

Abstract
The present study was designed to investigate the possible role of magnesium (Mg2+) on activation of the peroxisome proliferator-activated receptor gamma (PPAR-γ) and inhibition of nuclear factor-KB (NFKB p65) in muscle to increase glucose transporter 4 (GLUT4) gene expression. Fifty rats were divided into five groups, namely non-diabetic control (NDC), Mg2+-treated non-diabetic control (Mg2+-NDC), chronic diabetic (CD), Mg2+-treated chronic diabetic (Mg2+-CD), and insulin-treated chronic diabetic (Ins-CD). Diabetes was induced with streptozotocin (STZ) injection. The Mg2+-CD and Mg2+-NDC groups received 10 g/l of magnesium sulfate (MgSO4) added to drinking water and Ins-CD group received 2.5 U/kg of insulin. The blood glucose level and body weight were measured every week. After 16 weeks, intraperitoneal glucose tolerance test (IPGTT) was done and then animals were decapitated, blood samples were taken to determine the plasma levels of Mg2+ and gastrocnemius muscle legs were isolated for both PPAR-γ and NFKB (p65) genes and proteins expression. Administration of MgSO4 improved IPGTT, lowered blood glucose levels and increased PPAR-γ gene and protein expression. Diabetes increased NFKB gene and protein expression. Although Mg2+ therapy could not decrease NFKB (p65) gene expression, the protein decreased by Mg2+ therapy. Insulin decreased NFKB (p65) gene and protein expression, without any effect on PPAR-γ gene and protein expression. According to our findings it seems that suppressing NFKB (p65) protein synthesis and increases in PPAR-γ gene and protein expression could help Mg2+ administration to decreases blood glucose levels. But decreasing in NFKB (p65) gene and protein expression help insulin to decrease blood glucose level.
Keywords: Diabetes, insulin, magnesium, PPAR-γ, NFKB (p65)
Introduction
There is evidence suggesting that peroxisome proliferator-activated receptors (PPARs) may have a role in chronic diseases like diabetes, obesity and atherosclerosis [1]. There are three types of PPARs including PPAR-α, PPAR-β/δ and PPAR-γ [2]. These nuclear receptors are expressed in adipose, muscle and liver tissues [3]. But each PPAR isoform has exclusive roles in vivo. PPAR-α is a major activator of fatty acid oxidation pathways and is the target of the hypolipidemic fibrate drugs [4]. However, in the key metabolic tissues such as skeletal muscle, liver and heart PPAR-δ/β has a crucial role in fatty acid oxidation. PPAR-γ is the most highly expressed in white adipose tissue and muscle [5] and its agonists cause to improve glycemic control, decrease plasma insulin levels, and increase insulin sensitivity [6,7]. PPAR-γ ligands improve insulin sensitivity by promoting fatty acid storage in fat tissues and expression of GLUT4 (a major glucose transporter in skeletal muscle) mRNA [8]. Some studies observed that PPAR-γ agonist receptor therapy could enhancement muscle glucose metabolism via activation of insulin cell signaling. Other studies demonstrated that PPAR-γ increases insulin-stimulated glucose uptake in insulin target cells via GLUT4 [8,9]. Several studies have shown that the PPAR-γ agonist can inhibit macrophage activation and inflammatory secretion such as nuclear factor-KB (NFKB) [10-12]. But its utility is limited due to undesired side-effects of PPAR-γ agonist. It has been recently implicated that NFKB may serve as an important agent in adipose tissue for developing insulin resistance and diabetes [12]. Prominently, magnesium seems to play an essential role in regulating the PPAR-mediated signaling pathways as a key cofactor in the protein phosphorylation. Thus, controlling magnesium level in the body is essential for defense against some diseases [13].
In our previous studies [14-16] we introduced magnesium as a new anti-diabetic agent that develops insulin sensitivity and reduces plasma glucose and blood pressure in diabetic animal model and type II diabetic patients. Our previous findings indicated that Mg2+ deficiency occurs after diabetes induction and oral magnesium sulfate (MgSO4) administration reduces blood sugar through increased GLUT4 gene expression, not insulin secretion [17]. Because the mechanism of this action is not clear, the present study was designed to investigate the possible effect of Mg2+ on activation of PPAR-γ receptor and inhibition of NFKB (p65) in skeletal muscle for increasing GLUT4 gene expression and translocation.
