b), up to day 3, when the thioester gels begin to dissolve. Interestingly, acellular thioester hydrogels were stable for greater than two weeks in culture medium and over 30 days in phosphate buffered saline (Supplementary Figure S3), suggesting there is likely a cell-mediated degradation of these networks. Further evidence for cell-mediated hydrolysis in the thioester hydrogels is provided by visible void volumes trailing migrating cells (Figure 4e – black arrows). A similar phenomenon has been observed in hydrogels containing hydrolytically susceptible ester bonds. Nuttelman et al. showed that the presence of hMSCs in hydrogels with polylactide blocks dramatically accelerated the degradation rate compared to hydrolysis alone [31]. In contrast, chondrocytes [32], osteoblasts [33], and fibroblasts [31] were not observed to display this behavior. Therefore, it appears that hMSCs express elevated levels of enzyme-mediated hydrolysis, and as such, these other cell types may be better suited for long-term culture in thioester networks. These results indicate that hMSCs are able to remodel their environments in a similar manner to degradable matrices based on cleavable peptides. In thioester networks, however, hydrolysis yields a pendant thiol, the local production of which likely creates a viscoelastic environment that differs from that of the bulk gel. In this way, the thioester may serve as both a biodegradable and adaptable bond.
Figure 4. hMSCs encapsulated in thioester hydrogels are viable, acquire elongated morphologies and have increased proliferation relative to static networks.

a) Encapsulated cells were stained with Calcein-AM (green, live) and Ethidium homodimer-1 (red, dead) each day and visualized by confocal microscopy. Images shown at 72 h post-encapsulation. b) Quantified fraction of viable cells from confocal z-stacks. All conditions promote cell survival over three days of culture. c–d) Representative images of cell shape on day 3: cells in static networks remain rounded (c), while a subset of those in thioester networks take on a spread morphology. e) Transmitted light image of hMSC in (d) with visible cell tracks left behind (black arrows). f) Sphericity of the three-dimensional cell shapes was analyzed during culture within hydrogels. Thioester networks formed with excess thiols (blue squares) show dramatically increased spreading (i.e. lower sphericity) compared to static hydrogels within one day of encapsulation and this difference is retained throughout the experiment. Thioester networks formed with stoichiometric thiols begin to spread after one day in culture. d) hMSCs in thioester networks proliferate more than those in static hydrogels. Cells were incubated with EdU on day 2 to visualize dividing cells. Scale bars: 100 μm (a), 50 μm (c–e). Plots are shown as mean ± std. dev. (b&g), ± 95% conf. interval (f). *,**,*** indicate p < 0.05, 0.01, 0.001 respectively.
We next examined the morphology of hMSCs cultured in thioester hydrogels. Cell shape is a strong regulator of cell behavior in two- and three-dimensional culture. In 2D, it is well known that controlling cell shape through substrate adhesivity [34,35], cell confluency [36], or matrix stiffness [4] affects the proliferation and/or lineage commitment of adherent cells. The effect in 3D is less understood, largely because of the interdependence of the properties used to control cell spreading. For instance, changes in network connectivity not only change the stiffness, but can also affect the polymer density, mesh size, adhesive ligand spacing, and ability of cells to degrade the matrix. In general however, cells in covalent 3D matrices remain rounded unless some form of degradability (e.g., proteolysis) is included. In contrast, cells in covalent adaptable networks are able to deform their surroundings by applying forces through cytoskeletal machinery. In the present study, we have demonstrated that network reorganization can be controlled with minimal changes in network properties by changing the crosslinker structure and pendant thiol concentration (Supplementary Figure S4). Mooney and coworkers have recently shown that modifying the sress relaxation rate in ionically crosslinked alginate hydrogels, independent of stiffness, affects the spreading, differentiation capability, and proliferation of encapsulated cells [9]. We hypothesized that hMSCs in thioester networks would be able to spread in 3D by exerting traction forces on their surroundings in a similar manner.
To quantify the three-dimensional shape of encapsulated hMSCs, we visualized the cell cytoskeleton by immunohistochemistry and confocal imaging of individual cells. Sphericity, a measure of how closely an object resembles a perfect sphere, was used as a measure of the three-dimensional spreading (Figure 4f). In static, non-thioester crosslinked networks, hMSCs were unable to remodel their surroundings and retained a rounded shape (higher sphericity) typical of cells in covalent networks (Figure 4c). In contrast, cells in the thioester hydrogels with excess network thiols were able to spread within 24 h and acquire stellate morphologies (Figure 4d). In these networks, we observed a bimodal trend in cell shape, with a fraction of cells adopting elongated phenotypes and the remainder persisting in the spherical form. In a recent report of hMSCs in covalent adaptable networks with fibrillar components, this phenomenon was not observed [27]. Thus, it remains to be seen whether these two cell populations with distinct morphologies is unique to the thioester chemistry or amorphous adaptable networks. In thioester networks formed with an excess of alkenes, hMSCs remained rounded through the first 24 hours of culture, but were subsequently able to spread. This is in agreement with the observation of cell-mediated thioester hydrolysis: while this network is initially incapable of stress relaxation due to the lack of network thiols, thioester cleavage liberates free thiols and transitions this network to an adaptable one. Thus, cells are able to spread in both thioester networks, and the timing and extent are dependent on the presence or absence of pendant thiols in the initial network.
As cells spread and deform viscoelastic ECM mimics, cellular forces are transmitted to moveable RGD ligands in the network, leading to clustering of RGD/integrins, enhanced focal adhesion formation, and increased proliferation [9,37]. Indeed, the change in morphology in the thioester networks was accompanied by an increase in the fraction of proliferating cells. Using an EdU assay, cellular division was found to increase in both thioester formulations compared to the static hydrogels, corresponding to the increased stress relaxation and spreading observed in these gels (Figure 4g). Although cell shape may be correlated to, rather than causal of increased proliferation, it appears that cells in static hydrogels sense their inability to remodel their microenvironment and downregulate proliferation, as has been recently shown with chondrocytes in adaptable hydrogels [38]. The increased proliferation in these networks makes the thioester hydrogel a potential material choice for the transplantation or in vitro expansion of primary cells.
4. Conclusions
Thioester-containing hydrogels were photopolymerized using thiol-ene click chemistry to yield stable networks capable of reorganization at physiological conditions. The rate of network dynamics was tuned by changing the pH, stoichiometry, and crosslinker structure. Additionally, by uncaging the thioester exchange catalyst MPAA, viscoelastic properties could be mediated by exposure to light. Encapsulated hMSCs remained viable and were able to spread and remodel the network in a thioester-dependent manner. Furthermore, proliferation of hMSCs was increased in thioester hydrogels compared to static hydrogels. Due to the biological relevance and biocompatibility of thioester networks, this functionality is a useful addition to the growing collection of dynamic material chemistries for cell scaffold applications.
Supplementary Material
Acknowledgments
Funding for this work was provided by the NSF DMR (1408955) and the NIH NIDCR (DE016523). The authors also gratefully acknowledge fellowship support from the NSF GFRP (T.E.B and M.K.M), the NIH/CU Biophysics training program (T32 GM-065103) (T.E.B.), and the Arnold and Mabel Beckman Postdoctoral Fellowship (B.T.W).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingber DE. Mechanical control of tissue morphogenesis during embryological development. Int J Dev Biol. 2006;50:255–266. doi: 10.1387/ijdb.052044di. [DOI] [PubMed] [Google Scholar]
- 2.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown TE, Anseth KS. Spatiotemporal hydrogel biomaterials for regenerative medicine. Chem Soc Rev. 2017 doi: 10.1039/C7CS00445A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 5.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri O, Koshy ST, Branco da Cunha C, Shin J-W, Verbeke CS, Allison KH, Mooney DJ. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Mater. 2014;13 doi: 10.1038/nmat4009. [DOI] [PubMed] [Google Scholar]
- 7.Cameron AR, Frith JE, Cooper-White JJ. The influence of substrate creep on mesenchymal stem cell behaviour and phenotype. Biomaterials. 2011;32:5979–5993. doi: 10.1016/j.biomaterials.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 8.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Biophysically Defined and Cytocompatible Covalently Adaptable Networks as Viscoelastic 3D Cell Culture Systems. Adv Mater. 2014;26:865–872. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H, Lippens E, Duda GN, Mooney DJ. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat Mater. 2015;15:326–334. doi: 10.1038/nmat4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnell M, Young S, Gu L, Shah N, Lippens E, Weaver J, Duda G, Mooney D. Substrate Stress-Relaxation Regulates Scaffold Remodeling and Bone Formation In Vivo. Adv Healthc Mater. 2017;6:1601185. doi: 10.1002/adhm.201601185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Heilshorn SC. Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering. Adv Mater. 2015;27:3717–3736. doi: 10.1002/adma.201501558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosales AM, Anseth KS. The design of reversible hydrogels to capture extracellular matrix dynamics. Nat Rev Mater. 2016;1:15012. doi: 10.1038/natrevmats.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dankers PYW, Harmsen MC, Brouwer La, van Luyn MJa, Meijer EW. A modular and supramolecular approach to bioactive scaffolds for tissue engineering. Nat Mater. 2005;4:568–74. doi: 10.1038/nmat1418. [DOI] [PubMed] [Google Scholar]