Materials and methods
Animals
Animals were treated in accordance with standard guidelines [18]. The experimental protocol was approved by the Ethical Committee for Animal Care of Hormozgan University of Medical Science (Ethic number HUMS.REC.1395.88). Male Wistar rats, weighing 180-250 g were selected and maintained at a constant temperature of 22±2°C with a fixed 12:12-h light-dark cycle. Nutritionally balanced pellets and water were freely available. The animals were divided into five groups (10 rats in each group) that include non-diabetic control (NDC), Mg2+-treated non-diabetic control (Mg2+-NDC), chronic diabetic (CD), Mg2+-treated chronic diabetic (Mg2+-CD) and insulin-treated chronic diabetic (Ins-CD).
Diabetes was induced with a single I.P injection of 60 mg/kg STZ and 10 days after diabetes induction, fasting blood glucose level was measured and the presence of diabetes was confirmed by blood glucose levels of above 250 mg/dl. Animals with diabetes lasted for 16 weeks were defined as ‘chronic diabetic’. At 10 days after diabetes induction Mg2+-CD and Mg2+-NDC groups received 10 g/l of MgSO4 added to drinking water [14,15,17], for 16 weeks duration and water consumption was measured. Mg2+-treated chronic diabetic and Mg2+-treated non-diabetic control groups have significant lower water consumption compared to chronic diabetic group (43±3, 41±2 and 201±2.1 ml/24 h for Mg2+-treated chronic diabetic, Mg2+-treated non-diabetic control and chronic diabetic respectively). So the mean of exact dose of daily consumption of MgSO4 was 0.43 g/24 h.
The Ins-CD group received 2.5 U/kg of insulin (1/3 regular and 2/3 NPH) to keep the level of blood glucose in Ins-CD group around 134±4 mg/dl [17], 10 days after I.P induced diabetes for 16 weeks. Animals were monitored for blood glucose concentrations and body weight every week at 9 AM over the experiment. Blood glucose was measured with an Ascensia ELITE XL glucometer and Ascensia Elite blood glucose test strips while body weight of all rats recorded using a digital weighing scale.
Intraperitoneal glucose tolerance test (IPGTT)
For the purpose of conducting IPGTT, animals in all groups were fasted overnight for 15 h, and were given 1.5 g/kg glucose via I.P injection. Blood sample was taken from the tail vein at 0, 10, 20, 30, 60, 90 and 120 min after glucose administration [14,19].
Plasma magnesium measurement assay
Animals were anesthetized with ketamine HCl (50 mg/kg, I.P), before decapitating. Blood samples were taken from the neck vascular trunk in order to determine the plasma levels of magnesium by a Micro plate reader (Biotek, USA) and appropriate kits (Pars Azmun, Iran).
PPAR-γ and NFKB (p65) genes and proteins expression assay
Muscle was isolated under anesthesia for both PPAR-γ and NFKB (p65) genes and proteins expression via Real time PCR and immunohistochemistry methods, respectively.
RNA extraction
The total RNA was extracted from muscle tissue using RNeasy Fibrous Tissue mini Kit (Cat No.74704 QIAGEN, Germany) according to the manufacturer’s protocol. Briefly, RNA was treated with 1 µl DNase-I at 37°C for 30 min and then 1 µl EDTA was added. Then it was incubated at 65°C for 10 min for DNase inactivation. The extracted RNA was quantified by running on 1.5% agarose gel electrophoresis. The 18S and 28S RNA bands were visualized under a transillumiator. The yield was quantified spectro-photometrically at 260 and 280 ηm by Nano Drop 1000 (Thermo, USA). The extracted RNA had a ratio of 1.8 to 2.
cDNA synthesis and quantitative real-time PCR
The total RNA (2 µg) was reversely transcribed to cDNA using PrimeScriptTM RT Reagent Kit for RT-PCR (Cat. No.RR037A, Takara, Japan) by applying random hexamer primer following the manufacturer’s protocol. Quantitative RT-PCR was conducted on a real-time PCR (Corbett, Rotor-Gene 6000, Australia) and Syber Green (Takara, Japan) according to the manufacturer’s instructions. All reactions were carried out in a terminal volume of 20 µL containing 1 µL of cDNA sample, 1 μl of the primers, 10 μL Syber Green, 0/4 μL ROX and 7/6 μL of sterile double-distilled water for all β-actin, PPAR-γ or NFKB (p65). The thermal cycling status for β-actin was an initial denaturation at 95°C for 30 s followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 58°C for each primer for 15 s. For PPAR-γ the thermal cycling status was an initial denaturation at 95°C for 3 minutes followed by 40 cycles of denaturation at 95°C for 10 s and each primer was annealed for 30 s at 65°C. Also for NFKB (p65) the thermal cycling status was an initial denaturation at 95°C for 30 s followed by 40 cycles of denaturation at 95°C for 5 s and the 15 s annealing at 58°C for each primer. All the Standards and the samples were analyzed in duplicates. Melting curve analysis was used to characterize the specifications of each PCR product.
Expression levels of PPAR-γ and NFKB (p65) were normalized by β-actin expression as the house-keeping gene and calculated through the 2-ΔΔCT method. The sequence of specific primer sets for each gene, amplicon sizes and annealing temperature of the primers for real- time PCR was used (Table 1).
Table 1.
List of primer sequence used in Real Time-PCR
| Gene | Primer (5’→3’) | Amplicon Size (bp) | TM |
|---|---|---|---|
| β-actin | F: CAC ACC CGC CAC CAG TTCG | 165 | 60/5 |
| R: ACC CAT TCC CAC CAT CAC ACC | |||
| PPAR-γ | F: CTG TTC GCC AAG GTG CTC CA | 102 | 68/4 |
| R: GCT CAT ATC TGT CTC CGT CTT CTT | |||
| NFKB (p65) | F: TTA CGG GAG ATG TGA AGA TG | 94 | 59/9 |
| R: ATG ATG GCT AAG TGT AGG AC |
Immunohistochemistry
Five-micrometer sections of formalin-fixed paraffin-embedded tissue were cut and prepared for IHC staining. Before immunostaining the specimens were processed through xylene and a graded alcohol series and treated with 3% hydrogen peroxide to eliminate the endogenous peroxidase activity and blocked with 5% normal goat serum. These slides were then treated with mouse anti-rat PPAR-γ or NFKB Ab at 10 μg/ml (Santa Cruz Biotechnology, Santa Cruz, CA). After an overnight incubation, horse anti-mouse IgG-biotin secondary antibody (Vector Labs, Burlingame, CA) was added at a 1:200 dilution, followed by streptavidin-HRP (Jackson Labs, West Grove, PA) at 1:1000. Slides were developed with AEC Reagent (Zymed Laboratories, San Francisco, CA) and counterstained with hematoxylin (Sigma Co, St. Louis, MO). Cells were viewed and photographed under bright field.
Statistical analysis
Data was expressed as mean ± S.E.M and between group differences were evaluated by one-way and two-way ANOVA with the Tukey post-hoc test. The relative changes of gene expression were calculated by 2-ΔΔCt formula, where ΔCt = Ct (PPAR-γ or NFKB)-Ct (β-actin) and Ct demonstrates threshold cycle number. In addition, Spearman and Mann-Witney U-tests were used to investigate the expression pattern of target gene. Statistical analysis was carried out using SPSS-21, Microsoft Excel and Prism-5 software and in all instances P<0.05 was considered as an acceptable statistical significance.
Results
Changes in plasma glucose
Changes in feeding plasma glucose were measured in all groups (Figure 1A). Diabetes induction caused an increase in plasma glucose concentration (389.16±24.43 mg/dl) in one week and blood glucose continued to be elevated (597±1/91 mg/dl) 16 weeks after diabetes induction. Administration of MgSO4 or insulin for 16 weeks (from day 10) led to a decrease of plasma glucose concentrations in Mg2+-CD and Ins-CD groups (127/5±1.25 mg/dl, 90±16.74 mg/dl), respectively.