- 14.Highley CB, Rodell CB, Burdick JA. Direct 3D Printing of Shear-Thinning Hydrogels into Self-Healing Hydrogels. Adv Mater. 2015;27:5075–5079. doi: 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- 15.Rowley JA, Madlambayan G, Mooney DJ. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials. 1999;20:45–53. doi: 10.1016/S0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 16.Kloxin CJ, Scott TF, Adzima BJ, Bowman CN. Covalent Adaptable Networks (CANs): A Unique Paradigm in Crosslinked Polymers. Macromolecules. 2010;43:2643–2653. doi: 10.1021/ma902596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bracher PJ, Snyder PW, Bohall BR, Whitesides GM. The Relative Rates of Thiol-Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water. Orig Life Evol Biosph. 2011;41:399–412. doi: 10.1007/s11084-011-9243-4. [DOI] [PubMed] [Google Scholar]
- 18.Ghobril C, Charoen K, Rodriguez EK, Nazarian A, Grinstaff MW. A dendritic thioester hydrogel based on thiol-thioester exchange as a dissolvable sealant system for wound closure. Angew Chemie - Int Ed. 2013;52:14070–14074. doi: 10.1002/anie.201308007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Worrell BT, Bowman CN, et al. manuscript in preparation. [Google Scholar]
- 20.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kyburz KA, Anseth KS. Three-dimensional hMSC motility within peptide-functionalized PEG-based hydrogels of varying adhesivity and crosslinking density. Acta Biomater. 2013;9:6381–92. doi: 10.1016/j.actbio.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoyle CE, Bowman CN. Thiol-ene click chemistry. Angew Chem Int Ed Engl. 2010;49:1540–73. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 24.David RLA, Kornfield JA. Facile, efficient routes to diverse protected thiols and to their deprotection and addition to create functional polymers by thiol-ene coupling. Macromolecules. 2008;41:1151–1161. doi: 10.1021/ma0718393. [DOI] [Google Scholar]
- 25.Tang S, Wang M, Olsen BD. Anomalous Self-Diffusion and Sticky Rouse Dynamics in Associative Protein Hydrogels. J Am Chem Soc. 2015;137:3946–3957. doi: 10.1021/jacs.5b00722. [DOI] [PubMed] [Google Scholar]
- 26.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Bis-Aliphatic Hydrazone-Linked Hydrogels Form Most Rapidly at Physiological pH: Identifying the Origin of Hydrogel Properties with Small Molecule Kinetic Studies. Chem Mater. 2014;26:2382–2387. doi: 10.1021/cm5007789. [DOI] [Google Scholar]
- 27.Lou J, Stowers R, Nam S, Xia Y, Chaudhuri O. Stress relaxing hyaluronic acid-collagen hydrogels promote cell spreading, fiber remodeling, and focal adhesion formation in 3D cell culture. Biomaterials. 2017;154:213–222. doi: 10.1016/j.biomaterials.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Love DM, Kim K, Goodrich JT, Fairbanks BD, Worrell BT, Stoykovich MP, Musgrave CB, Bowman CN. Amine Induced Retardation of the Radical-Mediated Thiol–Ene Reaction via the Formation of Metastable Disulfide Radical Anions. J Org Chem. 2018 doi: 10.1021/acs.joc.8b00143. acs.joc.8b00143. [DOI] [PubMed] [Google Scholar]
- 29.Johnson ECB, Kent SBH. Insights into the mechanism and catalysis of the native chemical ligation reaction. J Am Chem Soc. 2006;128:6640–6646. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 30.Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 31.Nuttelman CR. Osteogenic Poly(ethylene glycol)-based Hydrogels for Three-Dimensional Human Mesenchymal Stem Cell Culture and Bone Regeneration. University of Colorado; 2005. [Google Scholar]
- 32.Bryant SJ, Anseth KS, Lee DA, Bader DL. Crosslinking density influences the morphology of chondrocytes photoencapsulated in PEG hydrogels during the application of compressive strain. J Orthop Res. 2004;22:1143–1149. doi: 10.1016/j.orthres.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315–23. doi: 10.1016/s0142-9612(02)00176-x. http://www.ncbi.nlm.nih.gov/pubmed/12219821. [DOI] [PubMed] [Google Scholar]
- 34.Folkman JJ, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 35.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–8. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 36.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/S1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif Sa, Weaver JC, Huebsch N, Mooney DJ. Substrate stress relaxation regulates cell spreading. Nat Commun. 2015;6:1–7. doi: 10.1038/ncomms7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HP, Gu L, Mooney DJ, Levenston ME, Chaudhuri O. Mechanical confinement regulates cartilage matrix formation by chondrocytes. Nat Mater. 2017;16:1243–1251. doi: 10.1038/nmat4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.