Figure 1.
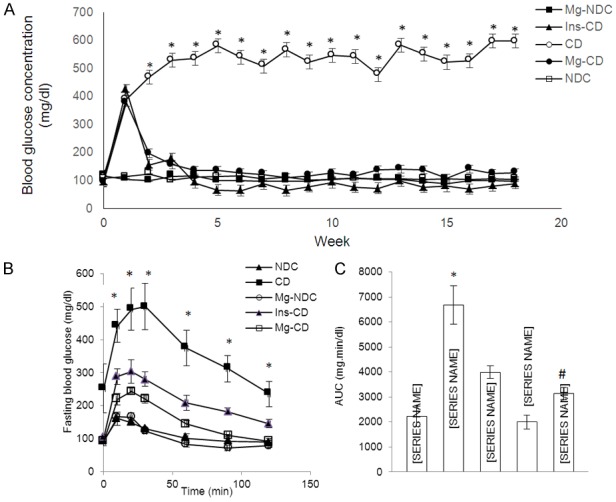
Comparison of fed blood glucose (A), intraperitoneal glucose tolerance test (IPGTT) (B) and the area under the glycemic curve (AUC) (C) in non-diabetic control (NDC), chronic diabetic (CD), Mg2+-treated non diabetic control (Mg2+-NDC), insulin -treated chronic diabetic (Ins-CD) and Mg2+-treated chronic diabetic groups (Mg2+-CD) groups (10 rats in each group, data are expressed as mean ± SEM). *Significant difference between CD and other groups (P<0.00001). #Significant difference between Mg2+-CD and other groups (P<0.0001).
Effects of MgSO4 on intraperitoneal glucose tolerance test (IPGTT)
Before administration of MgSO4 and STZ, the IPGTT patterns were similar for all groups and there was no significant difference between the groups when the glycemic response was expressed as the area under the curve (AUC) (data not shown). However, after 16 weeks, the CD group displayed severe glucose intolerance (Figure 1B), which was significantly improved in Mg2+-CD group (Figure 1C, AUC: NDC vs CD vs Mg2+-CD vs Ins-CD = 2228.33±408.4 vs 6674.8±765.1 vs 3137.83±235.4 vs 3990±246.3 P<0.0001).
Changes in the body weight and plasma magnesium levels
The body weight of CD animals was decreased insignificantly in comparison with NDC animals; increased body weight was significantly observed after administration of Mg2+ and insulin compared to CD animals (Figure 2A).
Figure 2.
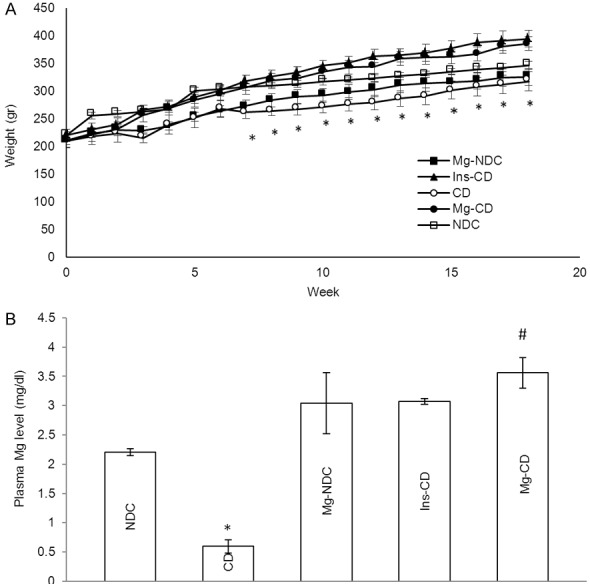
Comparison of body weight (A), plasma magnesium levels (B) in non-diabetic control (NDC), chronic diabetic (CD), Mg2+-treated non diabetic control (Mg2+-NDC), insulin-treated chronic diabetic (Ins-CD) and Mg2+-treated chronic diabetic (Mg2+-CD) groups (10 rats in each group, data are expressed as mean ± SEM). *Significant different between CD and other groups (P<0.001). #Significant difference between Mg2+-CD and Ins-CD (P<0.001).
To find the concentration as well as the duration of oral-administered Mg2+ in plasma, changes in plasma magnesium (Mg2+) level were measured in all groups. The plasma magnesium level in CD (0.59±0.11) significantly (P<0.001) decreased compared to the NDC group (2.2±0.06). Administration of Mg2+ and insulin for 16 weeks (from day 10) caused an in increase plasma magnesium concentrations in Mg2+-CD and Ins-CD groups (Mg2+-CD = 3.56±0.26, Ins-CD = 3.07±0.04) (Figure 2B).
Changes in PPAR-γ and NFKB (p65) gene expression
The mean level of Ct obtained from Real-time PCR in five groups of examined rats as well as the effect of MgSO4 and insulin in NFKB (p65) and PPAR-γ expression have been shown in Figures 3A, 4A, respectively. In magnesium treated diabetic rats, the PPAR-γ gene expression exhibited almost 1.5 fold increase (P<0.0001) in comparison with chronic diabetic rats (CD). But MgSO4 administration in diabetic rats could not reduce NFKB (p65) gene expression in comparison to CD group.
Figure 3.
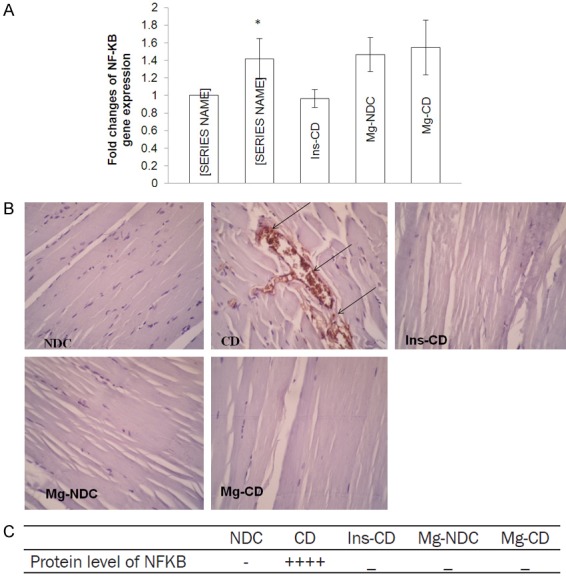
Comparison of the fold changes of NFKB gene expression (A), immunohistochemistry for NFKB (B and C) and protein level of NFKB in chronic diabetic (CD), Mg2+-treated non diabetic control (Mg2+-NDC), insulin-treated chronic diabetic (Ins-CD) and Mg2+-treated chronic diabetic (Mg2+-CD) groups (10 rats in each group, data are expressed as mean ± SEM). *Significant different between CD and Mg2+-CD or Ins-CD rats (P<0.01).
Figure 4.
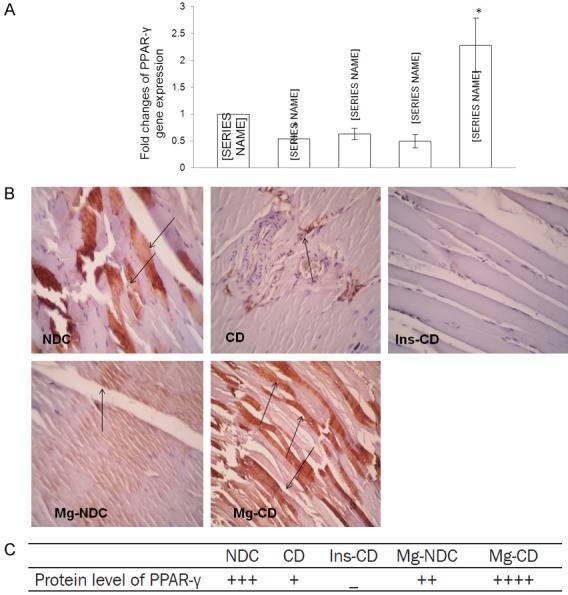
Comparison of the fold changes of PPAR-γ gene expression (A), immunohistochemistry for PPAR-γ (B and C) and protein level of PPAR-γ in non-diabetic control (NDC), chronic diabetic (CD), Mg2+-treated non diabetic control (Mg2+-NDC), insulin-treated chronic diabetic (Ins-CD) and Mg2+-treated chronic diabetic (Mg2+-CD) groups (10 rats in each group). *Significant different between Mg2+-CD and other groups (P<0.0001).
Although Insulin could not significantly up-regulate PPAR-γ, reduction in NFKB (p65) gene expression was observed in Ins-CD group in comparison with chronic diabetic (CD) animals.
Changes in PPAR-γ and NFKB (p65) protein level
Immunohistochemistry was done for all groups to observe NFKB (p65) and PPAR-γ protein levels and results were shown in Figures 3B, 3C and 4B, 4C respectively. According to our findings, NFKB (p65) protein level increased in CD group, however this result was not observed in the other groups. Interestingly, although 16 weeks of Mg2+ therapy in Mg2+-CD and Mg2+-NDC groups could not suppress NFKB mRNA gene expression, it seems that Mg2+ inhibited NFKB (p65) protein production in those groups. PPAR-γ protein level increased by Mg2+ therapy in diabetic rats, but this effect was not observed through insulin administration.
Discussion
It has been previously shown [14-17,20] that eight weeks use of oral magnesium in STZ-induced diabetic rats could decrease plasma glucose level and it can be mediated by increasing GLUT4 gene expression in skeletal muscle independent of insulin secretion. This study has tried to examine the possible role of PPAR-γ and NFKB (p65) gene expression in increased GLUT4 gene expression and improve glucose hemostasis following oral magnesium sulfate administration in skeletal muscle of STZ-induced diabetic rats.
The results of our study showed that magnesium therapy not only reduce blood glucose level, but also improves insulin resistance, although long term use of insulin reduces blood glucose concentration, but not able to improve insulin resistance. The previous study has shown that magnesium is more effective than insulin in GLUT4 translocation to the cell membrane [17].
GLUT4 is the major glucose transporter isoform in tissues that exhibits insulin-stimulated glucose uptake, such as adipose and skeletal muscle tissues. Eliciting the translocation of an intracellular pool of transporters to the plasma membrane, insulin stimulates GLUT4 expression primarily in adipose and muscle tissues [21]. It is described that fasting, high-fat feeding, obesity and diabetes initiate lowering the GLUT4 mRNA concentration in adipose and skeletal muscle tissue [22,23]. On the other hand, some researchers presented that PPAR-γ ligands play a role in metabolism of lipid, lipoprotein and glucose homeostasis [24] and decrease of insulin resistance [25]; this effect of PPAR-γ deletion was mediated by both GLUT4 and GLUT1 [6]. But undesired side-effects of PPAR-γ agonist have limited its utility.
Another study demonstrated that Mg2+ deficiency impairs PPAR-γ activity [26] and our findings in the present study showed post diabetic decrease of Mg2+ plasma level and PPAR-γ gene expression.
Interestingly, treatment with MgSO4 for 16 weeks in diabetic rats increased the level of PPAR-γ mRNA and its protein. According to our previous results [17], MgSO4 administration in STZ-induced diabetic rats increased the expression of GLUT4 mRNA about 23 percent. This’s why the results of this study showed that magnesium sulphate improved IPGTT and lowered blood glucose levels close to the normal range. The findings of the present research demonstrated that Mg2+ may induce GLUT4 gene expression via increasing PPAR-γ gene expression. To the best of our knowledge, no study has been done yet on the effect of magnesium on PPAR-γ gene expression. But our results showed that insulin administration in diabetic rats could not change both PPAR-γ gene expression and its protein level in skeletal muscle. However, studies showed that PPAR-γ expressed in muscle and adipose tissues and its level increases by insulin in adipose tissue [27] and it has been shown that PPAR-γ activation affects the insulin signaling cascade, through direct effects on the expression or phosphorylation of specific signaling molecules such as GLUT4 [9]. Although in another report no effect could be seen on GLUT4 expression in adipocytes of normal and diabetic animals by PPAR-γ ligands [28,29]. Our previous findings [17] showed that insulin increases the expression of GLUT4 gene by only 10% in insulin treated diabetic group. Although insulin could increase GLUT4 gene expression in skeletal muscle, according to our results in the present study it seems that PPAR-γ does not play a role in this achievement and it may involves other mechanisms.
It has been suggested that activation of nuclear factor-KB (NFKB) transcription may participate in diabetes and its complications [30]. A recent study demonstrated that hyperglycemia increases NFKB gene expression [31] and this pro-inflammatory agent can cause insulin resistance in adipose tissue [32]. Our findings in this study showed for the first time that insulin therapy could decrease NFKB gene expression and its protein in skeletal muscle. Although this effect was observed by Mg2+, administration of MgSO4 for 16 weeks resulted in reduced NFKB (p65) protein production. The previous documents revealed that NFKB promote insulin resistance and insulin administration in adipose and endothelial cells suppresses its expression and activity [33]. Mg2+ deficiency has been reported to be involved in NFKB up regulation [34]. NFKB (p65) protein was suppressed successfully by Mg2+ therapy, although its fortification of drinking water was not successful in inhibiting NFKB (p65) gene expression in diabetic animals. So it seems that 16 weeks administration of Mg2+ increased GLUT4 expression and decrease blood glucose level in diabetic rats by increasing PPAR-γ gene expression and decreasing NFKB (p65) protein.
Conclusions
PPAR-γ has emerged as a key regulation of glucose hemostasis and increased insulin sensitivity. Although the expression of NFKB (p65) gene was not changed by MgSO4 therapy, reduction of NFKB (p65) protein and PPAR-γ enhancement in muscle cells by Mg2+ could regulate plasma glucose level. Regarding the results of this study we may be able to consider Mg2+ as a PPAR-γ ligand in future as an agent to decrease insulin resistance.
Acknowledgements
This study was supported by the vice-Chancellor for research, Hormozgan University of Medical Science, Bandar Abbas, Iran. The grant number is 950095.
Disclosure of conflict of interest
None.
References
- 1.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 2.Habor A. Peroxisome proliferator activated receptors. Farmacia. 2010;58:1. [Google Scholar]
- 3.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 4.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 5.Quinn CE, Hamilton PK, Lockhart CJ, McVeigh GE. Thiazolidinediones: effects on insulin resistance and the cardiovascular system. Br J Pharmacol. 2008;153:636–645. doi: 10.1038/sj.bjp.0707452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olefsky J. Treatment of insulin resistance with peroxisome proliferator-activated receptor γ agonists. J Clin Invest. 2000;106:467–472. doi: 10.1172/JCI10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR. PPARγ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J Clin Invest. 1998;101:22–32. doi: 10.1172/JCI1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonardini A, Laviola L, Perrini S, Natalicchio A, Giorgino F. Cross-Talk between PPARgamma and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009:2009. doi: 10.1155/2009/818945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 11.Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 cytokine-induced downregulation of PPARγ in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology. 2005;41:1329–1338. doi: 10.1002/hep.20705. [DOI] [PubMed] [Google Scholar]
- 12.Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujii H. [Nuclear Receptor PPARs and magnesium] . Clin Calcium. 2005;15:52–64. [PubMed] [Google Scholar]
- 14.Soltani N, Keshavarz M, Minaii B, Mirershadi F, Asl SZ, Dehpour AR. Effects of administration of oral magnesium on plasma glucose and pathological changes in the aorta and pancreas of diabetic rats. Clin Exp Pharmacol Physiol. 2005;32:604–610. doi: 10.1111/j.0305-1870.2005.04238.x. [DOI] [PubMed] [Google Scholar]
- 15.Soltani N, Keshavarz M, Dehpour AR. Effect of oral magnesium sulfate administration on blood pressure and lipid profile in streptozocin diabetic rat. Eur J Pharmacol. 2007;560:201–205. doi: 10.1016/j.ejphar.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Solati M, Ouspid E, Hosseini S, Soltani N, Keshavarz M, Dehghani M. Oral magnesium supplementation in type II diabetic patients. Med J Islam Repub Iran. 2014;28:67. [PMC free article] [PubMed] [Google Scholar]
- 17.Solaimani H, Soltani N, MaleKzadeh K, Sohrabipour S, Zhang N, Nasri S, Wang Q. Modulation of GLUT4 expression by oral administration of Mg(2+) to control sugar levels in STZ-induced diabetic rats. Can J Physiol Pharmacol. 2014;92:438–444. doi: 10.1139/cjpp-2013-0403. [DOI] [PubMed] [Google Scholar]
- 18.CCAC. Guide to the Care and Use of Experimental Animals. Canadian Council on Animal Care. 1993;1:18–21. [Google Scholar]
- 19.Tavasoli R, Soltani N, Keshavarz M. Mg2+-induced adenosine-receptor mediated relaxations in mesenteric vascular beds of diabetic rats. Gen Physiol. 2012;31:409–413. doi: 10.4149/gpb_2012_050. [DOI] [PubMed] [Google Scholar]
- 20.Soltani N, Keshavarz M, Sohanaki H, Dehpour AR, Asl SZ. Oral magnesium administration prevents vascular complications in STZdiabetic rats. Life Sci. 2005;76:1455–1464. doi: 10.1016/j.lfs.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 21.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 22.Eckardt K, Taube A, Eckel J. Obesity-associated insulin resistance in skeletal muscle: role of lipid accumulation and physical inactivity. Rev Endocr Metab Disord. 2011;12:163–172. doi: 10.1007/s11154-011-9168-2. [DOI] [PubMed] [Google Scholar]
- 23.Tremblay F, Lavigne C, Jacques H, Marette A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein. Diabetes. 2001;50:1901–1910. doi: 10.2337/diabetes.50.8.1901. [DOI] [PubMed] [Google Scholar]
- 24.Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications-a review. Nutr J. 2014:13–17. doi: 10.1186/1475-2891-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribon V, Johnson JH, Camp HS, Saltiel AR. Thiazolidinediones and insulin resistance: peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc Natl Acad Sci U S A. 1998;95:14751–14756. doi: 10.1073/pnas.95.25.14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castiglioni S, Cazzaniga A, Maier JA. Potential interplay between NFκB and PPARγ in human dermal microvascular endothelial cells cultured in low magnesium. Magnes Res. 2014;27:86–93. doi: 10.1684/mrh.2014.0365. [DOI] [PubMed] [Google Scholar]
- 27.Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, Werman A, Pories WJ, Caro JF, Flier JS. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99:2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rieusset J, Auwerx J, Vidal H. Regulation of gene expression by activation of the peroxisome proliferator-activated receptor gamma with rosiglitazone (BRL 49653) in human adipocytes. Biochem Biophys Res Commun. 1999;265:265–271. doi: 10.1006/bbrc.1999.1657. [DOI] [PubMed] [Google Scholar]
- 29.Al-Khalili L, Forsgren M, Kannisto K, Zierath JR, Lönnqvist F, Krook A. Enhanced insulin-stimulated glycogen synthesis in response to insulin, metformin or rosiglitazone is associated with increased mRNA expression of GLUT4 and peroxisomal proliferator activator receptor gamma co-activator 1. Diabetologia. 2005;48:1173–1179. doi: 10.1007/s00125-005-1741-3. [DOI] [PubMed] [Google Scholar]
- 30.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Klöting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Häring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 31.Hattori Y, Hattori S, Sato N, Kasai K. Highglucose-induced nuclear factor kappaB activation in vascular smooth muscle cells. Cardiovasc Res. 2000;46:188–197. doi: 10.1016/s0008-6363(99)00425-3. [DOI] [PubMed] [Google Scholar]
- 32.de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shoelson SE, Lee J, Goldfine AB. Review series Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–18801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferrè S, Baldoli E, Leidi M, Maier JA. Magnesium deficiency promotes a pro-atherogenic phenotype in cultured human endothelial cells via activation of NFkB. Biochim Biophys Acta. 2010;1802:952–958. doi: 10.1016/j.bbadis.2010.06.016. [DOI] [PubMed] [Google Scholar]